

Abstract
Coxiella burnetii is an intracellular Gram-negative bacterium responsible for the important zoonotic disease Q fever. Improved genetic tools and the ability to grow this bacterium in host cell–free media has advanced the study of C. burnetii pathogenesis, but the mechanisms that allow it to survive inside the hostile phagolysosome remain incompletely understood. Previous screening of a transposon mutant library for replication within HeLa cells has suggested that nadB, encoding a putative l-aspartate oxidase required for de novo NAD synthesis, is needed for intracellular replication. Here, using genetic complementation of two independent nadB mutants and intracellular replication assays, we confirmed this finding. Untargeted metabolite analyses demonstrated key changes in metabolites in the NAD biosynthetic pathway in the nadB mutant compared with the WT, confirming the involvement of NadB in de novo NAD synthesis. Bioinformatic analysis revealed the presence of a functionally conserved arginine residue at position 275. Using site-directed mutagenesis to substitute this residue with leucine, which abolishes the activity of Escherichia coli NadB, and expression of WT and R275L GST-NadB fusion proteins in E. coli JM109, we found that purified recombinant WT GST-NadB has l-aspartate oxidase activity and that the R275L NadB variant is inactive. Complementation of the C. burnetii nadB mutant with a plasmid expressing this inactive R275L NadB failed to restore replication to WT levels, confirming the link between de novo NAD synthesis and intracellular replication of C. burnetii. This suggests that targeting this prokaryotic-specific pathway could advance the development of therapeutics to combat C. burnetii infections.
Keywords: bacterial pathogenesis, NAD biosynthesis, bacterial metabolism, metabolomics, lysosome, infectious disease, Coxiella burnetii, Q fever, virulence
Introduction
Coxiella burnetii is a Gram-negative intracellular bacterium that causes the zoonotic disease Q fever (1), which primarily spreads to humans from livestock such as cattle and sheep. In humans, infection can present as an acute disease with systemic signs including pneumonia and hepatitis. A chronic infection, characterized by endocarditis and chronic fatigue syndrome, may subsequently develop (1, 2). In 2007–2010 an outbreak in the Netherlands resulted in more than 4000 human cases, the culling of tens of thousands of animals, and cost in excess of 300 million Euros to control (3). C. burnetii primarily targets macrophages and enters via conventional phagocystosis (4). Once trafficked through the endocytic pathway, the bacterium switches from the small-cell variant form to the actively replicating large-cell variant form (5, 6) and activates the maturation of a phagolysosome-like compartment known as the Coxiella-containing vacuole (CCV),2 where it replicates freely (7–9).
The relatively recent development of axenic growth medium for C. burnetii (10) and improved techniques for genetic manipulation (11) facilitated identification of the Dot/Icm type 4 secretion system (T4SS) (responsible for translocating at least 130 effector proteins) as essential for replication inside cells (12–14). However, much less is known about the Dot/Icm type 4 secretion system–independent factors that are required for replication inside cells. Recent screening of a transposon mutant library (12) for replication within human HeLa cells demonstrated that ∼10% of transposon mutants were attenuated for growth, including three independent mutants with transposon insertions into nadB. The nadB gene encodes a putative l-aspartate oxidase that is essential for the canonical prokaryotic de novo NAD biosynthetic pathway, in which NadB converts aspartate to iminoaspartate, which is then converted to quinolinate and eventually NAD via downstream enzymes (15). NAD is an important cofactor for many core metabolic reactions (16, 17). Intracellular NAD+/NADH ratio is key to the maintenance of an adequate metabolic status and cell survival (18) and may be especially important for C. burnetii, because the phagolysosome is a highly oxidative environment.
In this study we genetically complemented two nadB mutants and performed quantitative and qualitative intracellular replication assays to confirm that nadB is required for intracellular replication of C. burnetii. We then used a combination of sophisticated biochemical and genetic techniques, including GC/MS and LC/MS, recombinant protein purification, enzymatic assays, and site-directed mutagenesis, to demonstrate that de novo NAD biosynthesis is required for C. burnetii replication inside human cells.
Results
nadB is required for efficient intracellular replication of C. burnetii and normal CCV biogenesis
Following the identification in our initial screen of nadB as a candidate gene required for intracellular replication, we aimed first to confirm this role. Two independent transposon mutants, nadB 7-G9::Tn and nadB 23-H5::Tn, with the transposon inserted 900 and 1080 bp into the ORF, respectively, were clonally isolated; the presence of the transposon was confirmed by PCR; and each mutant was complemented with plasmid expressing full-length 3xFLAG:NadB, with the expression of protein of the predicted molecular weight confirmed using anti-FLAG immunoblotting (Fig. S1). Intracellular growth assays in human HeLa CCL2 cells were performed, and both nadB mutants had significantly reduced growth, with only a 10-fold increase in genome equivalents at 7 days postinfection, in contrast to WT C. burnetii that increased by over 100-fold in the same period (Fig. 1). Complementation with the pJB-Kan:3xFLAG vector expressing 3xFLAG-NadB restored growth to WT levels (Fig. 1), demonstrating conclusively that nadB is required for efficient C. burnetii replication. Qualitative assays using immunofluorescence microscopy examined the effect of transposon insertion into nadB on the size and number of the CCVs. Both nadB mutants displayed much smaller CCVs (stained green) containing very few bacteria (stained red), whereas plasmid complementation of the nadB mutants restored the WT phenotype (Fig. 2). Because similar results were obtained for both nadB mutants, nadB 23-H5::Tn and the corresponding complemented strain were used for the remainder of experiments and are referred to as the nadB mutant and the complemented nadB mutant.
Figure 1.

nadB is essential for efficient intracellular replication of C. burnetii. Intracellular replication of C. burnetii NMII WT (closed circles), nadB::Tn 7-G9 (closed squares), nadB::Tn 7-G9 complemented strain (open squares), nadB::Tn 23-H5 (closed inverted triangles), and nadB::Tn 23-H5 complemented strain (open inverted triangles) was measured over 7 days. The fold increases in GEs relative to the inoculum were determined by ompA-specific qPCR and are represented here as the means ± S.D. of three independent infections at days 1, 3, 5, and 7 postinfection. Both nadB::Tn mutants were significantly reduced in growth compared with C. burnetii NMII at days 3, 5, and 7 (p < 0.05, paired t test).
Figure 2.
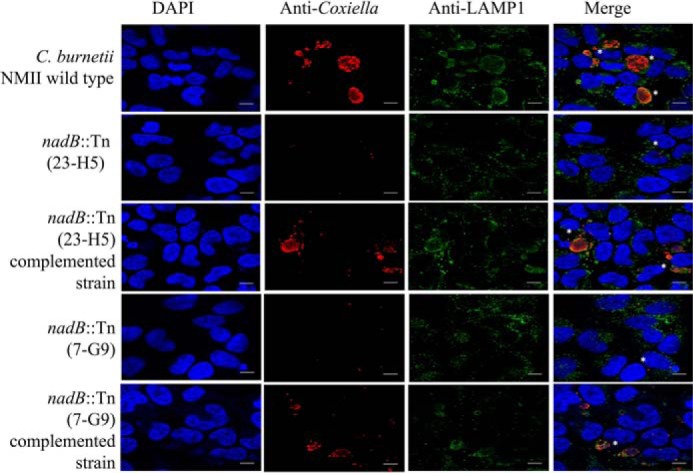
nadB is required for normal CCV formation. Shown are representative micrographs (three independent experiments) of HeLa cells infected for 3 days with C. burnetii NMII, the nadB::Tn 23-H5 mutant, the nadB::Tn (23-H5) complemented strain, the nadB::Tn 7-G9 mutant, and the nadB::Tn (7-G9) complemented strain. The cells were stained with anti-Coxiella antibody (red), anti-LAMP1 antibody (green), and DAPI (blue), and white asterisks indicate representative CCVs. Note that the CCVs containing the nadB mutant appear smaller and contain few bacteria compared with either the WT NMII WT strain or the complemented strains. The scale bars represent 10 μm.
nadB is required for normal NAD biosynthesis
To examine the effect of the loss of nadB in C. burnetii cultured in axenic medium, we performed GC/MS analysis to compare the abundance of metabolites within the de novo NAD biosynthetic pathway (Fig. 3A) between the nadB mutant, the complemented nadB mutant, and WT C. burnetii grown in axenic medium. Axenic cultures of C. burnetii were rapidly quenched, and polar metabolites were extracted, derivatized, and analyzed using GC/MS. Metabolites were identified from representative chromatograms by retention time (RT) and fragmentation pattern, and log transformation and median normalization were performed before the abundance of each metabolite was compared. As predicted, the loss of NadB resulted in a significantly increased abundance of aspartate in the nadB mutant, with a 4.1 log2 fold increase compared with WT and a 4.02 log2 fold increase compared with the complemented mutant (Fig. 3B). Furthermore, downstream metabolites were significantly decreased in the nadB mutant, with quinolate decreased by 2.04 log2 fold and 1.6 log2 fold in the mutant compared with WT and the complemented mutant, respectively (Fig. 3B). Similarly, nicotinic acid mononucleotide (NaMN) was decreased by 1.24 and 2.35 log2 fold in the nadB mutant, as compared with WT and the complemented mutant, respectively (Fig. 3B).
Figure 3.

A, the C. burnetii NAD biosynthetic pathway. B, disruption of nadB alters the level of de novo NAD biosynthetic pathway metabolites. Red, complemented nadB mutant; green, nadB mutant; blue, WT C. burnetii NMII. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (BH-adjusted t test). Fold difference is presented as log2 scale. C, disruption of nadB reduces the abundance of NAD-containing metabolites. NAD, NADH, and NADP were all significantly reduced in the nadB mutant compared with WT C. burnetii and the complemented nadB mutant. Metabolites were identified using LC/MS, data were median-normalized, and metabolites were identified by reference to authentic standards. *, p < 0.05; **, p < 0.01; ***, p < 0.001 (BH-adjusted t test). Fold difference is presented as log2 scale.
To examine whether the loss of nadB affected the abundance of the metabolites downstream of the predicted entry of nicotinate via a salvage pathway, namely NAD, NADP, and NADH (Fig. 3A), LC/MS analysis was performed. Metabolites were extracted from rapidly quenched axenically cultured C. burnetii and polar metabolites were analyzed by LC/MS, before median normalization and identification of metabolites using authentic standards. The relative abundance of each metabolite was compared. The nadB mutant displayed 1.32 and 2.07 log2 fold decreases in NAD levels relative to the WT and the complemented mutant strains, respectively (Fig. 3C). The NADH abundance in the nadB mutant was decreased by 1.06 and 2.58 log2 fold compared with the WT and the complemented strain, respectively (Fig. 3C). Similarly, NADP was decreased in the nadB mutant by 3.60 and 3.37 log2 fold compared with the WT and the complemented strain, respectively (Fig. 3C). Overall, our metabolite analysis demonstrated an effect on de novo NAD synthesis only in the absence of nadB, demonstrating that NadB is required for NAD biosynthesis and that salvage pathways are not sufficient, even in rich axenic medium, to restore the levels of NAD-containing metabolites in the nadB mutant to those present in WT C. burnetii.
Mutation of the critical amino acid residue Arg275 of NadB abolishes enzymatic function in vitro
To investigate whether the enzymatic function of NadB, and therefore NAD biosynthesis, was crucial to the ability of C. burnetii to replicate inside cells, we first investigated the function of NadB in vitro. Recombinant glutathione S-transferase (GST)-NadB (GST-NadB) was expressed in and purified from Escherichia coli JM109, with GST alone purified as a negative control. To confirm that NadB had the expected l-aspartate oxidase activity, a two-step in vitro enzyme assay was carried out. In vitro, in the absence of the downstream enzyme NadA, the iminoaspartate produced from l-aspartate by NadB catalysis spontaneously hydrolyzes to oxaloacetate (OAA) (19, 20). This OAA can then be detected by performing a second step, in which commercial malate dehydrogenase is used to reduce OAA to malate, and the change in absorbance at 340 nm (resulting from concurrent NADH oxidation during OAA reduction) is measured. The absorbance will only decrease in the second step of the assay if iminoaspartate is produced by the enzyme used in the first step of the assay.
When GST-NadB was used in the first step of the assay, a fall in absorbance at 340 nm to zero was observed in the second step (Fig. 4), indicating that NadB had l-aspartate oxidase activity (because it produced iminoaspartate in the first step). In contrast, when GST alone was used to catalyze the first step of the assay, no change in absorbance over time was observed in the second step of the assay.
Figure 4.

Recombinant GST-NadB possesses l-aspartate oxidase activity that is abolished by mutation of Arg275 to Leu. The graph depicts the second step of the two-step assay, demonstrating the change in absorbance at 340 nm resulting from oxidation of NADH and reduction of OAA in the presence of commercial malate dehydrogenase, if the first step of the assay produced iminoaspartate (which spontaneously hydrolyzes to OAA). A decrease in absorbance will only occur if the protein used in the first step possesses l-aspartate oxidase activity. Proteins used in the first step were GST-NadB (circles), GST (squares), and GST-R275L-NadB (triangles). The graph represents the mean ± S.D. of three independent experiments.
To link the enzymatic function of NadB with its role in intracellular replication and rule out a secondary, nonenzymatic role in intracellular replication, we aligned the amino acid sequence of C. burnetii NadB with that of E. coli NadB (Fig. 5) and identified a conserved arginine in position 275 (position 290 in E. coli). In E. coli NadB, mutation of this arginine to leucine abolishes the enzymatic activity (21). We performed site-directed mutagenesis, then expressed and purified recombinant C. burnetii GST-NadB-R275L (GST-R275L), and performed the two-step enzyme assay. When GST-R275L was used to catalyze the first step of the reaction, no change in absorbance was observed for the second step (Fig. 4), indicating that iminoaspartate was no longer produced in the first step of the assay, and thus that the mutant R275L protein had lost its l-aspartate oxidase function.
Figure 5.

Key amino acid residues are conserved between E. coli NadB and Coxiella NadB. Protein sequence alignment was performed using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/; Ref. 33). (Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.) The conserved amino acids were predicted to be involved substrate binding (His244, His336, and Arg371, highlighted green), and the catalytic site (Arg275, highlighted blue) was identified in Coxiella NadB. Numbers on the right side indicate position of amino acids for both organisms. Conserved regions are indicated by black shading and asterisks; regions of similarity are indicated by colons.
De novo NAD biosynthesis is required for intracellular replication of C. burnetii in both epithelial and macrophage-like cells
To confirm whether de novo NAD synthesis is required for intracellular replication of C. burnetii and to investigate whether the type of host cell affects this requirement, we complemented the nadB mutant with the pJB-Kan:3xFLAG plasmid expressing 3xFLAG-R275L-NadB and performed quantitative and qualitative replication assays using both HeLa cells and human macrophage-like THP-1 cells. In HeLa cells, complementation of the nadB mutant with this inactive R275L-NadB resulted in a similar phenotype to that of the original nadB mutant in HeLa cells, with a fold increase in genome equivalents of only 11.7 after 7 days (Fig. 6A). In contrast, as in our original experiments, complementation with the pJB-Kan:3xFLAG plasmid expressing WT 3xFLAG-NadB rescued the intracellular growth defect (Fig. 6A). Similarly, in the macrophage-like THP-1 cells, both WT and the complemented nadB mutant were more than 100-fold greater in number at day 7 than either the nadB mutant or the complemented strain expressing the inactive R275L-NadB (Fig. 6B). Expression of both NadB and R275L-NadB by the complemented strains during THP-1 infection was confirmed by immunoblotting (Fig. S2). Immunofluorescent microscopy revealed a similar CCV phenotype for the R257L complemented strain to that of the original nadB mutant, with very small CCVs containing few bacteria, in both HeLa cells and THP-1 cells (Fig. 7). Crucially, these results demonstrate that active NadB, and therefore a functional de novo NAD biosynthesis pathway, are required for intracellular replication of C. burnetii in both professional phagocytes and epithelial cells.
Figure 6.

Complementation of the nadB::Tn mutant with a plasmid expressing the inactive R275L-NadB did not rescue the intracellular growth defect in either HeLa cells (A) or THP-1 cells (B). Intracellular replication of C. burnetii was measured over 7 days. The fold increases in GEs relative to the inoculum were determined by ompA-specific qPCR and are represented here as the means ± S.D. of three independent infections at days 1, 3, 5, and 7 postinfection. Both the nadB mutant (closed squares) and the R275L complemented nadB mutant (closed triangles) were significantly reduced in growth in HeLa cells compared with C. burnetii NMII WT (closed circles) at days 5 and 7 (p < 0.05, paired t test) and at all time points in THP-1 cells (p < 0.05, paired t test). Open squares represent the original complemented nadB mutant.
Figure 7.

Enzymatically active NadB is required for normal CCV formation and C. burnetii growth inside cells. Representative micrographs (three independent experiments) of HeLa cells (A) and THP-1 cells (B) infected with C. burnetii strains for 3 days are shown. The cells were fixed and stained with anti-Coxiella (red), anti-LAMP1 (green), and DAPI (blue), and white asterisks indicate representative CCVs. Note that WT C. burnetii NMII and the complemented nadB mutant produced large CCVs containing numerous bacteria, whereas both the nadB mutant and the R275L complemented nadB mutant displayed smaller CCVs containing very few bacteria. The scale bars represent 10 μm.
The overall metabolic profile of the nadB mutant differs from that of WT and the complemented mutant
NAD and its derivatives are important cofactors in many metabolic reactions within cells. To investigate why the loss of de novo NAD synthesis may affect intracellular replication, we interrogated our GC/MS and LC/MS data to analyze the effect of the loss of NadB on the overall metabolite profile of the nadB mutant, as compared with the metabolite profiles of the WT and the complemented nadB mutant. In total, we identified 88 metabolites using LC/MS and 110 metabolites using GC/MS and performed pairwise comparisons of the abundance of each metabolite between WT and the nadB mutant and between the nadB mutant and the complemented strain. Metabolites were identified using standard retention time and mass spectra and by comparison with authentic standards.
Principal component analysis demonstrated good separation between the nadB mutant and the other two strains, with (as expected) overlap between the complemented mutant and WT (Fig. S3), demonstrating that the loss of nadB affected the overall metabolic profile. Pairwise comparison of metabolites from the GC/MS analysis identified statistically significant differences in the abundances of 20 metabolites between the nadB mutant and WT, and 27 metabolites differed between the nadB mutant and the complemented strain. (Fig. 8). Fold differences were similar, with the differences for the WT and mutant ranging from 4.1 to −2.36 log2 fold, and the differences for the complemented strain versus the mutant the fold differences varied by 4.02 to −2.35 log2 fold. In addition to the previously discussed differences in metabolites directly related to NAD biosynthesis, significant differences were seen in metabolites within glycolysis, the pentose phosphate pathway, the tricarboxylic acid (TCA) pathway, DNA synthesis, and amino acid metabolism.
Figure 8.

Metabolites differing significantly (p < 0. 05, BH-adjusted t test) in abundance between WT and the nadB mutant (A) and between the complemented nadB mutant and the nadB mutant (B) in the GC/MS analysis. Metabolites from all strains were identified from representative chromatograms using RTs. The data were log-transformed and median-normalized before analysis. The data represent the average log2 fold change from six biological repeats.
The TCA cycle intermediate citrate was elevated in abundance in both WT and the complemented mutant strain compared with the nadB mutant, with a relative elevation in isocitrate abundance in WT as well. The pentose phosphate pathway intermediate ribose 5-phosphate was relatively elevated in both WT and the complemented mutant strain. The amino acid abundances varied between the two comparisons, with differing patterns observed between WT and the complemented nadB mutant. Significant elevations in a number of nucleobases, nucleosides, and nucleotides were observed in WT and/or the complemented strain compared with the nadB mutant. Overall, disrupting NAD synthesis had significant effects on the levels of a number of metabolites, including those found in central carbon metabolic pathways.
Discussion
NAD and its phosphorylated and reduced derivatives such as NADP, NADPH, and NADH, are crucial cofactors in many core cellular metabolic reactions (16), including glycolysis and the TCA cycle. Broadly speaking, prokaryotes synthesize NAD via a three-step pathway from l-aspartate (Fig. 3A), although there are exceptions, such as Haemophilus influenzae, that lacks de novo synthesis and instead relies on salvage pathways. Most eukaryotes, including humans, synthesize NAD using a five-step pathway from tryptophan (16), suggesting that the bacterial NAD biosynthetic pathway could be a suitable therapeutic target. We have applied a combination of genetic and biochemical techniques to demonstrate the key role of this pathway in C. burnetii pathogenesis, supporting the identification of this pathway as a novel drug target.
A number of NAD salvage pathways exist in prokaryotes, with the most common converting nicotinamide to nicotinic acid (E. coli enzyme PncA) and then to NaMN (E. coli enzyme PncB) (16). Downstream of NaMN the de novo biosynthetic and salvage pathways converge, and NaMN is converted to nicotinic acid adenine dinucleotide (bacterial enzyme NadD) and then to NAD (bacterial enzyme NadE) (22). C. burnetii lacks the Pnc salvage pathway and instead appears to only possess cbu_1035, which encodes a putative nicotinate phosphoribosyltransferase (23) that is predicted to convert scavenged nicotinic acid to NaMN (Fig. 3A). In our study, we observed that the loss of NAD de novo synthesis can be overcome in vitro (Fig. S4), demonstrating that this salvage pathway must be active at least in vitro and that C. burnetii can scavenge nicotinic acid from the rich axenic medium used to culture the cells. Recent analysis of lysosomal metabolites (24) detected nicotinic acid within this cellular compartment, suggesting that it may be present within the CCV. However, our results demonstrate that this salvage pathway is not sufficient to support growth within the CCV in the absence of de novo NAD synthesis, indicating that the levels of nicotinic acid in the CCV are not high enough to provide sufficient NAD to support growth or that the salvage system is not active in vivo. We saw similar results in both human epithelial and human macrophage-like cells, suggesting that the specific host cell does not affect the requirement of C. burnetii for de novo NAD synthesis when inside the CCV. In contrast, another intracellular human pathogen, Mycobacterium tuberculosis, is fully virulent in an animal model even in the absence of de novo NAD synthesis (25). M. tuberculosis possesses the Pnc salvage system, and expression of this system is induced during mouse infection (25), suggesting that, in contrast to C. burnetii, it is salvage pathways rather than de novo synthesis that are crucial for M. tuberculosis virulence. This also suggests that the nutrient composition of the compartment that M. tuberculosis replicates in is different from that of the CCV. It is also possible that the observed differences may relate to the host species used, because the M. tuberculosis studies were carried out in mice, whereas our study used human cells, or the observed differences may be due to differences between animal and cell infection models.
Another intracellular human pathogen, Shigella flexneri, lacks the de novo NAD synthetic pathway because of mutations within both nadA and nadB and relies on the conversion of nicotinic acid to NaMN by PncB, resulting in an absolute requirement for nicotinic acid supplementation during growth in vitro. In contrast to C. burnetii, restoring de novo NAD synthesis reduced the ability of S. flexneri to invade and spread between host cells (26). This finding appeared to relate to the inhibition of cell-to-cell spread by quinolate, although the mechanisms by which this occurs is not yet known. Nonetheless, this observation further supports the idea that tight regulation of this pathway is crucial to the virulence of intracellular pathogens. The downstream enzymes, NadD and NadE, which are common to both the salvage pathway and the de novo synthetic pathway, have been shown to be essential in the model organism Mycobacterium smegmatis, because inhibition of these enzymes leads to metabolic catastrophe and bacterial death, because of the loss of essential NAD(H) cofactors, and these enzymes are therefore considered antimycobacterial drug targets (22). In humans, three isoforms of hsNMNAT carry out the physiologically equivalent reaction to that carried out by NadD in bacteria. Although there appear to be structural distinctions between the human and prokaryotic enzymes that would make selective targeting of bacterial enzymes such as NadD possible (27), our results suggest that further investigation of the role of de novo NAD synthesis in a variety of bacteria may reveal that targeting the prokaryotic-specific part of this pathway is a viable alternative therapeutic target for many pathogenic bacteria.
Loss of de novo synthesis reduced the levels of NAD-containing metabolites within axenically cultured C. burnetii, demonstrating that even in rich medium the salvage pathway is not sufficient to completely overcome the loss of de novo synthesis, although the nadB mutant grew normally in axenic medium (Fig. S4). Consistent differences in the steady-state metabolic profiles of the nadB mutant and both WT and the complemented strain were seen in the levels of nucleotides and related metabolites, and in TCA intermediates, with lower abundances of these metabolites in the nadB mutant. This may suggest a change in the flux through different key metabolic pathways when the levels of NAD-containing cofactors become limited. Furthermore, genome analysis suggests that C. burnetii lacks the oxidative arm of the pentose phosphate pathway, and it is not clear how NADPH, a major source of reducing equivalents for core metabolic reactions and antioxidant mechanisms, is regenerated (28). Therefore, C. burnetii and other bacteria that lack this pathway may be particularly vulnerable to inhibition of de novo NAD synthesis. We did observe some differences in changes in metabolite levels between the complemented nadB mutant and WT, when compared with the nadB mutant, which most likely relates to the overexpression of nadB in the complemented strain caused by the use of a plasmid for genetic complementation. Future studies employing stable isotope labeling would further define how the reduced levels of NAD-containing metabolites affect the overall cellular metabolism of C. burnetii.
Adequate levels of NAD-containing cofactors are crucial for cellular metabolism and maintenance of redox homeostasis. Furthermore, given the highly oxidative environment of the phagolysosome, it is expected that genes associated with redox balance are important to C. burnetii intracellular survival and replication. Here, we demonstrate conclusively that de novo NAD synthesis is critical for C. burnetii pathogenesis and suggest that further investigation of the importance of this prokaryotic-specific pathway in the virulence of a wider range of pathogens is warranted.
Experimental procedures
Cell strains and culture conditions
Plaque purified C. burnetii Nine Mile phase II (NMII) strain RSA 439 and derivatives were cultured in ACCM-2 liquid medium or ACCM-2–agarose plates at 37 °C with 5% CO2 and 2.5% O2. Chloramphenicol and kanamycin at final concentrations of 3 and 350 μg/ml, respectively, were used for selection of transposons and plasmids when required. E. coli JM109 was used for expression and purification of recombinant proteins and E. coli DH5a for routine cloning. E. coli XL1-Blue super competent cells were used for site-directed mutagenesis. E. coli was cultured in Luria–Bertani medium with ampicillin (100 μg/ml), chloramphenicol (25 μg/ml), and kanamycin (100 μg/ml) added as required for plasmid selection. HeLa CCL2 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) with 5 or 10% fetal calf serum (FCS) at 37 °C in 5% CO2.
Clonal isolation and complementation of the nadB mutants
The C. burnetii nadB transposon mutants from the previous screen (12) were clonally isolated as previously described (13). Briefly, clones were isolated by growth on two-layered ACCM-2 containing chloramphenicol. The plates were incubated for 6 days before single colonies were harvested using a sterile micropipette tip and expanded into 24-well plates. DNA was extracted from axenically cultured C. burnetii using the High Pure PCR template preparation kit as per the manufacturer's instructions (Roche). The cultures were screened by PCR using gene-specific and transposon primers to confirm the presence of the transposon within the nadB ORF.
To complement the nadB mutant, primers (Table 1) were used to amplify nadB with engineered SalI restriction sites to facilitate cloning into the C. burnetii complementation vector pJB-Kan:3xFLAG (29) to produce pFLAG-NadB. DNA sequencing was used to confirm the insertion of full-length nadB in the correct orientation. pFLAG-NadB was then introduced into the C. burnetii nadB mutant as described previously (12). Briefly, axenically cultured stationary phase C. burnetii were harvested by centrifugation (15,000 × g, 4 °C, 15 min) and resuspended in 20 ml of ice-cold 10% glycerol, before cells were harvested again and resuspended in 100 μl of ice-cold 10% glycerol. 10 μg of plasmid was then introduced via electroporation (18 kV, 500 Ω, and 25 microfarads). Following overnight recovery, kanamycin (350 μg/ml) was added, and transformants were selected on ACCM-2–agarose plates. The colonies were expanded to 24-well plates in ACCM-2 and screened for expression of FLAG-NadB using SDS-PAGE immunoblotting. Mouse anti-FLAG antibody was diluted (1/1000) in 1% (w/v) skim milk in PBS-T, and the secondary antibody (anti-mouse IgG HRP) was diluted to 1/3000 in 1% (w/v) skim milk in PBS-T for immunoblotting, before detection of signal using chemiluminescence.
Table 1.
Primers used in this study
| Name | Sequence (5′ to 3′) | Annealing temperature (°C) | Purpose |
|---|---|---|---|
| nadB F1 | AAAGTCGACATGCCCACCTATGATGTCCT | 62 | Complementation construct |
| nadB F2 | AAAGTCGACATATGCCCACCTATGATGTCCT | 61 | Cloning into pGEX-4T-1 |
| nadB R | AAAGTCGACCTAAAATTGCTTTTCGCCGATT | 62 | Cloning into pGEX-4T-1 or complementation construct |
| nadB (Internal) | CGTCTGTAAGGGCGCAGAGG | 55 | Sequencing within the inserted region |
| OmpA0008 F | CAGAGCCGGGAGTCAAGCT | 55 | Quantification of Coxiella genomes |
| OmpA0009 R | CTGAGTAGGAGATTTGAATCGC | 55 | Quantification of Coxiella genomes |
| pKM225 (5172) F | TGCTCACATGTTCTTTCCTGC | 55 | Transposon detection |
| pKM225 (5173) R | TGTGATGGCTTCCATGTCG | 55 | Transposon detection |
| pJB-Kan:3xFLAG F | GAGCTGTTGACAATTAATCATC | 55 | Sequencing complementation construct |
| pJB-Kan:3xFLAG R | GGATTCATCGACTGTGGCCG | 55 | Sequencing complementation construct |
| T7 | TAATACGACTCACTA | 55 | Sequencing |
| SP6 | TATTTAGGTGACACT | 55 | Sequencing |
| pGEX-4T-1 F | GGGCTGGCAAGCCACGTTTGGTG | 55 | Sequencing pGEX-nadB construct |
| pGEX-4T-1 R | CCGGGAGCTGCATGTGTCAGAGG | 55 | Sequencing pGEX-nadB construct |
| R275L F | GGAGATGGCCCCTCTAGACATTGTCGCTC | 55 | Site-directed mutagenesis |
| R275L R | GAGCGACAATGTCTAGAGGGGCCATCTCC | 55 | Site-directed mutagenesis |
Intracellular growth curves and immunofluorescence microscopy
The day prior to infection, 5 × 104 HeLa CCL2 cells were seeded into 24-well plates with or without coverslips. Stationary phase axenically grown C. burnetii strains were resuspended in DMEM + 5% FCS, and bacterial numbers were quantified by qPCR using ompA-specific primers (30). Bacteria were then diluted in DMEM + 5% FCS at 1 × 107 genome equivalents/ml to achieve a multiplicity of infection of 50. HeLa cells were infected for 4 h, and then the medium was removed, washed with PBS to remove uninfected bacteria, and incubated with fresh DMEM with 5% FBS. Infection lysate was collected at the time of infection (day 0) and 24 (day 1), 72 (day 3), 120 (day 5), and 168 (day 7) h after this initial time point. At each time point, the cells were lysed with H2O, samples were harvested, and the C. burnetii genome equivalents were determined using ompA-specific qPCR.
Replicate wells were also fixed with 4% paraformaldehyde at day 3 for subsequent immunofluorescent staining. The fixed cells were permeabilized and blocked with blocking buffer (PBS + 2% (w/v) BSA + 0.05% (v/v) saponin) for 1 h. The cells were then stained with rabbit anti-C. burnetii and mouse anti-LAMP1 monoclonal antibodies diluted in blocking buffer at 1/10,000 and 1/500, respectively. Secondary antibodies anti-rabbit Alexa Fluor 568 and anti-mouse 488 were used diluted at 1/3000 in blocking buffer. DAPI diluted 1/10,000 in PBS was added following removal of the secondary antibodies, and the cells washed were with PBS before mounting on glass slides using Dako fluorescent mounting medium. The images were acquired using Zeiss LSM700, Zeiss LSM710, and Nikon A1R confocal microscopes and analyzed using ImageJ software.
Three days prior to infection, THP-1 cells were seeded at 5 × 105/well in 24 well plates and treated with 10 nm phorbol 12-myristate 13-acetate to induce differentiation into macrophage-like cells. THP-1 cells were then infected at an multiplicity of infection of 25, and the intracellular replication assays were performed in a similar manner as for the HeLa cells. To confirm expression of FLAG-tagged proteins by C. burnetii strains during THP-1 infection, infected THP-1 cells were harvested 3 days after infection and subjected to SDS-PAGE. Mouse anti-FLAG antibody was diluted (1/1000) in 1% (w/v) skim milk in PBS-T and the secondary antibody (anti-mouse IgG HRP) diluted to 1/2000 in 1% (w/v) skim milk in PBS-T for immunoblotting, before detection of signal using chemiluminescence.
Site-directed mutagenesis
Sequence alignment using Geneious software was performed to identify conserved amino acids between the E. coli NadB protein (NP_417069.1, b2574) and C. burnetii NadB. C. burnetii nadB was amplified using gene-specific primers (Table 1) and cloned into pGEM-T (Promega) as per the manufacturer's instructions. To mutate Arg275 to Leu, primers were designed to introduce the desired mutation (Table 1) and site-directed mutagenesis using the QuikChange site-directed mutagenesis kit (Agilent Technologies) carried out following the manufacturer's instructions. The presence of the desired mutation in pGEM-R275L-NadB was verified by DNA sequencing. This plasmid was then used to subclone R275L-NadB into the C. burnetii complementation vector pJB-Kan:3xFLAG to produce pFLAG-R275L-NadB and as template for PCR amplification of R257L-nadB for cloning into pGEX-4T-1 to produce GST-R275L-NadB. The pFLAG-R275L-NadB construct was introduced into the C. burnetii nadB mutant as described above, with expression of full-length R275L-NadB confirmed by anti-FLAG immunoblotting, as for the original complementation construct (Fig. S1).
Expression and purification of recombinant proteins
Both nadB and R275L-NadB were PCR-amplified using gene-specific primers with engineered SalI sites (Table 1) and cloned into the SalI site of pGEX-4T-1 with an N-terminal GST tag. Correct insertion of the gene was confirmed by DNA sequencing. To purify recombinant proteins, overnight cultures of E. coli JM109 were diluted 1 in 100 in LB broth and grown to late logarithmic phase before induction of protein expression using 1 mm isopropyl-β-d-thiogalactopyranoside. Bacteria were harvested by centrifugation (7700 × g, 10 min, 4 °C) and incubated overnight in ice-cold PBS containing 1 mg/ml lysozyme and 1 mm phenylmethylsulfonyl fluoride. The cells were lysed by sonication, Triton X-100 was added to a final concentration of 1%, and the lysate was incubated for 30 min on ice. The soluble and insoluble fractions were separated by centrifugation. Recombinant proteins were purified from the soluble fraction using affinity chromatography on a GSH–Sepharose column according to the manufacturer's instructions (Amersham Pharmacia Biotech). Fractions containing the desired protein were pooled, and the purified protein was dialyzed against PBS using dialysis tubing cellulose membrane (Sigma) and protein concentration determined by Pierce BCA protein assay kit (Thermo Scientific) using BSA standards.
Determination of l-aspartate oxidase activity
A two-step enzyme assay was conducted to determine the l-aspartate oxidase activity of the recombinant proteins. In the first step, 100 mm bicine, pH 8.0, 2 μm FAD, 10 mm l-aspartate, 0.5 mg of BSA, 50 units of catalase, and 4.9 μg of recombinant protein (GST-NadB, GST-R275L-NadB, or GST) was incubated at 37 °C for 20 min and then stopped with perchloric acid. The denatured protein was removed by centrifugation (1000 × g, 3 min, RT). The supernatant was transferred to a new tube and neutralized with potassium hydroxide. For the second step, 0.1 mm NADH, 2.5 mm EDTA, and 100 mm bicine were added to the supernatant, and the reaction was started with 10 units of malate dehydrogenase (Sigma). The absorbance at 340 nm was measured at 1-min intervals for 8 min at 25 °C, and the average optical density of triplicate wells was calculated at each time point (following subtraction of the blank absorbance reading).
Quenching and extraction of polar metabolites
Six biological replicates of 20 ml of axenically cultured C. burnetii NMII strains were transferred into 50 ml of prechilled falcon tubes and immediately quenched to 0 °C in an ethanol–dry ice bath to stop metabolism. The quenched cultures were harvested by centrifugation (0 °C, 1000 × g, 20 min). To remove residual culture medium, the cell pellets were washed with ice-cold PBS (0 °C at 1000 × g, 20 min), resuspended in 1 ml of PBS, and transferred into prechilled 1.5-ml microcentrifuge tubes. Following centrifugation (0 °C at 23,000 × g, 15 min), the pellet was stored on ice. To lyse cells and extract metabolites, 400 μl of 3:1 (v/v) methanol:water containing 1 nmol of 13C6-sorbitol and 10 nmol of 13C5,15N-labeled valine as internal standards was added. Following vigorous mixing the cells were lysed using freeze-thaw cycles, and the reaction mixture was adjusted to a CHCl3:CH3OH:H2O ratio of 1:3:1 (v/v/v) by the addition of chloroform before mixing. After a 10-min incubation period, centrifugation (0 °C at 23,000 × g for 15 min) was performed to pellet cell debris and precipitated proteins. Phase separation was achieved after the supernatant was transferred to a fresh microcentrifuge tube and adjusted to CHCl3:CH3OH:H2O ratio of 1:3:3 (v/v/v) by addition of water. The upper aqueous phase containing polar metabolites was transferred to a fresh microcentrifuge tube and stored at −80 °C until drying. The samples were dried under vacuum at 37 °C with 30 μl of methanol added to the dried sample for the final drying stage. Polar metabolites were then derivatized and analyzed by GC/MS using a Shimadzu GC-TQ8040 triple quadrapole, as described previously (31, 32). Polar metabolites were also analyzed by HPLC–MS using an Agilent Technologies 1200 series HPLC system coupled to a 6545 Q-TOF, as described previously (31, 32).
Statistical analysis and comparison of metabolite profiles
Metabolites from all strains were identified from representative chromatograms by using an Agilent MSD Productivity Chemstation. Metabolites were identified by comparison of retention times and molecular masses with authentic standards. Peak integration was performed on the spectra from identified metabolites and collated into a targeted data matrix. The data were then missing value–imputed, median-normalized, and log-transformed and statistically analyzed by R-script using R-based statistical analysis package from Metabolomics Australia, as described previously (32) and the online tool MetaboAnalyst. Metabolite abundances between selected strain pairs were compared using unpaired Student's t test with the Benjamini–Hochberg adjustment to correct for false discoveries, with p < 0.05 considered significant.
Author contributions
M. A. B. data curation; M. A. B. and F. M. S. formal analysis; M. A. B. investigation; M. A. B., C. A. K., N. N., D. P. D. S., D. T., N. K. W., H. J. N., and F. M. S. methodology; M. A. B. and F. M. S. writing-original draft; D. P. D. S., D. T., N. K. W., and F. M. S. supervision; D. P. D. S., H. J. N., and F. M. S. writing-review and editing; H. J. N. and F. M. S. conceptualization; H. J. N. and F. M. S. funding acquisition; F. M. S. project administration.
Supplementary Material
Acknowledgments
Confocal microscopy was performed at the Biological Optical Microscopy Platform, The University of Melbourne.
This work was supported in part by Australian National Health and Medical Research Council Grants APP1063646 and APP1120344 (to H. J. N.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4.
- CCV
- Coxiella-containing vacuole
- RT
- retention time
- NaMN
- nicotinic acid mononucleotide
- GST
- glutathione S-transferase
- OAA
- oxaloacetate
- TCA
- tricarboxylic acid
- NMII
- Nine Mile phase II
- DMEM
- Dulbecco's modified Eagle's medium
- FCS
- fetal calf serum
- qPCR
- quantitative PCR
- DAPI
- 4′,6′-diamino-2-phenylindole.
References
- 1. Maurin M., and Raoult D. (1999) Q fever. Clin. Microbiol. Rev. 12, 518–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kazar J. (2005) Coxiella burnetii infection. Ann. N.Y. Acad. Sci. 1063, 105–114 10.1196/annals.1355.018 [DOI] [PubMed] [Google Scholar]
- 3. van Asseldonk M. A., Prins J., and Bergevoet R. H. (2013) Economic assessment of Q fever in the Netherlands. Prev. Vet. Med. 112, 27–34 10.1016/j.prevetmed.2013.06.002 [DOI] [PubMed] [Google Scholar]
- 4. Flannagan R. S., Jaumouillé V., and Grinstein S. (2012) The cell biology of phagocytosis. Annu. Rev. Pathol. 7, 61–98 10.1146/annurev-pathol-011811-132445 [DOI] [PubMed] [Google Scholar]
- 5. Coleman S. A., Fischer E. R., Howe D., Mead D. J., and Heinzen R. A. (2004) Temporal analysis of Coxiella burnetii morphological differentiation. J. Bacteriol. 186, 7344–7352 10.1128/JB.186.21.7344-7352.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Coleman S. A., Fischer E. R., Cockrell D. C., Voth D. E., Howe D., Mead D. J., Samuel J. E., and Heinzen R. A. (2007) Proteome and antigen profiling of Coxiella burnetii developmental forms. Infect. Immun. 75, 290–298 10.1128/IAI.00883-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Berón W., Gutierrez M. G., Rabinovitch M., and Colombo M. I. (2002) Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect. Immun. 70, 5816–5821 10.1128/IAI.70.10.5816-5821.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Howe D., and Mallavia L. P. (2000) Coxiella burnetii exhibits morphological change and delays phagolysosomal fusion after internalization by J774A.1 cells. Infect. Immun. 68, 3815–3821 10.1128/IAI.68.7.3815-3821.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Howe D., Melnicákova J., Barák I., and Heinzen R. A. (2003) Fusogenicity of the Coxiella burnetii parasitophorous vacuole. Ann. N.Y. Acad. Sci. 990, 556–562 10.1111/j.1749-6632.2003.tb07426.x [DOI] [PubMed] [Google Scholar]
- 10. Omsland A., Cockrell D. C., Howe D., Fischer E. R., Virtaneva K., Sturdevant D. E., Porcella S. F., and Heinzen R. A. (2009) Host cell-free growth of the Q fever bacterium Coxiella burnetii. Proc. Natl. Acad. Sci. U.S.A. 106, 4430–4434 10.1073/pnas.0812074106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beare P. A., Sandoz K. M., Omsland A., Rockey D. D., and Heinzen R. A. (2011) Advances in genetic manipulation of obligate intracellular bacterial pathogens. Front. Microbiol. 2, 97 10.3389/fmicb.2011.00097/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Newton H. J., Kohler L. J., McDonough J. A., Temoche-Diaz M., Crabill E., Hartland E. L., and Roy C. R. (2014) A screen of Coxiella burnetii mutants reveals important roles for Dot/Icm effectors and host autophagy in vacuole biogenesis. PLoS Pathog. 10, e1004286 10.1371/journal.ppat.1004286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carey K. L., Newton H. J., Lührmann A., and Roy C. R. (2011) The Coxiella burnetii Dot/Icm system delivers a unique repertoire of type IV effectors into host cells and is required for intracellular replication. PLoS Pathog. 7, e1002056 10.1371/journal.ppat.1002056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moffatt J. H., Newton P., and Newton H. J. (2015) Coxiella burnetii: turning hostility into a home. Cell. Microbiol. 17, 621–631 10.1111/cmi.12432 [DOI] [PubMed] [Google Scholar]
- 15. Brickman T. J., Suhadolc R. J., McKelvey P. J., and Armstrong S. K. (2017) Essential role of Bordetella NadC in a quinolinate salvage pathway for NAD biosynthesis. Mol. Microbiol. 103, 423–438 10.1111/mmi.13566 [DOI] [PubMed] [Google Scholar]
- 16. Gazzaniga F., Stebbins R., Chang S. Z., McPeek M. A., and Brenner C. (2009) Microbial NAD metabolism: lessons from comparative genomics. Microbiol. Mol. Biol. Rev. 73, 529–541 10.1128/MMBR.00042-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Osterman A. (2009) Biogenesis and homeostasis of nicotinamide adenine dinucleotide cofactor. EcoSal Plus 3, 3.6.3.10 10.1128/ecosalplus.3.6.3.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mesquita I., Varela P., Belinha A., Gaifem J., Laforge M., Vergnes B., Estaquier J., and Silvestre R. (2016) Exploring NAD+ metabolism in host–pathogen interactions. Cell. Mol. Life Sci. 73, 1225–1236 10.1007/s00018-015-2119-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marinoni I., Nonnis S., Monteferrante C., Heathcote P., Härtig E., Böttger L. H., Trautwein A. X., Negri A., Albertini A. M., and Tedeschi G. (2008) Characterization of l-aspartate oxidase and quinolinate synthase from Bacillus subtilis. FEBS J. 275, 5090–5107 10.1111/j.1742-4658.2008.06641.x [DOI] [PubMed] [Google Scholar]
- 20. Yang Z., Savchenko A., Yakunin A., Zhang R., Edwards A., Arrowsmith C., and Tong L. (2003) Aspartate dehydrogenase, a novel enzyme identified from structural and functional studies of TM1643. J. Biol. Chem. 278, 8804–8808 10.1074/jbc.M211892200 [DOI] [PubMed] [Google Scholar]
- 21. Tedeschi G., Ronchi S., Simonic T., Treu C., Mattevi A., and Negri A. (2001) Probing the active site of l-aspartate oxidase by site-directed mutagenesis: role of basic residues in fumarate reduction. Biochemistry 40, 4738–4744 10.1021/bi002406u [DOI] [PubMed] [Google Scholar]
- 22. Rodionova I. A., Schuster B. M., Guinn K. M., Sorci L., Scott D. A., Li X., Kheterpal I., Shoen C., Cynamon M., Locher C., Rubin E. J., and Osterman A. L. (2014) Metabolic and bactericidal effects of targeted suppression of NadD and NadE enzymes in mycobacteria. MBio 5, e00747–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seshadri R., Paulsen I. T., Eisen J. A., Read T. D., Nelson K. E., Nelson W. C., Ward N. L., Tettelin H., Davidsen T. M., Beanan M. J., Deboy R. T., Daugherty S. C., Brinkac L. M., Madupu R., Dodson R. J., et al. (2003) Complete genome sequence of the Q-fever pathogen Coxiella burnetii. Proc. Natl. Acad. Sci. U.S.A. 100, 5455–5460 10.1073/pnas.0931379100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abu-Remaileh M., Wyant G. A., Kim C., Laqtom N. N., Abbasi M., Chan S. H., Freinkman E., and Sabatini D. M. (2017) Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 358, 807–813 10.1126/science.aan6298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boshoff H. I., Xu X., Tahlan K., Dowd C. S., Pethe K., Camacho L. R., Park T. H., Yun C. S., Schnappinger D., Ehrt S., Williams K. J., and Barry C. E. 3rd. (2008) Biosynthesis and recycling of nicotinamide cofactors in Mycobacterium tuberculosis: an essential role for NAD in nonreplicating bacilli. J. Biol. Chem. 283, 19329–19341 10.1074/jbc.M800694200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Prunier A.-L., Schuch R., Fernández R. E., Mumy K. L., Kohler H., McCormick B. A., and Maurelli A. T. (2007) nadA and nadB of Shigella flexneri 5a are antivirulence loci responsible for the synthesis of quinolinate, a small molecule inhibitor of Shigella pathogenicity. Microbiology 153, 2363–2372 10.1099/mic.0.2007/006916-0 [DOI] [PubMed] [Google Scholar]
- 27. Sorci L., Pan Y., Eyobo Y., Rodionova I., Huang N., Kurnasov O., Zhong S., MacKerell A. D. Jr., Zhang H., and Osterman A. L. (2009) Targeting NAD biosynthesis in bacterial pathogens: structure-based development of inhibitors of nicotinate mononucleotide adenylyltransferase NadD. Chem. Biol. 16, 849–861 10.1016/j.chembiol.2009.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Omsland A., and Heinzen R. A. (2011) Life on the outside: the rescue of Coxiella burnetii from its host cell. Annu. Rev. Microbiol. 65, 111–128 10.1146/annurev-micro-090110-102927 [DOI] [PubMed] [Google Scholar]
- 29. Beare P. A. (2012) Genetic manipulation of Coxiella burnetii. In Coxiella burnetii: Recent Advances and New Perspectives in Research of the Q Fever Bacterium, pp. 249–271, Springer Springer-Verlag, New York Inc., New York [Google Scholar]
- 30. Jaton K., Peter O., Raoult D., Tissot J. D., and Greub G. (2013) Development of a high throughput PCR to detect Coxiella burnetii and its application in a diagnostic laboratory over a 7-year period. New Microbes New Infect. 1, 6–12 10.1002/2052-2975.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Best S. A., De Souza D. P., Kersbergen A., Policheni A. N., Dayalan S., Tull D., Rathi V., Gray D. H., Ritchie M. E., McConville M. J., and Sutherland K. D. (2018) Synergy between the KEAP1/NRF2 and PI3K pathways drives non-small-cell lung cancer with an altered immune microenvironment. Cell Metab. 27, 935–943.e4 10.1016/j.cmet.2018.02.006 [DOI] [PubMed] [Google Scholar]
- 32. Masukagami Y., De Souza D. P., Dayalan S., Bowen C., O'Callaghan S., Kouremenos K., Nijagal B., Tull D., Tivendale K. A., Markham P. F., McConville M. J., Browning G. F., and Sansom F. M. (2017) Comparative metabolomics of Mycoplasma bovis and Mycoplasma gallisepticum reveals fundamental differences in active metabolic pathways and suggests novel gene annotations. MSystems 2, e00055–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li W., Cowley A., Uludag M., Gur T., McWilliam H., Squizzato S., Park Y. M., Buso N., and Lopez R. (2015) The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 43, W580–W584 10.1093/nar/gkv279 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.