

Abstract
Mice depleted of hepatic stellate cells (HSCs) are protected from concanavalin A (ConA)-induced liver injury that is mediated by the activation of interferon regulatory factor 1 (IRF1). The aim of this study was to determine the mechanisms of ConA-mediated signaling and synthesis/release of mediators by HSCs that damage hepatocytes. Primary cultures of wildtype (WT) and IRF1-knockout (KO) HSCs and hepatocytes were used, and ConA-induced liver damage in interferon (IFN)αβ receptor-deficient (IFNαβR-KO) mice was determined. Specific binding of ConA to HSCs induced rapid activation of JAK2 and STAT1. ConA-induced expression of IRF1, IFNβ, tumor necrosis factor α, and CXCL1 was abrogated by selective inhibition of JAK2 and STAT1. Despite activating JAK2/STAT1, ConA failed to stimulate expression of inflammatory cytokines in HSCs from IRF1-KO mice. ConA-conditioned WT-HSC medium caused activation of JNK and caspase 3, and apoptosis of hepatocytes from WT but not from IRF1-KO or IFNαβR-KO mice. Conversely, ConA-conditioned medium of IRF1-KO HSCs failed to cause apoptosis of WT or IRF1-KO hepatocytes. IFNαβR-KO mice were protected from ConA-induced liver damage, and ConA-induced hepatic expression of IRF1 and pro-inflammatory cytokines and chemokines, and infiltration of neutrophils were significantly lower in IFNαβR-KO than in WT mice. These results demonstrate distinct roles of IRF1 in hepatic inflammation (HSCs) and injury (hepatocytes) and can be an important target for intervention in acute liver injury.
Keywords: liver injury, interferon, interferon regulatory factor (IRF), STAT transcription factor, hepatocyte, hepatic stellate cell (HSC), Janus kinase (JAK)
Introduction
The retinoid-storing hepatic stellate cells (HSCs),2 located in the perisinusoidal space, form up to 10% of the liver cell population and are physiologically quiescent. During liver injury, HSCs become activated, transdifferentiating into proliferating and contractile myofibroblast-like phenotypes. As a major source of excessive and abnormal extracellular matrix deposition during chronic liver injury, activated HSCs have been investigated extensively as a target for antifibrotic therapies (1–3). HSCs also regulate hepatic inflammation and immunological environment by producing an array of cytokines, chemokines, and growth mediators, by recruiting inflammatory and immune cells, and by influencing the characteristics of these and Kupffer cells (resident macrophages) (4–6). Thus, HSCs play a major role in hepatic inflammation and acute liver injury. Indeed, HSC-depleted mice were found to be protected from ischemia/reperfusion- and endotoxin-induced acute liver damage (7).
Concanavalin A (ConA; a mannose/glucose-binding plant lectin)-induced liver injury in mice is a widely used experimental model to understand mechanisms of immune cell-mediated acute hepatitis (8, 9). Previous studies implicated Kupffer cells (10–12), sinusoidal endothelial cells (13), NKT and CD4+ T cells (14, 15) in ConA-induced liver injury. Recently, we found that HSC-depleted mice are protected from ConA-induced liver injury (16). HSC depletion significantly reduced the infiltration of CD4+ T cells and neutrophils in ConA-challenged mice; in vitro, ConA-stimulated HSCs produced cytokines that induced apoptosis of hepatocytes. However, precise mechanisms of ConA-induced inflammatory response and coupled signaling pathway(s) in HSCs are unknown. In this investigation, we found that ConA stimulates JAK2/STAT1 signaling in HSCs that is coupled to increased nuclear translocation of IRF1 and expression of cytokines and chemokines including IFNβ. This effect was strongly mitigated in HSCs from IRF1-knockout (IRF1-KO) mice, and hepatocytes from IRF1-KO or IFNαβR-KO mice were protected from WT-HSC/ConA-induced apoptosis. The data demonstrate that IRF1, by stimulating JAK/STAT-mediated synthesis of IFNβ in HSCs and by causing IFNβ-induced hepatocyte injury, plays distinct roles in ConA-induced liver damage.
Results
ConA-induced intracellular signaling and synthesis of cytokines in HSCs
Fig. 1A shows the purity of HSCs stained for desmin and GFAP (glial fibrillary acidic protein). Binding of FITC-conjugated ConA (FITC-ConA) to the HSC membrane was followed by its internalization and distribution throughout the cytoplasm (Fig. 1B). ConA did not alter the expression of mannose 6-phosphate receptor (insulin-like growth factor 2 receptor; IGF2R) to which it binds (Fig. 1C). The specificity of ConA binding to HSCs was determined in incubations containing FITC-ConA and methyl-α-d-mannopyranoside (Me-αMan) or d-mannose. Me-αMan and d-mannose both markedly inhibited the binding of ConA, Me-αMan being more potent on a molar basis; excess unlabeled ConA strongly prevented the binding of FITC-ConA (Fig. 1D).
Figure 1.
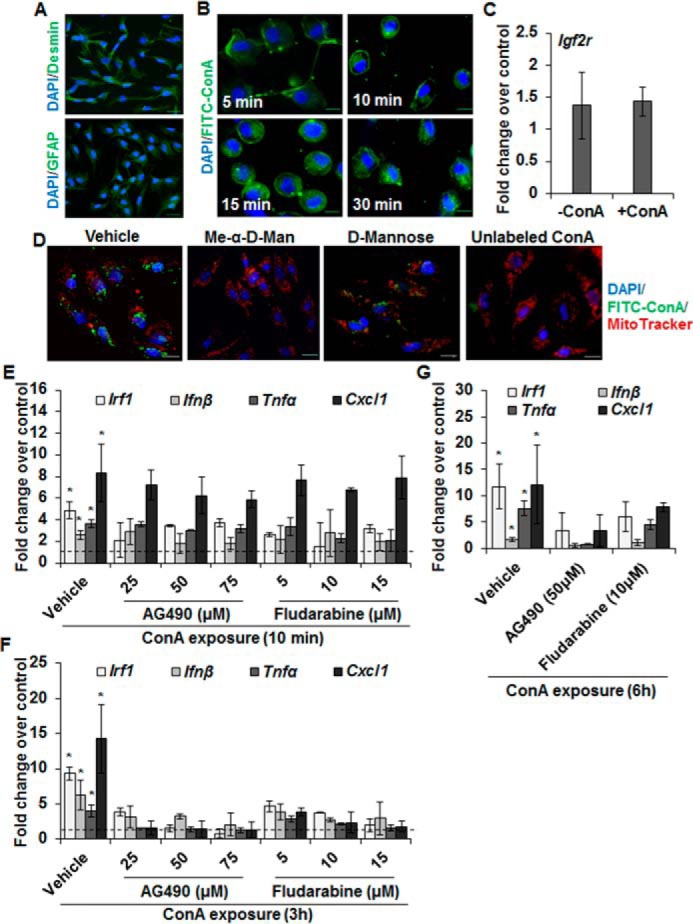
Characteristics of ConA binding and ConA-induced expression of cytokines in HSCs. A, purity of HSCs is demonstrated by staining for desmin or GFAP. B, immunofluorescence images of HSCs (day 2 of culture) incubated in medium containing FITC-ConA (12.5 μg/ml) for the indicated time points. Scale bars, 20 μm. C, mRNA expression of IGF2R in HSCs incubated in the presence of vehicle or ConA (50 μg/ml) for 6 h. D, HSCs were incubated in medium containing FITC-ConA (12.5 μg/ml) in the presence of 100 mm α-methyl-d-mannopyranoside or d-mannose or 500 μg/ml of ConA. Mitochondria and nuclei were stained with MitoTracker Red (100 nm) and DAPI (blue), respectively. Scale bars, 20 μm. E and F, HSCs from WT mice were preincubated in the presence of DMSO-vehicle or the indicated concentrations of JAK2 (AG490) or STAT1 (Fludarabine) inhibitors for 30 min, and then stimulated with ConA (50 μg/ml) for 10 min (E) or 3 h (F). G, HSCs from WT mice were preincubated in the presence of DMSO-vehicle, AG490 (50 μm), or Fludarabine (10 μm) for 30 min, then stimulated with ConA (50 μg/ml) for 6 h. Expression of the indicated mRNAs (E–G) was measured by qRT-PCR. The data are representative of three separate experiments, each performed in duplicate or triplicate. *, p < 0.05.
We reported that ConA increases expression of the transcription factor IRF1, inflammatory cytokines TNFα and IFNβ, and the neutrophil/monocyte chemoattractant CXCL1 in HSCs (16). To identify ConA-stimulated signaling pathway(s) coupled to the expression of these factors, HSCs were preincubated with inhibitors of various pathways and stimulated with ConA. ConA-induced mRNA expression of IRF1, TNFα, IFNβ, and CXCL1 in HSCs was remarkably abrogated by JAK2 (AG490) and STAT1 (fludarabine) inhibitors (Fig. 1, E–G) in a concentration- and time-dependent manner. Inhibitors of PI3K (LY294002), SRC kinase (PP2), p38-MAPK (SB202190), ERK1/2-MAPK (PD98059), JNK-MAPK (SP600125), AKT/PKB (GSK690693), AP1 (SR11302), and NF-κB (SN50 or BMS-345541) did not inhibit ConA-induced cytokine expression in HSCs (not shown). To ascertain that the effect of ConA is not restricted to mouse HSCs, we examined rat HSCs and found that ConA increased mRNA expression of IRF1, IFNβ, TNFα, and CXCL1 (Fig. S1A); the effect on mRNA expression was corroborated by increased accumulation of IFNβ and TNFα in the culture medium from ConA-stimulated HSCs (Fig. S1, B and C). Furthermore, AG490 and fludarabine prevented ConA-induced expression of IRF1, IFNβ, TNFα, and CXCL1 in rat HSCs (Fig. S1D).
Next, we examined the activation of JAK/STAT by ConA in WT-HSCs. Time course experiments showed rapid activation (phosphorylation) of JAK2 that peaked at 5 min and declined at 15 min but still was significantly higher than the basal level (Fig. 2A). STAT1 activation and nuclear translocation followed the same time course but remained increased at 15 min, and ConA-induced nuclear expression of IRF1 also showed a similar pattern as for STAT1. ConA-induced JAK2 and STAT1 activation was inhibited by AG490, whereas fludarabine inhibited ConA-induced STAT1 but not JAK2 activation (Fig. 2B). ConA did not induce JNK1/2 or caspase-3 activation suggesting that the viability of HSCs is not affected (Fig. 2C).
Figure 2.

ConA-induced intracellular signaling in HSCs. A, Western blotting shows expression of P-JAK2, P-STAT1, and IRF1 in cytosolic or nuclear protein lysates prepared from ConA (50 μg/ml)-stimulated WT-HSCs for the indicated time points. B, Western blotting shows expression of P-JAK2, P-STAT1, and IRF1 in total, cytosolic, or nuclear protein lysates prepared from WT-HSCs stimulated with ConA (50 μg/ml) for 10 min following 30 min preincubation with DMSO-vehicle, JAK2 (AG490; 50 μm), or STAT1 (Fludarabine; 10 μm) inhibitors. C, Western blots show expression of P-JNK1/2 and cleaved caspase 3 in HSCs stimulated with ConA for the indicated time points. Bar graphs (A–C) show relative densitometry values normalized to the respective internal controls. All data are representative of three independent experiments. Bar graphs show relative density of the signaling proteins normalized to the respective internal controls. *, p < 0.05.
IRF1 mediates ConA-induced cytokine expression in HSCs
Because ConA increased nuclear protein expression of IRF1 in HSCs, we determined its role in cytokine production using HSCs from IRF1-KO mice. The binding of FITC-ConA to IRF1-KO HSCs (Fig. S2A) was similar to WT-HSCs (Fig. 1B). We confirmed ConA-induced nuclear translocation of IRF1 in WT-HSCs (determined by Western blot analysis (Fig. 2, A and B)) via immunostaining (Fig. 3A). Next, we determined the expression of IRF1, IFNβ, TNFα, and CXCL1 in ConA-stimulated HSCs from IRF1-KO mice. ConA failed to stimulate IFNβ, TNFα, and CXCL1 expression in IRF1-KO HSCs (Fig. 3B) at all time points examined. Immunohistochemical analysis confirmed the failure of ConA to stimulate IFNβ synthesis in IRF1-KO HSCs unlike in WT-HSCs (Fig. S2B). However, ConA caused activation of JAK2 and STAT1 in IRF1-KO HSCs (Fig. 3C). Together, these data indicate that ConA-mediated JAK2/STAT1 activation and subsequent nuclear translocation of IRF1 is a major signaling mechanism of the synthesis of inflammatory cytokines/chemokines in HSCs.
Figure 3.
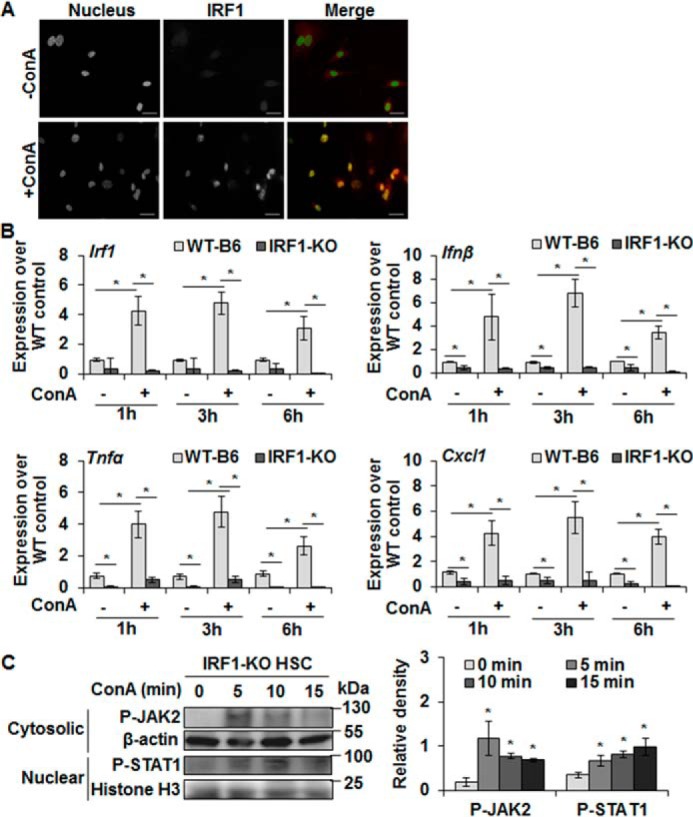
ConA does not induce expression of cytokines in IRF1-KO HSCs despite stimulating JAK/STAT signaling. A, HSCs from WT mice were stimulated with 50 μg/ml of ConA for 6 h and localization of IRF-1 (red) was determined by immunostaining with anti-IRF1 Ab. Nuclear IRF1 is strongly increased in ConA-stimulated HSCs. Nuclei are stained with DAPI (blue). Scale bars, 20 μm. B, mRNA expression of the indicated mediators in HSCs from WT or IRF1-KO mice stimulated with 50 μg/ml of ConA for 1, 3, or 6 h as measured via qRT-PCR. C, Western blotting shows P-JAK2 (cytosolic) and P-STAT1 (nuclear) expression in protein lysates prepared from ConA-stimulated IRF1-KO HSCs for the indicated time points. Bar graph shows the relative densitometry values normalized to respective internal controls. All data are representative of three independent experiments. *, p < 0.05.
IFNαβR-KO mice are protected from ConA-induced liver damage
Both type I and type II interferons are known to cause hepatocyte injury via IRF1 (16, 17). Although IFNγ has been implicated in ConA-induced liver injury (8, 15), HSCs do not produce IFNγ (18, 19). Therefore, we challenged IFNαβR-KO mice with ConA to ascertain the role of type I IFNs in ConA/HSC-mediated liver injury. Both WT-B6 and WT-BALB/c mice showed strong hepatic injury after ConA administration as determined by histopathology (Fig. 4A) and serum ALT levels (Fig. 4B), but IFNαβR-KO mice were protected (also see Fig. S3). Note that the injury to WT-BALB/c mice was of lower magnitude than to B6-WT mice, which is consistent with previously reported strain-specific sensitivity (20). Neutrophil infiltration in ConA-treated IFNαβR-KO mice was also lower than in WT mice (Fig. S4, A and B). Furthermore, the size of spleen increased in ConA-challenged WT-B6 and WT-BALB/c mice (indicating hepatic congestion), but not in IFNαβR-KO mice (Fig. S4C). Because nuclear translocation of IRF1 is an important component of ConA-induced hepatocyte damage, we examined its nuclear expression following ConA administration and found much lower translocation in IFNαβR-KO compared with the WT-B6 or WT-BALB/c livers (Fig. 4C). In this regard, it was interesting that ConA-induced hepatic expression of IRF1, TNFα, and CXCL1 was much lower in IFNαβR-KO mice than the WT mice (Fig. 4D) at all time points. However, this observation is not surprising as such diminished response due to type I IFN receptor deficiency has been reported (21). No change in the hepatic IFNβ expression in ConA-treated IFNαβR-KO mice at 6 or 8 h can be explained by its characteristic increase at early time points, followed by accelerated decline later compared with the WT mice (Fig. S4D). Such change in IFNβ expression was also observed in LPS-challenged rat HSCs (22).
Figure 4.

ConA-induced hepatic injury in IFNαβR-KO mice. A, mice were euthanized 6 (WT-B6) or 8 h (WT-BALB/c or IFNαβR-KO) after administering 20 mg/kg of ConA. Representative sections of the vehicle- or ConA-treated livers stained with hematoxylin/eosin (H/E) show protection in IFNαβR-KO mice compared with WT controls. Bar graph shows quantification of necrotic area as defined in the H/E-stained liver sections. Scale bars, 100 μm (left and middle panels) and 50 μm (right panels). B, mice were euthanized 1, 3, or 6 h after administering 20 mg/kg of ConA. ConA-treated IFNαβR-KO mice show significantly lower serum ALT levels as compared with the WT mice. Data are expressed as mean ± S.D. n = 4–6 mice/group. C, Western blotting of the nuclear lysates shows IRF1 in WT (B6 and BALB/c) and IFNαβR-KO mice. Bar graph shows the relative densitometry values normalized to TBP as an internal control (n = 4–6 mice/group). D, mRNA expression of the indicated mediators in ConA-challenged mice at 1, 3, or 6 h as measured via qRT-PCR. *, p < 0.05; **, p < 0.005; ***, p < 0.0005.
We next examined nuclear IRF1 in HSCs and hepatocytes of ConA-treated WT-B6, WT-BALB/c, and IFNαβR-KO mice. Immunohistochemical analysis showed a similar number of desmin-positive sinusoidal cells in WT-B6, WT-BALB/c, and IFNαβR-KO mice (Fig. S5). Following ConA challenge, the number of sinusoidal cells as well as hepatocytes positive for nuclear IRF1 increased in WT-B6 and WT-BALB/c mice; very small numbers of sinusoidal and parenchymal cells with IRF1-positive nuclei were observed in ConA-challenged IFNαβR-KO mice (Fig. S5). Although we cannot rule out that both HSCs and Kupffer cells might represent nuclear IRF1-positive cells in ConA-challenged mice, protection of HSC-depleted mice from ConA-induced liver damage (16) argues for HSC's role as the major orchestrator of this pathology. These data together indicate that type I IFNs released in ConA-challenged mice from HSCs play a critical role in hepatocyte injury by increasing nuclear IRF1.
Effects of ConA on IFNαβR-KO HSCs
We examined the effect of ConA directly on IFNαβR-KO HSCs. The binding/internalization of FITC-ConA in IFNαβR-KO HSCs was similar to that in WT-BALB/c HSCs (Fig. S6A). Furthermore, immunohistochemical analysis showed mitigated ConA-induced IFNβ accumulation in ConA-stimulated IFNαβR-KO HSCs compared with the BALB/c HSCs (Fig. S6B).
ConA-stimulated WT-HSCs do not induce oxidative stress, or activate JNK and caspase 3 in hepatocytes from IRF1-KO or IFNαβR-KO mice
To confirm the role of IRF1/IFNβ/IRF1 axis in ConA/HSC-induced hepatocyte damage, we used HSCs isolated from WT or IRF1-KO mice, and hepatocytes from these as well as IFNαβR-KO mice. 2′,7′-Dichlorofluorescin diacetate (DCFDA) analysis showed strong oxidative stress in WT hepatocytes incubated in ConA-conditioned WT-HSC medium, and was accompanied by rounding of hepatocytes, reduction in cell size with nuclear condensation, and loss of cytoskeletal definition with prominent cellular bleb formation (Fig. 5, A and E; Fig. S7A). ConA alone did not produce these effects. In contrast, ConA-conditioned medium from WT or IRF1-KO HSCs induced minimal oxidative stress and morphological changes in IRF1-KO (Fig. 5, B and E; Fig. S7B) and WT hepatocytes (Fig. 5, C and E; Fig. S7C), respectively. Furthermore, ConA-conditioned WT-HSC medium did not induce oxidative stress in IFNαβR-KO hepatocytes (Fig. 5, D and E; Fig. S7D).
Figure 5.
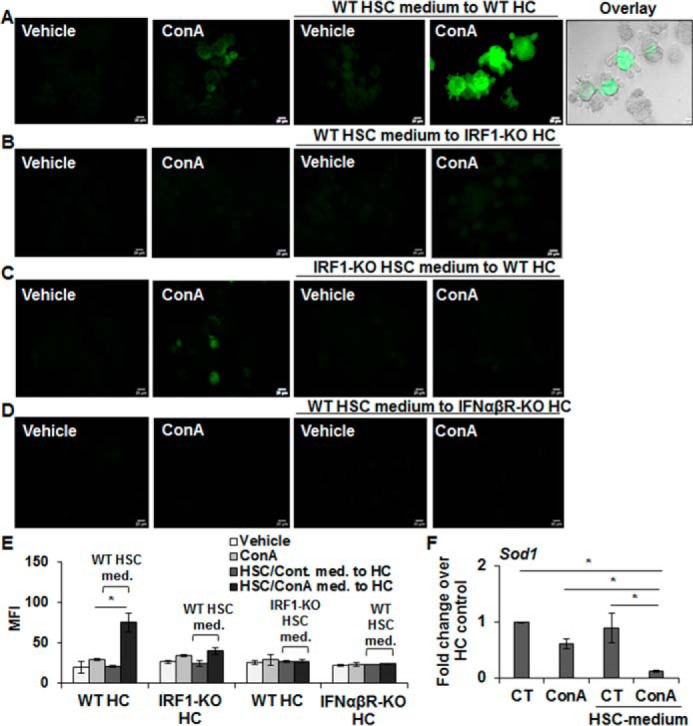
Hepatocytes from IRF1-KO and IFNαβR-KO are resistant to oxidative stress and apoptosis by ConA-stimulated WT-HSCs. A–D, hepatocytes (WT or IRF1-KO or IFNαβR-KO) were incubated in medium without or with 50 μg/ml of ConA or medium conditioned by HSCs (WT or IRF1-KO) for 8 h in the absence or presence of ConA. After 12 h of incubation, DCFDA staining was performed to determine oxidative stress in hepatocytes. An overlay of phase-contrast and DCF image in A shows ROS generation and bleb formation in WT hepatocytes treated with ConA/HSC medium from WT mice. Scale bars, 20 μm. Bar graph (E) shows mean fluorescence intensity (MFI) of combined values from three independent experiments. All images are representative of three separate experiments (see Fig. S7 for phase-contrast images). F, mRNA expression of SOD1 in WT hepatocytes incubated in medium without or with 50 μg/ml of ConA or medium conditioned by WT-HSCs. The data are representative of three sets of experiments. *, p < 0.05.
The basal level of constitutively generated intracellular reactive oxygen species is regulated by superoxide dismutase (SOD) that converts superoxide into hydrogen peroxide. We, therefore, asked whether altered SOD contributes to increased oxidative stress in hepatocytes incubated in ConA-conditioned HSC medium, and found a significant down-regulation of the expression of SOD1 in HSC/ConA-challenged hepatocytes (Fig. 5F).
Medium conditioned by WT-HSCs in the presence of ConA activated JNK and caspase 3, and caused apoptosis of WT hepatocytes (Fig. 6A; Fig. S8). However, this effect was not apparent in IRF1-KO hepatocytes challenged with ConA-conditioned medium from WT-HSCs, and in WT hepatocytes challenged with ConA-conditioned medium from IRF1-KO HSCs (Fig. 6A; Fig. S8). Also, ConA-conditioned WT-HSC medium failed to activate JNK or caspase 3 in hepatocytes isolated from IFNαβR-KO mice (Fig. 6A; Fig. S8). These data suggest a strong relationship between IRF1, in this case primarily induced by HSC-derived IFNβ, and pro-apoptotic signals in hepatocytes as has been shown in ischemia/reperfusion-induced liver injury (23).
Figure 6.

ConA-stimulated WT-HSCs do not stimulate JNK or caspase 3 activation in hepatocytes from IRF1-KO and IFNαβR-KO mice. A, WT, IRF1-KO, or IFNαβR-KO hepatocytes were incubated with ConA or medium conditioned by WT or IRF1-KO HSCs without/with ConA for 12 h. Protein lysates were prepared and subjected to Western blot analysis to detect P-JNK and cleaved caspase-3. The data are representative of three independent experiments. (See Fig. S8 for densitometry values.) B, WT hepatocytes were incubated for 12 h without/with ConA or medium conditioned by WT-HSCs in the absence or presence of ConA ± JNK inhibitor SP600125 (JNKi; 10 μg/ml) or anti-IFNβ Ab (2 μg/ml). SOD1 mRNA expression and SOD activity were determined. C, cytosolic and nuclear lysates prepared from hepatocytes in B were analyzed by Western blotting. Bar graph shows relative densitometry values normalized to the respective internal controls. All data are representative of three independent experiments. *, p < 0.05; **, p < 0.005.
Because activated JNK represses SOD1 expression (24, 25), we tested whether this may be a mechanism of reduced SOD1 expression in hepatocytes incubated in ConA-conditioned HSC medium. As shown in Fig. 6B, inhibitor of JNK activation prevented ConA/HSC-induced repression of SOD1 mRNA expression as well as SOD activity. Furthermore, ConA/HSC-induced repression of SOD1 expression/activity was prevented in incubations containing anti-IFNβ Ab (Fig. 6B), suggesting that IFNβ-induced JNK activation and subsequent repression of SOD activity contributes to the oxidative stress.
ConA is reported to induce hepatic activation of JAK2 and STAT1 via T cell-derived IFNγ (15, 26). Upon incubation of hepatocytes in ConA-conditioned HSC medium, we found increased nuclear expression of IRF1, and activation of JAK2, STAT1, and caspase 3 (Fig. 6C). Although the JNK inhibitor reduced caspase 3 activation, it did not affect nuclear IRF1 expression as well as JAK2 or STAT1 activation. However, the presence of anti-IFNβ Ab mitigated all of these ConA/HSC-induced effects in hepatocytes.
ConA increases IFNγ and reduces SOD1 expression in WT but not in IRF1-KO or IFNαβR-KO mice
Because ConA-induced liver damage in vivo is predominantly necrotic, whereas ConA-stimulated HSCs induce only apoptosis of hepatocytes in vitro (no LDH release indicative of membrane damage (16)), it was apparent that the injury in vivo involves multimediator action on hepatocytes. Increased expression of CXCL1 suggested that the infiltrating inflammatory cells as well as immune cells act in concert with HSCs in causing necrotic injury (e.g. see Fig. S4). We found increased expression of IFNγ in WT-B6 and WT-BALB/c mice but not in IRF1-KO mice, and the increase in this cytokine was significantly blunted in IFNαβR-KO mice following ConA treatment (Fig. 7A). Furthermore, SOD1 expression was down-regulated in the ConA-treated WT mice and but not in IRF1-KO or IFNαβR-KO mice (Fig. 7B). In fact, SOD1 mRNA expression increased in ConA-challenged IRF1-KO mice.
Figure 7.

ConA-induced hepatic expression of IFNγ and SOD1. Mice were euthanized 6 h (WT-B6 or IRF1-KO) or 8 h (WT-BALB/c or IFNαβR-KO) after administering 20 mg/kg of ConA. Hepatic mRNA expression of (A) IFNγ and (B) SOD1 is shown (n = 6–8 mice/group). *, p < 0.05; **, p < 0.005; ***, p < 0.0005.
Discussion
Understanding mechanisms of ConA-induced hepatocyte injury is important as this is an established model to study autoimmune hepatitis and other forms of liver damage involving immune cells (8). Because HSCs recruit and influence characteristics of immune cells (18, 27, 28), we previously investigated ConA-induced liver injury in HSC-depleted mice. Hepatocytes in HSC-depleted mice were found to be protected, and the injury was mediated, in part, by soluble mediators released by ConA-stimulated HSCs (16). The present investigation shows that binding of ConA to HSCs stimulates JAK2/STAT1-IRF1 signaling leading to the release of IFNβ that, in turn, causes apoptosis of hepatocytes by increasing nuclear translocation of IRF1.
ConA was shown to bind to the mannosyl sugar residues of insulin receptor in the liver (29), and binding of ConA to hepatocytes (30), endothelial cells, and Kupffer cells (14) is also reported. Quiescent HSCs express mannose 6-phosphate receptor IGF2R, but at a lower level than the activated HSCs (31). Here we found that FITC-ConA binds to quiescent HSC membrane and is internalized. The specificity is evident from inhibition of the FITC-ConA binding by competitive inhibitors d-mannose and Me-αMan as well as excess unlabeled ConA. Because hepatocytes do not internalize ConA (32), it is likely that HSCs may be an important site of its hepatic clearance.
Using specific inhibitors of several intracellular signaling pathways, we identified activation of JAK2 and STAT1 to be responsible for ConA-induced increased expression of IRF1, IFNβ, TNFα, and CXCL1 in mouse and rat HSCs. The rapid activation of JAK2 and STAT1, and inhibition of both by AG490 but only of STAT1 by fludarabine confirmed that STAT1 activation is stimulated by JAK2. Inhibition both by AG490 and fludarabine of ConA-induced expression and nuclear translocation of IRF1 indicated its requirement for the final response (cytokine production). Failure of ConA to induce the expression of IFNβ, TNFα, and CXCL1 in HSCs isolated from IRF1-KO mice, despite stimulating JAK2/STAT1, confirmed the essential role of IRF1 in cytokine/chemokine production by HSCs. In this regard, IRF1 has been shown to be a transcriptional regulator of TNFα (33) and CXCL1 (34). Previously, we have shown that LPS-stimulated cytokine/chemokine synthesis in HSCs is mediated by NF-κB (18, 35). However, NF-κB inhibition did not inhibit ConA-induced cytokine production by HSCs suggesting that the JAK2/STAT1/IRF1 pathway is an important mechanism of ConA-induced expression of inflammatory mediators in HSCs.
In contrast to the role of nuclear IRF1 in ConA-induced cytokine/chemokine synthesis in HSCs, nuclear translocation of IRF1 in hepatocytes led to their apoptosis. This effect is primarily mediated by type I IFNs as HSCs do not express IFNγ (18, 19). Although we found that ConA increased the expression of IFNβ (Fig. 1, E–G) as well as IFNα (not shown), the effect on hepatocytes appears to be mediated by IFNβ because anti-IFNβ antibody abrogated ConA/HSC-induced apoptosis of hepatocytes (16), and both mouse and rat hepatocytes respond very poorly to IFNα (36). Thus, the inability of ConA-conditioned WT-HSC medium to stimulate JNK and caspase 3 activation increase oxidative stress and induce apoptosis of IRF1-KO and IFNαβR-KO hepatocytes confirmed that stimulation of IFNαβR by ConA/HSC-derived IFNβ is required for these IRF1-mediated events. WT-BALB/c mice were also found to show less injury than B6 mice despite increased hepatic IFNγ and TNFα mRNA expression and plasma protein levels (20). Abrogation of ConA-induced liver injury both by anti-TNFα and anti-IFNγ Abs suggested that these cytokines use a common signaling pathway (20, 37, 38). Furthermore, IFNγ-KO mice were found to be protected from ConA-induced liver injury, and activation of CD4+ T cells and NKT cells by IFNγ was suggested to be critical in ConA-induced hepatitis (15). Our earlier work showed significantly lower influx of CD4+ T cells in ConA-challenged HSC-depleted mice and the extent of NKT cells did not change (16). Indeed, we have shown that HSCs play a critical role in recruitment of and cross-communication with circulating and resident immune cells (6, 18, 28, 39). Because, TNFα and type I and type II IFNs increase nuclear IRF1 (40, 41), based on our data, it can be concluded that IRF1 is that common pathway of liver cell injury. Our data showing protection of IRF1-KO and IFNαβR-KO mice from ConA-induced damage in vivo, and of IRF1-KO or IFNαβR-KO hepatocytes from apoptosis upon incubation in medium conditioned by ConA-stimulated WT-HSCs, support this contention.
It is reported that type I IFNs cause injury to hepatocytes by inhibiting SOD1, which is predominantly cytosolic, and SOD1-KO mice demonstrate exaggerated injury upon viral infection (42). Our findings suggest that ConA/HSC-induced inhibition of SOD1 in hepatocytes may be an important reason for increased oxidative stress. Whether increased nuclear IRF1 in ConA/HSC-challenged hepatocytes regulates SOD1, remains to be determined, but LPS-induced IRF1 has been implicated in SOD consumption and increased mitochondrial damage in macrophages (43). Consistent with the role of activated JNK in repressing SOD1 gene transcription/activity (24, 25), we found improved SOD activity in hepatocytes pretreated with JNK inhibitor or anti-IFNβ Ab. Because IFNβ stimulates nuclear IRF1 translocation, and IRF1-KO and IFNαβR-KO hepatocytes do not show JNK activation or oxidative stress, there appears to be a causal relationship between IRF1 and oxidative stress as well as JNK activation. We also found similar changes in SOD2 expression (as for SOD1) in ConA-challenged WT, IRF1-KO, and IFNαβR-KO mice (not shown), further supporting the critical role of IRF1 in regulating mitochondrial function and oxidative stress-induced hepatocyte injury. Indeed, SOD2 repression was observed upon mitochondrial JNK activation (44).
Reduced ConA-induced activation of CD4+ T cells and NKT cells, and amelioration of hepatitis in STAT1-KO mice was reported to be accompanied by much lower hepatic IRF1 expression as compared with the WT mice (15). Inability of ConA to increase P-STAT1 and IRF1 expression in IFNγ-KO mice suggested that IFNγ released by ConA-stimulated immune cells activated JAK2/STAT1 and subsequent IRF1-mediated apoptosis. In agreement with this report, STAT1-overexpressing mice demonstrated greater liver injury upon ConA administration, and anti-CD3/CD28-induced IFNγ production by splenic CD4+ T cells from STAT1-overexpressing mice was greater than the WT mice (26). Mizuhara et al. (20) found that B6- and BALB/c-splenic CD4+ T cells respond similarly to ConA in producing IFNγ and TNFα, but the hepatic expression of IFNγ in ConA-challenged BALB/c mice was lower than in B6 mice indicating that intrahepatic IFNγ is important in eliciting liver damage. Because HSCs do not produce IFNγ (18, 19, 28), it is apparent that IFNβ released by ConA-stimulated HSCs increase expression of IRF1 and its nuclear translocation in hepatocytes. We have previously shown that HSCs have an impressive ability to not only attract immune cells (CD4+ T cells, CD4+CD25+FoxP3+ regulatory (Treg) cells, and dendritic cells) but also modulate their characteristics skewing toward liver's immunotolerance, and the bidirectional interactions between co-cultured HSCs and immune cells influence the net synthesis of cytokines and chemokines (6, 18, 28). Because HSC-depleted mice are protected from ConA-induced injury (16), it is evident that HSCs play an important role in IFNγ production by CD4+ and NKT cells. Thus, in summary, our results demonstrate that JAK2/STAT1/IRF1 signaling plays distinct roles in HSCs (i.e. synthesis of type 1 IFNs) and hepatocytes (apoptosis by IFNβ). Such convergence of signaling to IRF1 in the two neighboring cell types (HSCs and hepatocytes) that eventually results in liver injury (summarized in Fig. 8), argues for stimulation of this pathway by both type I and type II IFNs to be a potential target to arrest/prevent liver damage.
Figure 8.

Schematic illustration of mechanism of ConA-induced liver injury. ConA binding to the mannose 6-phosphate receptor (Man-R) on HSCs induces JAK2 phosphorylation, which then phosphorylates STAT-1. The activated STAT1 translocates into the nucleus and induces IRF1 transcription. IRF1 produced this way stimulates IFNβ transcription. IFNβ protein released by HSCs binds to the IFNαβ receptor on hepatocytes and instigates JAK2/STAT1 activation, followed by IRF1 synthesis. IRF1 (through as yet unidentified mechanism) inhibits SOD expression leading to oxidative stress. Oxidative stress as well as IFNαβ stimulate JNK and caspase 3 activation, and cause apoptosis of hepatocytes.
Experimental procedures
Reagents
The reagents were obtained from the indicated sources: concanavalin A, d-mannose, Me-αMan, protease, Nycodenz, collagenase (C5138), and BMS-345541 (Sigma); FITC-conjugated ConA (FITC-ConA) (Vector Laboratories, Burlingame, CA); collagenase Type 4 (Worthington Biochemical Corp.); LY294002, AG490, PP2, SB202190, GSK690693, SR11302, and fludarabine (Tocris Bioscience, Minneapolis, MN); PD98059 and SP600125 (Calbiochem); SN50 (BioVision, Inc., Milpitas, CA); anti-P-JAK2 (Tyk2), anti-P-STAT1, anti-IRF1, anti-P-JNK1/2, anti-cleaved caspase 3, anti-Histone H3, and anti-TBP Abs (Cell Signaling, Danvers, MA); horseradish peroxidase-conjugated β-actin Ab (Santa Cruz Biotechnology, Inc.); rabbit anti-GFAP (Dako Cytomation, Carpinteria, CA); MitoTracker Red and anti-desmin monoclonal Ab (DE-U-10) (Life Technologies Corp.). A stock solution of ConA was prepared in saline and various inhibitors were dissolved in dimethyl sulfoxide (DMSO; Life Technologies Corp.). All solutions were stored at −20 °C, and diluted to the final concentration in fresh media before experiments.
Animals and treatment
The protocols were approved by the Institutional Animal Care and Use Committee according to the NIH guidelines (45). Male C57BL/6 (WT-B6), BALB/c (WT-BALB/c), and B6.129S2-Irf1tm1Mak/J (IRF1-KO) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Mice lacking IFNαβ receptor (IFNαβR-KO) on the BALB/c background were kindly provided by Dr. Gregory Tiao, Cincinnati Children's Hospital Medical Center, Cincinnati, OH.
Mice were administered 20 mg/kg of ConA intravenously and euthanized 1, 3, or 6 h later unless indicated otherwise. Blood was obtained, and livers were excised, washed in ice-cold PBS, and portions fixed in 10% buffered formalin or paraformaldehyde or snap-frozen in liquid nitrogen. Formalin-fixed, paraffin-embedded liver sections were stained with hematoxylin and eosin for histopathological examination. Serum alanine aminotransferase (ALT) was measured using a Kit from Pointe Scientific Inc., Canton, MI.
For immunohistochemical analysis, sections were deparaffinized with xylene, treated with a series of ethanol concentrations, followed by deionized water, then heated in a microwave oven in 10 mmol/liter of sodium citrate (pH 6) containing 0.05% Tween 20, and blocked with PBS containing 1% BSA, 10% goat serum, and 0.1% Triton X-100 for 1 h. Neutrophils were stained using rat monoclonal anti-neutrophil (NIMP-R14) antibody (Abcam, Cambridge, MA) (1:50) in PBS containing 1% BSA, 10% goat serum, and 0.1% Triton X-100. The sections were counterstained with hematoxylin after incubation with horseradish peroxidase-conjugated goat anti-rat IgG (ab97057; 1:400) and diaminobenzidine substrate (ab64238; Abcam).
Isolation and culture of HSCs and hepatocytes
HSCs were isolated by collagenase/protease digestion of the mouse (40–50 g) liver, purified using Nycodenz gradient, and cultured at a density of 0.4 × 106 cells/cm2 as described previously (18). The medium was renewed after overnight culture, and the cells were used on day 3 of culture interval. In some experiments, HSCs were incubated in a medium containing 50 μg/ml of ConA for up to 6 h. Cells and the culture supernatants (filtered through a sterile 0.22-μm syringe filter) were used for various determinations.
Mouse (20–25 g) hepatocytes were prepared by collagenase digestion of the liver, purified on Percoll gradient, and cultured as described previously (46, 47). The medium was renewed after a 3-h attachment period. After overnight culture, hepatocytes were incubated in control medium or medium conditioned by HSCs ± ConA; in some experiments, anti-IFNβ Ab (ab24324; 2 μg/ml) or SP600125 (JNKi; 10 μm) was added to the conditioned medium before transfer to hepatocytes; all incubations were terminated after 12 h for various assays.
Binding of ConA in HSCs in vitro
HSCs were washed and incubated in Dulbecco's modified Eagle's medium supplemented with 5% serum and containing 12.5 μg/ml of FITC-ConA for the indicated time points at 37 °C. In a competition binding assay, HSCs were incubated in medium containing FITC-ConA with/without 100 mm d-mannose or methyl-α-d-mannopyranoside, or excess unlabeled ConA (500 μg/ml) for 20 min. The cells were then fixed for 20 min in ice-cold methanol at −20 °C, washed once with PBS, and mounted with DAPI. Images were acquired with a Zeiss fluorescence microscope (Zeiss, Thornwood, NY).
Effects of inhibitors on the ConA-induced signaling in HSCs
HSCs were incubated in medium containing various inhibitors or the vehicle (DMSO) for 30 min before stimulation with ConA (50 μg/ml) for the indicated time points. Cells and the culture supernatant were used for various determinations.
ConA-induced cytokine/chemokine expression in HSCs
For mRNA determination, RNA was extracted using TRIzol® Reagent (Life Technologies) (48). 1.5 μg of RNA was reverse-transcribed into cDNA using High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA), and quantified via qRT-PCR using primers (Table 1) as described (16). β-Actin or GAPDH expression was used for normalization. Enzyme-linked immunosorbent assay (ELISA) kits were used to determine the concentrations of IFNβ (PBL Assay Science, Piscataway, NJ) and TNFα (eBioscience, San Diego, CA) in cell culture supernatants.
Table 1.
List of primers used for quantitative RT-PCR
| Direction | Sequence (5′ to 3′) | |
|---|---|---|
| Mouse | ||
| β-Actin | Forward | 5′-AGAGGGAAATCGTGCGTGAC-3′ |
| Reverse | 5′-CAATAGTGATGACCTGGCCGT-3′ | |
| Cxcl1 | Forward | 5′-CTGCACCCAAACCGAAGTC-3′ |
| Reverse | 5′-AGCTTCAGGGTCAAGGCAAG-3′ | |
| Gapdh | Forward | 5′-CATGGCCTTCCGTGTTCCTA-3′ |
| Reverse | 5′-CCTGCTTCACCACCTTCTTGAT-3′ | |
| Ifnα | Forward | 5′-TCTGATGCAGCAGGTGGG-3′ |
| Reverse | 5′-AGGGCTCTCCAGACTTCTGCTCTG-3′ | |
| Ifnβ | Forward | 5′-AGCTCCAAGAAAGGACGAACAT-3′ |
| Reverse | 5′-GCCCTGTAGGTGAGGTTGATCT-3′ | |
| Ifnγ | Forward | 5′-CTCTTCCTCATGGCTGTTTCT-3′ |
| Reverse | 5′-TTCTTCCACATCTATGCCACTT-3′ | |
| Igf2r | Forward | 5′-GGGAAGCTGTTGACTCCAAAA-3′ |
| Reverse | 5′-GCAGCCCATAGTGGTGTTGAA-3′ | |
| Irf1 | Forward | 5′-CAGAGGAAAGAGAGAAAGTCC-3′ |
| Reverse | 5′-CACACGGTGACAGTGCTGG-3′ | |
| Sod1 | Forward | 5′-CAGGACCTCATTTTAATCCTCAC-3′ |
| Reverse | 5′-TGCCCAGGTCTCCAACAT-3′ | |
| Tnfα | Forward | 5′-CCCAGGTATATGGGCTCATACC-3′ |
| Reverse | 5′-GCCGATTTGCTATCTCATACCAGG-3′ | |
| Rat | ||
| β-Actin | Forward | 5′-GAGACCTTCAACACCCCAGCC-3′ |
| Reverse | 5′-TCGGGGCATCGGAACCGCTCA-3′ | |
| Cxcl1 | Forward | 5′-GCACCCAAACCGAAGTCATA-3′ |
| Reverse | 5′-GGGACACCCTTTAGCATCTTT-3′ | |
| Ifnα | Forward | 5′-TCCCCTGACCCAGGAAGACTC-3′ |
| Reverse | 5′-CAGGCACAGGGGCTGTGTTTA-3′ | |
| Ifnβ | Forward | 5′-GGTGACATCCACGACTACTTTAG-3′ |
| Reverse | 5′-CCAGGCATAGCTGTTGTACTT-3′ | |
| Irf1 | Forward | 5′-CTCACCAAGAACCAGAGGAAAG-3′ |
| Reverse | 5′-AGATAAGGTGTCAGGGCTAGAA-3′ | |
| Tnfα | Forward | 5′-TCCCAACAAGGAGGAGAAGT-3′ |
Western blot analysis
Cell lysates were prepared in RIPA buffer (Abcam) containing protease inhibitors, and centrifuged at 10,000 × g for 10 min. Supernatants (20 μg of protein) were subjected to SDS-PAGE and separated proteins were transferred to polyvinylidene fluoride membrane. After immunoblotting overnight at 4 °C with primary Abs, washing and incubation with secondary anti-rabbit IgG (1:2000), blots were developed using Amersham BiosciencesTM ECL SelectTM detection reagent (GE Healthcare, Buckinghamshire, UK). β-Actin expression was determined as an internal control and the relative expression was quantified with ImageJ software (version 1.50i).
Nuclear extracts were prepared from the cultured cells using extraction reagents (Pierce-Endogen, Rockford, IL), and proteins (20 μg) were separated by 8% SDS-PAGE. Western blotting was performed using anti-P-STAT1 or anti-IRF1 Ab, and secondary anti-rabbit IgG (1:2000). Histone H3 or TBP expression was determined as an internal control.
Immunofluorescence analysis
Intracellular IFNβ was detected as described before (16, 22). Briefly, HSCs were incubated in medium containing 1 μl/ml of protein transport inhibitor brefeldin A (BD Biosciences, San Diego, CA) without or with ConA (50 μg/ml) for 6 h. The cells were washed with ice-cold PBS and fixed in 2% paraformaldehyde. After permeabilization and blocking with PBS containing 1% BSA, 10% goat serum, 0.3 mol/liter of glycine, and 0.1% Triton X-100, the cells were treated with anti-IFNβ (Abcam) and anti-desmin Abs overnight at 4 °C, washed, and incubated in Alexa Fluor® 488 anti-rabbit and Alexa Fluor® 594 anti-mouse (1:400) (Invitrogen Molecular Probes) for 1 h at room temperature. After incubation with secondary Abs and washing with PBS, the cells were mounted with DAPI mounting medium and imaged using fluorescence microscopy.
Fixed cells were incubated in PBS containing 1% BSA, 10% goat serum, and 0.1% Triton X-100 for 1 h for permeabilization and blocking nonspecific protein–protein interactions. The cells were then incubated with anti-IRF1 antibody overnight at 4 °C, washed twice with PBS, and incubated for 1 h in the dark with Alexa Fluor® 568 goat anti-rabbit secondary antibody (Invitrogen Molecular Probes). The cells were then washed with PBS, mounted with DAPI, and IRF1 was visualized via fluorescence microscopy.
Antigen-retrieved sections of the liver were blocked with PBS containing 1% BSA, 10% goat serum, and 0.1% Triton X-100 for 1 h. For nuclear IRF1 staining, sections were permeabilized with 0.25% Triton X-100 in PBS for 10 min before blocking. Following incubation with rabbit anti-desmin (Abcam) or rabbit anti-IRF1 Abs overnight at 4 °C, the sections were incubated with Alexa Fluor® 488 goat anti-rabbit secondary antibody (Invitrogen Molecular Probes) for 1 h in the dark. Sections were then washed with PBS, mounted with DAPI, and visualized via fluorescence microscopy.
Determination of oxidative stress
The cells were washed with PBS (3 times) and placed in PBS containing DCFDA (1 μmol/liter in DMSO; Invitrogen Molecular Probes) for 5 min before analysis. DCFDA is deacetylated by cellular esterase and oxidized by reactive oxygen species (ROS) to 2′,7′-dichlorofluorescin, which was detected by fluorescence microscopy with maximum excitation and emission spectra at 495 and 529 nm, respectively. Intracellular ROS levels were evaluated on the basis of mean DCF fluorescence intensity.
SOD activity assay
Superoxide dismutase (SOD) activity was measured in tissue homogenate or cell lysate using SOD Assay Kit (Cayman Chemicals, Ann Arbor, MI). The liver tissue was rinsed with PBS, homogenized in 5 ml/g of tissue cold buffer (20 mm HEPES, pH 7.2, containing 1 mm EGTA, 210 mm mannitol, and 70 mm sucrose), and centrifuged at 1,500 × g for 5 min at 4 °C. For cell lysate, cell pellets obtained by centrifugation at 1,000–2,000 × g for 10 min at 4 °C were sonicated in cold buffer and centrifuged at 1,500 × g for 5 min at 4 °C. 1,500 × g supernatants (obtained from tissue or cultured cells) were centrifuged at 10,000 × g for 15 min at 4 °C. The resulting supernatants were used for measuring the SOD activity according to the manufacturer's instructions. Protein concentrations in the homogenates or cell lysates were measured with a PierceTM BCA Protein Assay Kit (Pierce).
Statistical analysis
The results are expressed as mean ± S.D. Statistical significance between groups was determined by Student's t test using Prism 6 software (GraphPad Software, San Diego, CA). A p value <0.05 was considered statistically significant.
Author contributions
R. R., S. K., and C. G. data curation; R. R. formal analysis; R. R. validation; R. R., S. K., A. S., G. M. T., and C. G. investigation; R. R., S. K., A. S., S. K. M., B. D., and C. G. methodology; R. R. and C. G. writing-original draft; R. R., S. K. M., G. M. T., and C. G. writing-review and editing; C. G. conceptualization; C. G. resources; C. G. supervision; C. G. funding acquisition; C. G. project administration.
Supplementary Material
This work was supported by U.S. Department of Veterans Affairs (VA) Merit Review Award 1IO1BX001174 (to C. R. G.) and U.S. Department of Defense (DOD) Grant W81XWH-14-PRMRP-IIRA. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S8.
- HSC
- hepatic stellate cell
- Ab
- antibody
- ConA
- concanavalin A
- CXCL
- CXC chemokine ligand
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GFAP
- glial fibrillary acidic protein
- IFN
- interferon
- IFNαβR
- interferon α/β receptor
- IGF2R
- insulin-like growth factor 2 receptor
- IRF
- interferon regulatory factor
- JAK
- Janus kinase
- qRT
- quantitative reverse transcriptase
- ROS
- reactive oxygen species
- SOD
- superoxide dismutase
- STAT
- signal transducer and activator of transcription
- TNF
- tumor necrosis factor
- WT
- wildtype
- Me-αMan
- methyl-α-d-mannopyranoside
- ALT
- alanine aminotransferase
- DAPI
- 4′,6-diamidino-2-phenylindole
- DCFDA
- 2′,7′-dichlorofluorescin diacetate
- LPS
- lipopolysaccharide.
References
- 1. Friedman S. L. (2015) Hepatic fibrosis: emerging therapies. Dig. Dis. 33, 504–507 10.1159/000374098 [DOI] [PubMed] [Google Scholar]
- 2. Schuppan D. (2015) Liver fibrosis: common mechanisms and antifibrotic therapies. Clin. Res. Hepatol. Gastroenterol. 39, S51–S59 10.1016/j.clinre.2015.05.005 [DOI] [PubMed] [Google Scholar]
- 3. Trautwein C., Friedman S. L., Schuppan D., and Pinzani M. (2015) Hepatic fibrosis: concept to treatment. J. Hepatol. 62, S15–S24 10.1016/j.jhep.2015.02.039 [DOI] [PubMed] [Google Scholar]
- 4. Gandhi C. R. (2015) Stellate cells in hepatic immunological tolerance. in Stellate cells in health and disease (Gandhi C. R., and Pinzani M., eds) pp. 227–249, Elsevier Press, San Diego, CA [Google Scholar]
- 5. Gandhi C. R. (2015) Stellate cells in regulation of hepatocyte survival and function. in Stellate cells in health and disease (Gandhi C. R., and Pinzani M., eds) pp. 209–225, Elsevier Press, San Diego, CA [Google Scholar]
- 6. Kumar S., Wang J., Thomson A. W., and Gandhi C. R. (2017) Hepatic stellate cells increase the immunosuppressive function of natural Foxp3+ regulatory T cells via IDO-induced AhR activation. J. Leukoc. Biol. 101, 429–438 10.1189/jlb.2A0516-239R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stewart R. K., Dangi A., Huang C., Murase N., Kimura S., Stolz D. B., Wilson G. C., Lentsch A. B., and Gandhi C. R. (2014) A novel mouse model of depletion of stellate cells clarifies their role in ischemia/reperfusion- and endotoxin-induced acute liver injury. J. Hepatol. 60, 298–305 10.1016/j.jhep.2013.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tiegs G. (2007) Cellular and cytokine-mediated mechanisms of inflammation and its modulation in immune-mediated liver injury. Z. Gastroenterol. 45, 63–70 10.1055/s-2006-927397 [DOI] [PubMed] [Google Scholar]
- 9. Heymann F., Hamesch K., Weiskirchen R., and Tacke F. (2015) The concanavalin A model of acute hepatitis in mice. Lab. Anim. 49, 12–20 10.1177/0023677215572841 [DOI] [PubMed] [Google Scholar]
- 10. Schümann J., Wolf D., Pahl A., Brune K., Papadopoulos T., van Rooijen N., and Tiegs G. (2000) Importance of Kupffer cells for T-cell-dependent liver injury in mice. Am. J. Pathol. 157, 1671–1683 10.1016/S0002-9440(10)64804-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morita A., Itoh Y., Toyama T., Fujii H., Nishioji K., Kirishima T., Makiyama A., Yamauchi N., and Okanoue T. (2003) Activated Kupffer cells play an important role in intra-hepatic Th1-associated necro-inflammation in concanavalin A-induced hepatic injury in mice. Hepatol. Res. 27, 143–150 10.1016/S1386-6346(03)00206-7 [DOI] [PubMed] [Google Scholar]
- 12. Chen L., Xie X. J., Ye Y. F., Zhou L., Xie H. Y., Xie Q. F., Tian J., and Zheng S. S. (2011) Kupffer cells contribute to concanavalin A-induced hepatic injury through a Th1 but not Th17 type response-dependent pathway in mice. Hepatobiliary Pancreat. Dis. Int. 10, 171–178 10.1016/S1499-3872(11)60027-1 [DOI] [PubMed] [Google Scholar]
- 13. Knolle P. A., Gerken G., Loser E., Dienes H. P., Gantner F., Tiegs G., Meyer zum Buschenfelde K. H., and Lohse A. W. (1996) Role of sinusoidal endothelial cells of the liver in concanavalin A-induced hepatic injury in mice. Hepatology 24, 824–829 10.1002/hep.510240413 [DOI] [PubMed] [Google Scholar]
- 14. Tiegs G., Hentschel J., and Wendel A. (1992) A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J. Clin. Invest. 90, 196–203 10.1172/JCI115836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hong F., Jaruga B., Kim W. H., Radaeva S., El-Assal O. N., Tian Z., Nguyen V. A., and Gao B. (2002) Opposing roles of STAT1 and STAT3 in T cell-mediated hepatitis: regulation by SOCS. J. Clin. Invest. 110, 1503–1513 10.1172/JCI0215841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rani R., Tandon A., Wang J., Kumar S., and Gandhi C. R. (2017) Stellate cells orchestrate concanavalin A-induced acute liver damage. Am. J. Pathol. 187, 2008–2019 10.1016/j.ajpath.2017.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jaruga B., Hong F., Kim W. H., and Gao B. (2004) IFN-γ/STAT1 acts as a proinflammatory signal in T cell-mediated hepatitis via induction of multiple chemokines and adhesion molecules: a critical role of IRF-1. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G1044–G1052 10.1152/ajpgi.00184.2004 [DOI] [PubMed] [Google Scholar]
- 18. Dangi A., Sumpter T. L., Kimura S., Stolz D. B., Murase N., Raimondi G., Vodovotz Y., Huang C., Thomson A. W., and Gandhi C. R. (2012) Selective expansion of allogeneic regulatory T cells by hepatic stellate cells: role of endotoxin and implications for allograft tolerance. J. Immunol. 188, 3667–3677 10.4049/jimmunol.1102460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harvey S. A., Dangi A., Tandon A., and Gandhi C. R. (2013) The transcriptomic response of rat hepatic stellate cells to endotoxin: implications for hepatic inflammation and immune regulation. PLoS ONE 8, e82159 10.1371/journal.pone.0082159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mizuhara H., Kuno M., Seki N., Yu W. G., Yamaoka M., Yamashita M., Ogawa T., Kaneda K., Fujii T., Senoh H., and Fujiwara H. (1998) Strain difference in the induction of T-cell activation-associated, interferon γ-dependent hepatic injury in mice. Hepatology 27, 513–519 10.1002/hep.510270227 [DOI] [PubMed] [Google Scholar]
- 21. Goritzka M., Durant L. R., Pereira C., Salek-Ardakani S., Openshaw P. J., and Johansson C. (2014) α/β-Interferon receptor signaling amplifies early proinflammatory cytokine production in the lung during respiratory syncytial virus infection. J. Virol. 88, 6128–6136 10.1128/JVI.00333-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dangi A., Huang C., Tandon A., Stolz D., Wu T., and Gandhi C. R. (2016) Endotoxin-stimulated rat hepatic stellate cells induce autophagy in hepatocytes as a survival mechanism. J. Cell. Physiol. 231, 94–105 10.1002/jcp.25055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsung A., Stang M. T., Ikeda A., Critchlow N. D., Izuishi K., Nakao A., Chan M. H., Jeyabalan G., Yim J. H., and Geller D. A. (2006) The transcription factor interferon regulatory factor-1 mediates liver damage during ischemia-reperfusion injury. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G1261–G1268 10.1152/ajpgi.00460.2005 [DOI] [PubMed] [Google Scholar]
- 24. Afonso V., Santos G., Collin P., Khatib A. M., Mitrovic D. R., Lomri N., Leitman D. C., and Lomri A. (2006) Tumor necrosis factor-α down-regulates human Cu/Zn superoxide dismutase 1 promoter via JNK/AP-1 signaling pathway. Free Radic. Biol. Med. 41, 709–721 10.1016/j.freeradbiomed.2006.05.014 [DOI] [PubMed] [Google Scholar]
- 25. Riera H., Afonso V., Collin P., and Lomri A. (2015) A central role for JNK/AP-1 pathway in the pro-oxidant effect of pyrrolidine dithiocarbamate through superoxide dismutase 1 gene repression and reactive oxygen species generation in hematopoietic human cancer cell line U937. PLoS ONE 10, e0127571 10.1371/journal.pone.0127571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Siebler J., Wirtz S., Klein S., Protschka M., Blessing M., Galle P. R., and Neurath M. F. (2003) A key pathogenic role for the STAT1/T-bet signaling pathway in T-cell-mediated liver inflammation. Hepatology 38, 1573–1580 10.1016/j.hep.2003.09.020 [DOI] [PubMed] [Google Scholar]
- 27. Winau F., Hegasy G., Weiskirchen R., Weber S., Cassan C., Sieling P. A., Modlin R. L., Liblau R. S., Gressner A. M., and Kaufmann S. H. (2007) Ito cells are liver-resident antigen-presenting cells for activating T cell responses. Immunity 26, 117–129 10.1016/j.immuni.2006.11.011 [DOI] [PubMed] [Google Scholar]
- 28. Sumpter T. L., Dangi A., Matta B. M., Huang C., Stolz D. B., Vodovotz Y., Thomson A. W., and Gandhi C. R. (2012) Hepatic stellate cells undermine the allostimulatory function of liver myeloid dendritic cells via STAT3-dependent induction of IDO. J. Immunol. 189, 3848–3858 10.4049/jimmunol.1200819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Roth R. A., Cassell D. J., Maddux B. A., and Goldfine I. D. (1983) Regulation of insulin receptor kinase activity by insulin mimickers and an insulin antagonist. Biochem. Biophys. Res. Commun. 115, 245–252 10.1016/0006-291X(83)90996-8 [DOI] [PubMed] [Google Scholar]
- 30. Ferayorni L. S., McMillan P. N., Raines L., Gerhardt C. O., and Jauregui H. O. (1983) Lectin binding to adult rat liver in situ, isolated hepatocytes, and hepatocytes cultures. in Isolation, characterization, and use of hepatocytes (Harris R. A., and Cornell N. W., eds) pp. 271–276, Elsevier Science Publishing Co. Inc., New York [Google Scholar]
- 31. Scharf J. G., Knittel T., Dombrowski F., Müller L., Saile B., Braulke T., Hartmann H., and Ramadori G. (1998) Characterization of the IGF axis components in isolated rat hepatic stellate cells. Hepatology 27, 1275–1284 10.1002/hep.510270513 [DOI] [PubMed] [Google Scholar]
- 32. Leist M., and Wendel A. (1996) A novel mechanism of murine hepatocyte death inducible by concanavalin A. J. Hepatol. 25, 948–959 10.1016/S0168-8278(96)80301-1 [DOI] [PubMed] [Google Scholar]
- 33. Vila-del Sol V., Punzón C., and Fresno M. (2008) IFN-γ-induced TNF-α expression is regulated by interferon regulatory factors 1 and 8 in mouse macrophages. J. Immunol. 181, 4461–4470 10.4049/jimmunol.181.7.4461 [DOI] [PubMed] [Google Scholar]
- 34. Vandenbon A., Teraguchi S., Takeuchi O., Suzuki Y., and Standley D. M. (2014) Dynamics of enhancers in myeloid antigen presenting cells upon LPS stimulation. BMC Genomics 15, S4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thirunavukkarasu C., Watkins S. C., and Gandhi C. R. (2006) Mechanisms of endotoxin-induced nitric oxide, interleukin-6 and tumor necrosis factor-α production in activated rat hepatic stellate cells: role of p38MAPK. Hepatology 44, 389–398 10.1002/hep.21254 [DOI] [PubMed] [Google Scholar]
- 36. Radaeva S., Jaruga B., Hong F., Kim W. H., Fan S., Cai H., Strom S., Liu Y., El-Assal O., and Gao B. (2002) Interferon-α activates multiple STAT signals and down-regulates c-Met in primary human hepatocytes. Gastroenterology 122, 1020–1034 10.1053/gast.2002.32388 [DOI] [PubMed] [Google Scholar]
- 37. Mizuhara H., O'Neill E., Seki N., Ogawa T., Kusunoki C., Otsuka K., Satoh S., Niwa M., Senoh H., and Fujiwara H. (1994) T cell activation–associated hepatic injury: mediation by tumor necrosis factors and protection by interleukin 6. J. Exp. Med. 179, 1529–1537 10.1084/jem.179.5.1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Küsters S., Gantner F., Künstle G., and Tiegs G. (1996) Interferon γ plays a critical role in T cell–dependent liver injury in mice initiated by concanavalin A. Gastroenterology 111, 462–471 10.1053/gast.1996.v111.pm8690213 [DOI] [PubMed] [Google Scholar]
- 39. Kumar S., Wang J., Shanmukhappa S. K., and Gandhi C. R. (2017) Toll-like receptor 4-independent carbon tetrachloride-induced fibrosis and lipopolysaccharide-induced acute liver injury in mice: role of hepatic stellate cells. Am. J. Pathol. 187, 1356–1367 10.1016/j.ajpath.2017.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dhupar R., Klune J. R., Evankovich J., Cardinal J., Zhang M., Ross M., Murase N., Geller D. A., Billiar T. R., and Tsung A. (2011) Interferon regulatory factor-1 mediates acetylation and release of high mobility group box-1 from hepatocytes during murine liver ischemia-reperfusion injury. Shock 35, 293–301 10.1097/SHK.0b013e3181f6aab0 [DOI] [PubMed] [Google Scholar]
- 41. Kanda N., Shimizu T., Tada Y., and Watanabe S. (2007) IL-18 enhances IFN-γ-induced production of CXCL9, CXCL10, and CXCL11 in human keratinocytes. Eur. J. Immunol. 37, 338–350 10.1002/eji.200636420 [DOI] [PubMed] [Google Scholar]
- 42. Bhattacharya A., Hegazy A. N., Deigendesch N., Kosack L., Cupovic J., Kandasamy R. K., Hildebrandt A., Merkler D., Kühl A. A., Vilagos B., Schliehe C., Panse I., Khamina K., Baazim H., Arnold I., et al. (2015) Superoxide dismutase 1 protects hepatocytes from type I interferon-driven oxidative damage. Immunity 43, 974–986 10.1016/j.immuni.2015.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Deng S. Y., Zhang L. M., Ai Y. H., Pan P. H., Zhao S. P., Su X. L., Wu D. D., Tan H. Y., Zhang L. N., and Tsung A. (2017) Role of interferon regulatory factor-1 in lipopolysaccharide-induced mitochondrial damage and oxidative stress responses in macrophages. Int. J. Mol. Med. 40, 1261–1269 10.3892/ijmm.2017.3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chambers J. W., and LoGrasso P. V. (2011) Mitochondrial c-Jun N-terminal kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation. J. Biol. Chem. 286, 16052–16062 10.1074/jbc.M111.223602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Committee for the Update of the Guide for the Care and Use of Laboratory Animals (2011) National Research Council Guide for the Care and Use of Laboratory Animals, 8 Ed., National Academic Press, Washington, D. C. [Google Scholar]
- 46. Uemura T., and Gandhi C. R. (2001) Inhibition of DNA synthesis in cultured hepatocytes by endotoxin-conditioned medium of activated stellate cells is transforming growth factor-β- and nitric oxide-independent. Br. J. Pharmacol. 133, 1125–1133 10.1038/sj.bjp.0704151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thirunavukkarasu C., Uemura T., Wang L. F., Watkins S. C., and Gandhi C. R. (2005) Normal rat hepatic stellate cells respond to endotoxin in LBP independent manner to produce inhibitor(s) of DNA synthesis in hepatocytes. J. Cell. Physiol. 204, 654–665 10.1002/jcp.20366 [DOI] [PubMed] [Google Scholar]
- 48. Kumar S., Wang J., Rani R., and Gandhi C. R. (2016) Hepatic deficiency of augmenter of liver regeneration exacerbates alcohol-induced liver injury and promotes fibrosis in mice. PLoS ONE 11, e0147864 10.1371/journal.pone.0147864 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.