

Abstract
The endoplasmic reticulum (ER) is composed of flattened sheets and interconnected tubules that extend throughout the cytosol and makes physical contact with all other cytoplasmic organelles. This cytoplasmic distribution requires continuous remodeling. These discrete ER morphologies require specialized proteins that drive and maintain membrane curvature. The GTPase atlastin is required for homotypic fusion of ER tubules. All atlastin homologs possess a conserved domain architecture consisting of a GTPase domain, a three-helix bundle middle domain, a hydrophobic membrane anchor, and a C-terminal cytosolic tail. Here, we examined several Drosophila-human atlastin chimeras to identify functional domains of human atlastin-1 in vitro. Although all chimeras could hydrolyze GTP, only chimeras containing the human C-terminal tail, hydrophobic segments, or both could fuse membranes in vitro. We also determined that co-reconstitution of atlastin with reticulon does not influence GTPase activity or membrane fusion. Finally, we found that both human and Drosophila atlastin hydrophobic membrane anchors do not span the membrane, but rather form two intramembrane hairpin loops. The topology of these hairpins remains static during membrane fusion and does not appear to play an active role in lipid mixing.
Keywords: transmembrane domain, cell compartmentalization, membrane fusion, GTPase, endoplasmic reticulum (ER), intramembrane hairpin loop, SPG3A, FRET, phospholipid vesicle, neurodegeneration
Introduction
Organelle morphology is critical for correct localization and functionality. This is readily evident for the endoplasmic reticulum (ER),3 whose morphology consists of a dynamic network of sheetlike cisternae and tubular elongations that spread throughout the cytosol, all within a continuous membrane system. Disruption of ER morphology is often associated with cellular dysfunction (1, 2). Specialized structural proteins form and maintain ER architecture, and mutation of genes encoding these structural proteins can lead to human disease (3, 4).
Membrane fusion is required to maintain ER luminal continuity and allow ER dynamics. ER homotypic membrane fusion is mediated in animals by the fusion GTPase atlastin (5–8). Atlastins have a conserved domain structure consisting of a large GTPase, a three-helix bundle middle domain, two tandem transmembrane domains (TMDs), and a short cytoplasmic C-terminal tail (reviewed by McNew et al. (6)). Membrane-shaping proteins called reticulons and REEPs are also required to form ER tubules and the edges of ER sheets (9, 10). Reticulons consist of a variable N-terminal domain followed by two short hairpin TMDs, separated by a long hydrophilic loop, and a C-terminal domain. The membrane domains of reticulons primarily reside in the outer leaflet of the ER membrane to introduce curvature and generate tubules (10–13). An additional ER resident called lunapark (14–17) works with the REEP/reticulon family and atlastin to maintain the exquisite morphology of the ER network.
Humans express three atlastin homologs: atlastin-1, atlastin-2, and atlastin-3. Mutations in human atlastin-1 (SPG3A) cause a disease called hereditary spastic paraplegia (HSP) (18, 19). Whereas mutations in many other loci can also cause HSP (18, 20), an emerging theme is that disruption of ER function may underlie many HSP phenotypes (21, 22). Atlastin-1 is found primarily in neurons, whereas atlastin-2 and atlastin-3 are more ubiquitously expressed. Mutations in atlastin-3 have recently been shown to cause a form of hereditary sensory and autonomic neuropathy (23).
Humans express four reticulon (24) and six REEP genes, (25), each with multiple spliced isoforms. Mutations in REEP1 (SPG31) and reticulon-2 (SPG72) also cause HSP (18, 26, 27). In contrast to many species, Drosophila melanogaster has a single atlastin homolog (dAtl) (28) and one ubiquitously expressed reticulon (reticulon-like-1, Rtnl1) (29).
Several pieces of evidence suggest that atlastin and REEP/reticulons physically and functionally interact in the ER membrane. Defects in synaptic transmission in Drosophila due to loss of dAtl can be partially rescued by the additional loss of Rtnl1, strongly suggesting functional interaction in vivo (30). Co-immunoprecipitation experiments from rat brain extracts have also revealed an association between rat atlastin-1 and rat reticulon4a and reticulon3c. This work also suggested that the interaction between atlastin and reticulon was likely through the hydrophobic membrane–spanning segments (9).
Whereas the putative transmembrane domains of atlastin serve to anchor it in the ER membrane, experiments replacing the hydrophobic sequences with sequences from other ER-resident proteins led to loss of lipid-mixing activity (31), suggesting an additional role of this domain. Here, we show that the atlastin membrane anchor is an intramembrane hairpin rather than a bilayer-spanning transmembrane domain, and this topology is critical for membrane fusion.
Results
Biochemical analysis of human atlastin-1 and human-Drosophila atlastin chimeras
Whereas Drosophila atlastin is an efficient fusogen in vitro (5, 31–35), the human atlastin proteins have been resistant to in vitro fusion analysis (36). We have confirmed that human atlastin-1 does not significantly promote membrane fusion in vitro (Fig. 1, black bars), although it maintains a robust GTPase activity (Fig. 1, blue bars). In an effort to identify potential differences between Atl-1 and dAtl, we constructed chimeras that subdivide the protein(s) into four primary domains: the GTPase domain, the three-helical bundle middle domain, the transmembrane domains, and the C-terminal tail. Whereas dAtl with the human Atl-1 tail (Fig. 1, chimera I) was functional, as previously reported (32), the only additional chimera that drove in vitro fusion was the inclusion of the human transmembrane domains (Fig. 1, chimera II). The remaining three chimeras tested (chimeras III, IV, and V) were incapable of membrane fusion while maintaining at least 50% of native Drosophila GTPase activity, suggesting that species-specific interactions between the GTPase domain and the middle domain are important for fusion.
Figure 1.

Biochemical analysis of Drosophila atlastin and human atlastin-1 chimeras. Left, schematic diagram of atlastin domains. Green, Drosophila atlastin; red, human Atl-1. Right, lipid-mixing activity (black histogram, mean ± S.E. (error bars)) and GTPase activity (blue histogram, mean ± S.E.) expressed as a percentage of Drosophila atlastin.
Co-reconstitution of reticulon with atlastin does not influence the GTPase activity or membrane fusion propensity of atlastin
Atlastin has been shown to physically interact directly or indirectly with several proteins in the ER membrane, including reticulons, REEPs, spastin, and lunapark (6, 7, 17, 25, 37). Given the structural nature of the REEP/reticulon family (24, 38, 39) and the relatively large abundance of these proteins, the most likely interactions with atlastin that may regulate function are with the reticulons. To examine a potential regulatory role of reticulon, we analyzed the effects of Rtnl1 in dAtl-mediated in vitro fusion. Co-reconstitution of Rtnl1 and dAtl into preformed PC/PS liposomes was done by detergent-assisted insertion (5). Unincorporated protein was separated from proteoliposomes by flotation in a discontinuous Nycodenz gradient. We varied the amount of atlastin while keeping the reticulon concentration fixed to yield varying molar ratios of the two membrane proteins. Fig. 2D shows a Coomassie Blue–stained SDS-PAGE of proteoliposomes containing similar amounts of reticulon and decreasing concentrations of atlastin.
Figure 2.

Atlastin-mediated fusion and GTPase activity are not affected by co-reconstitution of reticulon. A, kinetic lipid mixing assay. Filled symbols, atlastin alone; open symbols, atlastin co-reconstituted with reticulon at varying molar ratios (1:1 (blue), 1:2 (green), and 1:4 (red)). B, histogram of average maximum fusion at 60 min normalized to atlastin alone. Each symbol represents an individual replicate. C, GTPase activity (μm phosphate/min/μm protein) of atlastin containing proteoliposomes with or without equimolar amounts of reticulon. D, Coomassie-stained SDS-polyacrylamide gel of gradient-purified proteoliposomes of atlastin with and without reticulon. Shown are the top layer (T), middle layer (M), and bottom layer (B) of a discontinuous Nycodenz gradient. Proteoliposome floats to the top, whereas unincorporated protein remains in the load at the bottom. E, atlastin reconstitution efficiency was quantified by densitometry and normalized to atlastin-alone proteoliposomes.
The influence of Rtnl1 on dAtl-mediated in vitro fusion was then assayed by lipid mixing (5, 32, 33). Three ratios of atlastin/reticulon were used, 1:1, 1:2, and 1:4, keeping Rtnl1 at a constant protein/lipid molar ratio of ∼1:400. The reconstitution of atlastin was only modestly affected by the presence of reticulon (Fig. 2E). When membrane fusion was examined, both the rate (Fig. 2A) and extent (Fig. 2B) of lipid mixing were unaffected when normalized to atlastin reconstitution efficiency. These results suggest that reticulon does not influence atlastin's ability to promote membrane fusion. Similarly, atlastin GTPase activity was also largely unaffected by reticulon (2.8 versus 2.5 μmol/min/μmol of protein) (Fig. 2C). Finally, we co-reconstituted Drosophila Rtnl1 with human atlastin-1 to determine whether the presence of reticulon influenced lipid mixing activity of the human protein. Whereas human Atl-1 was reconstituted with similar efficiency as dAtl when reticulon was included (Fig. S1), no improvement in lipid mixing activity was observed (data not shown).
Our results suggest that reticulons do not have a direct effect on the biochemical activities of atlastin under our conditions; however, this result does not preclude a role for atlastin–reticulon interactions in localization to specific subdomains of the ER, such as within tubules or to the edges of ER sheets. Finally, the inability of human Atl-1 to promote fusion in vitro is unlikely to be due to the lack of a specific binding partner in the ER membrane, because chimera II, which contains the human transmembrane domains, is functional for fusion (Fig. 1), and inclusion of an abundant likely binding partner, reticulon, did not rescue activity.
The membrane anchor of atlastin forms two intramembrane hairpin loops
The hydrophobic membrane anchors of atlastin are similar in sequence across many species (Fig. S2), and our data show that their function in membrane fusion is also conserved between Drosophila and humans (Fig. 1). The putative transmembrane segments also have some unique physical characteristics that distinguish them from other, conventional transmembrane domains. In particular, whereas the N terminus of “TM1” and the C terminus of “TM2” both begin and end with a membrane-delimiting charge (mostly arginine, although lysine is also seen in the atlastin-2 isoforms), the presence of charge within the putative ER luminal loop that would define the end of “TM1” and start of “TM2” is highly variable across species (Fig. S2). Whereas atlastin-1 and atlastin-3 contain a positively charged aspartate or glutamate that could terminate TM1, dAtl and atlastin-2 isoforms do not possess any formal charge in this location (Fig. S2). These observations suggest that the hydrophobic segment could play an active role rearranging lipids during membrane fusion, rather than just acting as a passive membrane anchor.
It has been largely assumed that the dAtl hydrophobic domains traverse the ER membrane twice and have an approximately 5-residue luminal loop (see Fig. 4A (a)) (10, 31, 33, 40). Our interest in understanding whether the atlastin hydrophobic domains actively move lipids during membrane fusion led us to determine the topology of the hydrophobic segments. The conventional membrane topology model (Fig. 4A (a)) places a short loop inside the ER lumen or inside the proteoliposome in our reconstituted system. This topology predicts that the loop residues are protected from modification by reagents added to the outside of the liposome. We targeted the accessibility of phenylalanine 448, which resides between hydrophobic segments in the center of the ∼5-amino acid loop (Fig. 4A and Fig. S2) by expressing a mutation at this location that allows for covalent modification by copper-free click chemistry (strain-promoted azide-alkyne cycloaddition). To this end, we expressed dAtl in bacteria that contains an amber stop codon (TAG) in place of the codon for phenylalanine at position 448 and a suppressor tRNA that codes for the nonnative amino acid p-azidophenylalanine (AzF) (41). The ability of AzF-448 to be labeled by reagents added to the outside of intact proteoliposome will determine whether this residue is exposed to the outside of the membrane or inside the lumen. Atlastin containing this modified amino acid was used to probe the location of phenylalanine 448 relative to the phospholipid bilayer. The location of this residue was determined by coupling the azido functionality of AzF with the membrane-impermeant strained alkyne, dibenzocycloocyl (DBCO), conjugated to a fluorescent carbocyanine dye, Cy5.5, utilizing copper-free click chemistry. This covalent attachment was performed with dAtl (F448AzF) reconstituted into proteoliposomes, whereas maximum labeling was achieved by protein labeling in detergent micelles. Unreacted AzF was quenched by the addition of NaN3, and excess DBCO-Cy5.5 was removed by flotation of proteoliposomes in a discontinuous density gradient. DBCO-Cy5.5–bound atlastin in proteoliposomes was analyzed by SDS-PAGE, and the fluorescence intensity of atlastin-coupled Cy5.5 was determined in gel using fluorescence imaging (Fujifilm LAS-4000).
Figure 4.

The atlastin membrane domain is a dual-hairpin loop that does not span the lipid bilayer. A, schematic representation of the putative conformations of atlastin and reticulon membrane domain. A (a), two transmembrane segments that traverse the lipid bilayer. A (b), two hairpin loops that are inserted into the outer bilayer. Phenylalanine 448 is represented as a red sphere, and the other residues in the membrane domain are shown as black spheres. A (c), schematic representation of reticulon homology domain with valine 69, phenylalanine 106, and glycine 170 represented with red spheres. B, proteoliposomes labeling of atlastin and reticulon azidophenylalanine mutants with DBCO-Cy5.5 by copper-free click chemistry. Following SDS-PAGE, Cy5.5 fluorescence in each band was analyzed and normalized to its corresponding Coomassie-stained band. Samples were then also normalized to detergent-solubilized proteoliposomes. Filled symbols, proteoliposomes following isolation by flotation in a density gradient; open symbols, proteoliposomes quenched with excess NaN3. C, representative SDS-PAGE samples of each mutant in proteoliposomes (− TX-100) or detergent micelles (+ TX-100) visualizing Cy5.5 fluorescence (top) and total protein by Coomassie staining (bottom). TX-100, Triton X-100. Error bars indicate S.E.
Fig. 3 examines the rate and extent of labeling of F448AzF followed by quenching unreacted DBCO with excess NaN3. Extensive labeling (∼40% for dAtl (F448AzF) and ∼70% chimera II (I450AzF)) is seen at the earliest time point (2 min), which increases to ∼55 and 90%, respectively, over the next 2 h.
Figure 3.
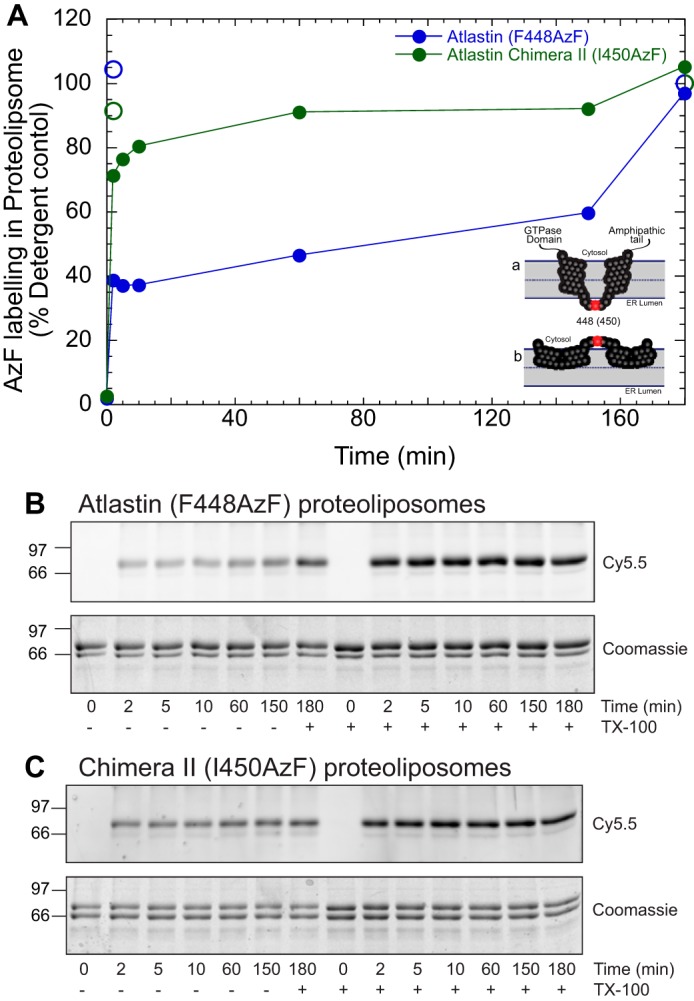
Kinetics of azidophenylalanine labeling in proteoliposome. A, kinetic trace of clicked proteoliposomes quenched at different time points with 5 mm sodium azide with 1% Triton X-100 (TX-100; open circles) and without (solid circles). Proteoliposomes containing atlastin (F448AzF) are shown in filled blue circles, and chimera II (I150AzF) is shown in filled green circles. At 150 min, 1% Triton X-100 was added to obtain a maximum labeling intensity. Each time point is normalized to labeling in the presence of detergent. Alternative atlastin topologies are shown as an inset. B and C, SDS-PAGE gel analyzed by Cy5.5 fluorescence and Coomassie staining for atlastin (F448AzF) (B) and chimera II (I450AzF) (C) proteoliposomes quenched at different time points with 5 mm sodium azide with and without 1% Triton X-100.
Fig. 4 shows a summary of AzF-448 labeling in liposomes over several experiments relative to labeling in detergent. Reconstituted dAtl(F448AzF) labeled to ∼54%, suggesting very strongly the Phe-448 is exposed to the outside of the proteoliposome rather than residing inside the proteoliposome lumen (Fig. 4B, blue circle). A similar extent of labeling was seen when a residue known to reside in a cytoplasmic loop (10, 11) of Rtnl1 (Phe-106) was interrogated in a similar manner (Fig. 4, A (c) and B (red squares)). The inability of DBCO-Cy5.5 to label residues inside proteoliposomes was confirmed using AzF labels in two locations within the Rtnl1 reticulon homology domain, specifically Val-69 and Gly-170 (Fig. 4A (c)). Whereas proteins containing V69AzF and G170AzF were efficiently labeled in detergent micelles (Fig. 4C, +TX-100), they were not significantly modified when reconstituted into proteoliposomes (Fig. 4, B (V69AzF (blue squares) and G170AzF (green squares)) and C), suggesting the DBCO-Cy5.5 dye does not have access to the interior of the proteoliposomes. Additionally, the speed of labeling shown in Fig. 3 and the inability of the much smaller molecule dithionite to leak into liposomes (Fig. S3) strongly argue against the possibility that internally labeling occurs. Overall, these data suggest an alternative domain topology in which the presumed luminal loop is facing the cytosol and the putative transmembrane domains form two intramembrane hairpins that do not fully traverse the lipid bilayer (Fig. 4A (b)).
The intramembrane hairpin topology or “W” conformation of atlastin hydrophobic segments is conserved in human atlastin-1. When the human-Drosophila chimera II protein (Fig. 1) was engineered to code for a modifiable amino acid in a similar position (I450AzF), DBCO-Cy5.5 labeling was even stronger than with the endogenous Drosophila membrane anchor (Figs. 3 and 4B (green circle)). In this case, labeling in proteoliposomes was as efficient as in detergent micelles (98%). This difference in labeling between WT dAtl and human-Drosophila chimera II may be attributed to differences in dye access to the modifiable residue. Additionally, differential association states of the recombinant proteins in the plane of the bilayer may alter labeling. Regardless, our data support a model that the atlastin membrane domain can adopt a dual intramembrane hairpin loop topology.
Atlastin-mediated fusion does not require conformational alterations of the membrane anchor for lipid mixing
The physical mechanism of lipid movement during membrane fusion is still a poorly understood process. Viral fusion proteins utilize a bilayer-penetrating fusion peptide for membrane perturbation (42–46), whereas SNARE proteins are thought to move lipids as rigid transmembrane helices are pulled together during zippering of the SNARE four-helix bundle (47–51). We have previously shown that one way that atlastin drives membrane destabilization during fusion is by using a C-terminal amphipathic α-helix that embeds in the membrane (32). Given the unconventional properties of the atlastin transmembrane segment, we considered the possibility that this region of the protein may also aid membrane destabilization and lipid mixing by changing conformations during fusion. The relatively few charges in the loop between hydrophobic segments (Fig. S2) raised the possibility that the topology of the membrane anchor could switch from the conventional “V” to the newly identified “W” topology (Fig. 4A (a) versus Fig. 3A (b)). To test for the possibility that movement of the small loop across the bilayer is part of the mechanism of atlastin-mediated fusion, we designed mutations that would stabilize the C-terminal end of the first hydrophobic segment (N450K), the N-terminal end of the second hydrophobic segment (Y466K), or both (N450K/Y466K). These mutations should prevent transbilayer movement of the intervening loop sequence, regardless of the initial topology (“W” or “V”). When proteins containing these mutations were expressed and reconstituted into proteoliposomes, no significant difference in lipid-mixing activity was observed (Fig. 5A), suggesting that the location of the loop remains unchanged during fusion. Additionally, we performed lipid-mixing assays with dAtl(F448AzF) labeled in proteoliposomes prior to the fusion reaction. As shown in Fig. 4, the method results in atlastin that contains a bulky, charged moiety on the outside of the proteoliposomes. DBCO-Cy5.5–conjugated atlastin (F488AzF) fused as efficiently as the AzF-containing protein that remained unlabeled (Fig. 5B). The ability of this covalently labeled atlastin to continue to drive membrane fusion further suggests that the “W” topology does not change during the fusion cycle.
Figure 5.

The atlastin membrane domain is a static membrane hairpin loop during membrane fusion. A, charge introduction mutants fuse normally. Shown is a histogram of average maximum fusion at 60 min normalized to WT atlastin for atlastin (Y446K) (green), atlastin (N450K) (red), and atlastin (Y446K/N450K) (blue). Each symbol represents an individual replicate. B, atlastin covalently modified with DBCO-Cy5.5 fuses normally. Shown is a histogram of average maximum fusion at 60 min normalized to WT atlastin for unmodified atlastin (F488AzF) (green) and covalently linked atlastin (F488AzF-DBCO-Cy5.5 (red). Each symbol represents an individual replicate. C, schematic representation of the of atlastin with two hairpin loops that are inserted into the outer bilayer (Ca) and an atlastin–reticulon chimera with the first RHD of Rtnl1 substituted for the atlastin hydrophobic membrane domain (Cb). D, an atlastin-Rtnl1 (TM1) chimera is nonfunctional. Histogram of average maximum fusion at 60 min normalized to WT atlastin (black) for an atlastin-Rtnl1 (TM1) chimera (green). Error bars indicate S.E.
The intramembrane hairpin topology of atlastin is important for function
Previous studies have shown that replacing the hydrophobic anchor of atlastin with a single transmembrane domain or the two transmembrane domains of the resident ER phosphatase SacI resulted in a nonfunctional protein, suggesting that the atlastin hydrophobic anchor has unique properties (31). Next, we chose to examine another hydrophobic membrane anchor that is known to reside in a similar ER subdomain of atlastin, namely reticulon. We expressed an atlastin–reticulon chimera in which the dAtl hydrophobic anchor was replaced with the first hydrophobic segment of the reticulon homology domain (amino acids 48–88) of Rtnl1 (Fig. 5C (b)). Although this chimera was expressed and reconstituted similarly to WT atlastin, it was unable to promote membrane fusion (Fig. 5D). Taken together, these results suggest that the unique “W” topology of the atlastin membrane anchor is functionally important for membrane fusion driven by atlastin.
Atlastin can form ER-like tubular networks in vitro
The observation that the atlastin membrane anchor likely occupies the outer leaflet of the ER membrane is consistent with the studies localizing atlastin primarily to ER tubules or the edges of ER sheet (i.e. areas of high membrane curvature) (16, 52, 53). Additionally, the “W” topology suggests that atlastin may also play a structural role in shaping ER membranes, similar to the defined role of the REEP/reticulon family (10–13). Recent work has demonstrated that ER-like networks can be formed in vitro using Xenopus egg extracts in an atlastin- and GTP-dependent manner (14). Additionally, proteoliposomes containing Drosophila atlastin can generate ER-like networks (54). We have been able to recapitulate an ER-like tubular network with atlastin proteoliposomes attached to coverslips with polylysine (Fig. 6). These results demonstrate that atlastins have the capacity to generate membrane curvature and produce stable tubular structures. The intramembrane hairpin loop topology described in this work is consistent with the capacity to wedge bilayer structure to generate curvature.
Figure 6.
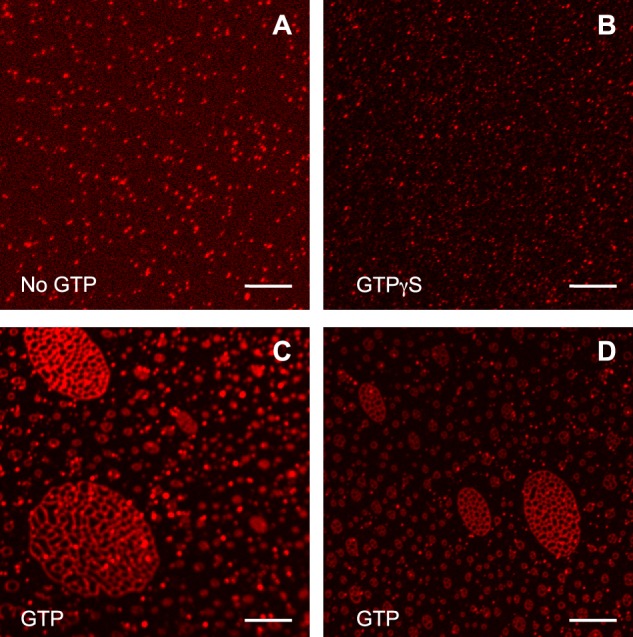
Drosophila atlastin can form tubular networks in vitro. Atlastin donor proteoliposomes (NBD/rhodamine) were incubated in nonfusogenic (no GTP (A) and GTPγS (B)) and fusogenic (5 mm GTP at 37 °C (C and D)) conditions and then bound to a poly-l-lysine–coated coverslip and imaged by fluorescent microscopy detecting the rhodamine lipid signal.
Discussion
Membrane fusion relies on protein machines to exert force on the membrane to allow for lipid and content mixing. SNARE-mediated fusion as well as viral fusion utilize the energy from protein folding to accomplish the task of membrane merger (7, 42–47, 49–51). The third class of membrane fusion proteins typified by atlastin and mitofusin are fusion GTPases (2, 5, 6) that use the chemical energy of GTP hydrolysis to drive conformational changes necessary to fuse membranes. Recent work has provided mechanistic insights into the conformational changes that occur during GTP hydrolysis (31, 34, 55–58), yet the role of the required hydrophobic membrane anchor remains relatively unexplored. Additionally, several proteins have been shown to interact with atlastin, primarily through the hydrophobic membrane anchor, including reticulon/REEP proteins, lunapark, and spastin (6, 7, 17, 25, 37).
Whereas membrane fusion by Drosophila atlastin has been characterized extensively in vitro (5, 31–35), other species of atlastins, including human, have been difficult to characterize in vitro (36). We generated protein chimeras between dAtl and HAtl-1 to identify potential functional regions of the human homolog. We found that the hydrophobic membrane anchor and cytoplasmic tail from HAtl-1 were functional when grafted into the Drosophila atlastin protein. The remaining chimeras were capable of cleaving GTP to at least 50% of WT, but none were competent for membrane fusion (Fig. 1).
One potential reason for the inability of human atlastin to promote membrane fusion is the absence of a required binding partner. We addressed the role of the most likely binding partner, reticulon, by co-reconstituting with atlastin. The presence of reticulon in the membrane in stoichiometric proportions did not influence the ability of dAtl (Fig. 2) or Atl-1 (Fig. S1) (data not shown) to drive membrane fusion or cleave GTP.
Our interest in probing an active role for the atlastin membrane anchor in the fusion process prompted us to examine the topology of the atlastin hydrophobic segments. Using copper-free click chemistry, we found that the predicted luminal loops between presumptive membrane spans one and two were, in fact, exposed to the outside of proteoliposomes (Fig. 4). This observation strongly suggests that the atlastin membrane anchor is not transmembrane, but rather two intramembranous hairpins that primarily occupy the outer leaflet of the membrane, forming a “W” topology rather than a conventional “V” (Fig. 4A, a versus b). This property is also conserved in Atl-1, suggesting that this topology may be a general feature of this class of proteins.
Regardless of the ultimate topology model, we wanted to explore the possibility that the transmembrane anchor was mobile, changing its conformation during the fusion cycle. To test this possibility, we expressed mutants that contained membrane-delimiting charges to define the boundaries of the hydrophobic segments. These mutations should prevent bilayer passage of lesser hydrophilic loops in their absence. However, we found that these mutants were virtually WT for membrane fusion (Fig. 5). Similarly, covalent attachment of the large, charged DBCO-Cy5.5 moiety to the loop between hydrophobic segments on the outside of proteoliposomes did not affect membrane fusion propensity, confirming that the “W” topology does not need to convert to a “V” topology as part of the fusion mechanism. The newly identified “W” topology of the atlastin membrane anchor appears to be a required part of the molecule. Attachment of atlastin to the membrane with a reticulon-spanning domain results in a nonfunctional protein (Fig. 5).
Finally, the “W” topology may also serve to shape membrane, similar to REEP/reticulon proteins. We have confirmed a published report (54) that Drosophila atlastin alone can produce an ER-like tubular network in vitro under conditions that drive membrane fusion (Fig. 6).
In summary, we report that both human and Drosophila atlastin contain a novel membrane anchor that forms two intramembranous hairpins that primarily occupy the outer leaflet of the membrane, forming a stable “W” topology that is a requirement for atlastin to function as a membrane fusion protein.
Experimental procedures
Molecular biology
The dAtl construct, pJM681, previously described (5), with an N-terminal GST tag and C-terminal eight-histidine tag in a modified pGEX-4-T3 bacterial expression vector was used for making recombinant dAtl. All dAtl point mutants or chimeras used this plasmid as a backbone. All point mutants of dAtl were made by PCR using a QuikChange II site-directed mutagenesis kit (Agilent Technologies). The mutant dAtl (Y446K), pJM1141, was made using the primer (CTTTGGATTGGTGGGTCTCAAGACGTTCGCCAACTTCTGC) and its complementary primer. In the same manner, dAtl (N450K), pJM1142, was made with (GTCTCTATACGTTCGCCAAGTTCTGCAATCTGATTATG) and its corresponding complementary primer; additionally, the double mutant dAtl (Y446K/N450K), pJM1143, was made by introducing this same mutation in pJM1142. The mutant dAtl (F448AzF), pJM1139, was made with a primer coding for an amber stop codon (TAG) (GGTGGGTCTCTATACGTAGGCCAACTTCTGCAATCTG) and its corresponding complementary primer.
Rtnl1 (PB isoform) and any corresponding point mutants were expressed from a bacterial expression pGEX-4T-3 vector with an N-terminal GST-SUMO tag, pJM1115. All point mutations were made by PCR using a QuikChange II site-directed mutagenesis kit. Rtnl1 (V69AzF), pJM1158, was made with the primer (CATCTCCAGCTTCTCGTAGATCAGCGTGTTCGCC) and its complementary primer. In the same manner, Rtnl1 (G106AzF), pJM1163, was made with CGAGGGTCACCCCTAGAAGGATTACCTGG, and Rtnl1 (G170AzF), pJM1159, was made with GTGCCTGGTTCAATTAGATGACTCTGGTC and their respective complementary primers.
The dAtl-Rtnl1 (TM1), pJM1160, chimera was constructed using a manufactured gene fragment G-block (IDT) containing the sequence for dAtl(354–421)-Rtnl1(48–88)-dAtl(477). Our dAtl in pGEX was digested with SacI and AflII, and this fragment was then ligated to the G-block by Gibson assembly.
Chimera I, pJM866, was constructed with Hs-Atl1 C-terminal tail generated by PCR from pJM693, GST-Hs-Atl1-H8 in pGEX4T3, using primers BspEI-Hs-Atl1-c-tail (ATTCCGGAGAATACCGAGAGCTGG) and Hs-Atl1-XhoI (TTCTCGAGCATTTTTTTCTTTTC). The PCR product was digested with BspEI and XhoI ligated into pJM681 cut with the same enzymes.
Chimera II, pJM1007, was constructed by generating Hs-Atl1 transmembrane and C-terminal tail by PCR from pJM693 using primers Hs-Atl1-NotI-forward (ATGCGGCCGCTCGTACCCCAGCCACACTG) and Hs-Atl1-XhoI-reverse (TTCTCGAGCATTTTTTTCTTTTC). The PCR product was digested with NotI and XhoI ligated into pJM999 (pJM681 modified to add NotI site) cut with the same enzymes. The mutant coding for the mutation chimera II (I450AzF), pJM1155, was made by the QuikChange technique using the primers (CATTGGTTTGGACATCTAGGCTAGCCTATGCAATATG) and its complementary primer.
Chimera III, pJM918, Hs-Atl1 middle domain was generated by PCR from pJM693 using primers Hs-Atl1–3HB-SphI (GAGGCATGCCATGTTACAGG) and Hs-Atl1-TMD-SacI reverse (CGCGAGCTCCCGGATATATGCCCAAGTG). The PCR product was digested with NcoI and SacI and ligated into pJM681 cut with the same enzymes.
Chimera IV, pJM917, was constructed by generating Hs-Atl1 middle domain with C-terminal domain by PCR from pJM693 using primers 3HB-SphI-forward (GAGGCATGCCCATGTTACAGGCCACAGC) and Hs-Atl1-XhoI-reverse (TTCTCGAGCATTTTTTTCTTTTC). The PCR product was digested with SphI and XhoI and ligated into pJM681 cut with the same enzymes.
Chimera V, pJM908, was constructed by generating the Hs-Atl1 GTPase domain by PCR from pJM693 (GST-Hs-Atl1-H8 in pGEX4T-3) using primers NcoI-Hs-Atl1-forward (ACCACCATGGCCAAGAACCGCAGG) and SphI-Hs-Atl1-reverse (CGCGCATGCGGATTTGGGATGTGG). The PCR product was digested with NcoI and SphI and ligated into pJM681 cut with the same enzymes.
Protein expression and purification
Protein expression
Recombinant proteins were purified with an N-terminal GST tag and an eight-histidine tag, as described previously (5), or a GST-SUMO tag, as described (33), with the following modifications. A 200-ml Luria-Bertani medium with 100 μg/ml ampicillin preculture was inoculated with 5 ml of transformed BL21 (DE3) Escherichia coli (Agilent Technologies) and incubated with shaking overnight at 25 °C. The culture was then pelleted at 2000 rcf for 10 min, using an Allegra tabletop centrifuge at 25 °C. The supernatant was removed, and the pellet was resuspended and inoculated to a 4-liter Luria-Bertani medium with 100 μg/ml ampicillin. The culture was allowed to reach an A600 of 0.4–0.5 at 25 °C before inducing with 0.2 mm isopropyl 1-thio-β-d-galactopyranoside, followed by an overnight shaking incubation at 16 °C.
Azidophenylalanine mutant proteins were expressed in a similar manner as the other tagged proteins as described (59) with the following modifications. BL21 DE3 cells were transformed with the corresponding mutant protein plasmid and pEVOL-AzF (Addgene entry 31186) (41). Bacterial cultures were grown with 100 μg/ml ampicillin and 35 μg/ml chloramphenicol in M9 minimal medium supplemented with 0.2% glucose and 0.1% casamino acids. A 2-liter culture was allowed to reach an A600 of 0.4–0.5 before inducing with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside, 0.06% arabinose, and 1.5 mm p-azidophenylalanine (Bachem); after induction, the culture was incubated overnight with shaking at 16 °C.
Lysate preparation
Cells were harvested by pelleting for 15 min at 7500 rcf at 4 °C on an Avanti JHC centrifuge with a JS-5.0 rotor. The supernatant was discarded, and cells were resuspended in A200 (25 mm HEPES (pH 7.4) and 200 mm KCl). Cells were then centrifuged for 5 min at 4 °C at 10,950 rcf in a JA-10 rotor. Cells were then resuspended in 40 ml of breaking buffer (A200 plus 10% glycerol, 2 mm 2-mercaptoethanol, 4% Triton X-100, 40 mm imidazole, and one Complete protease inhibitor mixture tablet (Roche Applied Science)). GST-SUMO–tagged proteins omitted imidazole. Cells were then passed three times through an EmulsiFlex C3 high-pressure homogenizer (Avastin) at 15,000–20,000 p.s.i. Cell extracts were then cleared by centrifugation at 125,000 rcf at 4 °C using a Type 45-Ti rotor in an Optima LE-80K ultracentrifuge. Cleared lysate was then filtered through a 0.45-μm pore cellulose nitrate sterile membrane filter (Whatman).
GST-H8–tagged proteins
dAtl proteins were tagged with an N-terminal GST tag and an eight-histidine tag. Lysates were passed through an equilibrated (A100 (25 mm HEPES (pH 7.4) and 100 mm KCl) + 10% glycerol, 2 mm 2-mercaptoethanol, 1% Triton X-100, 40 mm imidazole) HiTrap Chelating HP column (GE Healthcare) in an Akta prime LC system (Amersham Biosciences). The column was then washed with 25 ml of A100 plus 10% glycerol, 2 mm 2-mercaptoethanol, 0.1% Anapoe X-100, 40 mm imidazole. Protein was then eluted in a 30-ml linear gradient of imidazole from 40 to 500 mm and a final 5-ml wash at 500 mm. Peak fractions were pooled together and incubated for 1 h at 4 °C with 70 mg of equilibrated swollen GSH-agarose beads. Beads were pelleted, and unbound protein was removed by aspiration. Beads were then transferred to 10-ml Polyprep columns (Bio-Rad) and washed with 25 ml of A100 with 10% glycerol, 2 mm 2-mercaptoethanol, 0.1% Anapoe X-100, and 1 mm EDTA. Protein was then eluted with 1.5 ml of wash buffer (pH 7.4) supplemented with 10 mm reduced GSH. Protein was then aliquoted, plunged into liquid nitrogen, and stored at −80 °C. Purity was analyzed by SDS-PAGE and Coomassie staining and quantified by Amido black (60).
GST-SUMO–tagged proteins
Reticulon and its mutants were purified with a GST-SUMO tagged as described (33). Lysates were incubated for 3 h at 4 °C with 70 mg of equilibrated swollen GSH-agarose beads. Beads were pelleted, and unbound protein was removed by aspiration. Beads were then transferred to 10-ml Polyprep columns (Bio-Rad) and washed with 10 ml of A100 with 10% glycerol, 2 mm 2-mercaptoethanol, 1% Triton X-100, and 1 mm EDTA. This was followed by a 20-ml wash of A100 with 10% glycerol, 2 mm 2-mercaptoethanol, 0.1% Anapoe X-100, and 1 mm EDTA. The proteins were then cleaved off the beads by resuspending with 1 ml of wash buffer supplemented with 2.1 μg of GST-SENP2(365–589) overnight at 4 °C. Protein was eluted by centrifugation, aliquoted, plunged in liquid nitrogen, and stored at −80 °C. Protein was also analyzed by SDS-PAGE and Coomassie staining for purity and quantified by Amido black.
GST-SENP2(365–589) was expressed, and its lysate was prepared in the same way as described previously for GST-H8–tagged proteins. Purification was done as described (33) with the following modifications. The lysate was incubated for 1 h at 4 °C in 70 mg of swollen GSH-agarose beads equilibrated in A100 with 10% glycerol and 2 mm 2-mercaptoethanol. Beads were pelleted, and the supernatant was removed by aspiration. Beads were then transferred to a Polyprep column and washed with 20 ml of equilibration buffer. Protein was then eluted with 3 ml of equilibration buffer supplemented with 10 mm GSH. The elution was then diluted with 25 mm HEPES (pH 7.4) buffer to bring down the KCl concentration to 40 mm. The sample was then passed through a SP-Sepharose column (GE Healthcare) on an Akta prime LC system. Using its cation-exchange template, the protein was eluted off the column over a salt gradient from 40 mm to 1 m KCl. Peak fractions were pooled and stored in 50% glycerol at −20 °C. Protein concentration was determined by Bradford protein assay, and purity was analyzed by SDS-PAGE and Coomassie staining.
Liposome production and reconstitution
Liposome production and reconstitution were done as reported previously (32) with the following modifications. Unlabeled liposomes consisted of POPC/DOPS (85:15 molar ratio). Labeled liposomes consisted of POPC/DOPS/Rh-DPPE/NBD-DPPE (82:15:1.5:1.5 molar ratio). All liposomes contained trace amounts of [3H]1-palmitoyl 2-palmitoylphosphatidylethanolamine (DPPE) (American Radiolabeled Chemicals) for concentration determination by scintillation counting. Lipid mixes, in chloroform, were dried by a nitrogen stream and resuspended to 10 mm in A100 with 10% glycerol, 2 mm 2-mercaptoethanol, and 1 mm EDTA. Lipid mixtures were freeze-thawed 10 times in liquid nitrogen and then passed 19 times through a 100-nm pore polycarbonate membrane (Avanti).
Atlastin, atlastin mutants, and Rtnl1 were reconstituted by the detergent-assisted insertion method (61, 62). Protein in detergent, Triton X-100, was incubated for 1 h at 4 °C at a 1:400 protein/lipid ratio, unless otherwise stated, and an effective detergent/lipid ratio of ∼0.7, determined by Reff = Dtotal − Dwater/[lipid], where Dtot is the total detergent concentration and Dwater is the monomeric detergent concentration (0.18 mm) with lipids. The solution was then added to swollen SM2 biobeads (1 g per 70 mg of Triton X-100) to remove the Triton X-100 and incubated overnight at 4 °C. Unincorporated protein was pelleted by centrifugation at 16,000 rcf for 10 min. Proteoliposome concentration was then measured by scintillation counting.
Click chemistry and proteoliposome flotation
Labeling of azidophenylalanine mutants with DBCO-Cy5.5 was done to proteoliposomes at a concentration of 150 μm at 4 °C for 2.5 h. A sample of the proteoliposomes was solubilized by adding 1% Triton X-100; this was used as the positive control. This was followed by a quenching reaction with 500 μm NaN3 for 2.5 h at 4 °C. Proteoliposomes were then floated in a Nycodenz gradient to separate free protein and unclicked dye using a three-layer discontinuous gradient as described previously (63) with the following modifications. A stock solution of 80% Nycodenz in A100 was diluted with the proteoliposomes in A100 with 10% glycerol, 2 mm 2-mercaptoethanol, and 1 mm EDTA, to 40% Nycodenz, to a final volume of 300 μl. This was added to a 5 × 41-mm Ultra-Clear tube (Beckman). A 30% Nycodenz solution was layered on top. Finally, 50 μl of the same buffer with no glycerol was layered at the top. The gradient was then centrifuged in a SW55-Ti rotor for 4 h at 48,000 rpm at 4 °C. The resulting layers were harvested and analyzed by SDS-PAGE.
Gel imaging was done on a LAS4000 luminescent imager (Fuji), using red light and Cy5 filter for Cy5.5 fluorescence, and the gel was then stained with Coomassie Blue and imaged in the same instrument with the diascopic illumination settings. Densitometry of the gel bands was done using Multi Gauge version 3.X analysis software.
To determine the kinetics of azide click in proteoliposomes, we clicked proteoliposomes with 150 μm DBCO-Cy5.5 and quenched the reaction at different time points with 5 mm sodium azide. To determine a zero time point, the dye was mixed with sodium azide before mixing with the proteoliposomes. At the final time point, 150 min, 1% Triton X-100 was added and incubated for another 30 min, to determine its maximum labeling. Additionally, we had solubilized proteoliposomes in 1% Triton X-100 clicked and quenched in the same time points (only 0 and 180 min are shown). Samples were then analyzed by SDS-PAGE and quantified in the same manner as described.
In vitro lipid mixing assays
Fusion was measured by lipid-mixing assays based on the method described previously (5). Labeled and unlabeled proteoliposomes were brought to a final concentration of 0.15 mm in A100 with 10% glycerol, 2 mm 2-mercaptoethanol, and 1 mm EDTA supplemented with 5 mm MgCl2. After a 5-min incubation and mixing at 37 °C, fusion was induced with 5 mm GTP. NBD fluorescence was then monitored for 1 h using a TECAN M200 plate reader. After 1 h, 2.5% (w/v) n-dodecyl β-d-maltoside was added to solubilize the proteoliposomes and measure maximum NBD fluorescence; this was measured every minute for 15 min.
Pi release assays
GTPase activity was analyzed by Pi release as described previously (5). Purified protein in detergent and proteoliposomes solubilized in A100 with 10% glycerol, 2 mm 2-mercaptoethanol, 0.1% Anapoe X-100, 1 mm EDTA, and 0.5 mm MgCl2 were assayed for GTPase activity using the EnzChek phosphate release assay kit (Molecular Probes). Protein was incubated at 37 °C for 5 min before adding 0.5 mm GTP. Absorbance at 360 nm was recorded every 30 s for 30 min.
In vitro networks on polylysine-coated coverslips
Glass coverslips were spotted with 1 mg/ml poly-l-lysine hydrobromide (Sigma-Aldrich) for 1 h at room temperature in a biological hood. The polylysine solution was then removed, and the coverslips were washed twice with deionized water. The coverslips were dried overnight in the biological hood and stored.
Approximately 150 μm proteoliposomes were incubated at 37 °C for 30 min in fusogenic conditions (50 mm GTP and 50 mm MgCl2) or nonfusogenic conditions (no GTP or 50 mm GTPγS, and 50 mm MgCl2). The reaction was then diluted 1:10 in A100 buffer and spotted into a polylysine-coated coverslip, placed in a glass slide, and sealed with nail polish. The liposome networks were then imaged in a Nikon A1-rsi confocal microscope using the settings to image rhodamine.
Author contributions
M. A. B.-S. and J. A. M. conceptualization; M. A. B.-S. formal analysis; M. A. B.-S. and T. D. investigation; M. A. B.-S. and T. D. methodology; M. A. B.-S. and J. A. M. writing-original draft; M. A. B.-S. and J. A. M. writing-review and editing; J. A. M. funding acquisition; J. A. M. project administration.
Supplementary Material
Acknowledgments
We thank Dr. Mike Stern and Dr. Joseph Faust for comments and ideas for this project, Dr. Jun Ohata and Dr. Emily Thomas for guidance in noncanonical residue mutagenesis and click chemistry, and Alexandra Rangel and Calvin Su for help with the molecular biology.
The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S3.
- ER
- endoplasmic reticulum
- TMD
- transmembrane domain
- HSP
- hereditary spastic paraplegia
- PC
- phosphatidylcholine
- PS
- phosphatidylserine
- AzF
- p-azidophenylalanine
- DBCO
- dibenzocycloocyl
- SNARE
- soluble NSF attachment protein receptor(s)
- SUMO
- small ubiquitin-like modifier
- rcf
- relative centrifugal force
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- DOPS
- 1,2-dioleoyl-sn-glycero-3-phospho-l-serine
- DPPE
- [3H]1-palmitoyl 2-palmitoylphosphatidylethanolamine
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate.
References
- 1. English A. R., Zurek N., and Voeltz G. K. (2009) Peripheral ER structure and function. Curr. Opin. Cell Biol. 21, 596–602 10.1016/j.ceb.2009.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pendin D., McNew J. A., and Daga A. (2011) Balancing ER dynamics: shaping, bending, severing, and mending membranes. Curr. Opin. Cell Biol. 23, 435–442 10.1016/j.ceb.2011.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Noreau A., Dion P. A., and Rouleau G. A. (2014) Molecular aspects of hereditary spastic paraplegia. Exp. Cell Res. 325, 18–26 10.1016/j.yexcr.2014.02.021 [DOI] [PubMed] [Google Scholar]
- 4. Fink J. K. (2014) Hereditary spastic paraplegia: clinical principles and genetic advances. Semin. Neurol. 34, 293–305 10.1055/s-0034-1386767 [DOI] [PubMed] [Google Scholar]
- 5. Orso G., Pendin D., Liu S., Tosetto J., Moss T. J., Faust J. E., Micaroni M., Egorova A., Martinuzzi A., McNew J. A., and Daga A. (2009) Homotypic fusion of ER membranes requires the dynamin-like GTPase Atlastin. Nature 460, 978–983 10.1038/nature08280 [DOI] [PubMed] [Google Scholar]
- 6. McNew J. A., Sondermann H., Lee T., Stern M., and Brandizzi F. (2013) GTP-dependent membrane fusion. Annu. Rev. Cell Dev. Biol. 29, 529–550 10.1146/annurev-cellbio-101512-122328 [DOI] [PubMed] [Google Scholar]
- 7. Moss T. J., Daga A., and McNew J. A. (2011) Fusing a lasting relationship between ER tubules. Trends Cell Biol. 21, 416–423 10.1016/j.tcb.2011.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hu J., and Rapoport T. A. (2016) Fusion of the endoplasmic reticulum by membrane-bound GTPases. Semin. Cell Dev. Biol. 60, 105–111 10.1016/j.semcdb.2016.06.001 [DOI] [PubMed] [Google Scholar]
- 9. Hu J., Shibata Y., Zhu P. P., Voss C., Rismanchi N., Prinz W. A., Rapoport T. A., and Blackstone C. (2009) A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell 138, 549–561 10.1016/j.cell.2009.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Voeltz G. K., Prinz W. A., Shibata Y., Rist J. M., and Rapoport T. A. (2006) A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell 124, 573–586 10.1016/j.cell.2005.11.047 [DOI] [PubMed] [Google Scholar]
- 11. Zurek N., Sparks L., and Voeltz G. (2011) Reticulon short hairpin transmembrane domains are used to shape ER tubules. Traffic 12, 28–41 10.1111/j.1600-0854.2010.01134.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shibata Y., Voss C., Rist J. M., Hu J., Rapoport T. A., Prinz W. A., and Voeltz G. K. (2008) The reticulon and DP1/Yop1p proteins form immobile oligomers in the tubular endoplasmic reticulum. J. Biol. Chem. 283, 18892–18904 10.1074/jbc.M800986200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Voeltz G. K., and Prinz W. A. (2007) Sheets, ribbons and tubules: how organelles get their shape. Nat. Rev. Mol. Cell Biol. 8, 258–264 10.1038/nrm2119 [DOI] [PubMed] [Google Scholar]
- 14. Wang S., Tukachinsky H., Romano F. B., and Rapoport T. A. (2016) Cooperation of the ER-shaping proteins atlastin, lunapark, and reticulons to generate a tubular membrane network. Elife 5, e18605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen S., Desai T., McNew J. A., Gerard P., Novick P. J., and Ferro-Novick S. (2015) Lunapark stabilizes nascent three-way junctions in the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 112, 418–423 10.1073/pnas.1423026112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shemesh T., Klemm R. W., Romano F. B., Wang S., Vaughan J., Zhuang X., Tukachinsky H., Kozlov M. M., and Rapoport T. A. (2014) A model for the generation and interconversion of ER morphologies. Proc. Natl. Acad. Sci. U.S.A. 111, E5243–E5251 10.1073/pnas.1419997111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen S., Novick P., and Ferro-Novick S. (2012) ER network formation requires a balance of the dynamin-like GTPase Sey1p and the Lunapark family member Lnp1p. Nat Cell Biol. 14, 707–716 10.1038/ncb2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fink J. K. (2006) Hereditary spastic paraplegia. Curr. Neurol. Neurosci. Rep. 6, 65–76 10.1007/s11910-996-0011-1 [DOI] [PubMed] [Google Scholar]
- 19. Zhao X., Alvarado D., Rainier S., Lemons R., Hedera P., Weber C. H., Tukel T., Apak M., Heiman-Patterson T., Ming L., Bui M., and Fink J. K. (2001) Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat. Genet. 29, 326–331 10.1038/ng758 [DOI] [PubMed] [Google Scholar]
- 20. Klebe S., Stevanin G., and Depienne C. (2015) Clinical and genetic heterogeneity in hereditary spastic paraplegias: from SPG1 to SPG72 and still counting. Rev. Neurol. 171, 505–530 10.1016/j.neurol.2015.02.017 [DOI] [PubMed] [Google Scholar]
- 21. Blackstone C. (2012) Cellular pathways of hereditary spastic paraplegia. Annu. Rev. Neurosci. 35, 25–47 10.1146/annurev-neuro-062111-150400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Blackstone C. (2018) Hereditary spastic paraplegia. Handb. Clin. Neurol. 148, 633–652 10.1016/B978-0-444-64076-5.00041-7 [DOI] [PubMed] [Google Scholar]
- 23. Kornak U., Mademan I., Schinke M., Voigt M., Krawitz P., Hecht J., Barvencik F., Schinke T., Giesselmann S., Beil F. T., Pou-Serradell A., Vílchez J. J., Beetz C., Deconinck T., Timmerman V., et al. (2014) Sensory neuropathy with bone destruction due to a mutation in the membrane-shaping atlastin GTPase 3. Brain 137, 683–692 10.1093/brain/awt357 [DOI] [PubMed] [Google Scholar]
- 24. Yang Y. S., and Strittmatter S. M. (2007) The reticulons: a family of proteins with diverse functions. Genome Biol. 8, 234 10.1186/gb-2007-8-12-234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Park S. H., Zhu P. P., Parker R. L., and Blackstone C. (2010) Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J. Clin. Invest. 120, 1097–1110 10.1172/JCI40979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Montenegro G., Rebelo A. P., Connell J., Allison R., Babalini C., D'Aloia M., Montieri P., Schüle R., Ishiura H., Price J., Strickland A., Gonzalez M. A., Baumbach-Reardon L., Deconinck T., Huang J., et al. (2012) Mutations in the ER-shaping protein reticulon 2 cause the axon-degenerative disorder hereditary spastic paraplegia type 12. J. Clin. Invest. 122, 538–544 10.1172/JCI60560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Salinas S., Proukakis C., Crosby A., and Warner T. T. (2008) Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 7, 1127–1138 10.1016/S1474-4422(08)70258-8 [DOI] [PubMed] [Google Scholar]
- 28. Lee Y., Paik D., Bang S., Kang J., Chun B., Lee S., Bae E., Chung J., and Kim J. (2008) Loss of spastic paraplegia gene atlastin induces age-dependent death of dopaminergic neurons in Drosophila. Neurobiol. Aging 29, 84–94 10.1016/j.neurobiolaging.2006.09.004 [DOI] [PubMed] [Google Scholar]
- 29. Wakefield S., and Tear G. (2006) The Drosophila reticulon, Rtnl-1, has multiple differentially expressed isoforms that are associated with a sub-compartment of the endoplasmic reticulum. Cell Mol. Life Sci. 63, 2027–2038 10.1007/s00018-006-6142-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Summerville J. B., Faust J. F., Fan E., Pendin D., Daga A., Formella J., Stern M., and McNew J. A. (2016) The effects of ER morphology on synaptic structure and function in Drosophila melanogaster. J. Cell Sci. 129, 1635–1648 10.1242/jcs.184929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu T. Y., Bian X., Sun S., Hu X., Klemm R. W., Prinz W. A., Rapoport T. A., and Hu J. (2012) Lipid interaction of the C terminus and association of the transmembrane segments facilitate atlastin-mediated homotypic endoplasmic reticulum fusion. Proc. Natl. Acad. Sci. U.S.A. 109, E2146–E2154 10.1073/pnas.1208385109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Faust J. E., Desai T., Verma A., Ulengin I., Sun T. L., Moss T. J., Betancourt-Solis M. A., Huang H. W., Lee T., and McNew J. A. (2015) The atlastin C-terminal tail is an amphipathic helix that perturbs bilayer structure during endoplasmic reticulum homotypic fusion. J. Biol. Chem. 290, 4772–4783 10.1074/jbc.M114.601823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moss T. J., Andreazza C., Verma A., Daga A., and McNew J. A. (2011) Membrane fusion by the GTPase atlastin requires a conserved C-terminal cytoplasmic tail and dimerization through the middle domain. Proc. Natl. Acad. Sci. U.S.A. 108, 11133–11138 10.1073/pnas.1105056108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bian X., Klemm R. W., Liu T. Y., Zhang M., Sun S., Sui X., Liu X., Rapoport T. A., and Hu J. (2011) Structures of the atlastin GTPase provide insight into homotypic fusion of endoplasmic reticulum membranes. Proc. Natl. Acad. Sci. U.S.A. 108, 3976–3981 10.1073/pnas.1101643108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morin-Leisk J., Saini S. G., Meng X., Makhov A. M., Zhang P., and Lee T. H. (2011) An intramolecular salt bridge drives the soluble domain of GTP-bound atlastin into the postfusion conformation. J. Cell Biol. 195, 605–615 10.1083/jcb.201105006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wu F., Hu X., Bian X., Liu X., and Hu J. (2015) Comparison of human and Drosophila atlastin GTPases. Protein Cell 6, 139–146 10.1007/s13238-014-0118-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Evans K., Keller C., Pavur K., Glasgow K., Conn B., and Lauring B. (2006) Interaction of two hereditary spastic paraplegia gene products, spastin and atlastin, suggests a common pathway for axonal maintenance. Proc. Natl. Acad. Sci. U.S.A. 103, 10666–10671 10.1073/pnas.0510863103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hübner C. A., and Kurth I. (2014) Membrane-shaping disorders: a common pathway in axon degeneration. Brain 137, 3109–3121 10.1093/brain/awu287 [DOI] [PubMed] [Google Scholar]
- 39. Yalçin B., Zhao L., Stofanko M., O'Sullivan N. C., Kang Z. H., Roost A., Thomas M. R., Zaessinger S., Blard O., Patto A. L., Sohail A., Baena V., Terasaki M., and O'Kane C. J. (2017) Modeling of axonal endoplasmic reticulum network by spastic paraplegia proteins. Elife 6, e23882 10.7554/eLife.23882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhu P. P., Patterson A., Lavoie B., Stadler J., Shoeb M., Patel R., and Blackstone C. (2003) Cellular localization, oligomerization, and membrane association of the hereditary spastic paraplegia 3A (SPG3A) protein atlastin. J. Biol. Chem. 278, 49063–49071 10.1074/jbc.M306702200 [DOI] [PubMed] [Google Scholar]
- 41. Chin J. W., Santoro S. W., Martin A. B., King D. S., Wang L., and Schultz P. G. (2002) Addition of p-azido-l-phenylalanine to the genetic code of Escherichia coli. J. Am. Chem. Soc. 124, 9026–9027 10.1021/ja027007w [DOI] [PubMed] [Google Scholar]
- 42. Moss B. (2016) Membrane fusion during poxvirus entry. Semin. Cell Dev. Biol. 60, 89–96 10.1016/j.semcdb.2016.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Blijleven J. S., Boonstra S., Onck P. R., van der Giessen E., and van Oijen A. M. (2016) Mechanisms of influenza viral membrane fusion. Semin. Cell Dev. Biol. 60, 78–88 10.1016/j.semcdb.2016.07.007 [DOI] [PubMed] [Google Scholar]
- 44. White J. M., and Whittaker G. R. (2016) Fusion of enveloped viruses in endosomes. Traffic 17, 593–614 10.1111/tra.12389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. White J. M. (1995) Membrane fusion: the influenza paradigm. Cold Spring Harb. Symp. Quant. Biol. 60, 581–588 10.1101/SQB.1995.060.01.062 [DOI] [PubMed] [Google Scholar]
- 46. Hernandez L. D., Hoffman L. R., Wolfsberg T. G., and White J. M. (1996) Virus-cell and cell-cell fusion. Annu. Rev. Cell Dev. Biol. 12, 627–661 10.1146/annurev.cellbio.12.1.627 [DOI] [PubMed] [Google Scholar]
- 47. McNew J. A. (2008) Regulation of SNARE-mediated membrane fusion during exocytosis. Chem. Rev. 108, 1669–1686 10.1021/cr0782325 [DOI] [PubMed] [Google Scholar]
- 48. McNew J. A., Weber T., Parlati F., Johnston R. J., Melia T. J., Söllner T. H., and Rothman J. E. (2000) Close is not enough: SNARE-dependent membrane fusion requires an active mechanism that transduces force to membrane anchors. J. Cell Biol. 150, 105–117 10.1083/jcb.150.1.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rizo J., and Xu J. (2015) The synaptic vesicle release machinery. Annu. Rev. Biophys. 44, 339–367 10.1146/annurev-biophys-060414-034057 [DOI] [PubMed] [Google Scholar]
- 50. Südhof T. C., and Rothman J. E. (2009) Membrane fusion: grappling with SNARE and SM proteins. Science 323, 474–477 10.1126/science.1161748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rothman J. E. (2014) The principle of membrane fusion in the cell (Nobel lecture). Angew. Chem. Int. Ed. Engl. 53, 12676–12694 10.1002/anie.201402380 [DOI] [PubMed] [Google Scholar]
- 52. Campelo F., McMahon H. T., and Kozlov M. M. (2008) The hydrophobic insertion mechanism of membrane curvature generation by proteins. Biophys. J. 95, 2325–2339 10.1529/biophysj.108.133173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kozlov M. M., Campelo F., Liska N., Chernomordik L. V., Marrink S. J., and McMahon H. T. (2014) Mechanisms shaping cell membranes. Curr. Opin. Cell Biol. 29, 53–60 10.1016/j.ceb.2014.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Powers R. E., Wang S., Liu T. Y., and Rapoport T. A. (2017) Reconstitution of the tubular endoplasmic reticulum network with purified components. Nature 543, 257–260 10.1038/nature21387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. O'Donnell J. P., Byrnes L. J., Cooley R. B., and Sondermann H. (2018) A hereditary spastic paraplegia-associated atlastin variant exhibits defective allosteric coupling in the catalytic core. J. Biol. Chem. 293, 687–700 10.1074/jbc.RA117.000380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. O'Donnell J. P., Cooley R. B., Kelly C. M., Miller K., Andersen O. S., Rusinova R., and Sondermann H. (2017) Timing and reset mechanism of GTP hydrolysis-driven conformational changes of atlastin. Structure 25, 997–1010.e4 10.1016/j.str.2017.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Byrnes L. J., Singh A., Szeto K., Benvin N. M., O'Donnell J. P., Zipfel W. R., and Sondermann H. (2013) Structural basis for conformational switching and GTP loading of the large G protein atlastin. EMBO J. 32, 369–384 10.1038/emboj.2012.353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Byrnes L. J., and Sondermann H. (2011) Structural basis for the nucleotide-dependent dimerization of the large G protein atlastin-1/SPG3A. Proc. Natl. Acad. Sci. U.S.A. 108, 2216–2221 10.1073/pnas.1012792108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu T. Y., Bian X., Romano F. B., Shemesh T., Rapoport T. A., and Hu J. (2015) Cis and trans interactions between atlastin molecules during membrane fusion. Proc. Natl. Acad. Sci. U.S.A. 112, E1851–E1860 10.1073/pnas.1504368112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schaffner W., and Weissmann C. (1973) A rapid, sensitive, and specific method for the determination of protein in dilute solution. Anal. Biochem. 56, 502–514 10.1016/0003-2697(73)90217-0 [DOI] [PubMed] [Google Scholar]
- 61. Rigaud J. L., and Lévy D. (2003) Reconstitution of membrane proteins into liposomes. Methods Enzymol. 372, 65–86 10.1016/S0076-6879(03)72004-7 [DOI] [PubMed] [Google Scholar]
- 62. Rigaud J. L., Pitard B., and Levy D. (1995) Reconstitution of membrane proteins into liposomes: application to energy-transducing membrane proteins. Biochim. Biophys. Acta 1231, 223–246 10.1016/0005-2728(95)00091-V [DOI] [PubMed] [Google Scholar]
- 63. Scott B. L., Van Komen J. S., Liu S., Weber T., Melia T. J., and McNew J. A. (2003) Liposome fusion assay to monitor intracellular membrane fusion machines. Methods Enzymol. 372, 274–300 10.1016/S0076-6879(03)72016-3 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.