

Abstract
Halogenated phenol and nitrophenols are toxic compounds that are widely accumulated in the environment. Enzymes in the had operon from the bacterium Ralstonia pickettii DTP0602 have the potential for application as biocatalysts in the degradation of many of these toxic chemicals. HadA monooxygenase previously was identified as a two-component reduced FAD (FADH−)–utilizing monooxygenase with dual activities of dehalogenation and denitration. However, the partner enzymes of HadA, that is, the flavin reductase and quinone reductase that provide the FADH− for HadA and reduce quinone to hydroquinone, remain to be identified. In this report, we overexpressed and purified the flavin reductases, HadB and HadX, to investigate their functional and catalytic properties. Our results indicated that HadB is an FMN-dependent quinone reductase that converts the quinone products from HadA to hydroquinone compounds that are more stable and can be assimilated by downstream enzymes in the pathway. Transient kinetics indicated that HadB prefers NADH and menadione as the electron donor and acceptor, respectively. We found that HadX is an FAD-bound flavin reductase, which can generate FADH− for HadA to catalyze dehalogenation or denitration reactions. Thermodynamic and transient kinetic experiments revealed that HadX prefers to bind FAD over FADH− and that HadX can transfer FADH− from HadX to HadA via free diffusion. Moreover, HadX rapidly catalyzed NADH-mediated reduction of flavin and provided the FADH− for a monooxygenase of a different system. Combination of all three flavin-dependent enzymes, i.e. HadA/HadB/HadX, reconstituted an effective dehalogenation and denitration cascade, which may be useful for future bioremediation applications.
Keywords: flavin, flavin adenine dinucleotide (FAD), enzyme mechanism, enzyme kinetics, reductase, bioremediation, biotechnology, flavoenzyme, halogenated phenol, nitrophenol
Introduction
Halogenated phenols (HPs)2 and nitrophenols (NPs) are toxic chemicals that are widespread and have accumulated in the environment. These compounds belong to a persistent class of toxic compounds that can cause serious health problems (1, 2). HPs and NPs are derived from agrochemicals, pesticides, and disinfectant agents such as 2,4-dichlorophenoxyacetate (2,4-D), profenofos, or parathion. Although detoxification of these compounds can be carried out by physical or chemical treatment (3, 4), these methods are not efficient because they are costly and several generate toxic byproducts. Biodegradation processes using microbes (5–7) or engineered cells that contain clusters of enzymes capable of degrading these toxic compounds are effective and more sustainable methods for decontamination.
Several aerobic bacteria are capable of converting HPs or NPs to compounds that are ready to be assimilated via central metabolic pathways (5, 6, 8–15). Among these bacteria, the had operon from Ralstonia pickettii DTP0602 is particularly interesting because one of the key enzymes that catalyze a crucial step in detoxification, HadA monooxygenase, has recently been reported to have dual activities in catalyzing hydroxylation and group elimination of HPs (dehalogenation) and NPs (denitration) to generate a quinone product (16). The resulting quinone needs to be further reduced to hydroquinone, which can be further metabolized by other enzymes in the had operon as shown in Fig. 1. In previous experiments, ascorbic acid was used as a reductant to generate hydroquinone products in vitro (16, 17). It is still unknown how quinone is converted into hydroquinone in vivo. The reaction of HadA, therefore requires the activity of a quinone reductase that has not yet been identified (Fig. 1).
Figure 1.

Proposed pathway for phenol degradation by the had operon. Enzyme abbreviations: HadA, dechlorinating and denitrating flavin-dependent monooxygenase; HadC, hydroxyquinol 1,2-dioxygenase; HadD, maleylacetate reductase. *, the second hydroxylation and group elimination by HadA can occur if Y is halide or nitro groups.
HadA is a flavin-dependent monooxygenase that uses reduced FAD (FADH−) and oxygen as substrates for catalyzing the monooxygenation of aromatic phenols. Recent mechanistic studies of HadA have shown that the enzyme catalyzes its reaction via formation of a C4a-hydroperoxyflavin as a reactive intermediate. Furthermore, the deprotonation of phenolic substrates in the reaction is the key step that controls the overall reaction of hydroxylation and the group elimination steps (16, 17). As HadA belongs to the class of two-component flavin-dependent monooxygenases, a flavin reductase is required to supply reduced flavin to the system (18). A flavin reductase catalyzes electron transfer from NADH or NADPH to an enzyme-bound flavin (FAD or FMN) to generate reduced flavin. The resulting reduced flavin generally does not bind tightly to the reductase, and thus dissociates from the enzyme to bind to and act as a substrate for the monooxygenase (18–20). Previously, the reductase component of the p-hydroxyphenylacetate hydroxylase system (C1) (19, 20) was used as a flavin reductase to supply FADH− for HadA (16, 17). The partner reductase that was co-evolved with HadA for catalyzing dehalogenation and denitration with HadA is currently unknown (Fig. 1).
Therefore, we used bioinformatics tools to identify candidates that could serve as the flavin and quinone reductases for the HadA reaction. As the had operon consists of two separate gene clusters, hadRXABC and hadSYD (10), individual genes were analyzed for their potential to function as flavin and quinone reductases. The sequence of HadA was also used for analysis by the STRING database to identify proteins that are commonly found to be co-overexpressed with HadA and their orthologs (22). Results indicate that HadA is commonly found to be co-overexpressed with HadX, HadD, HadB, and HadC (from high to low association scores, Fig. S1). More detailed analysis of the protein sequences indicated that both HadX and HadB are flavoproteins in which HadX belongs to the class of flavin reductases that have the function of generating reduced flavin for two-component monooxygenases, whereas HadB belongs to the class of FMN-containing NAD(P)H nitroreductases (NR), which catalyzes the reduction of nitro to amino groups (Fig. S2). The HadC sequence is closely associated with catechol 1,2-dioxygenase, which catalyzes the further step of aromatic ring-cleavage, whereas HadD is likely an iron-containing alcohol dehydrogenase that catalyzes the reduction of maleylacetate to β-ketoadipate (Fig. 1) (10, 23). Therefore, HadX and HadB were identified as candidates that may function as the flavin reductase and quinone reductase for the HadA reaction, respectively.
HadB belongs to a family of reductases that have diverse substrate and functional specificities. Based on the initial sequence analysis using the Pfam database from the European Bioinformatics Institute (EMBL-EBI) (24), the HadB sequence is similar to the sequences of enzymes in the NR family, which can use NADH or NADPH as a reductant to reduce a variety of nitroaromatic compounds (25). Examples of NRs identified in the same family as HadB (Fig. S2A) include the enzymes from Escherichia coli, NfsA and NfsB, which can reduce nitroaromatic compounds and showed promiscuous activity with quinone derivatives, giving comparable catalytic turnover numbers (26–28). Based on sequence analysis, TcpB, the enzyme that has been proposed to be involved as a quinone reductase in the degradation pathway of 2,4,6-trichlorophenol (2,4,6-TCP) has the closest relationship with HadB (75% identity) (29). TcpB is encoded in the tcp cluster from Ralstonia eutropha JMP134.
HadX belongs to the family of flavin reductases that generate reduced flavin for their monooxygenase partners. The analysis in Fig. S2B indicates that HadX has a close relationship with TftC from Burkholderia cepacia AC1100 (51% identity) (9, 30) and TcpX from R. eutropha JMP134 (56% identity) (29). Both of these enzymes were isolated in the apoenzyme form, lacking a bound flavin prosthetic group. Mechanistic investigation of these enzymes has not been carried out. HadX was also classified in the same group as various reductases of two-component flavin-dependent monooxygenases, such as ActVB reductase from Streptomyces coelicolor (24% identity) (31–35), RebF from Lechevalieria aerocolonigenes (29% identity) (36), C1 from Acinetobacter baumannii (19, 20) (16% identity), and LuxG from Photobacterium leiognathi TH1 (9% identity) (37–39). Previous investigations have shown that these reductases can generate reduced flavin and pass it to their oxygenases without a requirement of protein–protein interaction (21). It would be of interest to investigate whether HadX is a flavin reductase and whether it can function similarly to other flavin reductases in the same class.
In the work reported here, we overexpressed, purified, and investigated the functional and catalytic properties of HadB and HadX. The results showed that HadB is a quinone reductase that can reduce quinone products from the HadA reaction to generate hydroquinone compounds. Furthermore, the HadB enzyme does not exhibit nitroreductase activity. HadX is indeed a flavin reductase that can efficiently generate FADH− for the HadA reaction and can pass on FADH− to HadA without requiring protein–protein interaction. Functional integration of these three flavin-dependent enzymes provides a complete and efficient system for the detoxification of HPs and NPs, which are important for future applications in bioremediation processes for detoxification of HPs and NPs.
Results
Biophysical properties of purified HadB and HadX
Recombinant HadB and HadX were overexpressed in E. coli BL21 (DE3) and purified following the protocols described under “Experimental procedures” to yield 360 and 5 mg of purified HadB and HadX from 1 liter of culture, respectively. Absorption properties of both enzymes indicated that they bind flavin prosthetic groups (Fig. S3), which are different from biophysical properties of their homologs, TftC, TcpX, and TcpB, which were previously isolated as apoenzymes (9, 29, 30). Analysis by TLC indicates that HadB binds oxidized FMN, whereas HadX binds oxidized FAD. Subunit molecular masses of HadB and HadX judged from SDS-PAGE analysis were ∼21 and ∼19 kDa, respectively. Molar absorption coefficients of HadB-bound FMN and HadX-bound FAD at 456 nm (ϵ456 nm) were calculated as 11.3 and 11.8 mm−1 cm−1, respectively. Comparison of the biophysical properties of HadB and HadX and other previously known reductases are summarized in Tables 1 and 2, respectively.
Table 1.
Comparison of catalytic properties of HadB with other quinone reductases
| Enzyme | Organism | Cofactor | Reaction | Kinetic parameters |
Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| Km,NAD(P)H | Km,acceptor | kcat | kred | kox | |||||
| μm | s−1 | ||||||||
| HadB | R. pickettii | FMN | Ping Pong | 31 ± 13 | 38 ± 10a | 31 | >350 s−1 | >350 s−1 | This study |
| TcpB | R. eutropha | FMN | –b | 231 ± 67 | 85 ± 17a | 351 ± 29 | – | – | 29 |
| NDH–2 | C. thermarum | FAD | Ping–pong ternery–complex | 29 ± 2 | 34 ± 4a | ∼880 | ∼200 s−1 | ∼200 s−1 | 43 |
| WrbA | E. coli | FMN | Ping–pong | 19 ± 2 | 37 ± 4c | 370 ± 2 | – | – | 49, 50 |
| NsfA | E. coli | FMN | Ping–pong | – | 11 ± 2c | 62 ± 6 | >400 s−1 | – | 28 |
| ChrR | P. putida | FMN | Ping–pong | – | 110c | 930 | – | – | 51 |
| Lot6p | S. cerevisiae | FMN | Ping–pong | – | 2.7 ± 0.4c | 21 ± 1 | 311 ± 16 m −1 s−1 | 1.03 × 107 m−1 s−1 | 52, 53 |
| NQO–1 | Rattus sp. | FAD | Ping–pong | 61 ± 1 | 5 ± 1d | 883 ± 12 | 1.5 × 107 m−1 s−1 | 1.5 × 103 m−1 s−1 | 54, 55 |
| PA1225 | P. aeruginosa | FAD | Ping–pong | 82 ± 5 | 5.9 ± 0.1b | 10.1 ± 0.3 | 10.4 s−1 | – | 56 |
a Electron acceptor is menadione.
b Dash (–) indicates none reported.
c Electron acceptor is PQQ.
d Electron acceptor is 1,4–benzoquinone.
Table 2.
Comparison of catalytic properties of HadX with other flavin reductases
| Enzyme | Organism | Cofactor | Oxygenase partner | Kinetic parameters |
CT complex formation | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| kred | kox with O2 | KD, flox | KD, flred | k, flred transfer | ||||||
| s−1 | m−1 s−1 | μm | s−1 | |||||||
| HadX | R. pickettii | FAD bound | HadA | 104 ± 1 | 4.3 ± 0.1 × 103 | 0.38 ± 0.02 | 1.7 ± 0.6 | 7.0 ± 0.6 | Yes | This study |
| TcpX | R. eutropha | FAD | TcpA | –a | – | – | – | – | – | 29 |
| TftD | B. cepacia | FAD | TftD | – | – | 1.8 ± 0.1 | – | – | – | 9, 30 |
| C1 (C1–HPA) | A. baumannii | FMN bound | C2 | 11.6 ± 0.3 (300) | 820 (820) | 0.006 ± 0.001 (0.038 ± 0.007) | – (≫0.038 ± 0.007) | ∼0.35 (∼80) | Yes | 19–21, 44, 46 |
| ActVB | S. coelicolor | FMN bound | ActVA | – | – | 4.4 ± 0.6 | 6.6 ± 0.6 | 5.0 ± 0.4 | Yes | 31, 34, 35 |
| LuxG | P. leiognathi TH1 | FMN | LuxAB | 68 ± 6 | – | Not bind | – | ∼5.0 | Yes | 36–38 |
| HpaC | E. coli | FAD | HpaB | – | – | 5.8 ± 0.3 | – | – | – | 65 |
| PheA2 | B. thermoglucosidasius A7 | FAD bound | PheA1 | – | – | Tight | – | – | – | 66 |
| StyB | Pseudomanas sp. | FAD | StyA | – | – | 2.3 ± 0.3 | – | – | – | 67 |
a Dash (–) indicates none reported.
Catalytic function and mechanistic properties of HadB
The substrate specificity and reaction kinetics of HadB were investigated using multiple turnover reactions, and steady-state and transient kinetics to assign the functional role of HadB.
HadB is a quinone reductase
To identify whether HadB has quinone reductase activity, multiple turnover reactions of HadA using 2,4-dichlorophenol (2,4-DCP), 2,4,5-trichlorophenol (2,4,5-TCP), and 2,4,6-TCP to produce unstable quinone products, chlorohydroquinone (CHQ), 2,5-dichlorohydroquinone (2,5-DCHQ), and 2,6-dichlorohydroquinone (2,6-DCHQ), respectively, were carried out in the absence and presence of HadB. C1, the pure flavin reductase, in addition of FAD is used as FADH− providing a system to avoid nonspecific quinone reductase activity. Conversion of quinone to hydroquinone was monitored by absorption changes at longer wavelengths due to the increase or decrease of polymerized quinone (40). Spectra shown in Fig. 2 reveal that in the absence of HadB, the increase of absorbance at the longer wavelength regions were observed in all substrate reactions, indicating that quinone products were polymerized into aggregated polymer. On the contrary, the reaction in the presence of HadB did not show any sign of polymerized products and only resulted in changes in the UV region due to the increase of hydroquinone products. These data reveal that HadB can reduce quinone and prevent quinone polymerization.
Figure 2.

HadB can prevent quinone polymerization. A–F, absorption spectra of multiple turnover reactions of HadA in the absence (A–C) and presence of HadB (1 μm) (D–F) using 2,4-DCP, 2,4,5-TCP, and 2,4,6-TCP as substrates. The FADH− generating system employed consisted of C1 reductase (1 μm), FAD (10 μm), Glc-6-P dehydrogenase (0.5 unit/ml), and Glc-6-P (1 mm).
To investigate whether HadB is also a nitroreductase, reactions of 2-NP, 3-NP, and 4-NP in the presence of HadB with NADH regenerating system were performed. No reduction of the nitro group was observed, indicating that HadB does not contain nitroreductase activity (data not shown).
Kinetics of HadB reduction by NADH and NADPH
The quinone reduction reaction catalyzed by HadB requires a reductant to reduce the enzyme-bound FMN during the reductive half-reaction. Thus, transient kinetic experiments using stopped-flow techniques were used to monitor and identify the preferred reductant for HadB. A solution of oxidized HadB (HadBFMN, 12.5 μm) was mixed with various concentrations of NADH or NADPH (0.025–1.6 mm) under anaerobic conditions to avoid autoxidation of FMNH− during the measurements. Concentrations indicated are the final concentrations after mixing. Reactions were performed in 50 mm NaPi, pH 7.0, at 4 °C. The absorbance at 456 nm was followed to monitor the reduction of HadBFMN. In the presence of NADH (Fig. 3A), the kinetic traces of flavin reduction showed three observable phases. The first phase had the highest amplitude (∼85% of the total HadB-bound enzyme) and had occurred mostly during the dead-time period of the instrument (∼0.002 s after mixing). Due to the inability to collect data during the dead-time, the observed rate constants for this phase could not be determined; however, it can be approximated that the observed rate constant of HadB reduction by NADH are >350 s−1 (t½ <0.002 s). The second (∼7% of total amplitude) and third phases (∼8% of total amplitude) showed slower kinetics of flavin reduction, with rate constants of 0.34 and 0.03 s−1, respectively, and are independent of NADH concentration. Detection using a diode-array detector did not indicate the presence of any long wavelength species such as charge-transfer complexes or flavin semiquinones (Fig. S4).
Figure 3.
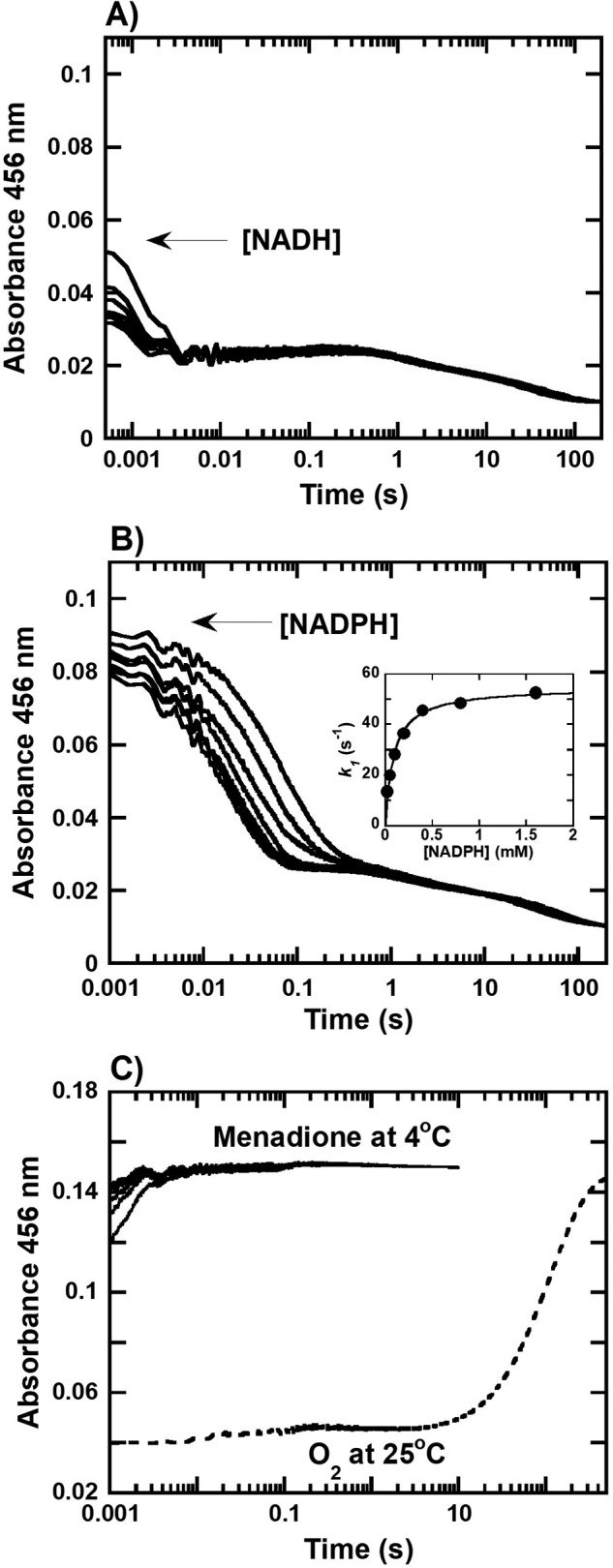
Transient kinetics of the reductive and oxidative half-reaction of HadB. A and B, kinetic traces of the reactions of anaerobic HadBFMN (12 μm) mixing with (A) NADH or (B) NADPH (0.05–3.2 mm) in 50 mm NaPi, pH 7.0, at 4 °C. Inset in B shows a plot between the observed rate constant (kobs) of the first phase of the reaction versus NADPH concentrations which yields a hyperbolic relationship. C, kinetic traces of the reactions of anaerobic HadBFMNH− (12.5 μm) mixing with various concentrations of menadione (50–200 μm) in 50 mm NaPi, pH 7.0, at 4 °C (solid lines) and reactions of HadBFMNH− (12.5 μm) mixed with O2 (0.13 mm) in 50 mm NaPi, pH 7.0, at 25 °C (dashed lines) were obtained by monitoring the absorbance of the reactions at 456 nm.
To further investigate the kinetics of flavin reduction of HadB, NADPH was utilized as a reductant similar to the experiments previously described for NADH. The results from NADPH reduction (Fig. 3B) also showed three observable phases with the amplitudes of each phase being similar to those of the corresponding phases in the reduction by NADH. Flavin reduction of the first phase also accounted for ∼80% of the total enzyme concentration similar to the first phase observed by NADH reduction, but occurred around 0.002–0.1 s. This was slower than the corresponding step in the reduction by NADH (Fig. 3A), indicating that NADH is the more preferred reductant over NADPH. Flavin reductions during the second (∼11% of total amplitude) and third (∼9% of total amplitude) phases were independent of the NADPH concentration, with observed rate constants of 0.59 and 0.02 s−1, respectively.
A plot of the observed rate constants of the first phase of flavin reduction versus NADPH concentrations yielded a hyperbolic relationship (Fig. 3B, inset), indicating that the majority of HadB reduction by NADPH occurs via a two-step process in which the first step is binding of NADPH to form a binary complex (HadBFMN−NADPH in Fig. 4) followed by a two-electron transfer flavin reduction to achieve complete reduction (HadBFMNH− in Fig. 4). Analysis of the data using Equation 1 (21) indicates that the dissociation constant (KDNADPH) for NADPH binding was 90 ± 6 μm, whereas the flavin reduction rate constant (kred) was 55 ± 1 s−1. A model describing flavin reduction by NADH, which is much faster than by NADPH can be assumed to be similar to the HadB reduction by NADPH (Fig. 4).
| (Eq. 1) |
Figure 4.

Proposed reaction mechanism of HadB quinone reductase. kred and k*red represent the rate constants of HadBFMN and HadBFMN* reduction by NAD(P)H, respectively. kox represents the rate constant of electron transfer from HadBFMNH− to an electron acceptor such as menadione.
The multiphase reduction of HadB is likely due to the presence of a mixture of enzyme populations in which ∼80% of the total species is reduced faster than the rest of the populations (Fig. 3, A and B). This explanation was confirmed experimentally by using another reductant to reduce HadB, i.e. sodium dithionite (Na2S2O4). Kinetic traces of the HadB reduction by dithionite were also multiphasic, with amplitudes similar to those for the reduction by NADH or NADPH, ∼78, ∼16, and ∼6% for first, second, and third phases, respectively (Fig. S5). Only the first and second phases were dependent upon the concentrations of Na2S2O4. As the mechanism of flavin reduction by dithionite is different from that of NAD(P)H, the data indicate that the multiphasic reduction (first and second phase) is indeed due to the nature of mixed protein populations (HadBFMN and HadBFMN* in Fig. 4). The third phase may be due to the release of NAD(P)+. The kinetic model describing the reductive half-reaction of HadB is shown in Fig. 4.
Kinetics of HadB-bound flavin oxidation by various electron acceptors
During the oxidative half-reaction, the reduced FMN bound to HadB transfers two electrons to an acceptor. The kinetics of the oxidative half-reaction of HadB was investigated to identify the best electron acceptor for HadB using stopped-flow spectrometry. A solution of reduced HadB (HadBFMNH−, 12.5 μm) was mixed with various concentrations of menadione (a stable quinone compound, 50–200 μm) or oxygen in 50 mm NaPi, pH 7.0, at 4 °C inside the stopped-flow spectrophotometer, and the oxidation state of the flavin was monitored by following the absorbance change at 456 nm. The kinetic traces (solid lines in Fig. 3C) showed that oxidation of HadBFMNH− by menadione occurred very rapidly during the dead-time of the instrument, and finishing within 0.003 s, corresponding to a rate constant of >350 s−1 (t½ < 0.002 s). Observed rate constants were dependent upon the menadione concentration, suggesting that the oxidation involves a two-step process in which a HadBFMNH−–menadione complex forms prior to complete electron transfer to menadione (Fig. 4). In contrast, flavin reoxidation by oxygen occurred quite slowly (9.8 ± 0.1 m−1 s−1 at 25 °C) (dashed line in Fig. 3C), clearly indicating that HadB is an O2–insensitive quinone reductase that prefers to use a quinone derivative as an electron acceptor.
Steady-state kinetics of HadB
Data from the previous section have shown that HadB prefers NADH and menadione as the electron donor and acceptor, respectively. Steady-state assays of HadB (10 nm) with various concentrations of both NADH (25–200 μm) and menadione (0–300 μm) were performed in 50 mm NaPi, pH 7.0, at 4 °C. NADH oxidation was observed by monitoring the decrease in absorbance at 340 nm. Direct plots of the rates of NADH consumption versus menadione and NADH concentrations and double reciprocal plots of e/v versus 1/[menadione] or 1/[NADH] concentrations are shown in Fig. S6, A and B. The reciprocal plots in Fig. S6B showed a parallel line pattern, a Ping Pong Bi Bi mechanism. These data indicates that HadB catalyzes quinone reduction via similarity of HadB to other quinone reductases from various organisms (Table 1) (41, 42). Analysis of the data in Fig. S6B using Dalziel's equation for a Ping Pong mechanism (Equation 2) indicates φNADH of 1.0 ± 0.4 × 10−3 mm·s, φmenadione of 1.2 ± 0.3 × 10−3 mm·s, and φ0 of 3.2 ± 0.1 × 10−2 s, respectively (Fig. S6B, inset). These values can be used for calculation of KmNADH, Kmmenadione, and kcat as 31 ± 13 μm, 38 ± 10 μm, and 31 s−1, respectively. Based on all the kinetic data, the kinetic mechanism of HadB reductase can be summarized as in Fig. 4.
| (Eq. 2) |
HadX is a flavin reductase
Multiple turnover reactions of HadA coupled with HadX were carried out to identify whether HadX can generate FADH− for the HadA reaction. The activities of HadA in hydroxylation and -NO2 group elimination from 4-NP were detected by a decrease in absorbance at 400 nm. The results indicated that HadX can generate FADH− for HadA (Fig. S7A). These data suggest that HadX is a flavin reductase that can provide FADH− for HadA monooxygenase.
Kinetics of flavin reduction on HadX
Transient kinetic experiments with HadX were performed using a stopped-flow apparatus with single wavelength and diode-array detectors to monitor the kinetics of flavin reduction by NADH. A solution of anaerobic oxidized HadX (HadXFAD) (15 μm) was mixed with solutions of anaerobic NADH (0.05–3.2 mm) in 50 mm NaPi, pH 7.0, at 4 °C. The results (Fig. 5 and Fig. S8) showed a decrease in flavin absorbance at 456 nm and an increase in absorption in the 500–700 nm region. The decrease of flavin absorption is caused by electron transfer from NADH to the oxidized flavin moiety, whereas formation of the species that absorbs at long wavelengths is caused by formation of a charge-transfer complex (HadXFADH−–NAD+) similar to what has been observed during the reductive half-reactions of many flavin reductases (21, 31, 35, 43). When similar experiments using NADPH were carried out, we could not detect any reduction of the enzyme-bound flavin by NADPH within 30 s (data not shown), indicating that HadX is only specific for using NADH as a reductant.
Figure 5.
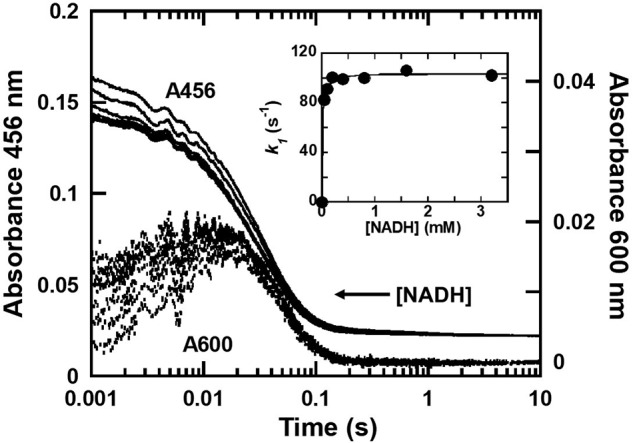
Transient kinetics of the reductive half-reaction of HadX. Kinetic traces of reactions of anaerobic HadXFAD (15 μm) mixed with various concentrations of NADH (0.05–3.2 mm) in 50 mm NaPi, pH 7.0, at 4 °C were obtained by monitoring the absorbance at 456 (solid lines) and 600 nm (dashed lines) to detect flavin reduction of HadB and formation of the charge-transfer complex, respectively. The inset is a plot between the observed rate constants of the first phase versus NADH concentration.
Analysis of the kinetic traces shown in Fig. 5 indicates that the flavin reduction on HadX occurred in three phases. The first phase occurred during the dead-time of the instrument (<2 ms) until 0.015 s. This phase shows a concomitant decrease in absorbance at 456 nm (A456) (∼50% of the total amplitude) due to flavin reduction, and an increase in absorbance at 600 nm (A600) due to formation of a charge-transfer complex (HadXFADH−–NAD+ in Fig. 6). A plot of the observed rate constants of this phase versus NADH concentrations yielded a hyperbolic relationship (Fig. 5, inset). Analysis based on Equation 3 (21) yielded a binding constant of NADH KD,1NADH and the rate constant for the charge-transfer (CT) complex formation or hydride transfer (kred,1) of 13 ± 2 μm and 104 ± 1 s−1, respectively. These data indicate that HadX reduction is a two-step reaction in which a binary complex of HadXFAD-NADH is formed prior to a hydride transfer or flavin reduction step. The charge-transfer complex is formed after the enzyme-bound flavin is reduced (Fig. 6).
| (Eq. 3) |
Figure 6.

Proposed reaction mechanism of HadX flavin reductase. kred,1 and kred,2 represent the rate constants of HadXFAD and HadXFAD* reduction by NADH, respectively. KD,NADH1 and KD,NADH2 represent dissociation constants for bindings of NADH to HadXFAD and HadXFAD*, respectively. kox,1, kox,2, and kox,3 represent the rate constants of free FADH−, HadXFADH−, and HadXFADH−* reacting with oxygen, respectively.
The second phase occurring around 0.015–0.2 s showed a decrease in A456 due to reduction of rest of flavin species (∼50% amplitude) with the observed rate constant of 33 ± 2 s−1 (kred,2). This kinetic phase occurred without formation of the long wavelength species and independent of NADH concentrations. We propose that HadX contains two species of populations in which the first one (HadXFAD in Fig. 6) is reduced faster and forms the charge-transfer complex, whereas the second population (HadXFAD* in Fig. 6) is reduced slower and does not form any charge-transfer species. The independence of the reduction rate constant of the second flavin population on NADH concentration may be due to the very low value of dissociation constant for the binding (KD,2NADH) (much lower than 0.05 mm, which is the lowest concentration of NADH used) (Fig. 5).
The last phase was the decrease of absorbance at long wavelengths due to the decay of a charge-transfer complex resulting from the first flavin reduction phase, consistent with the rate constant of 22 ± 1 s−1 (kdecay). The overall reaction of HadA reduction by NADH can be summarized as in Fig. 6.
Kinetics of flavin reoxidation on HadX
The kinetics of oxidation of HadX-bound reduced flavin was investigated by mixing a solution of reduced HadX (HadXFADH−) with various concentrations of oxygen (0.13, 0.31, and 0.61 mm) equilibrated in 50 mm NaPi, pH 7.0, at 25 °C. The kinetics of flavin reoxidation shows three observable phases, which are all dependent on the oxygen concentrations (Fig. 7). Plots of the observed rate constants of each phase versus oxygen concentration yielded linear relationships with bimolecular rate constants of 2.5 ± 0.2 × 104 m−1 s−1 (kox,1), 4.3 ± 0.1 × 103 m−1 s−1 (kox,2), and 1.2 ± 0.1 × 103 m−1 s−1 (kox,3), respectively (Fig. 7, inset). As the kinetics of the first phase of the flavin oxidation appears to be similar to the kinetics of free FADH− reacting with oxygen (2.7 × 104 m−1 s−1), the results indicate that the first phase is the reaction of free FADH− that rapidly diffuses out of HadXFADH− to react with oxygen. The second and third phases are likely the reaction of HadX-bound FADH− with oxygen, which reacts slower. A small perturbation signal between the second and third phases (∼1– 10 s) was proposed to be the re-binding of FAD from the first phase to apoHadX. The proposed model for HadX oxidation by molecular oxygen is shown in Fig. 6.
Figure 7.
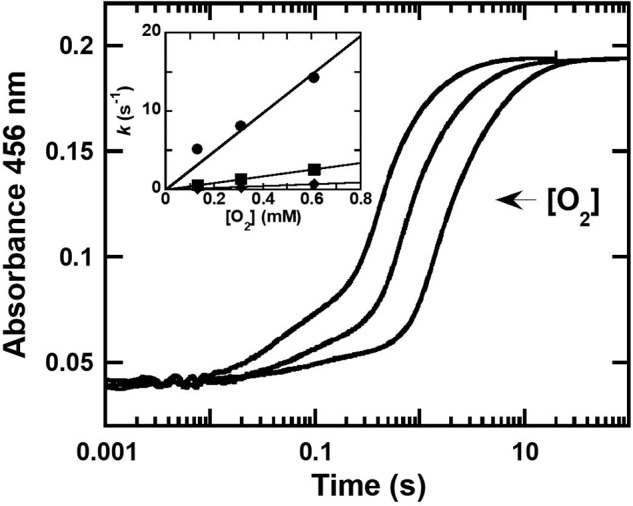
Transient kinetics of the reaction of HadXFADH− and oxygen. Kinetic traces of anaerobic HadXFADH− (15 μm) mixed with various concentrations of oxygen (0.13, 0.31, and 0.61 mm) in 50 mm NaPi, pH 7.0, at 25 °C were obtained by monitoring the absorbance at 456 nm. Inset shows a plot of the observed rate constants of the 1st phase (circle line), 2nd phase (square line), and 3rd phase (diamond line) versus oxygen concentration, yielding linear relationships with bimolecular rate constants of 2.5 ± 0.2 × 104, 4.3 ± 0.1 × 103, and 1.2 ± 0.1 × 103 m−1 s−1, respectively.
Binding affinity of HadX to FAD and FADH
One of the key properties of the flavin reductases that provide reduced flavin for the monooxygenase is that these enzymes bind tightly to oxidized flavin, but loosely to reduced flavin (21). We thus measured the binding affinity of HadX to FAD and FADH− to assess whether HadX also behaves similarly to the other reductases of the same class. Equilibrium binding of HadX to FAD was measured using an ultrafiltration method (“Experimental procedures”). The thermodynamic dissociation constant of HadX and FAD (KDFAD) was calculated as 0.38 ± 0.02 μm, indicating that HadX binds tightly to oxidized FAD. As the experiments described above in the previous section could measure the amount of free FADH− and HadX-bound FADH− due to their different kinetics in reacting with oxygen (Fig. 7), a dissociation constant for binding of HadX and FADH− (KDFADH−) can be calculated from the amplitude of the first phase, which represents free FADH− and the second phase, which represents the HadX-FADH− complex. Experiments similar to those in Fig. 7 but with variable concentrations of total HadX were carried out to calculate the amount of free and HadX-bound FADH− at various HadX concentrations (Fig. S9). Based on these data, the KDFADH− was calculated as 1.7 ± 0.6 μm. The KDFAD and KDFADH− values indicate that HadX is similar to other flavin reductases of the same class in that it binds to FADH− weaker than to FAD.
FADH− is transferred from HadX to HadA via free diffusion
HadX can provide FADH− to HadA and C2 monooxygenase
If HadX and HadA catalyze the reaction via free diffusion transfer of FADH−, the transfer of reduced flavin between HadX and HadA does not require specific protein interactions. Based on this principle, various reductases can be used to provide reduced flavin to the same oxygenase component, whereas the same flavin reductase can also provide reduced flavin to various monooxygenases.
The ability of HadX to provide FADH− for HadA was compared with C1 (the reductase of the p–hydroxyphenylacetate monooxygenase system). Multiple turnover reactions of HadA coupled with HadX or C1 (at the same concentrations) catalyzing elimination of an –NO2 group from 4-NP were carried out. The results indicate that HadX can generate FADH− for HadA with a 4-NP consumption rate of 2.8 μm/min which is higher than the rate of 4-NP consumed in the presence of C1 (0.4 μm/min, Fig. S7, A and B). These results indicate that HadX, which is a reductase encoded in the same operon as HadA can provide FADH− for the HadA reaction more effectively than C1, which is a reductase from a different system.
We next investigated whether HadX can also be used for providing free FADH− to C2, which is an oxygenase from a different system. C2 is an oxygenase component of p-hydroxyphenylacetate hydroxylase from A. baumannii, which can use FMNH− and FADH− as substrates equally well (44). The results (Fig. S10, A and B) indicate that HadX and also be used to provide FADH− for C2 efficiently. Altogether, these data clearly indicate that HadX functions as a flavin reductase, which can provide free FADH− for the HadA system via a free diffusion mechanism without requiring any specific protein–protein interactions.
Free FADH− from HadX can serve as a substrate for HadA to form C4a-hydroperoxy-FAD
In this experiment, we investigated whether FADH− generated from HadX can be bound to HadA and form C4a-hydroperoxy-FAD, a reactive intermediate required for the monooxygenation reaction. A solution of anaerobic HadXFADH− (15 μm) was mixed with air-saturated HadA (0.1 mm) in 50 mm NaPi, pH 7.0, at 25 °C in a stopped-flow spectrophotometer. If FADH− can dissociate from HadX and bind to HadA, we would observe formation of the C4a-hydroperoxy-FAD intermediate. The kinetic traces obtained from this experiment showed clear formation of C4a-hydroperoxy-FAD, as reflected by the increase in absorbance at 380 nm (observed rate constant of 7.0 s−1), which occurred before the flavin reoxidation detected at 456 nm (solid line in Fig. 8, A and B). A control experiment using free FADH− (dashed line in Fig. 8, A and B) also resulted in similar kinetics of intermediate formation (observed rate constants of 6.8 s−1). The spectra of the C4a-hydroperoxy-FAD of HadX and free FADH− reactions were similar (Fig. 8B, inset), indicating that the FADH− from HadA can dissociate and bind to HadA. The reactions of free FADH− and FADH− dissociated from HadX only differ in the last step of oxidized flavin formation (10–100 s). For free FADH−, the observed rate constant of the last step of FADH− oxidation was 0.007 s−1, whereas this step in the presence of HadX had an observed rate constant of 0.003 s−1. The differences in the kinetics of this step is due to the re-binding of oxidized FAD back to apoHadX in the reaction with HadX. The overall model describing the transfer of FAD and FADH− between HadX and HadA is shown in Fig. 9.
Figure 8.
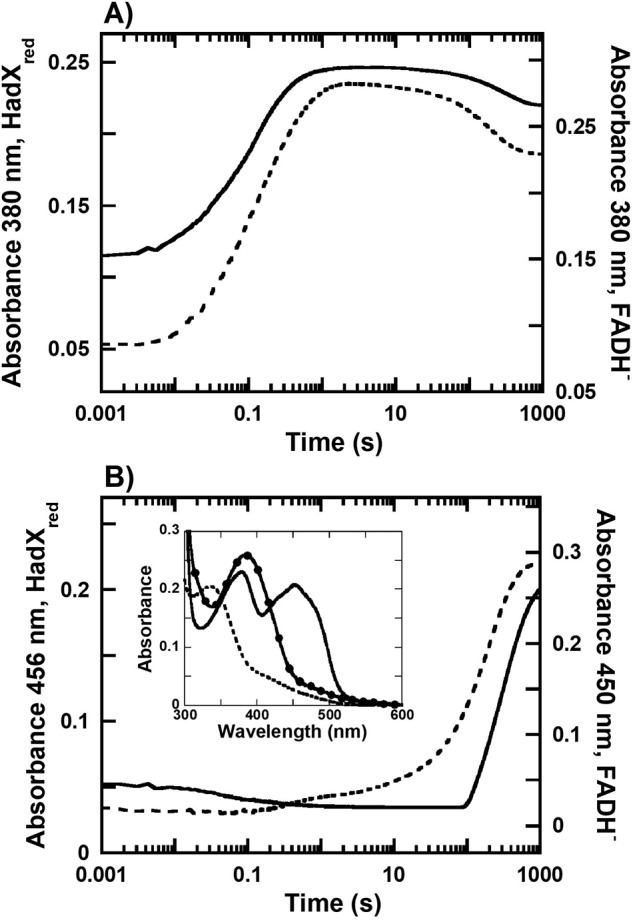
Kinetics of the transfer of FADH− from HadX to HadA. Anaerobic solutions of HadXFADH− (15 μm) were mixed against aerobic HadA (0.1 mm) in 50 mm NaPi, pH 7.0, at 25 °C. Kinetic traces (solid line) detected at wavelengths of (A) 380 and (B) 456 nm are compared with kinetic traces of aerobic HadA mixed with free FADH− (dashed line) previously reported from Ref. 17. Inset in B are the spectra of three flavin species including the HadXFADH− (dashed line), the intermediate obtained at 10 s after the reaction started (solid line with filled circles), and a mixture of HadA and HadXFADH− (solid line).
Figure 9.

Transfer of FADH− from HadX reductase to HadA monooxygenase.
Measurement of the rate of FADH− dissociation from HadXFADH− using formation of C4a-hydroperoxy-FAD bound to C2 as an indicator
The rate constants of FADH− transfer from HadX to C2 are the result of two steps of the reaction: dissociation of FADH− from HadX and binding of FADH− to C2 according to Equation 4. As the rate constant for binding of FADH− to C2 (second step) is very high (≫700 s−1) (45), the observed rate constant of FADH− transfer is limited by the rate constant of FADH− dissociation from HadX (first step).
| (Eq. 4) |
The overall process of FADH− transferred to C2 can be monitored by following the formation of C4a-hydroperoxy-FAD as reflected by an increase in absorption at 380 nm (A380) (21, 38, 46). In this experiment, a solution of HadXFADH− (15 μm) and C2 (25 μm) were mixed anaerobically in the first mixing step of a double-mixing stopped-flow spectrophotometer. The reaction was incubated for various time periods before a second mixing to add oxygen (0.61 mm). The increase in A380 (where Absmax of C4a-hydroperoxy-FAD occurs) at t = 0.02 s was used as the signal indicating the formation of C4a-hydroperoxy-FAD, which is directly controlled by the dissociation of FADH− from HadX. Higher absorbance values at longer incubation times indicated more complete transfer of FADH− to C2 because FADH− that is not transferred to C2 would not generate absorbance during this time period. Results in Fig. 10 indeed show that A380 at 0.02 s was increased when the incubation time was longer, indicating that the longer the incubation time, the more complete the FADH− transfer to C2. A plot of ΔA380 at t = 0.02 s versus the age time showed an exponential increase, which is consistent with the observed rate constant of FADH− dissociation from HadX of 7.0 ± 0.6 s−1 (Fig. 10, inset).
Figure 10.
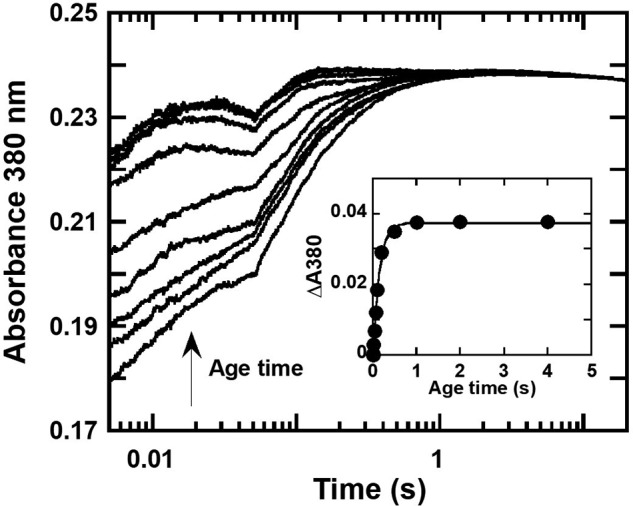
Determination of a rate constant of FADH− transfer from HadX to C2 monooxygenase. Double-mixing stopped-flow experiments were carried out by mixing a solution of HadXFADH− (15 μm) with C2 (25 μm) under anaerobic conditions in the first mixing. The mixing was prolonged at different age times (0.01–4 s) prior to mixing with a buffer containing oxygen (0.61 mm) under the second mixing. Absorption changes at wavelength 380 nm were measured to monitor the formation of C4a-hydroperoxy-FAD. Inset is a plot of ΔA380 at t = 0.02 s (the amount of C4a-hydroperoxy-FAD intermediate formed) versus age times to obtain the rate constant of the FADH− transfer from HadXFADH− to C2 monooxygenase as 7.0 ± 0.6 s−1.
Integrative three enzymatic reaction cascades of HadA, HadB, and HadX for efficient biodegradation of HPs and NPs
To demonstrate that by having the complete enzymatic system of all three flavin-dependent enzymes, HadA/HadB/HadX, the detoxification processes of HPs and NPs can occur efficiently, multiple turnover reactions of these enzymes using 4-NP and 4-CP as substrates were carried out. The rates of 4-NP bioconversion were monitored by measuring the change in UV-visible absorption (Fig. S7C). The results showed that a combination of the three enzymes yielded a rate of 4-NP consumption of 4.7 μm/min, which is ∼1.3-fold faster than that of the reaction in the absence of HadB and ∼1.7-fold faster than the reaction using C1 as the flavin reductase (Fig. S7, A and B).
When the amount of product was analyzed by HPLC/DAD, the data indicated that in both reactions of 4-NP and 4-CP (Fig. 11, A and B), almost 100% yield of hydroquinone (HQ) was observed in the reaction containing HadA/HadB/HadX (red squares lines in Fig. 11), whereas only ∼80% of HQ was observed in the reaction of HadA/HadX without HadB (blue circles lines in Fig. 11). This was probably due to the loss of unstable benzoquinone (BQ) during the turnovers. The data also indicated that the reaction without HadB and HadX could not remove substituents from 4-NP or 4-CP (purple triangle lines in Fig. 11). To compare the quinone reductase activity of HadB with chemical reductants, such as ascorbic acid, products from the reactions in the presence of ascorbic acid (green diamond lines in Fig. 11) or HadB were analyzed. The results indicated that both reactions could generate the same amount of product with similar rates (red square lines and green diamond lines in Fig. 11). Therefore, reduction of benzoquinone products by HadB or ascorbic acid is required for maintaining a high rate of product formation (Fig. 11) and HadB is responsible for in vivo production of hydroquinone. Altogether, these results demonstrate that the combination of HadA monooxygenase, HadX flavin reductase, and HadB quinone reductase can perform the most efficient bioconversion of 4-NP and 4-CP to HQ.
Figure 11.
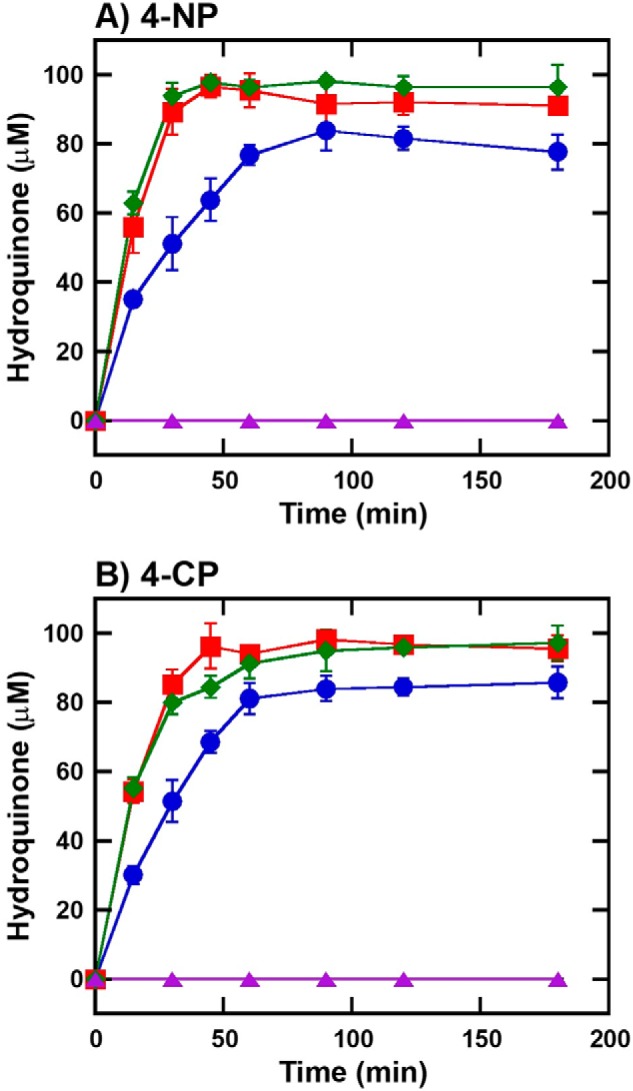
Biodegradation of 4-NP and 4-CP by cascade reactions of HadA monooxygenase, HadX and HadB reductases. Multiple turnover reactions of HadA consisted of (A) 4-NP (100 μm) or (B) 4-CP (100 μm), Glc-6-P (1 mm), Glc-6-P dehydrogenase (0.5 unit/ml), NAD+ (5 μm), HadA (10 μm), with different combinations of reductases in 50 mm NaPi, pH 8.0, at room temperature. Reactions were quenched at various times by adding an equal volume of acetonitrile. Samples were diluted 3-fold in 50 mm NaPi, pH 8.0, and ascorbic acid (0.5 mm) to convert all BQ products to HQ for more convenient analysis by HPLC/DAD. Blue circle lines are the reactions containing HadX (1 μm). Red square lines are the reactions containing HadX (1 μm) and HadB (1 μm). Green diamond lines are the reactions containing HadX (1 μm) and ascorbic acid (1 mm). Purple triangle lines are the reactions without any reductase.
The had biodegradation system
Based on all data, the enzymatic had system for HPs/NPs degradation by the three flavin-dependent enzymes, HadA monooxygenase coupled with HadX flavin reductase and HadB quinone reductase, can be summarized as in Fig. 12. Starting from reduction of HadXFAD by NADH and transfer of FADH− from HadXFADH− to HadA via free diffusion. The HadA-FADH− complex reacts with molecular oxygen to form a C4a-hydroperoxy-FAD intermediate prior to phenol substrate binding. HadA catalyzes hydroxylation in combination with group elimination to produce C4a-hydroxy-FAD and quinone as the product (16, 17). The flavin intermediate eliminates H2O to form FAD that is transferred back to HadX to re-form HadXFAD. For the quinone product route, quinone was released and reduced to hydroquinone by HadB. Rate constants from transient kinetic experiments indicate that the reaction of HadA (not HadX or HadB) is the rate-limiting step for the overall biodegradation system (16, 17). Understanding the complete metabolic pathway for the biodegradation of toxic aromatic compounds is useful for future development and optimization to increase the efficiency of the system.
Figure 12.

Overall mechanisms and steps involved in the detoxification of halogenated phenols and nitrophenols by three flavin-dependent enzymes in the had operon.
Discussion
The work reported herein is the first investigation of the functional and mechanistic properties of the two flavoenzymes, quinone and flavin reductases (HadB and HadX), which are required for generating FADH− and for reducing the quinone product of HadA monooxygenase. HadB and HadX are necessary for efficient detoxification by HadA monooxygenase especially under in vivo conditions in which an external reductant is not available.
HadB is the first FMN-dependent quinone reductase of the aromatic compound biodegradation pathway to have been investigated in-depth for its catalytic properties. Previously, a quinone reductase in the tcp clusters, TcpB, from R. eutropha JMP134 was shown to enhance the rate of its monooxygenase partner, TcpA, in degrading 2,4,6-TCP. However, the reaction mechanism of TcpB has not been explored (29). In the pentachlorophenol (PCP) degradation pathway of Sphingobium chlorophenolicum, a different type of quinone reductase was identified. PcpF in the pcp cluster was identified as S-glutathionyl-hydroquinone reductase that catalyzes GSH-dependent reduction of BQ to HQ via a two-step mechanism including a first spontaneous reaction of benzoquinone and GSH condensation to form the GS-HQ adduct followed by liberation of GS− and HQ (47). For other operons that can degrade CPs such as tft (9), or cph from Anthrobacter chlorophenolicus A6 (8) or 4-NP degradation systems such as npc from Rhodococcus opacus SAO101 (11), nph and nps from Rhodococcus sp. PN1 (13, 14), and npd from Arthrobacter sp. JS443 (12), the quinone reductases of these systems have not been identified. For the actinorhodin biosynthesis pathway, a quinone reductase has not been identified, but a flavin reductase, ActVB, was shown to be able to reduce dihydrokalafungin (DHKox) to the hydroquinone form product (DHKred) (33).
Generally, quinone reductases function to prevent oxidative stress inside the cell by preventing accumulation of quinone, which can be transformed to a semiquinone radical that can then convert molecular oxygen (O2) to a superoxide radical (O2˙̄) (42, 48). Therefore, HadB may also have another physiological function to prevent accumulation of high levels of quinone and semiquinone. The catalytic properties of HadB and other quinone reductases that are important to preventing cellular oxidative stress such as type II NADH:quinone reductase from Caldalkalibacillus thermarum (NDH-2) (43), quinone reductase WrbA, and nitroreductases NfsA from E. coli (26, 28, 49, 50), chromate-reducing enzyme (ChrR) from Pseudomonas putida (51), quinone reductase Lot6p from Saccharomyces cerevisiae (52, 53), NADH:quinone oxidoreductase type I (NQO1 or DT-diaphorase) from mammalian (54, 55), and PA1225 NADPH:quinone reductase from Pseudomonas aeruginosa PAO1 (56) are compared in Table 1. HadB is similar to most of these enzymes in that it utilizes a Ping Pong Bi Bi mechanism to catalyze the transfer of two electrons from NAD(P)H to quinone. We also noted that the rate constants of HadB reduction by NADH and HadB oxidation by menadione in HadB are very fast compared with other quinone reductases such as Lot6p (53), NDH-2 (43), and PA1225 (56) (Table 1).
Another interesting catalytic feature of HadB is its very low activity toward oxygen (Fig. 3C). This property allows HadB to function efficiently as the quinone reductase without having interference from oxygen during HadA turnovers such as under the conditions employed in Figs. 2 and 11. As HadA is a monooxygenase in which the oxygen is required as a substrate and always presents in the reaction, quinone reductases with low oxygen reactivity such as HadB prevents oxygen from competing with quinone in the HadB reaction. As the HadB structure is not known, the origin of the low oxygen reactivity of HadB cannot be pinpointed. However, the mechanism of oxygen activation has been an active area of research in other flavin-dependent enzymes. The mechanism of oxygen activation was found to be controlled by the gating pathway and angle for oxygen entrance (57–59), the electrostatic environment around the flavin C4a and N5 loci (60–62), and the active site environment to facilitate proton-coupled electron transfer (63, 64). Future studies into the HadB X-ray crystal structure should be helpful in understanding the mechanism controlling electron transfer to an acceptor substrate in HadB.
HadX is the first flavin reductase in the degradation pathway of HPs and NPs that has been characterized in-depth. The functional and catalytic properties of HadX and other known flavin reductases including TftC from Burkholderia cepacia (9, 30), TcpX from R. eutropha (29), C1 from A. baumannii (19, 20, 46), ActVB from S. coelicolor (33–35), LuxG from P. leiognathi TH1 (36–38), HpaC from E. coli (65), PheA2 from Bacillus thermoglucosidasius A7 (66), and StyB from Pseudomanas sp. (67) are compared in terms of their general properties and kinetic parameters in Table 2. HadX is similar to these flavin reductases, which function to provide reduced flavin as a substrate for monooxygenases in that they prefer to bind oxidized, but not reduced flavin (21) (Table 2). Our results in Fig. 8 and Fig. S10 indicate that FADH− generated by HadX can be transferred to a monooxygenase without requiring any specific protein–protein interaction and HadX can also supply FADH− to other enzyme systems such as to C2 monooxygenase. Features of the FADH− free diffusion transfer found in HadX-HadA system are similar to those of the C1-C2, LuxG-LuxAB, ActVB-ActVA, and HpaC-HpaB systems (20, 34, 38, 46, 65).
HadX has some unique properties different from other flavin reductases of two-component monooxygenase systems. Table 2 indicates that HadX is the fastest flavin reductase that is constitutively active (kred,1 of 104 s− 1 and kred,2 of 33 s− 1). Although the rate constant of flavin reduction in C1 (kred of 300 s−1) is faster than that of HadX, this condition is only effective when the effector (p-hydroxyphenylacetate or HPA) is present. The rate of flavin reduction in C1 reductase is quite slow in the absence of HPA (kred of 12 s−1). This is due to an autoinhibitory function of a MarR-like regulatory domain (20). Therefore, HadX should be an efficient reductase for supplying FADH− to any enzymatic systems that require the use of FADH−. HadX is also different from the other reductases of the biodegradation systems of HPs or NPs in that it is the only reductase that was isolated with an enzyme-bound FAD. Flavin reductases of six degradation pathways including TftC (30), TcpX (29), NpcB (11), NphA2 (13), NpsA2 (14), and NpdA1 (12), were isolated as apoenzymes. This can be explained by the KDFAD values; HadX binds to oxidized FAD with a KDFAD of 0.38 ± 0.02 μm, whereas the KDFAD of TftC is 1.8 ± 0.1 μm (30).
Another interesting point to note is that the highest efficiency in degradation of 4-CP and 4-NP can be obtained when all flavin-dependent enzymes, HadA/HadX/HadB, are present (Figs. 11 and 12). HadA and HadX work together as a two-component flavin-dependent monooxygenase, whereas HadB may stabilize the product in the HQ form (Figs. 11 and 12). We propose that the enhancement of the product formation rate may be due to the faster rate of product release by HadA. Based on structure modeling, the active site of HadA is quite hydrophobic. Product accumulation in the form of BQ may lead to product inhibition. In the presence of HadB or ascorbic acid, BQ was rapidly reduced to HQ. As HQ has greater hydrophilicity, it may diffuse out of the HadA active site faster than BQ, speeding up the generation of HadA that is ready to bind a new substrate for the next catalytic cycle. It should be noted that HPs and NPs in the environment may exist under aerobic and anaerobic conditions. In addition to oxidative dehalogenases, reductive dehalogenases such as a cobalamin (B12)–dependent reductive dehalogenase, RdhA from Nitratireductor pacificus (68, 69), can also take part in the detoxification process of organic halides. Oxidative and reductive dehalogenases may be employed in bioremediation applications to maximize the efficiency of biodegradation by microbes.
In conclusion, our work has identified and elucidated functional and catalytic properties of a novel quinone reductase, HadB, and a flavin reductase, HadX. Both of these function in cooperation with HadA monooxygenase to hydroxylate and eliminate halide and nitro group substituents of HPs and NPs. Mechanistic understanding of HadB and HadX reported herein is provides beneficial insight for future development of bioremediation processes.
Experimental procedures
Preparation of purified enzymes
Synthetic hadB and hadX genes from GenScript (NJ) were cloned into a pET-11a expression vector. Both constructed hadB–pET-11a and hadX–pET-11a plasmids were transformed into E. coli BL21(DE3) and grown overnight on LB agar plate containing 50 μg/ml of ampicillin at 37 °C. Large scale productions of recombinant proteins were overexpressed under autoinduction medium containing 50 μg/ml of ampicillin at 25 °C for 16 h (16, 70). Cells were harvested by centrifugation and cell paste was kept at −80 °C until used.
Purification of HadB
Cell paste was suspended in 50 mm sodium phosphate (NaPi), pH 8.0, containing 5 mm EDTA, 100 μm PMSF, and 1 mm DTT. Ultrasonication was utilized to lyse the cells. Cell debris was discarded by centrifugation at 27,000 × g for 1 h. Polyethyleneimine (0.1% w/v) was added into the soluble part to remove nucleic acids. Precipitant was removed by centrifugation at 27,000 × g for one-half hour. Ammonium sulfate ((NH4)2SO4) precipitation (0–20%, w/v) was performed to remove the remaining debris and other contaminants. The soluble yellow protein was precipitated by adding 20–40% (w/v) (NH4)2SO4. The resulting protein paste was suspended in 50 mm NaPi, pH 8.0, and was dialyzed against 4 liters of the same buffer overnight. Soluble protein was loaded onto an Q-SepharoseTM fast-flow (GE healthcare) anion exchange column (volume ∼200 ml), which was pre-equilibrated with 10 column volumes of 50 mm NaPi, pH 8.0. The column was washed with 5 column volumes of 50 mm NaPi, pH 8.0. Fractions of intense yellow protein in the flow-through and washed fractions that were not bound to the column were collected. Fractions of purified HadB were pooled and concentrated. The protein solution was passed through a SephadexTM G-25 (GE Healthcare) column to remove unbound flavin. Glycerol (15%, v/v) (final concentration) was added to HadB before it was stored at −80 °C until used.
Purification of HadX
Cell paste was lysed with the same method utilized for HadB as described in the previous section. Nucleic acid was removed by adding 0.5% (w/v) polyethyleneimine. (NH4)2SO4 (0–15% (w/v) precipitation) was performed to remove other contaminant proteins. HadX was precipitated by adding 15–40% (NH4)2SO4 (w/v). The protein pellet was resuspended in 50 mm NaPi, pH 6.5, and was dialyzed against the same buffer overnight. The resulting protein solution was loaded onto a DEAE-SepharoseTM fast-flow (GE Healthcare) anion exchange column (volume ∼200 ml) that was pre-equilibrated with 50 mm NaPi, pH 6.5. The column was washed with 5 column volumes of 50 mm NaPi, pH 6.5. HadX does not bind to the column and was collected from flow-through fractions. Yellow flow-through solutions of HadX were pooled, concentrated, and loaded onto a second DEAE-SepharoseTM fast-flow (GE healthcare) anion exchange column (volume ∼200 ml) that was pre-equilibrated with 50 mm NaPi, pH 8.0. HadX bound to the column was eluted out using a linear gradient of 0–300 mm NaCl in 50 mm NaPi, pH 8.0. Purified yellow fractions of HadX were pooled and concentrated. The protein solution was exchanged to 50 mm NaPi, pH 8.0, containing 15% (v/v) glycerol using a SephadexTM G-25 (GE Healthcare) column. The purified HadX was stored at −80 °C until used.
For both HadB and HadX, all purification processes were performed at 4 °C. Total protein amount was calculated by a Bradford assay. Purity and subunit molecular mass of proteins were analyzed by SDS-PAGE with Coomassie Brilliant Blue staining and compared with standard protein markers (Enzmart Biotech, Thailand).
Characterizations of purified HadB and HadX
Flavin cofactors of HadB and HadX were identified using the protocol previously described (44). Briefly, purified HadB and HadX were denatured by adding 1% (w/v) TCA. The resulting precipitated protein and yellowish solution containing flavin were separated by centrifugation. The yellow solution was analyzed by TLC (Silica Gel 60 F254 Merck) using n-butanol/glacial acetic acid/water (4:5:1, v/v) as a mobile phase. The migration of flavin from HadB and HadX was compared with the standard FAD, FMN, and riboflavin.
Molar absorption coefficients of flavin-bound HadB and HadX were calculated following the protocol previously reported for other flavoenzymes (71, 72). Spectra of flavin-bound HadB and HadX were recorded. SDS (0.1% w/v) was added to obtain a solution of free flavin in which their spectra were recorded. The concentration of released free flavin was calculated based on the molar absorption coefficient values of the standard FMN (ϵ446 = 12.2 mm−1 cm−1) or standard FAD (ϵ450 = 11.3 mm−1 cm−1). Molar absorption coefficient of flavin-bound HadB and HadX were calculated by comparing the absorption before and after free flavin was released.
Enzyme assay
For reductase activity assays, reactions were performed in 50 mm NaPi, pH 7.0. NADH was utilized as an electron donor, whereas menadione and FAD were used as electron acceptors for HadB and HadX, respectively. Absorption decrease at 340 nm due to NADH oxidation (ϵ340 = 6.2 mm−1 cm−1) was monitored. Enzyme activity was measured in triplicate to obtain the average value.
For HadA multiple turnover assays, reaction mixtures consisted of 4-NP (100 μm), glucose 6-phosphate (Glc-6-P) (1 mm), glucose-6-phosphate dehydrogenase (Glc-6-P dehydrogenase) (0.5 unit/ml), NAD+ (5 μm), and HadA (10 μm) in 50 mm NaPi, pH 7.5, were mixed. HadX (1 μm), HadB (1 μm), or ascorbic acid (1 mm) was added as a reductant system. 4-NP consumption was indicated by a decrease of absorbance at 400 nm monitored by UV-visible spectrophotometry. Product formation was analyzed by HPLC/DAD. For reactions using 4-CP as substrate, substrate consumption and product formation were analyzed by HPLC/DAD described below.
Product analysis of HadA reaction
Samples were prepared and analyzed based on the protocol described previously (16, 17). Briefly, reactions were quenched by adding an equal volume of acetonitrile. The resulting denatured proteins were removed by centrifugation at 13,800 × g for 30 min. Supernatants were diluted 3-fold in working buffer including ascorbic acid before being passed through an Amicon® Ultra-0.5 centrifugal filter device with a molecular mass cutoff 10 kDa (Millipore). Filtrates were analyzed by HPLC/diode array detector (Agilent) using a Nova-Pak® (Waters) C18 reverse phase column with a 4-μm particle size and 3.9 × 150-mm column size as the stationary phase. Mobile phase was a gradient of water/methanol (10–50% (v/v) for 4-NP or 10–70% (v/v) for 4-CP) containing 0.1% (v/v) formic acid. The HPLC system was run at ambient temperature with a flow rate of 0.5 ml/min.
Determination of a dissociation constant for binding of HadX and FAD
The thermodynamic binding constant of HadX and oxidized FAD (KDFAD) was determined by an ultrafiltration method. A solution of HadX (10 μm, 11 ml) in 50 mm NaPi, pH 7.0, was centrifuged through Centriprep® YM-10 filter (10 kDa cut-off) at 2,500 rpm, 25 °C for 5 min to collect an unbound FAD solution (whereas a HadX-bound FAD fraction was retained in the retentate). A free FAD solution in the filtrate (1 ml) recorded its fluorescence intensity (excitation wavelength at 450 nm and emission wavelength at 480 nm) and its concentration was determined by comparing the signal with those of standard FAD concentrations. Concentrations of free FAD and bound-FAD fractions were used to calculate a KDFAD value for the binding of HadX and FAD.
Transient kinetic experiments
Single mixing and double-mixing stopped-flow spectrophotometers models SF-61SX (TgK Scientific, Bradford-on-Avon, UK) were used to study transient kinetics. The flow path of the instrument was flushed with a solution of glucose/glucose oxidase overnight to make it anaerobic. Before performing the experiment, the flow path was rinsed with anaerobic buffer to remove excess solution of glucose/glucose oxidase. For the reductive half-reaction (reduction of enzyme-bound flavin) of both reductases, anaerobic HadBFMN or HadXFAD were prepared by passing enzyme solutions through a PD-10 desalting column (GE Healthcare) that was pre-equilibrated with anaerobic 50 mm NaPi, pH 7.0, inside an anaerobic glove box (Belle Technology UK Ltd., UK). The fully attained anaerobic solutions of enzymes were placed in an anaerobic tonometer. Anerobic solutions of electron donors (NADH or NADPH) were prepared by bubbling the solution with N2 gas (99.99% purity) before use. Solutions of anaerobic reductase and electron donor were mixed in the stopped-flow apparatus. The decrease of absorbance at 456 nm was monitored indicating the reduction of the flavin moieties in both HadB and HadX.
For investigating the oxidative half-reaction, solutions of anaerobic HadBFMNH− or HadXFADH− were prepared by adding stoichiometric amounts of NADH in anaerobic enzyme solutions inside an anaerobic glove box. Spectra of reduced enzymes were monitored to ensure that the enzyme was fully reduced before being placed in the anaerobic tonometer. Solutions of anaerobic HadBFMNH− or HadXFADH− were mixed against solutions of electron acceptors including molecular oxygen (O2), flavin, or quinone derivatives in 50 mm NaPi, pH 7.0. Absorbance at 456 nm was monitored to track the redox state of HadB and HadX. Analysis of kinetic traces and substrate dependence was carried out using KaleidaGraph software (Synergy Software). Observed rate constants were analyzed using ProgramA software (developed by C. J. Chiu, R. Chang, J. Diverno, and D. P. Ballou, at the University of Michigan, Ann Arbor, MI) or Kinetic Studio software (TgK Scientific, Bradford-on-Avon, UK).
Author contributions
P. P. and P. C. conceptualization; P. P. data curation; P. P. and P. C. formal analysis; P. P. and P. C. investigation; P. P. methodology; P. P. and P. C. writing-original draft; P. P. and P. C. writing-review and editing; P. C. resources; P. C. supervision; P. C. funding acquisition; P. C. project administration.
Supplementary Material
This work was supported by The Thailand Research Fund Grant RTA5980001, a grant from Vidyasirimedhi Institute of Science and Technology (VISTEC) (to P. C.), and Development and Promotion of Science and Technology Talents Project (DPST) Scholarship and postdoctoral fellowship from VISTEC (to P. P.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S10.
- HPs
- halogenated phenol
- HadA
- two-component FADH− utilizing monooxygenase
- HadB
- a quinone reductase encoded in the had operon
- HadX
- a flavin reductase encoded in the had operon
- NPs
- nitrophenols
- FADH−
- reduced flavin adenine dinucleotide
- FMNH−
- reduced flavin mononucleotide
- 4-NP
- 4-nitrophenol
- 4-CP
- 4-chlorophenol
- HQ
- hydroquinone
- BQ
- benzoquinone
- C1
- the flavin reductase component of p-hydroxyphenylacetate 3-hydroxylase from A. buamannii
- C2
- the oxygenase component of p-hydroxyphenylacetate 3-hydroxylase from A. buamannii
- 2,4-DCP
- 2,4-dichlorophenol
- 2,4,5-TCP
- 2,4,5-trichlorophenol
- 2,4,6-TCP
- 2,4,6-trichlorophenol
- CHQ
- chlorohydroquinone
- 2,5-DCHQ
- 2,5-dichlorohydroquinone
- 2,6-DCHQ
- 2,6-dichlorohydroquinone
- Glc-6-P
- glucose 6-phosphate
- DAD
- diode array detector
- PCP
- pentachlorophenol
- HPA
- p-hydroxyphenylacetate
- NR
- nitroreductases.
References
- 1. Olaniran A. O., and Igbinosa E. O. (2011) Chlorophenols and other related derivatives of environmental concern: properties, distribution and microbial degradation processes. Chemosphere 83, 1297–1306 10.1016/j.chemosphere.2011.04.009 [DOI] [PubMed] [Google Scholar]
- 2. Igbinosa E. O., Odjadjare E. E., Chigor V. N., Igbinosa I. H., Emoghene A. O., Ekhaise F. O., Igiehon N. O., and Idemudia O. G. (2013) Toxicological profile of chlorophenols and their derivatives in the environment: the public health perspective. Scientific World J. 2013, 1–11 10.1155/2013/460215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pera-Titus M., Garcia-Molina V., Banos M. A., Gimenez J., and Esplugas S. (2004) Degradation of chlorophenols by means of advanced oxidation processes: a general review. Appl. Catal. B-Environ 47, 219–256 10.1016/j.apcatb.2003.09.010 [DOI] [Google Scholar]
- 4. Karci A. (2014) Degradation of chlorophenols and alkylphenol ethoxylates, two representative textile chemicals, in water by advanced oxidation processes: the state of the art on transformation products and toxicity. Chemosphere 99, 1–18 10.1016/j.chemosphere.2013.10.034 [DOI] [PubMed] [Google Scholar]
- 5. Arora P. K., and Bae H. (2014) Bacterial degradation of chlorophenols and their derivatives. Microb. Cell Fact. 13, 31–47 10.1186/1475-2859-13-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arora P. K., Srivastava A., and Singh V. P. (2014) Bacterial degradation of nitrophenols and their derivatives. J. Hazard Mater. 266, 42–59 10.1016/j.jhazmat.2013.12.011 [DOI] [PubMed] [Google Scholar]
- 7. Harms H., Schlosser D., and Wick L. Y. (2011) Untapped potential: exploiting fungi in bioremediation of hazardous chemicals. Nat. Rev. Microbiol. 9, 177–192 10.1038/nrmicro2519 [DOI] [PubMed] [Google Scholar]
- 8. Cho S. Y., Kwean O. S., Yang J. W., Cho W., Kwak S., Park S., Lim Y., and Kim H. S. (2017) Identification of the upstream 4-chlorophenol biodegradation pathway using a recombinant monooxygenase from Arthrobacter chlorophenolicus A6. Bioresour. Technol. 245, 1800–1807 10.1016/j.biortech.2017.05.006 [DOI] [PubMed] [Google Scholar]
- 9. Gisi M. R., and Xun L. (2003) Characterization of chlorophenol 4-monooxygenase (TftD) and NADH:flavin adenine dinucleotide oxidoreductase (TftC) of Burkholderia cepacia AC1100. J. Bacteriol. 185, 2786–2792 10.1128/JB.185.9.2786-2792.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hatta T., Fujii E., and Takizawa N. (2012) Analysis of two gene clusters involved in 2,4,6-trichlorophenol degradation by Ralstonia pickettii DTP0602. Biosci. Biotechnol. Biochem. 76, 892–899 10.1271/bbb.110843 [DOI] [PubMed] [Google Scholar]
- 11. Kitagawa W., Kimura N., and Kamagata Y. (2004) A novel p-nitrophenol degradation gene cluster from a Gram-positive bacterium, Rhodococcus opacus SAO101. J. Bacteriol. 186, 4894–4902 10.1128/JB.186.15.4894-4902.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Perry L. L., and Zylstra G. J. (2007) Cloning of a gene cluster involved in the catabolism of p-nitrophenol by Arthrobacter sp. strain JS443 and characterization of the p-nitrophenol monooxygenase. J. Bacteriol. 189, 7563–7572 10.1128/JB.01849-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Takeo M., Murakami M., Niihara S., Yamamoto K., Nishimura M., Kato D., and Negoro S. (2008) Mechanism of 4-nitrophenol oxidation in Rhodococcus sp. strain PN1: characterization of the two-component 4-nitrophenol hydroxylase and regulation of its expression. J. Bacteriol. 190, 7367–7374 10.1128/JB.00742-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yamamoto K., Nishimura M., Kato D., Takeo M., and Negoro S. (2011) Identification and characterization of another 4-nitrophenol degradation gene cluster, nps, in Rhodococcus sp. strain PN1. J. Biosci. Bioeng. 111, 687–694 10.1016/j.jbiosc.2011.01.016 [DOI] [PubMed] [Google Scholar]
- 15. Xun L., Topp E., and Orser C. S. (1992) Confirmation of oxidative dehalogenation of pentachlorophenol by a Flavobacterium pentachlorophenol hydroxylase. J. Bacteriol. 174, 5745–5747 10.1128/jb.174.17.5745-5747.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pimviriyakul P., Thotsaporn K., Sucharitakul J., and Chaiyen P. (2017) Kinetic mechanism of the dechlorinating flavin-dependent monooxygenase HadA. J. Biol. Chem. 292, 4818–4832 10.1074/jbc.M116.774448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pimviriyakul P., Surawatanawong P., and Chaiyen P. (2018) Oxidative dehalogenation and denitration by a flavin-dependent monooxygenase is controlled by substrate deprotonation. Chem. Sci. 9, 7468–7482 10.1039/C8SC01482E [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Romero E., Gómez Castellanos J. R., Gadda G., Fraaije M. W., and Mattevi A. (2018) Same substrate, many reactions: oxygen activation in flavoenzymes. Chem. Rev. 118, 1742–1769 10.1021/acs.chemrev.7b00650 [DOI] [PubMed] [Google Scholar]
- 19. Sucharitakul J., Chaiyen P., Entsch B., and Ballou D. P. (2005) The reductase of p-hydroxyphenylacetate 3-hydroxylase from Acinetobacter baumannii requires p-hydroxyphenylacetate for effective catalysis. Biochemistry 44, 10434–10442 10.1021/bi050615e [DOI] [PubMed] [Google Scholar]
- 20. Phongsak T., Sucharitakul J., Thotsaporn K., Oonanant W., Yuvaniyama J., Svasti J., Ballou D. P., and Chaiyen P. (2012) The C-terminal domain of 4-hydroxyphenylacetate 3-hydroxylase from Acinetobacter baumannii is an autoinhibitory domain. J. Biol. Chem. 287, 26213–26222 10.1074/jbc.M112.354472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sucharitakul J., Tinikul R., and Chaiyen P. (2014) Mechanisms of reduced flavin transfer in the two-component flavin-dependent monooxygenases. Arch. Biochem. Biophys. 555, 33–46 [DOI] [PubMed] [Google Scholar]
- 22. Szklarczyk D., Morris J. H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N. T., Roth A., Bork P., Jensen L. J., and von Mering C. (2017) The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362–D368 10.1093/nar/gkw937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hatta T., Nakano O., Imai N., Takizawa N., and Kiyohara H. (1999) Cloning and sequence analysis of hydroxyquinol 1,2-dioxygenase gene in 2,4,6-trichlorophenol-degrading Ralstonia pickettii DTP0602 and characterization of its product. J. Biosci. Bioeng. 87, 267–272 10.1016/S1389-1723(99)80030-9 [DOI] [PubMed] [Google Scholar]
- 24. Finn R. D., Bateman A., Clements J., Coggill P., Eberhardt R. Y., Eddy S. R., Heger A., Hetherington K., Holm L., Mistry J., Sonnhammer E. L., Tate J., and Punta M. (2014) Pfam: the protein families database. Nucleic Acids Res. 42, D222–230 10.1093/nar/gkt1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pitsawong W., Hoben J. P., and Miller A. F. (2014) Understanding the broad substrate repertoire of nitroreductase based on its kinetic mechanism. J. Biol. Chem. 289, 15203–15214 10.1074/jbc.M113.547117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jarrom D., Jaberipour M., Guise C. P., Daff S., White S. A., Searle P. F., and Hyde E. I. (2009) Steady-state and stopped-flow kinetic studies of three Escherichia coli NfsB mutants with enhanced activity for the prodrug CB1954. Biochemistry 48, 7665–7672 10.1021/bi900674m [DOI] [PubMed] [Google Scholar]
- 27. Rau J., and Stolz A. (2003) Oxygen-insensitive nitroreductases NfsA and NfsB of Escherichia coli function under anaerobic conditions as lawsone-dependent Azo reductases. Appl. Environ. Microbiol. 69, 3448–3455 10.1128/AEM.69.6.3448-3455.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Valiauga B., Williams E. M., Ackerley D. F., and Čėnas N. (2017) Reduction of quinones and nitroaromatic compounds by Escherichia coli nitroreductase A (NfsA): characterization of kinetics and substrate specificity. Arch. Biochem. Biophys. 614, 14–22 10.1016/j.abb.2016.12.005 [DOI] [PubMed] [Google Scholar]
- 29. Belchik S. M., and Xun L. (2008) Functions of flavin reductase and quinone reductase in 2,4,6-trichlorophenol degradation by Cupriavidus necator JMP134. J. Bacteriol. 190, 1615–1619 10.1128/JB.01697-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Webb B. N., Ballinger J. W., Kim E., Belchik S. M., Lam K. S., Youn B., Nissen M. S., Xun L., and Kang C. (2010) Characterization of chlorophenol 4-monooxygenase (TftD) and NADH:FAD oxidoreductase (TftC) of Burkholderia cepacia AC1100. J. Biol. Chem. 285, 2014–2027 10.1074/jbc.M109.056135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Filisetti L., Fontecave M., and Niviere V. (2003) Mechanism and substrate specificity of the flavin reductase ActVB from Streptomyces coelicolor. J. Biol. Chem. 278, 296–303 10.1074/jbc.M209689200 [DOI] [PubMed] [Google Scholar]
- 32. Filisetti L., Valton J., Fontecave M., and Nivière V. (2005) The flavin reductase ActVB from Streptomyces coelicolor: characterization of the electron transferase activity of the flavoprotein form. FEBS Lett. 579, 2817–2820 10.1016/j.febslet.2005.04.019 [DOI] [PubMed] [Google Scholar]
- 33. Valton J., Fontecave M., Douki T., Kendrew S. G., and Niviere V. (2006) An aromatic hydroxylation reaction catalyzed by a two-component FMN-dependent monooxygenase: the ActVA-ActVB system from Streptomyces coelicolor. J. Biol. Chem. 281, 27–35 [DOI] [PubMed] [Google Scholar]
- 34. Valton J., Mathevon C., Fontecave M., Nivière V., and Ballou D. P. (2008) Mechanism and regulation of the two-component FMN-dependent monooxygenase ActVA-ActVB from Streptomyces coelicolor. J. Biol. Chem. 283, 10287–10296 10.1074/jbc.M709730200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Valton J., Filisetti L., Fontecave M., and Niviere V. (2004) A two-component flavin-dependent monooxygenase involved in actinorhodin biosynthesis in Streptomyces coelicolor. J. Biol. Chem. 279, 44362–44369 10.1074/jbc.M407722200 [DOI] [PubMed] [Google Scholar]
- 36. Yeh E., Garneau S., and Walsh C. T. (2005) Robust in vitro activity of RebF and RebH, a two-component reductase/halogenase, generating 7-chlorotryptophan during rebeccamycin biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 102, 3960–3965 10.1073/pnas.0500755102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nijvipakul S., Wongratana J., Suadee C., Entsch B., Ballou D. P., and Chaiyen P. (2008) LuxG is a functioning flavin reductase for bacterial luminescence. J. Bacteriol. 190, 1531–1538 10.1128/JB.01660-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nijvipakul S., Ballou D. P., and Chaiyen P. (2010) Reduction kinetics of a flavin oxidoreductase LuxG from Photobacterium leiognathi (TH1): half-sites reactivity. Biochemistry 49, 9241–9248 10.1021/bi1009985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tinikul R., Pitsawong W., Sucharitakul J., Nijvipakul S., Ballou D. P., and Chaiyen P. (2013) The transfer of reduced flavin mononucleotide from LuxG oxidoreductase to luciferase occurs via free diffusion. Biochemistry 52, 6834–6843 10.1021/bi4006545 [DOI] [PubMed] [Google Scholar]
- 40. Zaborina O., Daubaras D. L., Zago A., Xun L., Saido K., Klem T., Nikolic D., and Chakrabarty A. M. (1998) Novel pathway for conversion of chlorohydroxyquinol to maleylacetate in Burkholderia cepacia AC1100. J. Bacteriol. 180, 4667–4675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ross D. (2004) Quinone reductases multitasking in the metabolic world. Drug. Metab. Rev. 36, 639–654 10.1081/DMR-200033465 [DOI] [PubMed] [Google Scholar]
- 42. Deller S., Macheroux P., and Sollner S. (2008) Flavin-dependent quinone reductases. Cell. Mol. Life. Sci. 65, 141–160 10.1007/s00018-007-7300-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Blaza J. N., Bridges H. R., Aragão D., Dunn E. A., Heikal A., Cook G. M., Nakatani Y., and Hirst J. (2017) The mechanism of catalysis by type-II NADH:quinone oxidoreductases. Sci. Rep. 7, 40165–40176 10.1038/srep40165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chaiyen P., Suadee C., and Wilairat P. (2001) A novel two-protein component flavoprotein hydroxylase. Eur. J. Biochem. 268, 5550–5561 10.1046/j.1432-1033.2001.02490.x [DOI] [PubMed] [Google Scholar]
- 45. Sucharitakul J., Chaiyen P., Entsch B., and Ballou D. P. (2006) Kinetic mechanisms of the oxygenase from a two-component enzyme, p-hydroxyphenylacetate 3-hydroxylase from Acinetobacter baumannii. J. Biol. Chem. 281, 17044–17053 [DOI] [PubMed] [Google Scholar]
- 46. Sucharitakul J., Phongsak T., Entsch B., Svasti J., Chaiyen P., and Ballou D. P. (2007) Kinetics of a two-component p-hydroxyphenylacetate hydroxylase explain how reduced flavin is transferred from the reductase to the oxygenase. Biochemistry 46, 8611–8623 10.1021/bi7006614 [DOI] [PubMed] [Google Scholar]
- 47. Lam L. K., Zhang Z., Board P. G., and Xun L. (2012) Reduction of benzoquinones to hydroquinones via spontaneous reaction with glutathione and enzymatic reaction by S-glutathionyl-hydroquinone reductases. Biochemistry 51, 5014–5021 10.1021/bi300477z [DOI] [PubMed] [Google Scholar]
- 48. Bolton J. L., Trush M. A., Penning T. M., Dryhurst G., and Monks T. J. (2000) Role of quinones in toxicology. Chem. Res. Toxicol. 13, 135–160 10.1021/tx9902082 [DOI] [PubMed] [Google Scholar]
- 49. Kishko I., Harish B., Zayats V., Reha D., Tenner B., Beri D., Gustavsson T., Ettrich R., and Carey J. (2012) Biphasic kinetic behavior of E. coli WrbA, an FMN-dependent NAD(P)H:quinone oxidoreductase. PloS One 7, e43902 10.1371/journal.pone.0043902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Patridge E. V., and Ferry J. G. (2006) WrbA from Escherichia coli and Archaeoglobus fulgidus is an NAD(P)H:quinone oxidoreductase. J. Bacteriol. 188, 3498–3506 10.1128/JB.188.10.3498-3506.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gonzalez C. F., Ackerley D. F., Lynch S. V., and Matin A. (2005) ChrR, a soluble quinone reductase of Pseudomonas putida that defends against H2O2. J. Biol. Chem. 280, 22590–22595 10.1074/jbc.M501654200 [DOI] [PubMed] [Google Scholar]
- 52. Sollner S., Nebauer R., Ehammer H., Prem A., Deller S., Palfey B. A., Daum G., and Macheroux P. (2007) Lot6p from Saccharomyces cerevisiae is a FMN-dependent reductase with a potential role in quinone detoxification. FEBS J. 274, 1328–1339 10.1111/j.1742-4658.2007.05682.x [DOI] [PubMed] [Google Scholar]
- 53. Sollner S., Deller S., Macheroux P., and Palfey B. A. (2009) Mechanism of flavin reduction and oxidation in the redox-sensing quinone reductase Lot6p from Saccharomyces cerevisiae. Biochemistry 48, 8636–8643 10.1021/bi900734a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tedeschi G., Chen S., and Massey V. (1995) DT-diaphorase. Redox potential, steady-state, and rapid reaction studies. J. Biol. Chem. 270, 1198–1204 10.1074/jbc.270.3.1198 [DOI] [PubMed] [Google Scholar]
- 55. Miseviciene L., Anusevicius Z., Sarlauskas J., and Cenas N. (2006) Reduction of nitroaromatic compounds by NAD(P)H:quinone oxidoreductase (NQO1): the role of electron-accepting potency and structural parameters in the substrate specificity. Acta Biochim. Polonica. 53, 569–576 [PubMed] [Google Scholar]
- 56. Flores E., and Gadda G. (2018) Kinetic characterization of PA1225 from Pseudomonas aeruginosa PAO1 reveals a new NADPH:quinone reductase. Biochemistry 57, 3050–3058 10.1021/acs.biochem.8b00090 [DOI] [PubMed] [Google Scholar]
- 57. Baron R., Riley C., Chenprakhon P., Thotsaporn K., Winter R. T., Alfieri A., Forneris F., van Berkel W. J., Chaiyen P., Fraaije M. W., Mattevi A., and McCammon J. A. (2009) Multiple pathways guide oxygen diffusion into flavoenzyme active sites. Proc. Natl. Acad. Sci. U.S.A. 106, 10603–10608 10.1073/pnas.0903809106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chaiyen P., Fraaije M. W., and Mattevi A. (2012) The enigmatic reaction of flavins with oxygen. Trends Biochem. Sci. 37, 373–380 10.1016/j.tibs.2012.06.005 [DOI] [PubMed] [Google Scholar]
- 59. Leferink N. G., Fraaije M. W., Joosten H. J., Schaap P. J., Mattevi A., and van Berkel W. J. (2009) Identification of a gatekeeper residue that prevents dehydrogenases from acting as oxidases. J. Biol. Chem. 284, 4392–4397 10.1074/jbc.M808202200 [DOI] [PubMed] [Google Scholar]
- 60. Zhao G., Bruckner R. C., and Jorns M. S. (2008) Identification of the oxygen activation site in monomeric sarcosine oxidase: role of Lys265 in catalysis. Biochemistry 47, 9124–9135 10.1021/bi8008642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. McDonald C. A., Fagan R. L., Collard F., Monnier V. M., and Palfey B. A. (2011) Oxygen reactivity in flavoenzymes: context matters. J. Am. Chem. Soc. 133, 16809–16811 10.1021/ja2081873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gadda G. (2012) Oxygen activation in flavoprotein oxidases: the importance of being positive. Biochemistry 51, 2662–2669 10.1021/bi300227d [DOI] [PubMed] [Google Scholar]
- 63. Wongnate T., Surawatanawong P., Visitsatthawong S., Sucharitakul J., Scrutton N. S., and Chaiyen P. (2014) Proton-coupled electron transfer and adduct configuration are important for C4a-hydroperoxyflavin formation and stabilization in a flavoenzyme. J. Am. Chem. Soc. 136, 241–253 10.1021/ja4088055 [DOI] [PubMed] [Google Scholar]
- 64. Visitsatthawong S., Chenprakhon P., Chaiyen P., and Surawatanawong P. (2015) mechanism of oxygen activation in a flavin-dependent monooxygenase: a nearly barrierless formation of C4a-hydroperoxyflavin via proton-coupled electron transfer. J. Am. Chem. Soc. 137, 9363–9374 10.1021/jacs.5b04328 [DOI] [PubMed] [Google Scholar]
- 65. Louie T. M., Xie X. S., and Xun L. (2003) Coordinated production and utilization of FADH2 by NAD(P)H-flavin oxidoreductase and 4-hydroxyphenylacetate 3-monooxygenase. Biochemistry 42, 7509–7517 10.1021/bi034092r [DOI] [PubMed] [Google Scholar]
- 66. Kirchner U., Westphal A. H., Müller R., and van Berkel W. J. (2003) Phenol hydroxylase from Bacillus thermoglucosidasius A7, a two-protein component monooxygenase with a dual role for FAD. J. Biol. Chem. 278, 47545–47553 10.1074/jbc.M307397200 [DOI] [PubMed] [Google Scholar]
- 67. Otto K., Hofstetter K., Röthlisberger M., Witholt B., and Schmid A. (2004) Biochemical characterization of StyAB from Pseudomonas sp. strain VLB120 as a two-component flavin-diffusible monooxygenase. J. Bacteriol. 186, 5292–5302 10.1128/JB.186.16.5292-5302.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Payne K. A., Quezada C. P., Fisher K., Dunstan M. S., Collins F. A., Sjuts H., Levy C., Hay S., Rigby S. E., and Leys D. (2015) Reductive dehalogenase structure suggests a mechanism for B12-dependent dehalogenation. Nature 517, 513–516 10.1038/nature13901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Collins F. A., Fisher K., Payne K. A. P., Gaytan Mondragon S., Rigby S. E. J., and Leys D. (2018) NADPH-driven organohalide reduction by a nonrespiratory reductive dehalogenase. Biochemistry 57, 3493–3502 10.1021/acs.biochem.8b00255 [DOI] [PubMed] [Google Scholar]
- 70. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 10.1016/j.pep.2005.01.016 [DOI] [PubMed] [Google Scholar]
- 71. Luanloet T., Sucharitakul J., and Chaiyen P. (2015) Selectivity of substrate binding and ionization of 2-methyl-3-hydroxypyridine-5-carboxylic acid oxygenase. FEBS J. 282, 3107–3125 10.1111/febs.13220 [DOI] [PubMed] [Google Scholar]
- 72. Sucharitakul J., Medhanavyn D., Pakotiprapha D., van Berkel W. J., and Chaiyen P. (2016) Tyr217 and His213 are important for substrate binding and hydroxylation of 3-hydroxybenzoate 6-hydroxylase from Rhodococcus jostii RHA1. FEBS J. 283, 860–881 10.1111/febs.13636 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.