

Abstract
Cancer is a set of diseases characterized by uncontrolled cell growth. In certain cancers of the gastrointestinal tract, the adenomatous polyposis coli (APC) tumor suppressor gene is altered in either germline or somatic cells and causes formation of risk factors, such as benign colonic or intestinal neoplasia, which can progress to invasive cancer. APC is a key component of the WNT pathway, contributing to normal GI tract development, and APC alteration results in dysregulation of the pathway for production of polyamines, which are ubiquitous cations essential for cell growth. Studies with mice have identified nonsteroidal anti-inflammatory drugs (NSAIDs) and difluoromethylornithine (DFMO), an inhibitor of polyamine synthesis, as potent inhibitors of colon carcinogenesis. Moreover, gene expression profiling has uncovered that NSAIDs activate polyamine catabolism and export. Several DFMO–NSAID combination strategies are effective and safe methods for reducing risk factors in clinical trials with patients having genetic or sporadic risk of colon cancer. These strategies affect cancer stem cells, inflammation, immune surveillance, and the microbiome. Pharmacotherapies consisting of drug combinations targeting the polyamine pathway provide a complementary approach to surgery and cytotoxic cancer treatments for treating patients with cancer risk factors. In this Minireview, we discuss the role of polyamines in colon cancer and highlight the mechanisms of select pharmacoprevention agents to delay or prevent carcinogenesis in humans.
Keywords: polyamine, cancer chemoprevention, molecular pharmacology, colon cancer, anticancer drug, GI tract, DFMO, NSAID, Wnt signaling, adenomatous polyposis
Background
Polyamines were described as early as the late 17th century with their discovery credited to Van Leeuwenhoek as discussed in Ref. 1. Their importance as targets for cancer treatment has only become apparent since the 1960s, as highlighted in the timeline shown in Fig. 1. In several seminal papers, Dykstra and Herbst (2) and Raina et al. (3) reported strong associations between concentrations of specific polyamines and tissue growth in rodents. Russell and Snyder (4) extended these original findings to other species and tumor models. In addition, they demonstrated that the activity of ornithine decarboxylase 1 (ODC1) was rapidly induced by growth stimuli. They also found that the enzyme had an exceedingly short half-life (∼10 min), suggesting that ODC1 was under strict regulatory control. By the mid-1970s, O'Brien et al. (5) showed that a variety of tumor promoters of different classes had similar abilities to induce both ODC1 enzyme activity and skin tumor formation. These reports were all important original findings but did not provide evidence of cause–effect relationships between polyamines and growth.
Figure 1.
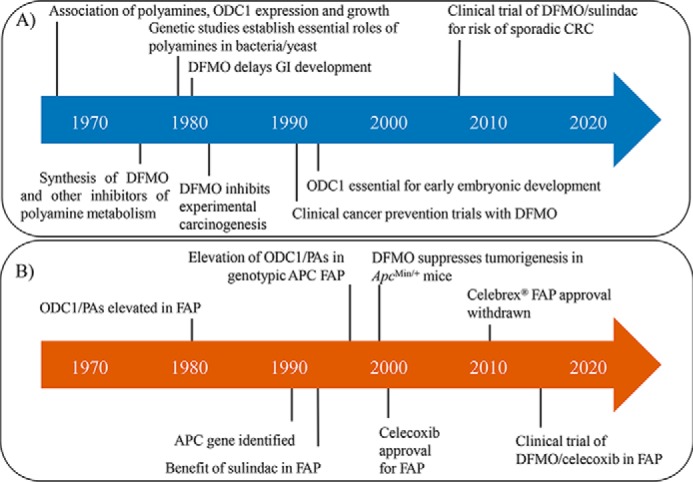
Timeline of research findings linking the polyamine pathway to cancer development. A, key observations and accomplishments (discussed in text) that established the polyamine pathway as an integral aspect of carcinogenesis and a target for treating cancer risk factors. B, specific findings and clinical trials in patients with FAP that supported the rationale for developing a combination drug product consisting of DFMO and an NSAID.
Herbert Tabor, for whom this issue of minireviews is dedicated on the anniversary of his 100th birth year, and colleagues at the National Institutes of Health used genetic methods to address the question of causality. They established that polyamines were not essential for growth of bacteria in general (6, 7), but the lack of enzymes to produce polyamines did compromise bacterial growth under conditions that suggested polyamines were acting by a mechanism affecting protein translation (8). In collaboration with his wife Celia, Herbert Tabor showed that polyamines were essential for growth in specific strains of yeast (9, 10).
In this same time frame, Metcalf et al. (11) at the Merrell Research Institute in Strasbourg, France (known by its French name Centre de Recherche Merrell International-CRMI), reported the synthesis of difluoromethylornithine (DFMO),2 a highly targeted drug whose mechanism involved enzyme activation and irreversible inhibition of ODC1, in 1978. Scientists at CRMI quickly reported the growth inhibitory and anti-tumor effects of DFMO and other ODC1 inhibitors (12–14). These early investigations found that the profound growth inhibitory effects of DFMO were not accompanied by cytotoxicity. Slaga and co-workers (15) used DFMO to show that inhibition of polyamine synthesis could inhibit skin carcinogenesis in mouse models and that the effect of the drug was on specific features of tumor promotion. Kingsnorth et al. (16) were the first group to show DFMO inhibited colon carcinogenesis in a rodent model of colon carcinogenesis, the dimethylhydrazine-treated rat.
Clinical trials of high-dose intravenous DFMO as a therapy for advanced cancers were conducted in the 1980s and were generally negative from both a safety and efficacy perspective (17). Clinical trials in patients with conditions other than cancer were also conducted, and these trials led to regulatory approval of high-dose DFMO administered intravenously for treatment of patients with a form of African Sleeping Sickness in 1990 (18) and a topical form of DFMO for female hirsutism in 2000 (19). Achieving regulatory approval of an oral dosage form of DFMO has never been accomplished for any medical indication and is a current major challenge for development of this and related drugs to treat cancer risk factors.
A major motivation for the work described in this Minireview was identification of risk factors for leading causes of disease and death in the United States and the understanding that some of these factors are associated with the risk of cancer (20). Many common risk factors, such as diet, tobacco use, high body-mass index, air pollution, and low physical activity, are not amenable to interventions with pharmacotherapies. However, some cancer-specific risk factors can be managed by pharmacotherapies to reduce risk of disease and mortality analogous to targeting high cholesterol in patients with a risk of cardiovascular disease using pharmacoprevention strategies (e.g. statins) (21). Cancer-specific risk factors that could be targets for pharmacoprevention strategies include intraepithelial neoplasia (IEN) (22). Colorectal adenomas (CRA) are an example of an IEN risk factor for colorectal cancer (CRC). Failure to remove CRAs is associated with an increase in CRC in humans (23). More recent studies indicate that screening for CRAs, which is associated with the removal of large/advanced CRAs, is strongly associated with a reduction in mortality (24). A significant challenge in the field of oncology is to determine whether managing cancer risk factors, such as CRAs, with pharmacotherapies can prevent or delay cancer and reduce cancer disease burden and deaths.
The major goal of the work summarized in this Minireview was to understand the mechanistic basis of pharmacoprevention agents and determine whether intervening in the polyamine pathway could be used successfully to delay/prevent carcinogenesis in humans.
Polyamines as mediators of colon carcinogenesis
Following the earlier observations of the association of polyamines and growth, Luk et al. (25) took advantage of the new inhibitor of this pathway, DFMO, to address the importance of polyamines in gut development in rodent models. They reported that DFMO could delay gut development in fetal rats and recovery from chemotherapy-induced gut injury in adult rats (25). This finding led Luk et al. (25) to ask whether the expression of ODC1 and polyamines, which appeared to be important in normal gut mucosal development, was altered in the apparently normal gut mucosa of patients with familial adenomatous polyposis (FAP), a genetic syndrome associated with near 100% risk of development of colon cancer. They discovered that both ODC1 and polyamines are elevated in the apparently normal colonic mucosa of FAP patients and appeared to identify genotypic individuals (26). Following the identification of the APC gene in humans (27), the elevation of ODC1 enzyme activity and polyamine contents in apparently normal colonic mucosa of genotypic FAP patients was established (28).
Fig. 2 depicts the signaling of ODC1 and the polyamine pathway in patients with FAP and in normal individuals. This depiction is based on studies in humans and mouse models. Multiple intestinal neoplasia in the ApcMin/+ mouse model of FAP is caused by a mutation in the murine homolog of the human APC gene (29). Expression of ODC1 and other genes in the polyamine pathway, including the gene encoding the ornithine decarboxylase inhibitory protein antizyme (OAZ), are influenced by the mutant APC-encoding gene in the mouse model (30). ODC1 RNA levels were increased in both intestinal and colonic mucosa, whereas OAZ RNA levels are decreased especially in the intestinal mucosa of these mice.
Figure 2.
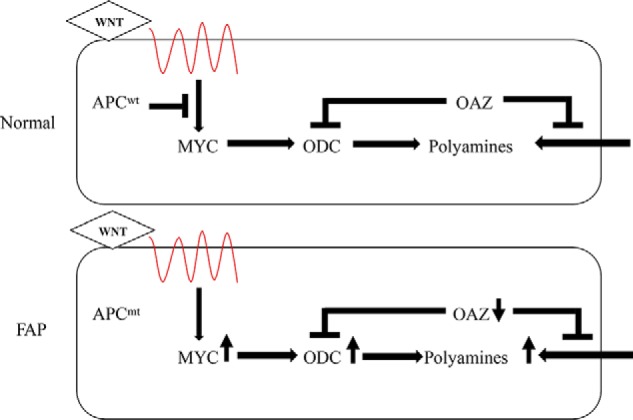
Role of the APC tumor suppressor gene in signaling expression of genes regulating the polyamine pathway. Top (Normal), in individuals with normal APC (APCWT), the canonical WNT pathway controls MYC expression (in part regulated by APC) and MYC target genes, including ODC and other polyamine metabolic genes. Bottom (FAP), in genotypic patients with FAP, ODC activity and polyamine contents in apparently normal mucosa are elevated, compared with nongenotypic family members. In the ApcMin/+ mouse model, mutant APC is associated with an increase in ODC, a decrease in antizyme (OAZ) RNA, and a consequent increase in intestinal tumor and normal tissue polyamines. OAZ interacts with ODC 3A2 to initiate ODC degradation and inhibits polyamine transport.
Mechanistic studies in human cells support the pathway depiction shown in Fig. 2. He et al. (31) showed that c-MYC is transcriptionally activated by the APC signaling pathway, and ODC1 was known to be a transcriptional target of c-MYC (32). Conditional expression of WT APC in human colon cancer cells containing mutant APC demonstrated that WT APC was restrictive for ODC1 expression (33). This suppression depended on the presence of canonical MYC-binding sites in the ODC1 promoter. Tissue-specific knockdown of c-MYC in ApcMin/+ mice established that MYC was involved in APC-dependent intestinal and colonic carcinogenesis (34) and that treatment of ApcMin/+ mice with DFMO reduced both intestinal and colonic carcinogenesis (30, 35).
Polyamines may be involved in colon carcinogenesis due to disruption in pathways other than the APC/MYC pathway. Green and Hudson (36) have reviewed the roles of several signaling pathways implicated in the development of colon cancers. KRAS-dependent tumorigenesis is inhibited by DFMO in human Caco-2 xenografts (37), and colon carcinogenesis in transforming growth factor β (TGFβ)–deficient mice is associated with changes in the polyamine and other metabolic pathways in the gut microbiome of these mice (38). The role of the microbiome will be discussed further in a subsequent section of this Minireview.
Drug combinations targeting the polyamine pathway to inhibit carcinogenesis
DFMO was an effective but incomplete inhibitor of experimental carcinogenesis in the ApcMin/+ mouse (30) and other models (39). Sporn (40) was an early advocate for using combinations of agents as a means to increase efficacy and reduce toxicity of treatments to suppress carcinogenesis. The nonsteroidal anti-inflammatory drug (NSAID) sulindac reduced colonic and rectal polyps in patients with FAP in a statistically significant but incomplete manner in a randomized placebo-controlled trial (41). The efficacy of combinations of DFMO with other NSAIDs, including paroxysm (42), indomethacin (43), and aspirin (44), was reported for several models of experimental carcinogenesis.
The precise mechanism by which NSAIDs inhibit carcinogenesis remains elusive. Whereas NSAIDs are generally considered to work via their effects on cyclooxygenases and prostaglandin metabolism, noncyclooxygenase mechanisms have been reported (45). Indomethacin suppresses the tumor-promoter induction of ODC1 in experimental skin carcinogenesis (46). To understand potential mechanisms of action of sulindac, patterns of gene expression resulting from treatment with sulindac sulfone, a sulindac metabolite lacking cyclooxygenase inhibitory activity, were measured in human colon tumor-derived cells (47). Sulindac sulfone inhibited cell growth and induced apoptosis and the expression of the spermidine/spermine N-acetyltransferase (SAT1), a polyamine catabolic gene product implicated in polyamine export (48). The sulindac sulfone induction of SAT1 gene expression was shown to occur via the cyclooxygenase-independent transcriptional activation of SAT1 by a peroxisomal proliferator-activated receptor γ (PPARγ)–dependent mechanism acting at a specific PPARγ-responsive element in the SAT1 gene. Treatment of cells with sulindac sulfone induces SAT1 and stimulates polyamine export. Other NSAIDs also induce SAT1, and presumably polyamine export, but by unique mechanisms (49).
These results led us to hypothesize that combinations of DFMO and NSAIDs, such as sulindac, might be working via complementary mechanisms (depicted in Fig. 3) to suppress dysregulated and high levels of polyamines in neoplasia by inhibiting both polyamine synthesis and stimulating polyamine catabolism and export (50). Experimental studies in the ApcMin/+ mouse supported this hypothesis (DFMO and sulindac combination in cancer chemoprevention; United States patent no. 6,258,845, 2001) (51). A clinical trial of DFMO and sulindac in patients with sporadic risk of colorectal cancer showed dramatic efficacy to reduce both metachronous colorectal adenomas (52) and rectal mucosal polyamine but not prostaglandin E2 contents (53). Results from an international clinical trial of DFMO and the NSAID celecoxib in patients with FAP, and based on earlier preclinical data in the ApcMin/+ mouse (51), were reported in 2016 (54). This clinical trial found that the combined DFMO-NSAID treatment had a highly statistically significant 80% reduction on global measures of tumor burden from baseline, compared with the 30% reduction from baseline for the NSAID alone. This trial reported a difference in total polyp number from baseline in the two groups, but the difference was not statistically significant. A trial of DFMO combined with sulindac in patients with FAP is in progress and uses a composite primary end point closely related to global tumor burden (55).
Figure 3.
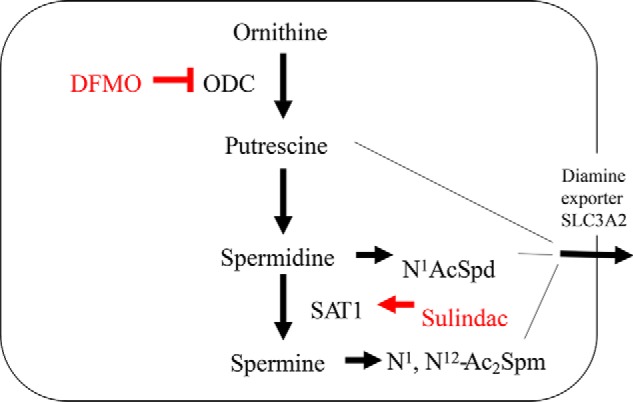
DFMO and sulindac reduce polyamines via dual mechanisms of action. DFMO is an enzyme-activated irreversible inhibitor of ODC. As discussed in the text, DFMO has been reported to have some activity against arginase. Sulindac and other NSAIDs can activate SAT1 by specific transcriptional mechanisms. The diamine putrescine and the SAT1 products N1-acetylspermidine and N1,N12-diacetylspermine are substrates for polyamine export mediated by the solute carrier transporter (SLC3A2). Thus, DFMO acts with sulindac and other NSAIDS in complementary ways to inhibit polyamine biosynthesis (DFMO) and activate polyamine export (NSAIDS).
Other drug combinations with DFMO are being investigated in a number of preclinical cancer models. Several groups have synthesized polyamine transport inhibitors (PTIs) (56–58) that suppress polyamine uptake as depicted in Fig. 2. PTIs appear to work best in concert with a polyamine synthesis inhibitor, like DFMO, to act as polyamine-blocking therapy (56, 59–61). PTIs are expected to begin clinical trial evaluations in the near future.
Relevance of polyamines to the extracolonic sequelae of APC mutations
The high penetrance of APC mutations in FAP results in nearly 100% of patients developing colon and rectal cancer if they retain their colons and rectums. Consequently, the standard of care for these patients in 2018 is colectomy with either proctectomy or rectum-preserving surgery, followed by close monitoring of any retained colonic/rectal tissue. Nonetheless, these patients still develop several types of extracolonic sequelae, including intestinal polyposis, especially in the duodenum, and other neoplasia, including desmoid tumors.
ApcMin/+ mice express increased levels of ODC1 RNA and polyamines in intestinal tissues, compared with normal littermates. Administration of DFMO alone is effective in suppressing carcinogenesis in the small intestines in these mice (30). Combinations of DFMO and NSAIDS are potent inhibitors of carcinogenesis in both the large and small intestines in this model of FAP (51, 62). Isobologram analysis of drug–drug interactions in human colon cancer-derived cells in culture indicates that DFMO and sulindac, or its metabolites, interact in at least an additive manner (63), supporting the conclusion depicted in Fig. 3 that the two agents are acting by complementary mechanisms. The grade of intestinal polyps is polyamine-dependent, and the anti-intestinal carcinogenic effects of sulindac in ApcMin/+ mice can be rescued by dietary putrescine (64). Together, these findings support a role for polyamines in intestinal carcinogenesis in ApcMin/+ mice and the rationale for combination DFMO and sulindac in therapy for this end point. A current clinical trial evaluating the DFMO sulindac combination for control of intestinal polyposis is in progress (55)
ApcMin/+ mice crossed with p53−/− mice show enhanced formation of desmoid tumors, which are another example of the extracolonic sequelae of loss of normal APC in germline. The combination of DFMO and the NSAID piroxicam exerted a moderate effect on development of desmoids in this model. This combination reduced formation of desmoids by nearly 50% (62), which was significant but less substantial than the effect this same combination exerted on intestinal carcinogenesis. These experimental findings suggest that the mechanisms depicted in Figs. 2 and 3 are likely operational in the colon, small intestine, fibroblasts (the source of desmoid tumors), and other tissues. A major unanswered question is whether DFMO alone or in combination with NSAIDs or other agents can be used effectively to manage desmoids in a clinically significant manner.
Mechanisms of cancer pharmacoprevention using drugs targeting the polyamine pathway
DFMO is a targeted therapy acting in a very selective manner to irreversibly inhibit a single enzyme (ODC1). There are limited data suggesting DFMO may have a modest effect on arginase (65). Sulindac is a much less selective drug, acting by both cyclooxygenase-dependent and -independent mechanisms. The anti-growth effects of DFMO are reversed by adding an extracellular source of polyamines, which can replete cellular pools via the transport processes depicted in Fig. 2. The anti-intestinal carcinogenic effects of sulindac can also be reversed by providing ApcMin/+ mice with putrescine in the drinking water (64), supporting the conclusion that sulindac suppresses tumor formation in these mice by a polyamine-dependent mechanism.
Although DFMO and NSAIDs appear to affect the levels of cell and tissue polyamines by influencing the activity and/or expression of specific polyamine metabolic proteins, the mechanisms by which polyamines affect carcinogenesis are more complicated. Evidence supporting some of these mechanisms are further discussed in terms of the Hallmarks of Cancer, as proposed by Hanahan and Weinberg (66, 67) and summarized in Fig. 4.
Figure 4.
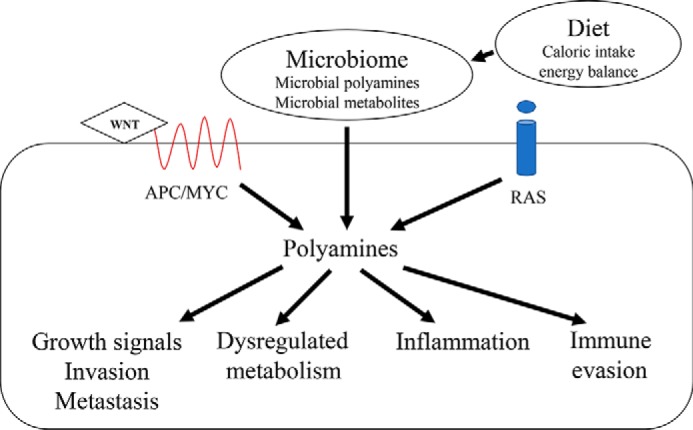
Role of polyamines in Hallmarks of Cancer. Polyamine metabolism is signaled by a number of pathways, including the APC-dependent mechanisms, as in FAP, dietary factors acting via RAS, and other pathways and the microbiome. Dysregulation of polyamine metabolism occurs when features of these pathways are altered (e.g. mutations in APC and RAS). The polyamines exert effects on a range of cell phenotypes, including Hallmarks of Cancer such as growth signals, invasion and metastasis, broad aspects of cell metabolism, inflammation, and immune responses.
Roles of polyamines in growth signaling, self-renewal, invasion, and metastasis
The original Hallmarks of Cancer proposed six capabilities that were general features of cancer. As seen in Figs. 2 and 4, unregulated expression of genes like MYC or RAS lead to the simple analogy of the “accelerator pedal stuck on,” where the polyamines are the accelerant. Mutation/deletion of genes like APC is loss of a “brake” on growth mediated by the polyamines.
Additionally, the polyamine metabolic genes ODC1 and adenosylmethionine decarboxylase (AMD1) have been implicated in the self-renewal of embryonic stem cells (ESCs) (68). AMD1 has been shown to be essential for differentiation of ESCs to neural precursor cells (69). Forced expression of either ODC1 or AMD1 is able to maintain patterns of ESC gene expression in the absence of inducers. Studies have not yet demonstrated a role for polyamines or polyamine metabolic genes in specific cancer stem cells. MYC is known to control the balance of self-renewal and differentiation in hematopoietic stem cells (70). Knockdown of c-MYC in the intestinal tract reduces intestinal carcinogenesis due to mutant APC (34), but intestinal homeostasis appears to occur in a MYC-independent manner (71). It remains an unanswered question whether known MYC transcriptional targets like the polyamine metabolic genes play any role in putative intestinal or colonic cancer stem cells.
Polyamines mediate other cancer hallmarks, including invasion (72) and metastasis (73). Dietary putrescine increases tissue polyamine contents and intestinal tumor grade when administered to ApcMin/+ mice (64). DFMO inhibits cell motility and migration and suppresses tumor-forming ability in colon tumor cells expressing an activated KRAS (37).
The distinction between “regulated” and “dysregulated” is an important nuance in interpreting the consequences of the polyamine pathway on cell/tissue phenotypes. The polyamines are associated with optimal growth in single and multicellular organisms, but their regulation is equally important. The polyamines are critical to good health in humans, and some diseases may be associated with age-related down-regulation of these molecules (74). In a different context, loss of regulation (or dysregulation) of this pathway, as depicted in Fig. 2 for patients with FAP, leads to severe pathological consequences, which are largely reversible by agents targeting the polyamine pathway in preclinical models (30, 51) and in initial clinical trials in humans (54). The distinction has significant consequences for possible therapeutic interventions. In the case of dysregulated metabolism, such as occurs in cancer, therapies will aim to reduce abnormally high polyamine levels. In the case of polyamine deficiencies associated with regulated metabolism, therapies will aim to increase abnormally low polyamines.
Roles of polyamines in deregulated metabolism
Deregulated cancer metabolism was added to the list of Hallmarks of Cancer by Hanahan and Weinberg in 2011 (67). Cancer metabolism has many facets (75), and polyamines appear to participate in several of these processes. Activation of SAT1 and polyamine acetylation suppresses tumor growth in the transgenic adenocarcinoma of the mouse prostate (TRAMP) model of prostate carcinogenesis (76), modulates polyamine metabolic flux, and leads to broad metabolic consequences in cell and rodent models (77, 78). Polyamine pools can affect the production of 5′-methylthioadenosine, via the S-adenosylmethionine salvage enzyme 5′-methylthioadenosine phosphorylase (MTAP) (79). MTAP has been implicated in several cancers, both as a tumor promoter (79) and suppressor (80). Inhibitors of MTAP suppress growth and metastasis of at least one model of human lung cancer (81).
Another facet of altered metabolism involving the polyamines is the LIN28/let-7 pathway, which is thought to play a key role in the regulation of self-renewal of both normal and cancer stem cells. LIN28 controls developmental timing in Caenorhabditis elegans (82) and glucose metabolism in mice (83). It cooperates with the WNT signaling pathway to promote invasive intestinal and colonic carcinogenesis (84). An unbiased screen of noncoding RNAs discovered that DFMO increased the steady-state levels of a number of microRNAs, including let-7, in human colon cancer–derived cells (85). The mechanism of this increase was due to DFMO-dependent decreases in cellular putrescine and spermidine pools and depletion of cellular amounts of hypusine-modified eukaryotic translation initiation factor 5A (eIF-5A). The hypusine modification uses spermidine as a substrate (see Minireview by Park and Wolff for details (108)). Genetic knockdown experiments indicated that eIF-5A was associated with expression of LIN28 protein, and depletion of eIF-5A isoforms was associated with decreases in LIN28 and increases in let-7 RNA. In neuroblastoma, let-7 is regulated by multiple mechanisms, including the MYC family member MYCN and LIN28. An unanswered question in this field is as follows: can low levels of let-7 RNA be increased using a small molecule like DFMO, or other drugs directed at the polyamine pathway, in clinically significant settings in humans?
Role of polyamines in inflammation
Inflammation is a process that may be essential for tumorigenesis (86). The polyamines have been implicated in inflammation in cancer in several ways. One mechanism involves polyamine oxidation with the generation of aldehydes and reactive oxygen species (ROS) (87–89). This topic is dealt with extensively in the Minireview by Casero and co-workers (109).
Ornithine, putrescine, and the polyamines are downstream products of arginine metabolism, which has been widely implicated in cancer and other inflammation-associated diseases (90). Arginases convert arginine to ornithine, and specific arginases have been implicated in H. pylori–associated gastric carcinogenesis by affecting host polyamine metabolism (91). DFMO inhibits arginine- and NOS2-dependent carcinogenesis in the ApcMin/+ mouse model of FAP (35).
A novel mechanism of polyamine action in inflammation involves the formation of neutrophil extracellular traps (NETs), which have been implicated in tumor cell migration and metastasis (92). NETs are composed of extracellular DNA, extruded by an active process, that coats pancreatic and other tumor cells. The tumor cells also produce and secrete the chemokine CXCL8. NETs capture, as a consequence of CXCL8, circulating neutrophils that also produce this chemokine and matrix metalloproteinases such as stromelysin. Together, these factors promote tumor invasion and metastasis. Polyamines are involved in the extrusion of DNA. Elevated polyamine levels lead to the formation of nuclear aggregates of polyamines, which are subsequently exported (93).
Treatment with the DNase of pancreatic cancer cells producing NETs suppresses measures of cell migration and motility. Similarly, treatment of mice injected with xenografts of pancreatic cancer cells producing NETs reduces the growth of the xenografts (92). It remains to be determined whether treatment of tumor xenografts in mouse models, which is inhibited by DFMO (37), does so by reducing the formation of NETs.
Roles of polyamines in anti-tumor immune responses
Polyamines are elevated in tumors, and inhibitors of polyamine metabolism exert their anti-tumor effects by acting on both tumor cells (intrinsic cancer cell mechanisms) and the tumor microenvironment (extrinsic mechanisms) (94). DFMO alone or in combination with the statin rosuvastatin reduced polyamines and enhanced natural killer cell activity associated with cancer prevention in the azoxymethane-induced model of colon carcinogenesis (95). Combination therapy with DFMO and a polyamine transport inhibitor reduced tumor cell polyamines and relieved immunosuppression in the microenvironment allowing activation of anti-tumor T-cells to reduce tumor growth in another mouse model of colon cancer (60). Polyamines have been broadly implicated in the function of normal immune cells (96). The effects of inhibitors of polyamine metabolism on tumor immunity and immune suppression may involve both cancer cell intrinsic and extrinsic mechanisms, as immune evasion may be tumor-induced in certain circumstances (97)
Role of the polyamines on the impact of diet and the microbiome on carcinogenesis
Microorganisms in the human GI tract (referred to here as the microbiome) are well established to impact colon carcinogenesis, and its role in other cancer types and diseases continues to be investigated (98). Of note, the microbiome is a rich source of exogenous polyamines and can contribute to the overall polyamine content in an organism in normal and disease settings. Through the microbiome and host sources, the GI is a rich source of polyamines to support tumor growth (99). For instance, the role of the microbiome in colon carcinogenesis has been shown in germ-free mice that fail to develop colon cancer in a TGFβ-deficient mouse model that is predisposed for cancer (100). Metabolomic analyses have identified increased levels of N1,N12-Ac2Spm in both cancer and normal host tissues of patients with bacterial biofilms (101). Treatment of patients with antibiotics indicates that levels of N1,N12-Ac2Spm are contributed by both the host and bacterial biofilm. Biofilms containing tumorigenic bacteria appear to affect colonic neoplasia at an early stage of carcinogenesis, based on studies of apparently normal colonic mucosa from patients with FAP (102). Urinary levels of N1,N12-Ac2Spm are highly statistically and significantly associated with occurrence of colon and breast cancer in humans (103).
Polyamines present in certain foods in the human diet have been estimated (104), and these estimates have been associated with risk of colon carcinogenesis in humans (105) and resistance to the DFMO-sulindac therapy for reducing metachronous colorectal adenomas in human clinical trials (106). It is unlikely, however, that polyamines in the diet act as sources for host tissue polyamine pools. Rather, the diet provides a source of a number of elements (e.g. energy, red meat) that impact the host via both direct metabolic mechanisms and indirect mechanisms mediated by the microbiome. The impact of diet on host polyamine metabolism, assessed by measuring urinary polyamine levels, indicated that these other factors, rather than the estimated dietary polyamine content, were more closely associated with urinary polyamine contents (107).
Conclusions
The polyamine pathway is an important contributor to both normal growth and development and specific pathologies, including neoplasia. Dysregulation of the polyamine pathway, as a consequence of tumor suppressor gene inactivation (e.g. APC in colon carcinogenesis) or oncogene activation (e.g. MYC in colon carcinogenesis or neuroblastoma), is causatively associated with carcinogenesis. Mechanisms of this association include the role of polyamines in normal and cancer stem cells, inflammation, immunity/immune evasion, the microbiome, and diet and affect both tumor and host. The relative importance of each of these mechanisms linking dysregulation of the polyamine pathway and carcinogenesis remains to be established in specific types of cancer. Clinical evidence indicates that such therapies can successfully reduce risk factors for certain cancers (e.g. colonic and intestinal polyposis for sporadic or genetic forms of colon cancer). A major challenge for the future is to determine whether these therapies can also reduce those specific cancers and decrease cancer-related deaths.
This article is part of a series on “Polyamines,” written in honor of Dr. Herbert Tabor's 100th birthday. E. W. G. is a co-founder, board member, and Chief Scientific Officer (CSO) of Cancer Prevention Pharmaceuticals (CPP), Inc. E. B. is Vice President for Drug Development, and A. C. is Chief Medical Officer at Cancer Prevention Pharmaceuticals.
- DFMO
- difluoromethylornithine
- APC
- adenomatous polyposis coli
- GI
- gastrointestinal
- NSAID
- nonsteroidal anti-inflammatory drug
- FAP
- familial adenomatous polyposis
- IEN
- intraepithelial neoplasia
- CRC
- colorectal cancer
- CRA
- colorectal adenoma
- OAZ
- ornithine decarboxylase inhibitory protein antizyme
- TGFβ
- transforming growth factor β
- PPARγ
- peroxisomal proliferator-activated receptor γ
- PTI
- polyamine transport inhibitor
- ESC
- embryonic stem cell
- MTAP
- 5′-methylthioadenosine phosphorylase
- NET
- neutrophil extracellular trap
- ODC
- ornithine decarboxylase.
References
- 1. Tabor C. W., and Tabor H. (1999) It all started on a streetcar in Boston. Annu. Rev. Biochem. 68, 1–32 10.1146/annurev.biochem.68.1.1 [DOI] [PubMed] [Google Scholar]
- 2. Dykstra W. G. Jr., and Herbst E. J. (1965) Spermidine in regenerating liver: relation to rapid synthesis of ribonucleic acid. Science 149, 428–429 10.1126/science.149.3682.428 [DOI] [PubMed] [Google Scholar]
- 3. Raina A., Jänne J., and Siimes M. (1966) Stimulation of polyamine synthesis in relation to nucleic acids in regenerating rat liver. Biochim. Biophys. Acta 123, 197–201 10.1016/0005-2787(66)90173-0 [DOI] [PubMed] [Google Scholar]
- 4. Russell D., and Snyder S. H. (1968) Amine synthesis in rapidly growing tissues: ornithine decarboxylase activity in regenerating rat liver, chick embryo, and various tumors. Proc. Natl. Acad. Sci. U.S.A. 60, 1420–1427 10.1073/pnas.60.4.1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. O'Brien T. G., Simsiman R. C., and Boutwell R. K. (1975) Induction of the polyamine-biosynthetic enzymes in mouse epidermis by tumor-promoting agents. Cancer Res. 35, 1662–1670 [PubMed] [Google Scholar]
- 6. Hafner E. W., Tabor C. W., and Tabor H. (1979) Mutants of Escherichia coli that do not contain 1,4-diaminobutane (putrescine) or spermidine. J. Biol. Chem. 254, 12419–12426 [PubMed] [Google Scholar]
- 7. Tabor H., Hafner E. W., and Tabor C. W. (1980) Construction of an Escherichia coli strain unable to synthesize putrescine, spermidine, or cadaverine: characterization of two genes controlling lysine decarboxylase. J. Bacteriol. 144, 952–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tabor H., Tabor C. W., Cohn M. S., and Hafner E. W. (1981) Streptomycin resistance (rpsL) produces an absolute requirement for polyamines for growth of an Escherichia coli strain unable to synthesize putrescine and spermidine [Δ(speA-speB)ΔspecC]. J. Bacteriol. 147, 702–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tabor C. W., and Tabor H. (1984) Polyamines. Annu. Rev. Biochem. 53, 749–790 10.1146/annurev.bi.53.070184.003533 [DOI] [PubMed] [Google Scholar]
- 10. Tabor C. W., Tabor H., Tyagi A. K., and Cohn M. S. (1982) The biochemistry, genetics, and regulation of polyamine biosynthesis in Saccharomyces cerevisiae. Fed. Proc. 41, 3084–3088 [PubMed] [Google Scholar]
- 11. Metcalf B. W., Bey P., Danzin C., Jung M. J., Cagara P., and Ververt J. P. (1978) Catalytic irreversible inhibition of mammalian ornithine decarboxylase (E.C. 4.1.1.17) by substrate and product analogues. J. Am. Chem. Soc. 100, 2551–2553 10.1021/ja00476a050 PMID not found. [DOI] [Google Scholar]
- 12. Mamont P. S., Duchesne M. C., Grove J., and Bey P. (1978) Anti-proliferative properties of dl-α-difluoromethyl ornithine in cultured cells. A consequence of the irreversible inhibition of ornithine decarboxylase. Biochem. Biophys. Res. Commun. 81, 58–66 10.1016/0006-291X(78)91630-3 [DOI] [PubMed] [Google Scholar]
- 13. Prakash N. J., Schechter P. J., Mamont P. S., Grove J., Koch-Weser J., and Sjoerdsma A. (1980) Inhibition of EMT6 tumor growth by interference with polyamine biosynthesis; effects of α-difluoromethylornithine, an irreversible inhibitor of ornithine decarboxylase. Life Sci. 26, 181–194 10.1016/0024-3205(80)90292-1 [DOI] [PubMed] [Google Scholar]
- 14. Bartholeyns J., Mamont P., and Casara P. (1984) Antitumor properties of (2R,5R)-6-heptyne-2,5-diamine, a new potent enzyme-activated irreversible inhibitor of ornithine decarboxylase, in rodents. Cancer Res. 44, 4972–4977 [PubMed] [Google Scholar]
- 15. Weeks C. E., Herrmann A. L., Nelson F. R., and Slaga T. J. (1982) α-Difluoromethylornithine, an irreversible inhibitor of ornithine decarboxylase, inhibits tumor promoter-induced polyamine accumulation and carcinogenesis in mouse skin. Proc. Natl. Acad. Sci. U.S.A. 79, 6028–6032 10.1073/pnas.79.19.6028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kingsnorth A. N., King W. W., Diekema K. A., McCann P. P., Ross J. S., and Malt R. A. (1983) Inhibition of ornithine decarboxylase with 2-difluoromethylornithine: reduced incidence of dimethylhydrazine-induced colon tumors in mice. Cancer Res. 43, 2545–2549 [PubMed] [Google Scholar]
- 17. Von Hoff D. D. (1998) There are no bad anticancer agents, only bad clinical trial designs–twenty-first Richard and Hinda Rosenthal Foundation Award Lecture. Clin. Cancer Res. 4, 1079–1086 [PubMed] [Google Scholar]
- 18. Nightingale S. L. (1991) From the food and drug administration. JAMA 265, 1229 10.1001/jama.1991.03460100029008 [DOI] [PubMed] [Google Scholar]
- 19. Balfour J. A., and McClellan K. (2001) Topical eflornithine. Am. J. Clin. Dermatol. 2, 197–201 [DOI] [PubMed] [Google Scholar]
- 20. US Burden of Disease Collaborators, Mokdad A. H., Ballestros K., Echko M., Glenn S., Olsen H. E., Mullany E., Lee A., Khan A. R., Ahmadi A., Ferrari A. J., Kasaeian A., Werdecker A., Carter A., Zipkin B., et al. (2018) The State of US Health, 1990–2016: Burden of diseases, injuries, and risk factors among US states. JAMA 319, 1444–1472 10.1001/jama.2018.0158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ford E. S., Ajani U. A., Croft J. B., Critchley J. A., Labarthe D. R., Kottke T. E., Giles W. H., and Capewell S. (2007) Explaining the decrease in U.S. deaths from coronary disease, 1980–2000. N. Engl. J. Med. 356, 2388–2398 10.1056/NEJMsa053935 [DOI] [PubMed] [Google Scholar]
- 22. O'Shaughnessy J. A., Kelloff G. J., Gordon G. B., Dannenberg A. J., Hong W. K., Fabian C. J., Sigman C. C., Bertagnolli M. M., Stratton S. P., Lam S., Nelson W. G., Meyskens F. L., Alberts D. S., Follen M., Rustgi A. K., et al. (2002) Treatment and prevention of intraepithelial neoplasia: an important target for accelerated new agent development. Clin. Cancer Res. 8, 314–346 [PubMed] [Google Scholar]
- 23. Winawer S. J., Zauber A. G., Ho M. N., O'Brien M. J., Gottlieb L. S., Sternberg S. S., Waye J. D., Schapiro M., Bond J. H., and Panish J. F. (1993) Prevention of colorectal cancer by colonoscopic polypectomy. The National Polyp Study Workgroup. N. Engl. J. Med. 329, 1977–1981 10.1056/NEJM199312303292701 [DOI] [PubMed] [Google Scholar]
- 24. Baxter N. N., Goldwasser M. A., Paszat L. F., Saskin R., Urbach D. R., and Rabeneck L. (2009) Association of colonoscopy and death from colorectal cancer. Ann. Intern. Med. 150, 1–8 10.7326/0003-4819-150-1-200901060-00306 [DOI] [PubMed] [Google Scholar]
- 25. Lux G. D., Marton L. J., and Baylin S. B. (1980) Ornithine decarboxylase is important in intestinal mucosal maturation and recovery from injury in rats. Science 210, 195–198 10.1126/science.6774420 [DOI] [PubMed] [Google Scholar]
- 26. Luk G. D., and Baylin S. B. (1984) Ornithine decarboxylase as a biologic marker in familial colonic polyposis. N. Engl. J. Med. 311, 80–83 10.1056/NEJM198407123110202 [DOI] [PubMed] [Google Scholar]
- 27. Groden J., Thliveris A., Samowitz W., Carlson M., Gelbert L., Albertsen H., Joslyn G., Stevens J., Spirio L., and Robertson M. (1991) Identification and characterization of the familial adenomatous polyposis coli gene. Cell 66, 589–600 10.1016/0092-8674(81)90021-0 [DOI] [PubMed] [Google Scholar]
- 28. Giardiello F. M., Hamilton S. R., Hylind L. M., Yang V. W., Tamez P., and Casero R. A. Jr. (1997) Ornithine decarboxylase and polyamines in familial adenomatous polyposis. Cancer Res. 57, 199–201 [PubMed] [Google Scholar]
- 29. Su L. K., Kinzler K. W., Vogelstein B., Preisinger A. C., Moser A. R., Luongo C., Gould K. A., and Dove W. F. (1992) Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 256, 668–670 10.1126/science.1350108 [DOI] [PubMed] [Google Scholar]
- 30. Erdman S. H., Ignatenko N. A., Powell M. B., Blohm-Mangone K. A., Holubec H., Guillén-Rodriguez J. M., and Gerner E. W. (1999) APC-dependent changes in expression of genes influencing polyamine metabolism, and consequences for gastrointestinal carcinogenesis, in the Min mouse. Carcinogenesis 20, 1709–1713 10.1093/carcin/20.9.1709 [DOI] [PubMed] [Google Scholar]
- 31. He T. C., Sparks A. B., Rago C., Hermeking H., Zawel L., da Costa L. T., Morin P. J., Vogelstein B., and Kinzler K. W. (1998) Identification of c-MYC as a target of the APC pathway. Science 281, 1509–1512 10.1126/science.281.5382.1509 [DOI] [PubMed] [Google Scholar]
- 32. Bello-Fernandez C., Packham G., and Cleveland J. L. (1993) The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc. Natl. Acad. Sci. U.S.A. 90, 7804–7808 10.1073/pnas.90.16.7804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fultz K. E., and Gerner E. W. (2002) APC-dependent regulation of ornithine decarboxylase in human colon tumor cells. Mol. Carcinog. 34, 10–18 10.1002/mc.10043 [DOI] [PubMed] [Google Scholar]
- 34. Ignatenko N. A., Holubec H., Besselsen D. G., Blohm-Mangone K. A., Padilla-Torres J. L., Nagle R. B., de Alboránç I. M., Guillen-R. J. M., and Gerner E. W. (2006) Role of c-Myc in intestinal tumorigenesis of the ApcMin/+ mouse. Cancer Biol. Ther. 5, 1658–1664 10.4161/cbt.5.12.3376 [DOI] [PubMed] [Google Scholar]
- 35. Yerushalmi H. F., Besselsen D. G., Ignatenko N. A., Blohm-Mangone K. A., Padilla-Torres J. L., Stringer D. E., Guillen J. M., Holubec H., Payne C. M., and Gerner E. W. (2006) Role of polyamines in arginine-dependent colon carcinogenesis in Apc(Min) (/+) mice. Mol. Carcinog. 45, 764–773 10.1002/mc.20246 [DOI] [PubMed] [Google Scholar]
- 36. Green J. E., and Hudson T. (2005) The promise of genetically engineered mice for cancer prevention studies. Nat. Rev. Cancer 5, 184–198 10.1038/nrc1565 [DOI] [PubMed] [Google Scholar]
- 37. Ignatenko N. A., Zhang H., Watts G. S., Skovan B. A., Stringer D. E., and Gerner E. W. (2004) The chemopreventive agent α-difluoromethylornithine blocks Ki-ras-dependent tumor formation and specific gene expression in Caco-2 cells. Mol. Carcinog. 39, 221–233 10.1002/mc.20008 [DOI] [PubMed] [Google Scholar]
- 38. Daniel S. G., Ball C. L., Besselsen D. G., Doetschman T., and Hurwitz B. L. (2017) Functional changes in the gut microbiome contribute to transforming growth factor β-deficient colon cancer. mSystems 2, e00065–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meyskens F. L. Jr., and Gerner E. W. (1999) Development of difluoromethylornithine (DFMO) as a chemoprevention agent. Clin. Cancer Res. 5, 945–951 [PubMed] [Google Scholar]
- 40. Sporn M. B. (1980) Combination chemoprevention of cancer. Nature 287, 107–108 10.1038/287107a0 [DOI] [PubMed] [Google Scholar]
- 41. Giardiello F. M., Hamilton S. R., Krush A. J., Piantadosi S., Hylind L. M., Celano P., Booker S. V., Robinson C. R., and Offerhaus G. J. (1993) Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N. Engl. J. Med. 328, 1313–1316 10.1056/NEJM199305063281805 [DOI] [PubMed] [Google Scholar]
- 42. Nigro N. D., Bull A. W., and Boyd M. E. (1986) Inhibition of intestinal carcinogenesis in rats: effect of difluoromethylornithine with piroxicam or fish oil. J. Natl. Cancer Inst. 77, 1309–1313 [PubMed] [Google Scholar]
- 43. Abou-el-Ela S. H., Prasse K. W., Farrell R. L., Carroll R. W., Wade A. E., and Bunce O. R. (1989) Effects of dl-2-difluoromethylornithine and indomethacin on mammary tumor promotion in rats fed high n-3 and/or n-6 fat diets. Cancer Res. 49, 1434–1440 [PubMed] [Google Scholar]
- 44. Li H., Schut H. A., Conran P., Kramer P. M., Lubet R. A., Steele V. E., Hawk E. E., Kelloff G. J., and Pereira M. A. (1999) Prevention by aspirin and its combination with α-difluoromethylornithine of azoxymethane-induced tumors, aberrant crypt foci and prostaglandin E2 levels in rat colon. Carcinogenesis 20, 425–430 10.1093/carcin/20.3.425 [DOI] [PubMed] [Google Scholar]
- 45. Gurpinar E., Grizzle W. E., and Piazza G. A. (2013) COX-independent mechanisms of cancer chemoprevention by anti-inflammatory drugs. Front. Oncol. 3, 181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Verma A. K., Ashendel C. L., and Boutwell R. K. (1980) Inhibition by prostaglandin synthesis inhibitors of the induction of epidermal ornithine decarboxylase activity, the accumulation of prostaglandins, and tumor promotion caused by 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 40, 308–315 [PubMed] [Google Scholar]
- 47. Babbar N., Ignatenko N. A., Casero R. A. Jr, and Gerner E. W. (2003) Cyclooxygenase-independent induction of apoptosis by sulindac sulfone is mediated by polyamines in colon cancer. J. Biol. Chem. 278, 47762–47775 10.1074/jbc.M307265200 [DOI] [PubMed] [Google Scholar]
- 48. Xie X., Gillies R. J., and Gerner E. W. (1997) Characterization of a diamine exporter in Chinese hamster ovary cells and identification of specific polyamine substrates. J. Biol. Chem. 272, 20484–20489 10.1074/jbc.272.33.20484 [DOI] [PubMed] [Google Scholar]
- 49. Babbar N., Gerner E. W., and Casero R. A. Jr. (2006) Induction of spermidine/spermine N1-acetyltransferase (SSAT) by aspirin in Caco-2 colon cancer cells. Biochem. J. 394, 317–324 10.1042/BJ20051298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gerner E. W., and Meyskens F. L. Jr. (2004) Polyamines and cancer: old molecules, new understanding. Nat. Rev. Cancer 4, 781–792 10.1038/nrc1454 [DOI] [PubMed] [Google Scholar]
- 51. Ignatenko N. A., Besselsen D. G., Stringer D. E., Blohm-Mangone K. A., Cui H., and Gerner E. W. (2008) Combination chemoprevention of intestinal carcinogenesis in a murine model of familial adenomatous polyposis. Nutr. Cancer 60, Suppl. 1, 30–35 10.1080/01635580802401317 [DOI] [PubMed] [Google Scholar]
- 52. Meyskens F. L., McLaren C. E., Pelot D., Fujikawa-Brooks S., Carpenter P. M., Hawk E., Kelloff G., Lawson M. J., Kidao J., McCracken J., Albers C. G., Ahnen D. J., Turgeon D. K., Goldschmid S., Lance P., et al. (2008) Difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: a randomized placebo controlled, double-blind trial. Cancer Prev. Res. 1, 32–38 10.1158/1940-6207.CAPR-08-0042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Thompson P. A., Wertheim B. C., Zell J. A., Chen W. P., McLaren C. E., LaFleur B. J., Meyskens F. L., and Gerner E. W. (2010) Levels of rectal mucosal polyamines and prostaglandin E2 predict ability of DFMO and sulindac to prevent colorectal adenoma. Gastroenterology 139, 797–805, 805.e1 10.1053/j.gastro.2010.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lynch P. M., Burke C. A., Phillips R., Morris J. S., Slack R., Wang X., Liu J., Patterson S., Sinicrope F. A., Rodriguez-Bigas M. A., Half E., Bulow S., Latchford A., Clark S., Ross W. A., et al. (2016) An international randomised trial of celecoxib versus celecoxib plus difluoromethylornithine in patients with familial adenomatous polyposis. Gut 65, 286–295 10.1136/gutjnl-2014-307235 [DOI] [PubMed] [Google Scholar]
- 55. Burke C. A., Dekker E., Samadder N. J., Stoffel E., and Cohen A. (2016) Efficacy and safety of eflornithine (CPP-1X)/sulindac combination therapy versus each as monotherapy in patients with familial adenomatous polyposis (FAP): design and rationale of a randomized, double-blind, Phase III trial. BMC Gastroenterol. 16, 87 10.1186/s12876-016-0494-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Burns M. R., Graminski G. F., Weeks R. S., Chen Y., and O'Brien T. G. (2009) Lipophilic lysine-spermine conjugates are potent polyamine transport inhibitors for use in combination with a polyamine biosynthesis inhibitor. J. Med. Chem. 52, 1983–1993 10.1021/jm801580w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gardner R. A., Delcros J. G., Konate F., Breitbeil F. 3rd., Martin B., Sigman M., Huang M., and Phanstiel O. 4th (2004) N1-substituent effects in the selective delivery of polyamine conjugates into cells containing active polyamine transporters. J. Med. Chem. 47, 6055–6069 10.1021/jm0497040 [DOI] [PubMed] [Google Scholar]
- 58. Muth A., Madan M., Archer J. J., Ocampo N., Rodriguez L., and Phanstiel O. 4th (2014) Polyamine transport inhibitors: design, synthesis, and combination therapies with difluoromethylornithine. J. Med. Chem. 57, 348–363 10.1021/jm401174a [DOI] [PubMed] [Google Scholar]
- 59. Samal K., Zhao P., Kendzicky A., Yco L. P., McClung H., Gerner E., Burns M., Bachmann A. S., and Sholler G. (2013) AMXT-1501, a novel polyamine transport inhibitor, synergizes with DFMO in inhibiting neuroblastoma cell proliferation by targeting both ornithine decarboxylase and polyamine transport. Int. J. Cancer 133, 1323–1333 10.1002/ijc.28139 [DOI] [PubMed] [Google Scholar]
- 60. Alexander E. T., Minton A., Peters M. C., Phanstiel O. 4th, and Gilmour S. K. (2017) A novel polyamine blockade therapy activates an anti-tumor immune response. Oncotarget 8, 84140–84152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gitto S. B., Pandey V., Oyer J. L., Copik A. J., Hogan F. C., Phanstiel O. 4th, and Altomare D. A. (2018) Difluoromethylornithine combined with a polyamine transport inhibitor is effective against gemcitabine resistant pancreatic cancer. Mol. Pharm. 15, 369–376 10.1021/acs.molpharmaceut.7b00718 [DOI] [PubMed] [Google Scholar]
- 62. Halberg R. B., Katzung D. S., Hoff P. D., Moser A. R., Cole C. E., Lubet R. A., Donehower L. A., Jacoby R. F., and Dove W. F. (2000) Tumorigenesis in the multiple intestinal neoplasia mouse: redundancy of negative regulators and specificity of modifiers. Proc. Natl. Acad. Sci. U.S.A. 97, 3461–3466 10.1073/pnas.97.7.3461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lawson K. R., Ignatenko N. A., Piazza G. A., Cui H., and Gerner E. W. (2000) Influence of K-ras activation on the survival responses of Caco-2 cells to the chemopreventive agents sulindac and difluoromethylornithine. Cancer Epidemiol. Biomarkers Prev. 9, 1155–1162 [PubMed] [Google Scholar]
- 64. Ignatenko N. A., Besselsen D. G., Roy U. K., Stringer D. E., Blohm-Mangone K. A., Padilla-Torres J. L., Guillen-R J. M., and Gerner E. W. (2006) Dietary putrescine reduces the anti-carcinogenic intestinal activity of sulindac in a murine model of familial adenomatous polyposis. Nutr. Cancer 56, 172–181 10.1207/s15327914nc5602_8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Selamnia M., Mayeur C., Robert V., and Blachier F. (1998) α-Difluoromethylornithine (DFMO) as a potent arginase activity inhibitor in human colon carcinoma cells. Biochem. Pharmacol. 55, 1241–1245 10.1016/S0006-2952(97)00572-8 [DOI] [PubMed] [Google Scholar]
- 66. Hanahan D., and Weinberg R. A. (2000) The hallmarks of cancer. Cell 100, 57–70 10.1016/S0092-8674(00)81683-9 [DOI] [PubMed] [Google Scholar]
- 67. Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 68. Zhao T., Goh K. J., Ng H. H., and Vardy L. A. (2012) A role for polyamine regulators in ESC self-renewal. Cell Cycle 11, 4517–4523 10.4161/cc.22772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang D., Zhao T., Ang H. S., Chong P., Saiki R., Igarashi K., Yang H., and Vardy L. A. (2012) AMD1 is essential for ESC self-renewal and is translationally down-regulated on differentiation to neural precursor cells. Genes Dev. 26, 461–473 10.1101/gad.182998.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wilson A., Murphy M. J., Oskarsson T., Kaloulis K., Bettess M. D., Oser G. M., Pasche A. C., Knabenhans C., Macdonald H. R., and Trumpp A. (2004) c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 18, 2747–2763 10.1101/gad.313104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bettess M. D., Dubois N., Murphy M. J., Dubey C., Roger C., Robine S., and Trumpp A. (2005) c-Myc is required for the formation of intestinal crypts but dispensable for homeostasis of the adult intestinal epithelium. Mol. Cell. Biol. 25, 7868–7878 10.1128/MCB.25.17.7868-7878.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wallon U. M., Shassetz L. R., Cress A. E., Bowden G. T., and Gerner E. W. (1994) Polyamine-dependent expression of the matrix metalloproteinase matrilysin in a human colon cancer-derived cell line. Mol. Carcinog. 11, 138–144 10.1002/mc.2940110304 [DOI] [PubMed] [Google Scholar]
- 73. Sunkara P. S., and Rosenberger A. L. (1987) Antimetastatic activity of dl-α-difluoromethylornithine, an inhibitor of polyamine biosynthesis, in mice. Cancer Res. 47, 933–935 [PubMed] [Google Scholar]
- 74. Madeo F., Eisenberg T., Pietrocola F., and Kroemer G. (2018) Spermidine in health and disease. Science 359, pii: eaan2788 10.1126/science.aan2788 [DOI] [PubMed] [Google Scholar]
- 75. Cantor J. R., and Sabatini D. M. (2012) Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2, 881–898 10.1158/2159-8290.CD-12-0345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kee K., Foster B. A., Merali S., Kramer D. L., Hensen M. L., Diegelman P., Kisiel N., Vujcic S., Mazurchuk R. V., and Porter C. W. (2004) Activated polyamine catabolism depletes acetyl-CoA pools and suppresses prostate tumor growth in TRAMP mice. J. Biol. Chem. 279, 40076–40083 10.1074/jbc.M406002200 [DOI] [PubMed] [Google Scholar]
- 77. Kramer D. L., Diegelman P., Jell J., Vujcic S., Merali S., and Porter C. W. (2008) Polyamine acetylation modulates polyamine metabolic flux, a prelude to broader metabolic consequences. J. Biol. Chem. 283, 4241–4251 10.1074/jbc.M706806200 [DOI] [PubMed] [Google Scholar]
- 78. Jell J., Merali S., Hensen M. L., Mazurchuk R., Spernyak J. A., Diegelman P., Kisiel N. D., Barrero C., Deeb K. K., Alhonen L., Patel M. S., and Porter C. W. (2007) Genetically altered expression of spermidine/spermine N1-acetyltransferase affects fat metabolism in mice via acetyl-CoA. J. Biol. Chem. 282, 8404–8413 10.1074/jbc.M610265200 [DOI] [PubMed] [Google Scholar]
- 79. Bistulfi G., Affronti H. C., Foster B. A., Karasik E., Gillard B., Morrison C., Mohler J., Phillips J. G., and Smiraglia D. J. (2016) The essential role of methylthioadenosine phosphorylase in prostate cancer. Oncotarget 7, 14380–14393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tang B., Lee H. O., An S. S., Cai K. Q., and Kruger W. D. (2018) Specific targeting of MTAP-deleted tumors with a combination of 2′-fluoroadenine and 5′-methylthioadenosine. Cancer Res. 78, 4386–4395 10.1158/0008-5472.CAN-18-0814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Basu I., Locker J., Cassera M. B., Belbin T. J., Merino E. F., Dong X., Hemeon I., Evans G. B., Guha C., and Schramm V. L. (2011) Growth and metastases of human lung cancer are inhibited in mouse xenografts by a transition state analogue of 5′-methylthioadenosine phosphorylase. J. Biol. Chem. 286, 4902–4911 10.1074/jbc.M110.198374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Moss E. G., Lee R. C., and Ambros V. (1997) The cold shock domain protein LIN-28 controls developmental timing in C. elegans and is regulated by the lin-4 RNA. Cell 88, 637–646 10.1016/S0092-8674(00)81906-6 [DOI] [PubMed] [Google Scholar]
- 83. Zhu H., Shyh-Chang N., Segrè A. V., Shinoda G., Shah S. P., Einhorn W. S., Takeuchi A., Engreitz J. M., Hagan J. P., Kharas M. G., Urbach A., Thornton J. E., Triboulet R., Gregory R. I., DIAGRAM Consortium, et al. (2011) The Lin28/let-7 axis regulates glucose metabolism. Cell 147, 81–94 10.1016/j.cell.2011.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tu H. C., Schwitalla S., Qian Z., LaPier G. S., Yermalovich A., Ku Y. C., Chen S. C., Viswanathan S. R., Zhu H., Nishihara R., Inamura K., Kim S. A., Morikawa T., Mima K., Sukawa Y., et al. (2015) LIN28 cooperates with WNT signaling to drive invasive intestinal and colorectal adenocarcinoma in mice and humans. Genes Dev. 29, 1074–1086 10.1101/gad.256693.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Paz E. A., LaFleur B., and Gerner E. W. (2014) Polyamines are oncometabolites that regulate the LIN28/let-7 pathway in colorectal cancer cells. Mol. Carcinog. 53, Suppl. 1, E96–E106 10.1002/mc.22051 [DOI] [PubMed] [Google Scholar]
- 86. Coussens L. M., and Werb Z. (2002) Inflammation and cancer. Nature 420, 860–867 10.1038/nature01322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Babbar N., Murray-Stewart T., and Casero R. A. Jr. (2007) Inflammation and polyamine catabolism: the good, the bad and the ugly. Biochem. Soc. Trans. 35, 300–304 10.1042/BST0350300 [DOI] [PubMed] [Google Scholar]
- 88. Babbar N., and Gerner E. W. (2011) Targeting polyamines and inflammation for cancer prevention. Recent Results Cancer Res. 188, 49–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Chaturvedi R., de Sablet T., Asim M., Piazuelo M. B., Barry D. P., Verriere T. G., Sierra J. C., Hardbower D. M., Delgado A. G., Schneider B. G., Israel D. A., Romero-Gallo J., Nagy T. A., Morgan D. R., Murray-Stewart T., et al. (2015) Increased Helicobacter pylori-associated gastric cancer risk in the Andean region of Colombia is mediated by spermine oxidase. Oncogene 34, 3429–3440 10.1038/onc.2014.273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gogoi M., Datey A., Wilson K. T., and Chakravortty D. (2016) Dual role of arginine metabolism in establishing pathogenesis. Curr. Opin. Microbiol. 29, 43–48 10.1016/j.mib.2015.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Hardbower D. M., Asim M., Murray-Stewart T., Casero R. A. Jr., Verriere T., Lewis N. D., Chaturvedi R., Piazuelo M. B., and Wilson K. T. (2016) Arginase 2 deletion leads to enhanced M1 macrophage activation and upregulated polyamine metabolism in response to Helicobacter pylori infection. Amino Acids 48, 2375–2388 10.1007/s00726-016-2231-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wen F., Shen A., Choi A., Gerner E. W., and Shi J. (2013) Extracellular DNA in pancreatic cancer promotes cell invasion and metastasis. Cancer Res. 73, 4256–4266 10.1158/0008-5472.CAN-12-3287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Brooks W. H. (2013) Increased polyamines alter chromatin and stabilize autoantigens in autoimmune diseases. Front. Immunol. 4, 91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Bassiri H., Benavides A., Haber M., Gilmour S. K., Norris M. D., and Hogarty M. D. (2015) Translational development of difluoromethylornithine (DFMO) for the treatment of neuroblastoma. Transl. Pediatr. 4, 226–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Janakiram N. B., Mohammed A., Bryant T., Zhang Y., Brewer M., Duff A., Biddick L., Singh A., Lightfoot S., Steele V. E., and Rao C. V. (2016) Potentiating NK cell activity by combination of rosuvastatin and difluoromethylornithine for effective chemopreventive efficacy against colon cancer. Sci. Rep. 6, 37046 10.1038/srep37046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Hesterberg R. S., Cleveland J. L., and Epling-Burnette P. K. (2018) Role of polyamines in immune cell functions. Med. Sci. 6, E22 10.3390/medsci6010022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Grivennikov S. I., Greten F. R., and Karin M. (2010) Immunity, inflammation, and cancer. Cell 140, 883–899 10.1016/j.cell.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Cani P. D. (2018) Human gut microbiome: hopes, threats and promises. Gut 67, 1716–1725 10.1136/gutjnl-2018-316723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Sarhan S., Knodgen B., and Seiler N. (1989) The gastrointestinal tract as polyamine source for tumor growth. Anticancer Res. 9, 215–223 [PubMed] [Google Scholar]
- 100. Engle S. J., Ormsby I., Pawlowski S., Boivin G. P., Croft J., Balish E., and Doetschman T. (2002) Elimination of colon cancer in germ-free transforming growth factor β1-deficient mice. Cancer Res. 62, 6362–6366 [PubMed] [Google Scholar]
- 101. Johnson C. H., Dejea C. M., Edler D., Hoang L. T., Santidrian A. F., Felding B. H., Ivanisevic J., Cho K., Wick E. C., Hechenbleikner E. M., Uritboonthai W., Goetz L., Casero R. A. Jr., Pardoll D. M., White J. R., et al. (2015) Metabolism links bacterial biofilms and colon carcinogenesis. Cell Metabolism 21, 891–897 10.1016/j.cmet.2015.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Dejea C. M., Fathi P., Craig J. M., Boleij A., Taddese R., Geis A. L., Wu X., DeStefano Shields C. E., Hechenbleikner E. M., Huso D. L., Anders R. A., Giardiello F. M., Wick E. C., Wang H., Wu S., et al. (2018) Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 359, 592–597 10.1126/science.aah3648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Hiramatsu K., Takahashi K., Yamaguchi T., Matsumoto H., Miyamoto H., Tanaka S., Tanaka C., Tamamori Y., Imajo M., Kawaguchi M., Toi M., Mori T., and Kawakita M. (2005) N(1),N(12)-Diacetylspermine as a sensitive and specific novel marker for early- and late-stage colorectal and breast cancers. Clin. Cancer Res. 11, 2986–2990 10.1158/1078-0432.CCR-04-2275 [DOI] [PubMed] [Google Scholar]
- 104. Zoumas-Morse C., Rock C. L., Quintana E. L., Neuhouser M. L., Gerner E. W., and Meyskens F. L. Jr. (2007) Development of a polyamine database for assessing dietary intake. J. Am. Diet. Assoc. 107, 1024–1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Vargas A. J., Wertheim B. C., Gerner E. W., Thomson C. A., Rock C. L., and Thompson P. A. (2012) Dietary polyamine intake and risk of colorectal adenomatous polyps. Am. J. Clin. Nutr. 96, 133–141 10.3945/ajcn.111.030353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Raj K. P., Zell J. A., Rock C. L., McLaren C. E., Zoumas-Morse C., Gerner E. W., and Meyskens F. L. (2013) Role of dietary polyamines in a phase III clinical trial of difluoromethylornithine (DFMO) and sulindac for prevention of sporadic colorectal adenomas. Br. J. Cancer 108, 512–518 10.1038/bjc.2013.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Vargas A. J., Ashbeck E. L., Thomson C. A., Gerner E. W., and Thompson P. A. (2014) Dietary polyamine intake and polyamines measured in urine. Nutr. Cancer 66, 1144–1153 10.1080/01635581.2014.949801 [DOI] [PubMed] [Google Scholar]
- 108. Park M. H., and Wolff E. C. (2018) Hypusine, a polyamine-derived amino acid critical for eukaryotic translation. J. Biol. Chem. 293, 18710–18718 10.1074/jbc.TM118.003341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Murray Stewart T., Dunston T. T., Woster P. M., and Casero R. A. Jr. (2018) Polyamine catabolism and oxidative damage. J. Biol. Chem. 293, 18736–18745 10.1074/jbc.TM118.003337 [DOI] [PMC free article] [PubMed] [Google Scholar]