

Abstract
The Myosin Binding Protein-C (MyBP-C) family is a group of sarcomeric proteins important for striated muscle structure and function. Comprising approximately 2% of the myofilament mass, MyBP-C has important roles in both contraction and relaxation. Three paralogs of MyBP-C are encoded by separate genes with distinct expression profiles in striated muscle. In mammals, cardiac MyBP-C is limited to the heart, and it is the most extensively studied owing to its involvement in cardiomyopathies. However, the roles of two skeletal paralogs, slow and fast, in muscle biology remain poorly characterized. Nonetheless, both have been recently implicated in the development of skeletal myopathies. This calls for a better understanding of their function in the pathophysiology of distal arthrogryposis. This review characterizes MyBP-C as a whole and points out knowledge gaps that still remain with respect to skeletal MyBP-C.
Keywords: Distal arthrogryposis, Myofilament, MYBPC, Sarcomere, Striated muscle
Introduction:
Movement of the body is produced through the contraction and relaxation of muscles. Three types of muscle responsible for this - striated, cardiac and smooth (Figure 1). Smooth muscle cells are found in the walls of organs and blood vessels and are under involuntary control. Cardiac muscle is located in the heart and acts to pump blood around the body. Skeletal muscle is the only muscle type under voluntary control and is responsible for multiple bodily functions such as postural maintenance and locomotion. The cardiac and skeletal muscles are considered striated muscles. The basic functional unit of striated muscle is the sarcomere (Figure 2). The sarcomere is a highly organized system comprised of interdigitating thick and thin filaments, and is bordered at its ends by Z-disks (Figure 2A). Muscle contraction occurs through the sliding of these thick and thin filaments across one another, thereby pulling the Z-disks closer together. This contraction is primarily driven by the cyclic interaction of myosin and actin. However, within the sarcomere, there are many additional accessory proteins within the sarcomere that regulate the rate, force, and timing of contraction (Figure 2B).
Figure 1. The three types of muscle.
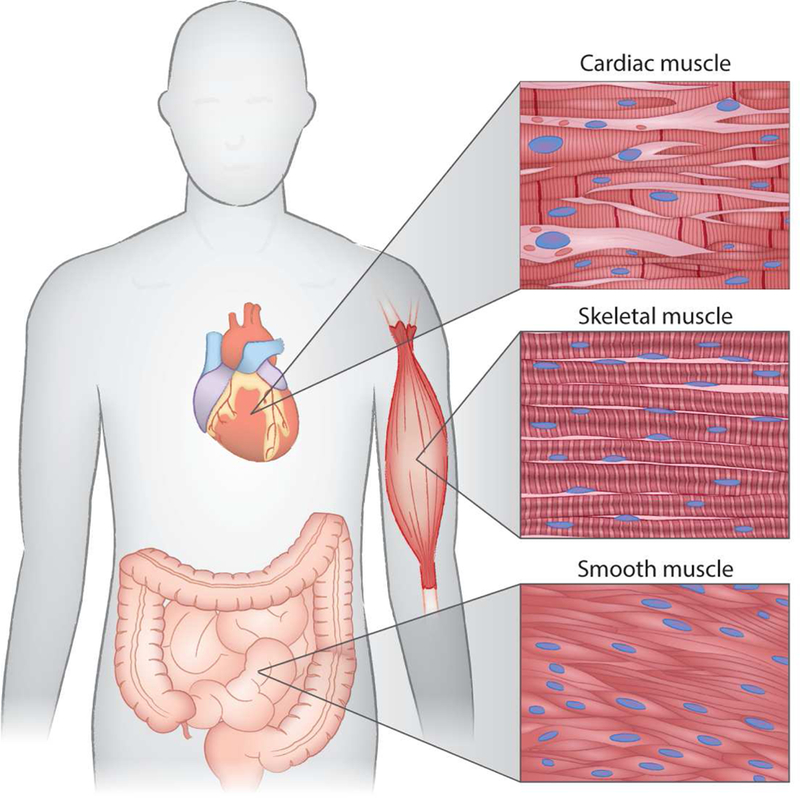
Striated cardiac muscle is expressed exclusively within the heart. Skeletal muscles are also striated, but under voluntary control. Smooth muscle lines the walls of hollow organs and vessels and is under the control of the autonomic nervous system
Figure 2. Striated muscle ultrastructure and the myofilament.
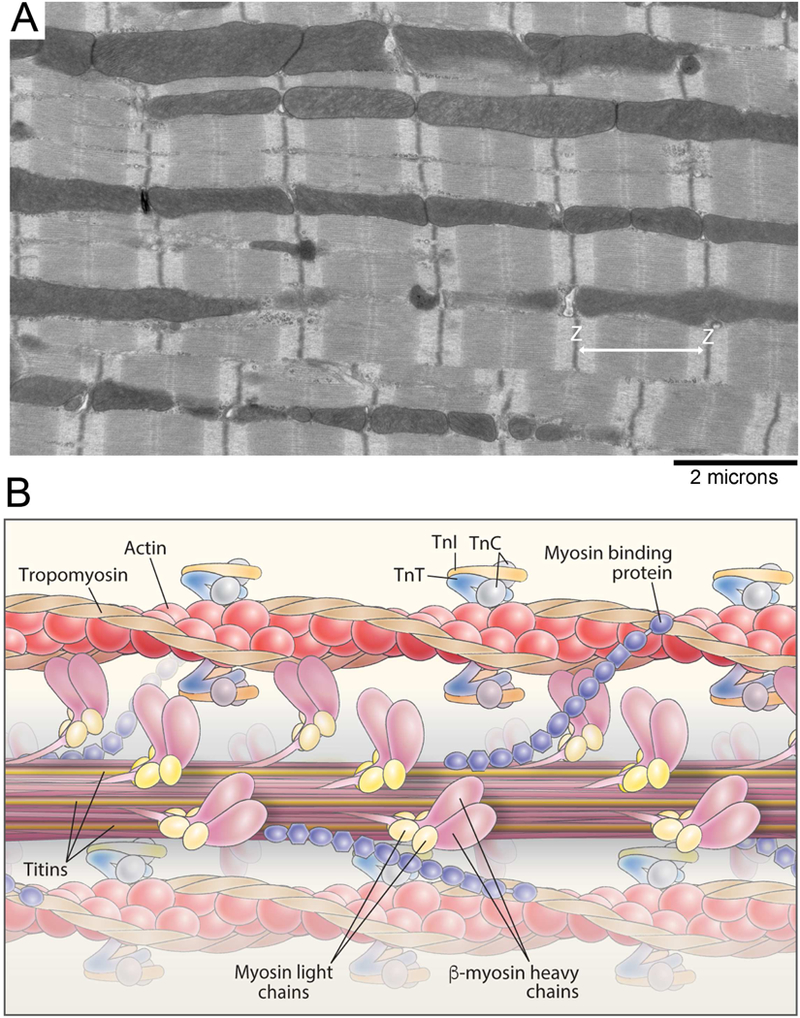
A, Electron micrograph of the cardiac myofilament. B, Schematic diagram illustrating the structure of the cardiac myofilament. Myofilament proteins, such as thick filament proteins (titin, myosin, myosin binding protein-C, light chains) and thin filament proteins (actin, tropomyosin, troponin I, C and T), are shown.
The Myosin Binding Protein-C (MyBP-C) family is a group of sarcomeric proteins known as regulators of myofilament contractility. Three paralogs of MyBP-C are encoded by separate genes with distinct expression profiles in striated muscle. Slow skeletal MyBP- C (ssMyBP-C) is encoded by MYBPC1, and fast skeletal MyBP-C (fsMyBP-C) is encoded by MYBPC2, while MYBPC3 encodes cardiac MyBP-C (cMyBP-C). These three paralogs share significant sequence homology, mainly comprised of immunoglobulin (Ig) and fibronectin-III domains termed C1 through C10 from the amino terminus to the carboxyl terminus (Figure 3). One primary divergence in the structure of cMyBP-C includes an additional amino terminal Ig domain, termed C0.
Figure 3. Domain structure of the three paralogs of MyBP-C.
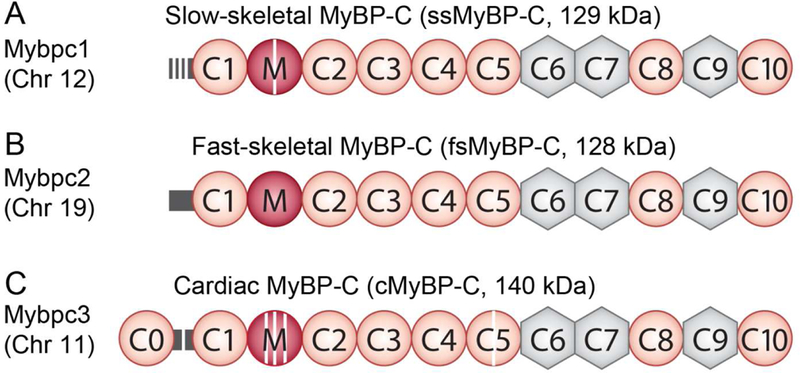
Immunoglobulin domains are shown in orange circles, while fibronectin-III domains are shown in grey hexagons. The proline-alanine (P/A)-rich linker is shown in yellow, and the M-domain is shown as a blue circle. Phosphorylation sites are shown as purple stripes. ssMyBP-C is shown in A with its three phosphorylation sites in the P/A and one within the M-domain. B shows the domain structure of fsMyBP-C. cMyBP-C has an additional N-terminal immunoglobulin domain (C0), four phosphorylation sites in the M-domain, one phosphorylation site in the P/A linker, and a 28 novel amino acid insertion in the C5 domain (red stripe), C.
The discovery of MyBP-C as a myofilament protein dates back to the early 1970s when Starr and Offer revealed it as a contaminant of myosin preparations via the use of polyacrylamide gel electrophoresis [1]. Interestingly, this initial discovery, as well as much of the subsequent work, was performed in skeletal muscle. Each impurity was designated a letter B through J, depending on its position relative to myosin heavy chain. This was how MyBP-C was initially given the name “C-Protein”, which is still widely used today [2]. It was later confirmed that MyBP-C is a component of the thick filament, most likely through its strong interaction with the myosin light meromyosin (LMM), which is the myosin C-terminal rod, important for the assembly of myosin thick filaments [3]. Electron microscopy studies determined that MyBP-C localized to 7 transverse stripes in the inner two thirds of each half A-band, a region fortuitously termed the C-zone that overlaps between the thick and thin filaments [4].
While cMyBP-C was subsequently discovered in cardiac muscle [5, 6], much of the initial work on MyBP-C was performed in predominately fast-type skeletal muscles. However, in 1995, two reports were simultaneously published [7, 8] describing mutations in the cMyBP-C gene, MYBPC3, in patients with hypertrophic cardiomyopathy. These discoveries drove a significant change in focus, and the urgency to understand the structure and function of cMyBP-C in disease pathogenesis left the skeletal MyBP-C paralogs largely unstudied for almost two decades.
Subsequently, an abundance of literature emerged that reported on cMyBP-C in both normal and disease physiology. Conversely, it was only recently that research has focused on regulation of skeletal muscle function by skeletal isoforms. This review summarizes what is known about the structure and function of MyBP-C with a particular focus on knowledge gaps in the context of skeletal muscle MyBP-Cs.
Differential expression of MyBP-C paralogs
The expression of each paralog seems to be tightly regulated. As may be expected, cMyBP-C is exclusively expressed within the cardiac sarcomere. It was previously shown that cMyBP-C is expressed in the developing skeletal muscles of chicken embryos [9, 10]. However, cMyBP-C was restricted to cardiac muscles in the case of mammalian embryos [11]. The expression profile of fsMyBP-C is less clear, but it is expected to be specifically restricted to the skeletal sarcomere. In fact, studies in the literature suggest that it is expressed at very low levels in slow type muscle [12, 13]. Strikingly, evidence suggests the possible ectopic expression of fsMyBP-C within the heart in some cardiac diseases [12], clearly deserving of further investigation. ssMyBP- C appears to be more ubiquitously expressed within skeletal muscles [14]. Reports have also demonstrated that ssMyBP-C may be expressed within a subset of myocardial cells within the right atrium and interatrial septum, but to the best of our knowledge, this has not been confirmed [15]. Furthermore, expression of the MYBPC1 transcript has been identified in a subset of frontal cortex neurons, hinting towards a possible role for ssMyBP-C outside the sarcomere. However, confirmation of ssMyBP-C protein expression in the nervous system is required [16, 17]. The underlying molecular mechanisms behind the tight regulation for each of these paralogs remain to be elucidated.
MYBPC1 undergoes a high level of splicing
A major distinguishing feature of MYBPC1 compared to MYBPC2 and MYBPC3 is the high level of splicing that its transcript may undergo [14]. In humans and mice, at least 14 protein coding transcripts have been described, resulting in protein products between roughly 126 and 132 kDa [14, 18]. Much of this alternative splicing occurs within the first six exons, encoding the proline/alanine-rich region. However, alternative splicing can also occur in regions encoding the M-domain, C7 domain, and carboxyl terminus [14, 19]. These variants appear to be differentially expressed, not just in different muscle groups, but within different myofibers of the same muscle [14, 19]. The exact function of each of these variants is yet to be revealed, and such determination would clearly require exhaustive study. However, we believe that the robust alternative splicing of MYBPC1 supports its importance for skeletal muscle structure and function.
Interaction with myosin
As the name aptly suggests, MyBP-C is a major binding partner of myosin. It is thought that the interaction between the C-terminal of MyBP-C and myosin light meromyosin (LMM) drives its localization to the C-zone [3, 20, 21]. The affinity between the C- terminal MyBP-C and myosin LMM is between 0.5 and 3.5 μM, depending on the MyBP-C paralog [21, 22]. Of note, it is likely that a specific tropism of MyBP-C C-termini exists for LMM such that skeletal MyBP-C binds more tightly to skeletal LMM, and cardiac MyBP-C shows higher affinity to cardiac myosin LMM [23].
The N-terminus of MyBP-C also has the ability to bind myosin heavy meromyosin (HMM), the catalytically active N-terminal product that results from α-chymotrypsin cleavage of myosin. The major binding site for MyBP-C to HMM is believed to reside within a proximal 126 amino acid region in the myosin subfragment-2 (S2). This process is modulated by MyBP-C phosphorylation [24, 25]. However, more recently, a weak interaction between the N-terminus of MyBP-C and the myosin regulatory light chain has been described. Residing in the C0 domain, this may represent a unique cardiac- specific interaction between myosin and MyBP-C [26, 27]. These interactions likely stabilize the myosin super-relaxed state, also known as the OFF state, thereby modulating muscle contractility [28-30]. While it is unknown whether skeletal MyBP-Cs regulate the super-relaxed state, its likelihood is supported by the binding of skeletal MyBP-C which stabilizes the coiled-coil structure of myosin S2 [31]. Importance of the MyBP-C:S2 binding site is further demonstrated by the fact that transgenic mice expressing a mutated cMyBP-C lacking three putative myosin binding residues develop significant cardiac hypertrophy [32].
Interaction with actin
MyBP-C has also long been known to interact with the thin filament [33], emphasizing the dynamic nature of these proteins. However, the interaction of actin and MyBP-C remained largely unexplored until a series of landmark studies by the Harris group [34- 36]. The C0C2 domains bind the thin filament in a phosphorylation-dependent manner, and incubation with these domains results in a large increase in force production at submaximal Ca2+ concentrations [35, 37, 38]. While the C0 domain can bind actin, it does not contribute to this phenomenon. Instead, it seems that the C1 domain can bind to actin in a position that allows it to activate the thin filament by shifting tropomyosin. However, a direct interaction between tropomyosin and the proline-alanine-rich region of cMyBP-C may also contribute to this activation [37, 39].
At the molecular level, MyBP-C fragments can act as viscous load in in vitro motility assays. In particular, unregulated actin filaments slow down when moving over the C- zone of native thick filaments [40, 41]. Calpain-mediated proteolysis of endogenous MyBP-C abolished this effect, indicating the role of the N-terminal domains of cMyBP-C in the regulation of contractility. The slowing of actin filaments over the C-zone is expected to arise from the binding of MyBP-C to actin causing a drag effect. However, it is also possible that MyBP-C may act to sequester myosin heads within the C-zone, but this mechanism is yet to be fully investigated.
The differences among the N-terminal regions of ssMyBP-C, fsMyBP-C, and cMyBP-C were recently explored [42]. The effect of these fragments was determined in permeabilized rat trabeculae with cMyBP-C having the greatest effect on muscle activation at submaximal Ca2+, followed by fsMyBP-C and then ssMyBP-C. Furthermore, native thin filament motility was differentially regulated by each fragment with C0C2 activating motility at low Ca2+, while inhibiting motility at higher Ca2+ concentrations. The fsMyBP-C paralog could not activate motility at low Ca2+, but it could inhibit motility at high Ca2+. Conversely, ssMyBP-C promoted activation at low Ca2+, but could not inhibit motility at higher Ca2+ concentrations. The exact physiological consequences of this differential modulation remain the subject for intensive study, and this study used only rat trabeculae to study the roles of these proteins. However, this phenomenon suggests nonredundant roles for each of these paralogs in striated muscle function.
Noncanonical binding partners of MyBP-C
MyBP-C is generally thought to be a structural and functional protein that regulates contractility through its differential binding of thick and thin filaments. However, a number of reports describe novel binding partners that may contribute to striated muscle function and homeostasis. As the field turns further towards up- and downstream regulators of myofilament proteins, we expect that many more unanticipated interactions will be discovered.
Four and a half LIM protein 1 (FHL1) has previously been demonstrated to colocalize and bind to the C-terminus of MyBP-C [43]. The LIM domain family of proteins is known to be a critical regulator of striated muscle development [44]. The binding of FHL1 to MyBP-C appears to be critical to its incorporation into the thick filament, and knockdown of FHL1 impaired the assembly of thick filaments in differentiating mouse myoblasts [43].
An interesting relationship has also been described between ssMyBP-C and muscle- type creatine kinase (MM-CK). An interaction between these two proteins was uncovered with the interaction localized to the C-terminal domains of ssMyBP-C [45]. It was hypothesized that ssMyBP-C acts as a recruiter for MM-CK to the myosin heads, thus connecting the major ATP consumer with an ATP regenerator to increase the efficiency of ATP regeneration. Whether this has any role in physiology is yet to be determined.
A calcium-dependent interact between MyBP-C and calmodulin has also been described at a 1:1 stoichiometric ratio [46]. It was further determined that a 23-residue sequence within the tri-helix bundle of M-domain was sufficient to maintain this interaction [46, 47]. It was hypothesized that this interaction represented a means to initiate rapid phosphorylation CaMKII targets within MyBP-C, or even promote myosin light chain kinase-mediated phosphorylation of the regulatory light chain [46], although this is yet to be shown. More recently, this calmodulin binding has been demonstrated in ssMyBP-C [48]. It remains to be determined whether this interaction drives any differential regulation of skeletal muscle function.
A fascinating variant of MYBPC1 with a unique C-terminal coding sequence has been described as a ligand for the striated muscle giant protein obscurin [49]. This variant protein, termed ssMyBP-C variant 1, contains an additional 26 amino acids following the usual C10 domain. Rather than localizing to the C-zone like most MyBP-Cs, this variant of ssMyBP-C seems to spend its time at the M-band where it interacts with the N- terminal Ig domains of obscurin. Knockdown of obscurin in myotubes resulted in the failure of ssMyBP-C variant 1 to localize to the M-band. This interaction was proposed to be critical for the proper formation of skeletal muscle M-bands during myofibrillogenesis.
More recently, an in-depth study was published outlining the interaction between cMyBP-C and the formin homology-2 domain-containing protein FHOD3 [50]. FHOD3 is an actin-binding protein thought to be important for sarcomeric organization [51]. Strikingly, this group showed a surprisingly strong kd of about 800nM between the C0 domain of cMyBP-C and FHOD3. This interaction is stronger than the interaction reported to occur between the N-terminal domains and both myosin S2 [24] and actin [36]. Loss of this interaction appeared to be detrimental to cardiac structure and function in mice overexpressing FHOD3 in a cMyBP-C null background. These mice were unable to survive more than 16 days after birth and displayed significant disruption to sarcomeric organization, particularly to the thick filaments.
Phosphorylation
Phosphorylation of cMyBP-C was first described in intact frog cardiac muscle with functional significance [52]. Isoproterenol treatment of intact frog myocardium showed a relationship between cMyBP-C phosphorylation and cardiac twitch kinetics [5]. In particular, increased phosphorylation of cMyBP-C correlated with increased tension generation, accelerating both contraction and relaxation rates. Multiple conserved phosphorylation sites within the M-domain have since been described, including serines 273, 282, 302, and 307 in mice [53, 54]. These sites are the target for many kinases, including protein kinase A (PKA), protein kinase C (PKC), and Ca2+/calmodulin dependent kinase-II (CaMKII), each site containing a different set of kinases with an ability to phosphorylate it [55]. Other phosphorylation sites have been described outside the M-domain, such as the GSK3β target serine 133 within the proline-alanine region [56].
Under baseline conditions, cMyBP-C is highly phosphorylated; however, in many cardiac diseases, including heart failure, atrial fibrillation, and HCM, cMyBP-C phosphorylation is significantly reduced, highlighting the physiological importance of cMyBP-C phosphorylation [57-59]. This importance is further supported by the constitutive phosphorylation of cMyBP-C, as expressed in a cMyBP-C null background which resulted in the restoration of cardiac structure and function, while constitutive dephosphorylation of cMyBP-C did not [60]. Furthermore, dephosphorylated cMyBP-C was subjected to proteolysis during ischemia [60, 61], while phosphorylated cMyBP-C was not. This protection from proteolysis might be explained by structural changes within the N-terminus of cMyBP-C masking a proteolytic recognition site [62, 63]. The 29kDa fragment that is released during ischemia has since been shown to be a useful and early marker for myocardial infarction [64, 65].
The functional effects of cMyBP-C phosphorylation were initially difficult to parse from other major β1-adrenergic targets, such as cardiac troponin I (cTnI) and phospholamban. Thus, the generation of transgenic models has been fundamental to furthering our understanding in this area. PKA phosphorylation of skinned murine ventricular samples increased both the rate of relaxation and force development, while similar treatment in cMyBP-C null samples had no effect, implicating that cMyBP-C is a principal mediator of increases in lusitropy and inotropy, as observed with β-adrenergic stimulation [66, 67]. This cMyBP-C phosphorylation shifted myosin heads closer to the actin filament, with no apparent contribution from cTnI phosphorylation, in a manner similar to that of myosin light chain kinase phosphorylation of myosin regulatory light chain [68, 69]. The cMyBP-C protein has also been described as a modulator of length- dependent activation of myocardium in a manner independent of cTnI phosphorylation [70, 71]. Using N-terminal fragments of cMyBP-C, it was reported that the serine-to- aspartate substitution widely used to mimic phosphorylation in transgenic mice is unable to recapitulate all aspects of PKA-mediated cMyBP-C phosphorylation [72]. This is a very important consideration in interpreting future results; however, to date, no other method has been able to isolate the functional roles of phosphorylation sites of endogenous proteins, particularly in vivo.
Recently, ssMyBP-C has been demonstrated as a target for both PKA and PKC in mouse skeletal muscles [13]. Specifically, serine-59 and −62 were demonstrated to be targeted by PKA, while PKC phosphorylates serine-83 and threonine-84. Moreover, serine-204 is targeted by both PKA and PKC [13]. These phosphorylation sites appear to be dynamically regulated in both health and disease with aged healthy mice displaying up to a 40% reduction in phosphorylation compared to young and healthy mice [73]. Furthermore, dystrophic mice display a significantly reduced ssMyBP-C phosphorylation at both young and old ages [73, 74]. Intriguingly, the same group previously reported an overall increase in ssMyBP-C phosphorylation in dystrophic tissues by 2-dimensional (2D) SDS-PAGE and ProQ Diamond staining, possibly indicating the presence of additional unreported phosphorylation sites upregulated in disease. It remains unknown how each of these phosphorylation sites may affect the function of ssMyBP-C, or even if they possess divergent functions in each ssMyBP-C isoform. Clearly, an in-depth systematic study is required to define the function of each site. To the best of our knowledge, no publications have defined a phosphorylation site in fsMyBP-C; however, this is not evidence that such modification does not exist.
Skeletal MyBP-C in disease pathogenesis
Arthrogryposis is a family of diseases in which mutations in MYBPC1 and MYBPC2 have been implicated to have an etiological role. Also known as arthrogryposis multiplex congenita (AMC), these diseases are defined by congenital joint contractures that involve at least two areas of the body [75, 76]. Present in up to 1 in 3,000 live births, AMC affects both upper and lower limbs [75-77]. While the exact etiology of arthrogryposis remains unknown, it is thought that any condition that induces fetal akinesia and reduced fetal mobility would result in such contractures.
Of the many subgroups of arthrogryposis, MYBPC1 and MYBPC2 mutations are most commonly associated with distal arthrogryposis (DA), comprising at least 10 subtypes. Mutations in a wide range of skeletal muscle myofilament genes have previously been implicated in DA, including myosin heavy chain (MYH3 and MYH8) [78, 79] and thin filament proteins troponin I [80], troponin T [81-83], and tropomyosin [84] (TNNI2, TNNT3, and TPM2, respectively). DA type 1 (DA1) is the most common subtype of congenital distal limb contracture syndromes, affecting approximately 1 in 10,000 live births [85, 86]. DA1 is an autosomal dominant disease, and patients present with contractures limited to the hands and feet. Patients may display clubfoot, vertical talus, and ulnar deviation of the fingers, among other defects [87].
The first report to implicate MYBPC1 in skeletal muscle myopathies described two families presenting with DA1 [87]. In these two families, a single nucleotide polymorphism (SNP) was traced to chromosome position 12q23.2, the location of the MYBPC1 gene. Sequencing of MYBPC1 confirmed that affected members from both families harbored mutations in this gene. The first family’s mutation was c.706T>C, corresponding to an arginine replacing a tryptophan at amino acid 236 (W236R), while the second family’s mutation corresponded to Y856H. Patient biopsies from skeletal muscle tissue demonstrated significant atrophy of type I muscle fibers with variable effects on the number and distribution of fiber types [87]. In vivo electroporation showed that these mutated MYBPC1 genes could incorporate correctly to the C-zone of the sarcomere, suggesting that they cause a poison polypeptide effect, rather than haploinsufficiency, as is the case with many HCM-causing MYBPC3 mutations [88, 89]. Further in vitro investigation of these mutations suggested that they negatively affected the binding of ssMyBP-C to its major binding partners, actin and myosin [90]. Furthermore, proteomic analysis of patient biopsies harboring these mutations showed that the Y856H mutation reduced total and phosphorylated ssMyBP-C, while no major effect resulted from the W236R mutation [74]. However, the biopsy sample for Y825H was taken from the abductor hallucis, while the W236R biopsy was taken from the gastrocnemius, making interpretation between the two groups difficult. This mutated tryptophan has recently been implicated as an important residue for ssMyBP-C calmodulin binding, discussed above [48]. While the physiological relevance of the interaction is yet to be determined, the W236R mutation may be detrimental to this interaction and contribute to disease progression.
Additional mutations in MYBPC1 have been identified in two Chinese families with DA type 2 (DA2) [91]. The DA2 subtype is similar to that of DA1, with camptodactyly and ulnar deviation, but with additional facial and pharyngeal involvement [92]. These two mutations, P319L and E359K, both occurred within the C2 domain, thus potentially affecting the critical interaction with myosin S2, which is required for normal muscle function [24]. It remains unclear why these mutations result in slightly different clinical outcomes compared to the DA1-causing mutations.
Mutations in MYBPC1 have also been linked to a more severe, neonatally lethal form of arthrogryposis, termed Lethal Congenital Contracture Syndrome-4 (LCCS-4) [93]. A homozygous SNP was identified in three affected individuals. This SNP resulted in the generation of a premature stop codon at amino acid 318 within the C2 Ig domain. Such a truncation mutation could be expected to undergo decay at either the transcript or protein level, suggesting that the complete loss of ssMyBP-C may cause lethality, as previously suggested [85].
A second, less severe homozygous mutation in MYBPC1, has more recently been described in a consanguineous Israeli-Druze family [94]. This mutation, E186K, occurs at the border of the C1- and M-domains, a region expected to contribute to both actin and myosin binding [95]. Unlike the other missense mutations described in MYBPC1 above [87, 91], individuals harboring a heterozygous mutation were unaffected, thus becoming the first non-lethal autosomal recessive MYBPC1 mutation. Excitingly, two novel mutations in MYBPC1 have recently been described, resulting in Y248H and E248K substitutions, respectively. These missense mutations were associated with myogenic tremors and muscle weakness [96, 97]. Strikingly, the E248K phenotype could be replicated in a CRISPR/Cas9-generated mouse model [96]. This is unique as these are the first reports associating MYBPC1 mutations with non-AMC disease, further expanding the spectrum of MYBPC1-related myopathies.
MYBPC1 mutations are not restricted to human diseases. A SNP in the promoter region of MYBPC1 has been associated with increased marbling, or amount of intramuscular fat, in Japanese Black steers [98]. This same variant was later associated with increased muscle size in the same breed, and increased MYBPC1 gene expression was also associated with steers developing more muscle mass [99], potentially implicating ssMyBP-C in muscle growth. Additionally, a de novo mutation was identified in a female calf presenting with a DA1-like phenotype [100]. This mutation caused a point mutation, L295R, within the M-domain, which was not present in either parent. This calf, which struggled to stand unassisted, showed increased muscle tone in the limbs without any apparent atrophy and hypermetria. Strikingly, this calf also presented with tremors and ataxia, suggesting that this mutation results in a more complex outcome than DA1.
MYBPC2 has also been implicated in the development of a severe and lethal form of unclassified DA [101]. A compound heterozygous or homozygous mutation in MYBPC2 was identified (T236I and S255T) concurrently with a homozygous truncating mutation in the G protein-coupled receptor 126 (GPR126). GPR126 encodes a member of the adhesion G protein-coupled receptor family [102], and it appears to be important for peripheral myelination. Mutations in GPR126 had previously been implicated in lethal forms of arthrogryposis, making it difficult to determine the contribution of these MYBPC2 mutations to the disease [103]. The details and locations of the above mutations are shown in Table 1 and Figure 4.
Table 1:
List of disease associated mutations in MYBPC1 and MYBPC2
| Gene | Mutation | Classification | Disease | Phenotype | Reference |
|---|---|---|---|---|---|
| MYBPC1 | Trp236Arg | Pathogenic | Distal arthrogryposis IB |
Bilateral Clubfoot, camptodactyly, ulnar deviation, vertical talus, fiber type I atrophy |
83, 86 |
| MYBPC1 | Tyr856His | Pathogenic | Distal arthrogryposis IB |
Bilateral vertical talus and clubfoot, hand contractures, fiber type I atrophy |
83, 87 |
| MYBPC1 | Val560Leu | Likely Pathogenic |
Distal arthrogryposis |
Not described | RCV000626822.1 |
| MYBPC1 | Leu259Pro | Likely Pathogenic |
MYBPC1 related condition |
Not described | RCV000626001.1 |
| MYBPC1 | Tyr247His | Not available | Autosomal dominant congenital myopathy |
Axial muscle weakness, high frequency postural and tongue tremor, calf hypertrophy, mild facial dysmorphism |
94 |
| MYBPC1 | Glu248Lys | Pathogenic | Not available | Muscle weakness, skeletal deformities, myogenic tremor, spinal rigidity |
93, RCV000408887.1 |
| MYBPC1 | Arg318Ter (homozygous) |
Pathogenic | Lethal congenital contracture syndrome 4 |
Postnatal lethality | 90 |
| MYBPC1 | Glu186Lys (Homozygous) |
Not available | Arthrogryposis multiplex congenita |
Hypotonia, contractures of distal limbs, camptodactyly, clinodactyly, flexed contracture of hips, knees and feet, scoliosis, postnatal lethality |
91 |
| MYBPC1 | Glu359Lys | Not available | Distal arthrogryposis 2 |
Ulnar deviation of fingers, adducted stiff thumb, camptodactyly, overlapping fingers, overextension contractures, prominent nasolabia1 fold, small mouth, pouting or pinched lips |
88 |
| MYBPC1 | Pro319Leu | Not available | Distal arthrogryposis 2 |
88 | |
| MYBPC1 | Leu295Arg* | Not available | Similar to distal arthrogryposis | Tachycardia, tachypnea, inability to stand unassisted, muscular tremors, ataxia, hypermetria, increased muscle tone |
97 |
| MYBPC2 | T236I | Not available | Unclassified arthrogryposis, Concurrent with GPR126 Arg7Ter |
Narrow thorax, polyhydramnios in utero, postnatal death |
98 |
| MYBPC2 | S255T | 98 | |||
Mutation determined in 2-week-old female calf.
Note: Where publication references are not available, accession numbers are provided.
Figure 4. Location of skeletal MyBP-C mutations.
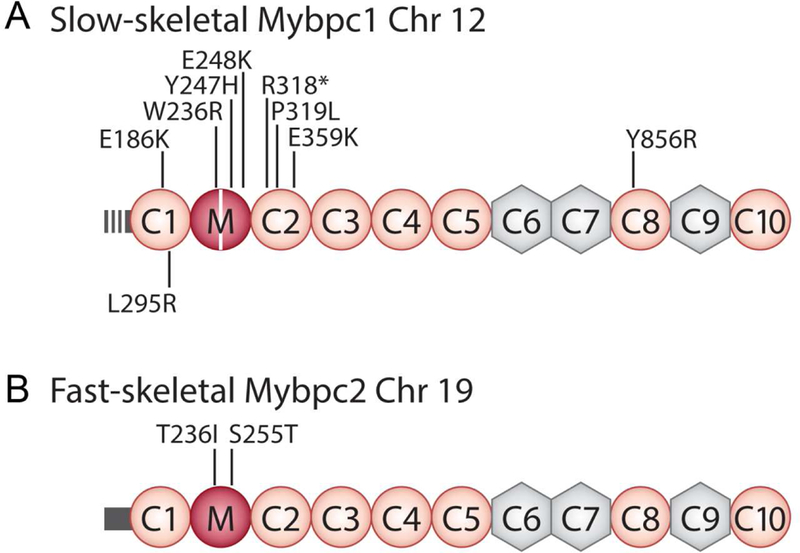
The many MYBPC1 mutations shown in A. Note that the L295R mutation was discovered in a 2-week-old female calf. The two co-discovered MYBPC2 mutations are shown in fsMyBP-C in B.
The association of MYBPC1 with skeletal muscle diseases outlined above shows the critical need for a better understanding of how MYBPC1 and MYBPC2 function in both normal and diseased physiology. For example, these proteins are constitutively expressed in all skeletal muscles, but we still do not know why the primary target for these mutations appears to be distal limbs. One approach toward understanding the role of these proteins has been to utilize zebrafish embryos (Danio rerio) as a model. Knockdown of MYBPC1 using an antisense morpholino oligonucleotide (MO) resulted in severe ventral body curvature, reduced survival, and, somewhat surprisingly, defects in eye, head and cardiac growth [104]. This phenotype could be reversed with the concurrent injection of MYBPC1 mRNA. The same study also determined the effect of the initial DA1-causing MYBPC1 mutations (W236R and Y845H) by overexpression of mutant mRNA. While some fish also demonstrated body curvature, only about 30% of fish were affected, suggesting incomplete penetrance of these mutations, even though overall survival was reduced compared to wild-type fish [104]. Strikingly, while motion appeared normal under baseline conditions, these mutant fish exhibited a reduced “touch-escape” response, possibly suggesting that these mutations required some form of stress to show their true function. Knockdown of fsMyBP-C in zebrafish also resulted in skeletal myopathy, albeit without the severe body curvature described in ssMyBP-C knockouts [105]. These fsMyBP-C null fish exhibited sarcomeric disorganization, reduced active force production, and an increase in markers for muscle atrophy. While the zebrafish is indeed an attractive model based on the relative ease and time required to establish the model, the skeletal muscle physiology is not the same in fish and mammals. Thus, mammalian models to study MYBPC1 and MYBPC2 are also required.
To understand the function(s) of MYBPC1 function in skeletal muscle, knockdown of Mybpc1 in the footpad of mice was achieved via in vivo electroporation of a CRISPR- mediated knockdown plasmid [19]. This plasmid primarily targeted the extensor digitorum brevis (FDB) and lumbricalis muscles. Strikingly, ssMyBP-C knockdown of about 60% in the FDB was accompanied by reduction in the number of thick filament proteins, including myosin and myomesin, without affecting thin filament protein expression or localization. Somewhat surprisingly, fsMyBP-C expression remained unchanged in FDB, and it was even slightly overexpressed in the lumbricalis. Given the loss of myosin, as well as the inconsistencies in fsMyBP-C antibodies, it remains unclear how fsMyBP-C, which is classically thought to localize to the sarcomere through its interaction with myosin, is upregulated in these muscles. Loss of ssMyBP-C also significantly reduced contractile kinetics and tension production in vitro and in vivo [19]. Overall, the excellent work from this group suggests that ssMyBP-C may be structurally required for the overall maintenance of adult skeletal muscle and that loss of this structural component leads to functional decline. However, the electroporation technique could only target a small group of muscles, and it was incomplete in its knockdown. Therefore, we suggest that more in-depth genetic approaches are also required to completely assess the in vivo role of ssMyBP-C.
While the work presented in this review is groundbreaking, the need for further study of skeletal MyBP-Cs is clear. As a result, we can expect an increasing number of disease- associated mutations in these paralogs. By necessity, many assumptions about these paralogs have been made based on the function of cMyBP-C, but further study may, in fact, invalidate them. It is, however, certain that much remains to be elucidated about the function of skeletal MyBP-Cs and that this portends the dawning of a newly emerging field in striated muscle physiology.
Footnotes
Disclosures
Dr. McNamara is supported by an American Heart Association postdoctoral fellowship (17P0ST33630095). Dr. Sadayappan has received support from National Institutes of Health grants (R01HL130356, R01HL105826, and K02HL114749), an American Heart Association catalyst award (17CCRG33671128), AstraZeneca, Inc., Merck and Amgen.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Starr R, Offer G, Polypeptide chains of intermediate molecular weight in myosin preparations, FEBS letters 15(1) (1971) 40–44. [DOI] [PubMed] [Google Scholar]
- [2].Offer G, Moos C, Starr R, A new protein of the thick filaments of vertebrate skeletal myofibrils. Extractions, purification and characterization, J Mol Biol 74(4) (1973) 653–76. [DOI] [PubMed] [Google Scholar]
- [3].Moos C, Offer G, Starr R, Bennett P, Interaction of C-protein with myosin, myosin rod and light meromyosin, J Mol Biol 97(1) (1975) 1–9. [DOI] [PubMed] [Google Scholar]
- [4].Craig R, Offer G, The location of C-protein in rabbit skeletal muscle, Proc R Soc Lond B Biol Sci 192(1109) (1976) 451–61. [DOI] [PubMed] [Google Scholar]
- [5].Hartzell HC, Titus L, Effects of cholinergic and adrenergic agonists on phosphorylation of a 165,000-dalton myofibrillar protein in intact cardiac muscle, J Biol Chem 257(4) (1982) 2111–20. [PubMed] [Google Scholar]
- [6].Yamamoto K, Moos C, The C-proteins of rabbit red, white, and cardiac muscles, J Biol Chem 258(13) (1983) 8395–401. [PubMed] [Google Scholar]
- [7].Bonne G, Carrier L, Bercovici J, Cruaud C, Richard P, Hainque B, Gautel M, Labeit S, James M, Beckmann J, Weissenbach J, Vosberg HP, Fiszman M, Komajda M, Schwartz K, Cardiac myosin binding protein-C gene splice acceptor site mutation is associated with familial hypertrophic cardiomyopathy, Nature Genet 11(4) (1995) 438–40. [DOI] [PubMed] [Google Scholar]
- [8].Watkins H, Conner D, Thierfelder L, Jarcho JA, MacRae C, McKenna WJ, Maron BJ, Seidman JG, Seidman CE, Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy, Nature Genet 11(4) (1995) 434–7. [DOI] [PubMed] [Google Scholar]
- [9].Kawashima M, Kitani S, Tanaka T, Obinata T, The earliest form of C-protein expressed during striated muscle development is immunologically the same as cardiac-type C-protein, J Biochem 99(4) (1986) 1037–47. [DOI] [PubMed] [Google Scholar]
- [10].Yasuda M, Koshida S, Sato N, Obinata T, Complete primary structure of chicken cardiac C-protein (MyBP-C) and its expression in developing striated muscles, J Mol Cell Cardiol 27(10) (1995) 2275–86. [DOI] [PubMed] [Google Scholar]
- [11].Fougerousse F, Delezoide AL, Fiszman MY, Schwartz K, Beckmann JS, Carrier L, Cardiac myosin binding protein C gene is specifically expressed in heart during murine and human development, Circ Res 82(1) (1998) 130–3. [DOI] [PubMed] [Google Scholar]
- [12].Lin B, Govindan S, Lee K, Zhao P, Han R, Runte KE, Craig R, Palmer BM, Sadayappan S, Cardiac Myosin Binding Protein-C Plays No Regulatory Role in Skeletal Muscle Structure and Function, PLoS ONE 8(7) (2013) e69671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ackermann MA, Kontrogianni-Konstantopoulos A, Myosin binding protein-C slow is a novel substrate for protein kinase A (PKA) and C (PKC) in skeletal muscle, J Proteome Res 10(10) (2011) 4547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ackermann MA, Kontrogianni-Konstantopoulos A, Myosin binding protein-C slow: a multifaceted family of proteins with a complex expression profile in fast and slow twitch skeletal muscles, Front Physiol 4 (2013) 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dhoot GK, Perry SV, Expression of slow skeletal myosin binding C-protein in normal adult mammalian heart, J Muscle Res Cell Motil 26(2–3) (2005) 143–8. [DOI] [PubMed] [Google Scholar]
- [16].French L, Ma T, Oh H, Tseng GC, Sibille E, Age-Related Gene Expression in the Frontal Cortex Suggests Synaptic Function Changes in Specific Inhibitory Neuron Subtypes, Front Aging Neurosci 9 (2017) 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang L, Huang J, Jiang M, Sun L, MYBPC1 computational phosphoprotein network construction and analysis between frontal cortex of HIV encephalitis (HIVE) and HIVE-control patients, Cell Mol neurobiol 31(2) (2011) 233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ackermann MA, Kontrogianni-Konstantopoulos A, Myosin binding protein-C slow: an intricate subfamily of proteins, Journal of biomedicine & biotechnology 2010 (2010) 652065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Geist J, Ward CW, Kontrogianni-Konstantopoulos A, Structure before function: myosin binding protein-C slow is a structural protein with regulatory properties, FASEB J (2018) fj201800624R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gilbert R, Kelly MG, Mikawa T, Fischman DA, The carboxyl terminus of myosin binding protein C (MyBP-C, C-protein) specifies incorporation into the A-band of striated muscle, J Cell Sci 109(1) (1996) 101–11. [DOI] [PubMed] [Google Scholar]
- [21].Okagaki T, Weber FE, Fischman DA, Vaughan KT, Mikawa T, Reinach FC, The major myosin-binding domain of skeletal muscle MyBP-C (C protein) resides in the COOH-terminal, immunoglobulin C2 motif, J Cell Biol 123(3) (1993) 619–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Miyamoto CA, Fischman DA, Reinach FC, The interface between MyBP-C and myosin: site-directed mutagenesis of the CX myosin-binding domain of MyBP-C, J Muscle Res Cell Motil 20(7) (1999) 703–15. [DOI] [PubMed] [Google Scholar]
- [23].Alyonycheva TN, Mikawa T, Reinach FC, Fischman DA, Isoform-specific interaction of the myosin-binding proteins (MyBPs) with skeletal and cardiac myosin is a property of the C- terminal immunoglobulin domain, J Biol Chem 272(33) (1997) 20866–72. [DOI] [PubMed] [Google Scholar]
- [24].Gruen M, Gautel M, Mutations in beta-myosin S2 that cause familial hypertrophic cardiomyopathy (FHC) abolish the interaction with the regulatory domain of myosin-binding protein-C, J Mol Biol 286(3) (1999) 933–49. [DOI] [PubMed] [Google Scholar]
- [25].Gruen M, Prinz H, Gautel M, cAPK-phosphorylation controls the interaction of the regulatory domain of cardiac myosin binding protein C with myosin-S2 in an on-off fashion, FEBS Lett 453(3) (1999) 254–9. [DOI] [PubMed] [Google Scholar]
- [26].Nag S, Trivedi DV, Sarkar SS, Adhikari AS, Sunitha MS, Sutton S, Ruppel KM, Spudich JA, The myosin mesa and the basis of hypercontractility caused by hypertrophic cardiomyopathy mutations, Nature Struct Biol 24(6) (2017) 525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ratti J, Rostkova E, Gautel M, Pfuhl M, Structure and interactions of myosin-binding protein C domain C0: cardiac-specific regulation of myosin at its neck?, J Biol Chem 286(14) (2011) 12650–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ababou A, Rostkova E, Mistry S, Le Masurier C, Gautel M, Pfuhl M, Myosin binding protein C positioned to play a key role in regulation of muscle contraction: structure and interactions of domain C1, J Mol Biol 384(3) (2008) 615–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].McNamara JW, Li A, Lal S, Bos JM, Harris SP, van der Velden J, Ackerman MJ, Cooke R, Dos Remedios CG, MYBPC3 mutations are associated with a reduced super-relaxed state in patients with hypertrophic cardiomyopathy, PLoS ONE 12(6) (2017) e0180064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].McNamara JW, Li A, Smith NJ, Lal S, Graham RM, Kooiker KB, van Dijk SJ, dos Remedios CG, Harris SP, Cooke R, Ablation of cardiac myosin binding protein-C disrupts the super-relaxed state of myosin in murine cardiomyocytes, J Mol Cell Cardiol 94 (2016) 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Singh RR, Dunn JW, Qadan MM, Hall N, Wang KK, Root DD, Whole length myosin binding protein C stabilizes myosin S2 as measured by gravitational force spectroscopy, Arch Biochem Biophys 638 (2018) 41–51. [DOI] [PubMed] [Google Scholar]
- [32].Bhuiyan MS, McLendon P, James J, Osinska H, Gulick J, Bhandary B, Lorenz JN, Robbins J, In vivo definition of cardiac myosin-binding protein C’s critical interactions with myosin, Pflugers Archiv 468(10) (2016) 1685–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Moos C, Mason CM, Besterman JM, Feng I, Nan M, Dubin JH, The binding of skeletal muscle C-protein to F-actin, and its relation to the interaction of actin with myosin subfragment-1., J Mol Biol 124(4) (1978) 571–586. [DOI] [PubMed] [Google Scholar]
- [34].Kensler RW, Shaffer JF, Harris SP, Binding of the N-terminal fragment C0-C2 of cardiac MyBP-C to cardiac F-actin, J Struct Biol 174(1) (2011) 44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Razumova MV, Bezold KL, Tu A-Y, Regnier M, Harris SP, Contribution of the myosin binding protein C motif to functional effects in permeabilized rat trabeculae, J Gen Physiol 132(5) (2008) 575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Shaffer JF, Kensler RW, Harris SP, The myosin-binding protein C motif binds to F-actin in a phosphorylation-sensitive manner, J Biol Chem 284(18) (2009) 12318–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Harris SP, Belknap B, Van Sciver RE, White HD, Galkin VE, C0 and C1 N-terminal Ig domains of myosin binding protein C exert different effects on thin filament activation, Proc Nat Acad Sci USA 113(6) (2016) 1558–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Herron TJ, Rostkova E, Kunst G, Chaturvedi R, Gautel M, Kentish JC, Activation of myocardial contraction by the N-terminal domains of myosin binding protein-C, Circ Res 98(10) (2006) 1290–8. [DOI] [PubMed] [Google Scholar]
- [39].Witayavanitkul N, Ait Mou Y, Kuster DW, Khairallah RJ, Sarkey J, Govindan S, Chen X, Ge Y, Rajan S, Wieczorek DF, Irving T, Westfall MV, de Tombe PP, Sadayappan S, Myocardial infarction-induced N-terminal fragment of cardiac myosin-binding protein C (cMyBP-C) impairs myofilament function in human myocardium, J Biol Chem 289(13) (2014) 8818–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Previs MJ, Beck Previs S, Gulick J, Robbins J, Warshaw DM, Molecular mechanics of cardiac myosin-binding protein C in native thick filaments, Science 337(6099) (2012) 1215–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Weith A, Sadayappan S, Gulick J, Previs MJ, Vanburen P, Robbins J, Warshaw DM, Unique single molecule binding of cardiac myosin binding protein-C to actin and phosphorylation-dependent inhibition of actomyosin motility requires 17 amino acids of the motif domain, J Mol Cell Cardiol 52(1) (2012) 219–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lin BL, Li A, Mun JY, Previs MJ, Previs SB, Campbell SG, Dos Remedios CG, Tombe PP, Craig R, Warshaw DM, Sadayappan S, Skeletal myosin binding protein-C isoforms regulate thin filament activity in a Ca(2+)-dependent manner, Sci Rep 8(1) (2018) 2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].McGrath MJ, Cottle DL, Nguyen MA, Dyson JM, Coghill ID, Robinson PA, Holdsworth M, Cowling BS, Hardeman EC, Mitchell CA, Brown S, Four and a half LIM protein 1 binds myosin-binding protein C and regulates myosin filament formation and sarcomere assembly, J Biol Chem 281(11) (2006) 7666–83. [DOI] [PubMed] [Google Scholar]
- [44].Vafiadaki E, Arvanitis DA, Sanoudou D, Muscle LIM Protein: Master regulator of cardiac and skeletal muscle functions, Gene 566(1) (2015) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chen Z, Zhao TJ, Li J, Gao YS, Meng FG, Yan YB, Zhou HM, Slow skeletal muscle myosin-binding protein-C (MyBPC1) mediates recruitment of muscle-type creatine kinase (CK) to myosin, Biochem J 436(2) (2011) 437–45. [DOI] [PubMed] [Google Scholar]
- [46].Lu Y, Kwan AH, Jeffries CM, Guss JM, Trewhella J, The motif of human cardiac myosin- binding protein C is required for its Ca2+-dependent interaction with calmodulin, J Biol Chem 287(37) (2012) 31596–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Michie KA, Kwan AH, Tung CS, Guss JM, Trewhella J, A Highly Conserved Yet Flexible Linker Is Part of a Polymorphic Protein-Binding Domain in Myosin-Binding Protein C, Structure 24(11) (2016) 2000–2007. [DOI] [PubMed] [Google Scholar]
- [48].Springer TIJ, Cable CW, Lin J, Sadayappan BL, Finley S, N.L., Calcium-Dependent Interaction Occurs between Slow Skeletal Myosin Binding Protein C and Calmodulin, Magnetochemistry 4(1) (2018). [Google Scholar]
- [49].Ackermann MA, Hu LY, Bowman AL, Bloch RJ, Kontrogianni-Konstantopoulos A, Obscurin interacts with a novel isoform of MyBP-C slow at the periphery of the sarcomeric M- band and regulates thick filament assembly, Mol Biol Cell 20(12) (2009) 2963–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Matsuyama S, Kage Y, Fujimoto N, Ushijima T, Tsuruda T, Kitamura K, Shiose A, Asada Y, Sumimoto H, Takeya R, Interaction between cardiac myosin-binding protein C and formin Fhod3, Proc Nat Acad Sci USA 115(19) (2018) E4386–e4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Taniguchi K, Takeya R, Suetsugu S, Kan OM, Narusawa M, Shiose A, Tominaga R, Sumimoto H, Mammalian formin fhod3 regulates actin assembly and sarcomere organization in striated muscles, J Biol Chem 284(43) (2009) 29873–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hartzell HC, Glass DB, Phosphorylation of C-protein in intact amphibian cardiac muscle. Correlation between 32P incorporation and twitch relaxation, J Gen Physiol 83(4) (1984) 563–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gautel M, Zuffardi O, Freiburg A, Labeit S, Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction?, EMBO J 14(9) (1995) 1952–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Jia W, Shaffer JF, Harris SP, Leary JA, Identification of novel protein kinase A phosphorylation sites in the M-domain of human and murine cardiac myosin binding protein-C using mass spectrometry analysis, J Proteome Res 9(4) (2010) 1843–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sadayappan S, Gulick J, Osinska H, Barefield D, Cuello F, Avkiran M, Lasko VM, Lorenz JN, Maillet M, Martin JL, Brown JH, Bers DM, Molkentin JD, James J, Robbins J, A critical function for Ser-282 in cardiac Myosin binding protein-C phosphorylation and cardiac function, Circ Res 109(2) (2011) 141–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kuster DW, Sequeira V, Najafi A, Boontje NM, Wijnker PJ, Witjas-Paalberends ER, Marston SB, Dos Remedios CG, Carrier L, Demmers JA, Redwood C, Sadayappan S, van der Velden J, GSK3beta phosphorylates newly identified site in the proline-alanine-rich region of cardiac myosin-binding protein C and alters cross-bridge cycling kinetics in human: short communication, Circ Res 112(4) (2013) 633–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].El-Armouche A, Pohlmann L, Schlossarek S, Starbatty J, Yeh Y-H, Nattel S, Dobrev D, Eschenhagen T, Carrier L, Decreased phosphorylation levels of cardiac myosin-binding protein-C in human and experimental heart failure, J Mol Cell Cardiol 43(2) (2007) 223–229. [DOI] [PubMed] [Google Scholar]
- [58].Sadayappan S, Gulick J, Osinska H, Martin LA, Hahn HS, Dorn GW, 2nd, R. Klevitsky, C.E. Seidman, J.G. Seidman, J. Robbins, Cardiac myosin-binding protein-C phosphorylation and cardiac function, Circ Res 97(11) (2005) 1156–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].van Dijk SJ, Dooijes D, dos Remedios C, Michels M, Lamers JMJ, Winegrad S, Schlossarek S, Carrier L, ten Cate FJ, Stienen GJM, van der Velden J, Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction, Circulation 119(11) (2009) 1473–1483. [DOI] [PubMed] [Google Scholar]
- [60].Sadayappan S, Osinska H, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, Seidman CE, Seidman JG, Robbins J, Cardiac myosin binding protein C phosphorylation is cardioprotective, Proc Nat Acad Sci USA 103(45) (2006) 16918–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Decker RS, Decker ML, Kulikovskaya I, Nakamura S, Lee DC, Harris K, Klocke FJ, Winegrad S, Myosin-binding protein C phosphorylation, myofibril structure, and contractile function during low-flow ischemia, Circulation 111(7) (2005) 906–12. [DOI] [PubMed] [Google Scholar]
- [62].Colson BA, Thompson AR, Espinoza-Fonseca LM, Thomas DD, Site-directed spectroscopy of cardiac myosin-binding protein C reveals effects of phosphorylation on protein structural dynamics, Proc Nat Acad Sci USA 113(12) (2016) 3233–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Previs MJ, Mun JY, Michalek AJ, Previs SB, Gulick J, Robbins J, Warshaw DM, Craig R, Phosphorylation and calcium antagonistically tune myosin-binding protein C’s structure and function, Proc Nat Acad Sci USA 113(12) (2016) 3239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Govindan S, Kuster DW, Lin B, Kahn DJ, Jeske WP, Walenga JM, Leya F, Hoppensteadt D, Fareed J, Sadayappan S, Increase in cardiac myosin binding protein-C plasma levels is a sensitive and cardiac-specific biomarker of myocardial infarction, Am J cardiovasc Dis 3(2) (2013) 60–70. [PMC free article] [PubMed] [Google Scholar]
- [65].Govindan S, McElligott A, Muthusamy S, Nair N, Barefield D, Martin JL, Gongora E, Greis KD, Luther PK, Winegrad S, Henderson KK, Sadayappan S, Cardiac myosin binding protein- C is a potential diagnostic biomarker for myocardial infarction, J Mol Cell Cardiol 52(1) (2012) 154–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Stelzer JE, Patel JR, Moss RL, Protein kinase A-mediated acceleration of the stretch activation response in murine skinned myocardium is eliminated by ablation of cMyBP-C, Circ Res 99(8) (2006) 884–90. [DOI] [PubMed] [Google Scholar]
- [67].Tong CW, Stelzer JE, Greaser ML, Powers PA, Moss RL, Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function, Circ Res 103(9) (2008) 974–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Colson BA, Locher MR, Bekyarova T, Patel JR, Fitzsimons DP, Irving TC, Moss RL, Differential roles of regulatory light chain and myosin binding protein-C phosphorylations in the modulation of cardiac force development, J Physiol 588(6) (2010) 981–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Colson BA, Patel JR, Chen PP, Bekyarova T, Abdalla MI, Tong CW, Fitzsimons DP, Irving TC, Moss RL, Myosin binding protein-C phosphorylation is the principal mediator of protein kinase A effects on thick filament structure in myocardium, J Moll Cell Cardiol 53(5) 609–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Kumar M, Govindan S, Zhang M, Khairallah RJ, Martin JL, Sadayappan S, de Tombe PP, Cardiac Myosin-binding Protein C and Troponin-I Phosphorylation Independently Modulate Myofilament Length-dependent Activation, J Biol Chem 290(49) (2015) 29241–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Mamidi R, Gresham KS, Verma S, Stelzer JE, Cardiac Myosin Binding Protein-C Phosphorylation Modulates Myofilament Length-Dependent Activation, Front Physiol 7 (2016) 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Kampourakis T, Ponnam S, Sun YB, Sevrieva I, Irving M, Structural and functional effects of myosin binding protein-C phosphorylation in heart muscle are not mimicked by serine-to- aspartate substitutions, J Biol Chem (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Ackermann MA, Kerr JP, King B, W.W. C, A. Kontrogianni-Konstantopoulos, The Phosphorylation Profile of Myosin Binding Protein-C Slow is Dynamically Regulated in Slow- Twitch Muscles in Health and Disease, Sci Rep 5 (2015) 12637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Ackermann MA, Ward CW, Gurnett C, Kontrogianni-Konstantopoulos A, Myosin Binding Protein-C Slow Phosphorylation is Altered in Duchenne Dystrophy and Arthrogryposis Myopathy in Fast-Twitch Skeletal Muscles, Sci Rep 5 (2015) 13235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Hall JG, Reed SD, Greene G, The distal arthrogryposes: delineation of new entities-- review and nosologic discussion, Am J Med Genet 11(2) (1982) 185–239. [DOI] [PubMed] [Google Scholar]
- [76].Ma L, Yu X, Arthrogryposis multiplex congenita: classification, diagnosis, perioperative care, and anesthesia, Front Med 11(1) (2017) 48–52. [DOI] [PubMed] [Google Scholar]
- [77].Darin N, Kimber E, Kroksmark AK, Tulinius M, Multiple congenital contractures: birth prevalence, etiology, and outcome, J Pediatr 140(1) (2002) 61–7. [DOI] [PubMed] [Google Scholar]
- [78].Toydemir RM, Rutherford A, Whitby FG, Jorde LB, Carey JC, Bamshad MJ, Mutations in embryonic myosin heavy chain (MYH3) cause Freeman-Sheldon syndrome and Sheldon-Hall syndrome, Nature Genet 38(5) (2006) 561–5. [DOI] [PubMed] [Google Scholar]
- [79].Veugelers M, Bressan M, McDermott DA, Weremowicz S, Morton CC, Mabry CC, Lefaivre JF, Zunamon A, Destree A, Chaudron JM, Basson CT, Mutation of perinatal myosin heavy chain associated with a Carney complex variant, N Engl J Med 351(5) (2004) 460–9. [DOI] [PubMed] [Google Scholar]
- [80].Shrimpton AE, Hoo JJ, A TNNI2 mutation in a family with distal arthrogryposis type 2B, Eur J Med Genet 49(2) (2006) 201–6. [DOI] [PubMed] [Google Scholar]
- [81].Ochala J, Thin filament proteins mutations associated with skeletal myopathies: defective regulation of muscle contraction, J Mol Med 86(11) (2008) 1197–204. [DOI] [PubMed] [Google Scholar]
- [82].Sung SS, Brassington AM, Grannatt K, Rutherford A, Whitby FG, Krakowiak PA, Jorde LB, Carey JC, Bamshad M, Mutations in genes encoding fast-twitch contractile proteins cause distal arthrogryposis syndromes, Am J Hu Genet 72(3) (2003) 681–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Zhao N, Jiang M, Han W, Bian C, Li X, Huang F, Kong Q, Li J, A novel mutation in TNNT3 associated with Sheldon-Hall syndrome in a Chinese family with vertical talus, Eur J Med Genet 54(3) (2011) 351–3. [DOI] [PubMed] [Google Scholar]
- [84].Tajsharghi H, Kimber E, Holmgren D, Tulinius M, Oldfors A, Distal arthrogryposis and muscle weakness associated with a beta-tropomyosin mutation, Neurology 68(10) (2007) 772–5. [DOI] [PubMed] [Google Scholar]
- [85].Geist J, Kontrogianni-Konstantopoulos A, MYBPC1, an Emerging Myopathic Gene: What We Know and What We Need to Learn, Front Physiol 7 (2016) 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Hall JG, Genetic aspects of arthrogryposis, Clin Orthop Relat Res (194) (1985) 44–53. [PubMed] [Google Scholar]
- [87].Gurnett CA, Desruisseau DM, McCall K, Choi R, Meyer ZI, Talerico M, Miller SE, Ju JS, Pestronk A, Connolly AM, Druley TE, Weihl CC, Dobbs MB, Myosin binding protein C1: a novel gene for autosomal dominant distal arthrogryposis type 1, Hum Mol Genet 19(7) (2010) 1165–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Marston S, Copeland O, Gehmlich K, Schlossarek S, Carrier L, How do MYBPC3 mutations cause hypertrophic cardiomyopathy?, J Muscle Res Cell Motil 33(1) (2012) 75–80. [DOI] [PubMed] [Google Scholar]
- [89].Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, Jalilzadeh S, Carballo S, Redwood C, Watkins H, Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency, Circ Res 105(3) (2009) 219–22. [DOI] [PubMed] [Google Scholar]
- [90].Ackermann MA, Patel PD, Valenti J, Takagi Y, Homsher E, Sellers JR, Kontrogianni- Konstantopoulos A, Loss of actomyosin regulation in distal arthrogryposis myopathy due to mutant myosin binding protein-C slow, FASEB J 27(8) (2013) 3217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Li X, Zhong B, Han W, Zhao N, Liu W, Sui Y, Wang Y, Lu Y, Wang H, Li J, Jiang M, Two novel mutations in myosin binding protein C slow causing distal arthrogryposis type 2 in two large Han Chinese families may suggest important functional role of immunoglobulin domain C2, PLoS ONE 10(2) (2015) e0117158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Bamshad M, Van Heest AE, Pleasure D, Arthrogryposis: A Review and Update, J Bone Joint Surg Am. American volume. 91(Suppl 4) (2009) 40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Markus B, Narkis G, Landau D, Birk RZ, Cohen I, Birk OS, Autosomal recessive lethal congenital contractural syndrome type 4 (LCCS4) caused by a mutation in MYBPC1, Hum Mutat 33(10) (2012) 1435–8. [DOI] [PubMed] [Google Scholar]
- [94].Ekhilevitch N, Kurolap A, Oz-Levi D, Mory A, Hershkovitz T, Ast G, Mandel H, Baris HN, Expanding the MYBPC1 phenotypic spectrum: a novel homozygous mutation causes arthrogryposis multiplex congenita, Clin Genet 90(1) (2016) 84–9. [DOI] [PubMed] [Google Scholar]
- [95].Barefield D, Sadayappan S, Phosphorylation and function of cardiac myosin binding protein-C in health and disease., J Mol Cell Cardiol 48(5) (2010) 866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Geist J, Stavusis J, Lace B, Wright N, Bonneman C, Ward C, Kontrogianni- Konstantopoulos A, Myosin Binding Protein-C Slow (sMyBP-C) Function, Regulation, and Disease Implications, Myofilament Meeting (2018). [Google Scholar]
- [97].Schäfer J, Saak A, Reichmann H, Jackson S, Kongenitale Myopathie ohne distale Arthrogrypose in einer Familie mit einer neuen Mutation im MYBPC1-Gen, Neurowoche 2018. (2018). [Google Scholar]
- [98].Tong B, Sasaki S, Muramatsu Y, Ohta T, Kose H, Yamashiro H, Fujita T, Yamada T, Association of a single-nucleotide polymorphism in myosin-binding protein C, slow-type (MYBPC1) gene with marbling in Japanese Black beef cattle, Anim Genet 45(4) (2014) 611–2. [DOI] [PubMed] [Google Scholar]
- [99].Tong B, Xing YP, Muramatsu Y, Ohta T, Kose H, Zhou HM, Yamada T, Association of expression levels in skeletal muscle and a SNP in the MYBPC1 gene with growth-related trait in Japanese Black beef cattle, J Genet 94(1) (2015) 135–7. [DOI] [PubMed] [Google Scholar]
- [100].Wiedemar N, Riedi AK, Jagannathan V, Drogemuller C, Meylan M, Genetic Abnormalities in a Calf with Congenital Increased Muscular Tonus, J Vet Intern Med 29(5) (2015) 1418–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Bayram Y, Karaca E, Coban Akdemir Z, Yilmaz EO, Tayfun GA, Aydin H, Torun D, Bozdogan ST, Gezdirici A, Isikay S, Atik MM, Gambin T, Harel T, El-Hattab AW, Charng WL, Pehlivan D, Jhangiani SN, Muzny DM, Karaman A, Celik T, Yuregir OO, Yildirim T, Bayhan IA, Boerwinkle E, Gibbs RA, Elcioglu N, Tuysuz B, Lupski JR, Molecular etiology of arthrogryposis in multiple families of mostly Turkish origin, J Clin Invest 126(2) (2016) 762–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Langenhan T, Aust G, Hamann J, Sticky signaling--adhesion class G protein-coupled receptors take the stage, Sci Signal 6(276) (2013) re3. [DOI] [PubMed] [Google Scholar]
- [103].Ravenscroft G, Nolent F, Rajagopalan S, Meireles AM, Paavola KJ, Gaillard D, Alanio E, Buckland M, Arbuckle S, Krivanek M, Maluenda J, Pannell S, Gooding R, Ong RW, Allcock RJ, Carvalho ED, Carvalho MD, Kok F, Talbot WS, Melki J, Laing NG, Mutations of GPR126 are responsible for severe arthrogryposis multiplex congenita, Am J Hum Genet 96(6) (2015) 955–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Ha K, Buchan JG, Alvarado DM, McCall K, Vydyanath A, Luther PK, Goldsmith MI, Dobbs MB, Gurnett CA, MYBPC1 mutations impair skeletal muscle function in zebrafish models of arthrogryposis, Hum Mol Genet 22(24) (2013) 4967–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Li M, Andersson-Lendahl M, Sejersen T, Arner A, Knockdown of fast skeletal myosin- binding protein C in zebrafish results in a severe skeletal myopathy, J Gen Physiol 147(4) (2016) 309–22. [DOI] [PMC free article] [PubMed] [Google Scholar]