

Abstract
Nonalcoholic fatty liver disease (NAFLD) is a global health problem characterized by excessive accumulation of fat in the liver without effect of other pathological factors including hepatitis infection and alcohol abuse. Current studies indicate that gene factors play important roles in the development of NAFLD. However, the molecular characteristics of differentially expressed genes (DEGs) and associated mechanisms with NAFLD have not been well elucidated. Using two microarray data associated with the gene expression profiling in liver tissues of NAFLD mice models, we identified and selected several common key DEGs that contributed to NAFLD. Based on bioinformatics analysis, we discovered that the DEGs were associated with a variety of biological processes, cellular components, and molecular functions and were also related to several significant pathways. Via pathway crosstalk analysis based on overlapping DEGs, we observed that the identified pathways could form large and complex crosstalk networks. Besides, large and complex protein interaction networks of DEGs were further constructed. In addition, many hub host factors with a high degree of connectivity were identified based on interaction networks. Furthermore, significant modules in interaction networks were found, and the DEGs in the identified modules were found to be enriched with distinct pathways. Taken together, these results suggest that the key DEGs, associated pathways, and modules contribute to the development of NAFLD and might be used as novel molecular targets for the treatment of NAFLD.
Key words: Nonalcoholic fatty liver disease (NAFLD), Mouse model, Bioinformatics analysis, Differentially expressed genes (DEGs), Interaction network
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is a global health problem characterized with lipid accumulation in liver tissue without the effect of other pathological factors including hepatitis infection and alcohol abuse1. Based on histologic characteristics, NAFLD is categorized into hepatic steatosis and nonalcoholic steatohepatitis (NASH). Hepatic steatosis is defined by the presence of simple steatosis in liver cells without hepatocellular injury. In addition, NASH is characterized by hepatic steatosis, inflammation, and hepatocyte damage with or without fibrosis2. Importantly, NAFLD not only increases the risk of developing cirrhosis and hepatocellular carcinoma (HCC) but also is related to type 2 diabetes and cardiovascular diseases2–4. The harmfulness of NAFLD highlights the importance of a clear understanding of the molecular mechanisms associated with this disease to find effective intervention and treatment strategies.
The development of NAFLD is considered to be mediated by many contributing factors such as genetics, environmental factors, and microbiota5–8. Because of limitations such as genetic heterogeneity of human population in various regions, the long time for the progression of the disease, and ethical constraints to obtain human liver tissues, it is difficult to study the disease in patients to obtain enough information to understand the pathogenesis of NAFLD9–11. Therefore, suitable animal models, especially mice models with a high-fat diet (HFD), have been developed to detect the pathogenetic mechanisms responsible for NAFLD12,13. Using the mice models, many important genetic factors, dietary factors, and important hypothesis, in particular the “two-hit” hypothesis, for the pathogenesis of NAFLD have been recognized and proposed14,15.
With the application of high-throughput technology, several differentially expressed genes (DEGs) associated with NAFLD in patients and animal models were discovered8,16,17. However, NAFLD is a dynamic and complex process, and the current published research is not enough to elucidate the exact mechanisms of this disease. Recently, systems biology methods, including the analyses of pathway crosstalk, protein–protein interaction (PPI), and molecular module, have been applied to elucidate the pathological mechanism and identify the potential therapeutic drugs for different diseases18–20. In order to better understand the molecular characteristics of NAFLD and find potential biomarkers or treatment targets for the disease, bioinformatics analysis based on systems biology approaches was used in this study to analyze two microarray data from liver tissues of C57BL/6(N) mice fed with HFD. In addition, the biological function of DEGs, associated pathways, interaction network, and module information associated with NAFLD were investigated. Our results provide further insight into the molecular characteristics of NAFLD. In addition, the results from this study could provide the groundwork for potential therapy targeting identified key genes, associated pathways, and modules for NAFLD.
MATERIALS AND METHODS
Microarray Availability
The gene expression profiling studies related to NAFLD mice models were retrieved in the Gene Expression Omnibus (GEO) database (www.ncbi.nlm.nih.gov/geo/). The data information from different contributors were screened and analyzed if the following conditions were met: 1) the mice were fed with HFD and the complete microarray raw data of liver tissues were available; 2) the same mouse type or relevant mouse subtypes were used; 3) the mice were fed with HFD at the same time; and 4) a comparison was conducted between NAFLD groups (mouse under HFD or NASH diet) and negative control (NC) groups (mouse under normal diet or regular diet). Finally, we chose GSE5274821 and GSE5742522 for our analysis. In GSE52748, the 14-week-old mice were housed in a temperature- and light-controlled room (22°C, 12-h light/dark cycle) and allowed free access to food and water. In addition, mice in the control group were fed with standard diet, and mice in the NAFLD group were fed with NASH-inducing diet, which were enriched with beef tallow (15%), pork lard (15%), palmitic acid (4%), cholesterol (0.2%), stearic acid (4%), and sucrose (30%)21. In GSE57425, the mice at 8 weeks of age were maintained with free access to chow and water in a temperature-controlled environment (21° ± 1°C) with 12-h light/dark cycle. Besides, mice in the control group were fed with normal diet, and mice in the NAFLD group were fed with HFD containing 20% kcal from protein, 60% kcal of fat, and 20% kcal from carbohydrates22. The two microarray data were measured by a different Affymetrix platform, and the basic information of these two microarray data is listed in Table 1. The study was approved by the ethics committee of Xuzhou Medical University.
Table 1.
Characteristics of Two Microarray Studies Selected From the GEO Database
Data Processing
The raw data of the two microarrays were preprocessed in expression console Microsoft (Affymetrix). The detailed manipulation was followed according to the manufacturer’s instruction. Briefly, using the Expression Console Microsoft, probe signal values of the raw data were converted to log2 values, and genes annotated by the probes were analyzed based on annotation files of Affymetrix Mouse Gene 1.1 ST Array and Affymetrix Mouse Genome 430 2.0 Array. Then data were further normalized through the Robust Multichip Average (RMA) algorithm via Microsoft of Expression Console. Next, the preprocessed data were further analyzed by the Transcriptome Analysis Console v4.0 Microsoft (Affymetrix), and the DEGs were identified by statistical analysis through one-way ANOVA. The threshold for the DEGs was set as fold change at 1.5 and a value of p = 0.05.
Gene Function, Pathway, and Pathway Crosstalk Analysis
The biological significance of DEGs in NAFLD was assessed by Gene Ontology (GO) enrichment analysis. The Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was used to evaluate the enrichment pathways of the identified DEGs. In addition, GO analysis was measured with the online tool g:Profiler23. KEGG pathway analysis was performed with the ConsensusPathDB database24, and with minimum overlap of input genes of 3; a value of p < 0.05 was considered significant. Furthermore, pathway crosstalk analysis based on overlapped annotation genes was measured with an online tool in the ConsensusPathDB database, and with overlap genes no less than 3; a value of p < 0.05 was considered significant.
Data, Protein–Protein Interaction Network, and Module Visualization
Bar graph and circular graph were made to visualize the data that were analyzed in Excel 2007. Venn diagram was performed with the Venny 2.0 online tool (http://bioinfogp.cnb.csic.es/tools/venny/index.html). PPI data were collected from STRING databases25. In addition, the interaction networks were visualized with Cytoscape 3.2.1 software26, and module analysis was performed using the Molecular Complex Detection (MCODE) plugin in Cytoscape software.
RESULTS
Identification and Selection of the Common Key DEGs in NAFLD Mice Models
As shown in the heat maps in Figure 1A, using the expression console Microsoft and Transcriptome Analysis Console v4.0 Microsoft, we identified 1,056 DEGs in the NAFLD group, compared to the control group in GSE52748. In addition, compared with the control group, 1,846 DEGs were found in the NAFLD group in GSE57425. Current studies show that NAFLDs are histologically categorized into hepatic steatosis and NASH2. In GSE52748, the phenotype of mice liver was in the status of NASH, which was characterized by steatosis, inflammation, hepatocellular damage, and fibrosis21. However, in GSE57425, the phenotype of the mice was only in the status of steatosis22. In order to identify the key genes that contribute to both steatosis and NASH in NAFLD, we analyzed the common DEGs between two microarray data, and a total of 379 common DEGs were identified (Fig. 1B). Furthermore, among these common DEGs, 293 DEGs were found to upregulate and 46 DEGs to downregulate in both the two microarray data, 16 DEGs upregulate in GSE52748 but downregulate in GSE57425, and 24 DEGs downregulate in GSE52748 but upregulate in GSE57425 (Table 2). We could not confirm whether these inconsistent expressions of the 40 identified common DEGs between two microarray data were caused by different microarray methods or mediated by distinct microenvironments with steatosis or NASH in two mice models of NAFLD. For the accuracy of the bioinformatics analysis, the DEGs of whose expression patterns in GSE52748 were consistent with these in GSE57425 were only selected for further investigation.
Figure 1.

The identification of common key differentially expressed genes (DEGs) from two different microarray data. (A) The DEGs in nonalcoholic fatty liver disease (NAFLD) groups compared to control groups from microarray data GSE52748 and GSE57425. (B) The identified common DEGs between GSE52748 and GSE57425 by Venny 2.0 online tool. NC, control groups; NAFLD, NAFLD groups.
Table 2.
The Information of Common Key DEGs in Two Microarrays
| Types of DEGs | Gene Names |
|---|---|
| DEGs upregulated in both GSE52748 and GSE57425 (n = 293) | Cidea, Cidec, Sprr1a, Ly6d, Rgs16, Abcd2, Gprc5b, Osbpl3, Anxa2, Plin4, Stap1, S100a11, Ccl5, Ppp1r3g, Tlr12, Themis, Mogat1, Gpnmb, Slc22a27, Vnn1, Gpc1, H2-Aa, Enc1, Mfsd2a, Apoa4, Cxcl9, Postn, Tubb2a, Lyve1, Tmem86a, Cd74, Mmp12, Serpina7, Cgref1, Lgals1, Mtnr1a, Klf6,F cer1g, Wee1, Cd68, Cybb, Lgals3, Ifi27l2b, Ccnd1, Pld4, Limk1, Tceal8, H2-Ab1, Ly86, Fabp4, Spon2, Anxa5, Mest, Vcam1, Clec7a, Sirpa, Laptm5, C1qc, Cd53, C1qb, Endod1, Cyba, H2-Eb1, Fabp2, Abhd2, Vim, Wfdc2, Inhbe, Samd9l, Pex11a, Serinc2, Ttc39a, Plscr4, Vldlr, Haus8, Nid1, Pls1, Cbr3, Ccdc80, Uap1l1, Axl, H2-DMa, Slc35f2, Jun, Ms4a6b, Clec4a3, Mki67, D17H6S56E-5, Pilra, Gck, Iqgap1, Dpt, Lamb3, Gdf15, Rgs2, Ano6, Crat, Nckap1l, Spc25, Cd300ld, Plekha1, Unc119, Bhlhb9, Capg, Ephb2, Il2rg, Slc15a3, Tnfrsf19, 8430408G22Rik, Ctss, Mthfd1l, Mcm6, Rac2, Cd52, Tbc1d31, Tm6sf1, Tyrobp, Myo1f, Alpl, Gpc6, Cyp17a1, Frzb, Col1a1, Mgll, Slc39a5, AB124611, Aldh3a2, Mfge8, Pparg, Rgs10, Acot9, Dock10, Morc4, Ermp1, 1810011O10Rik, Kbtbd11, Sh3bgrl3, Itgb2, Itgax, Pdzrn3, Ptprc, Tubb2b, Ehd4, Fstl1, Rps6ka1, Car2, Cd93, Col1a2, Ifi27l2a, Paqr7, Tmem184b, Cd44, Nrp2, Slc25a47, Fitm1, Cd5l, Col14a1, Rgs5, Rtn4, Wwtr1, Col3a1, Gk, Sod3, Csf1r, Hk2, Aqp4, B4galt6, Cyp2b13, Igfbp3, Oasl2, Sparc, Fam126a, Lipo1, Ubd, Crip1, Fbln5, Slc16a7, Marcks, Pla2g7, Tppp, Ccdc3, Adgre4, Prss23, Mad2l1, Mpeg1, Myo9b, Rgs19, Tmem43, Atp9a, Cd83, Prune, Vsig4, Hck, Ppt1, Tmem140, Tor3a, Igsf11, Mpc1, Pla2g6, Fos, Hn1, Pak1, Pnldc1, Tgfbi, Cd48, Emcn, Ivns1abp, Lrat, Slc5a6, Tsc22d1, Anxa3, Itgal, Retsat, Tmem71, Igfbp7, Mpp1, Tpm1, Ctgf, Rhbdf1, Rhpn2, Arsg, Fermt3, Itga6, Itpripl2, Mylip, Nt5e, Slamf8, Acat1, Lgals3bp, Ptp4a3, Serpinb6a, Srd5a3, Arrdc3, Clec1b, Gal3st1, P2ry14, Pea15a, Rab8b, Snai2, Gltp, Ifit2, Mtmr11, Pctp, Plscr2, Sulf2, Arpc1b, Msr1, Sorbs1, Adora1, Coro1a, Ctps2, Hykk, Mgst3, Tbc1d1, Chpt1, Cmtm7, Lyz2, Nt5c2, Pde4d, Sema6d, Tmem106a, Arhgap11a, Arpp19, Bdh1, Gss, Hacd4, Pcolce, Ralgps2, Slc25a4, Tax1bp3, Cyp8b1, Dhrs7b, Gpd2, Kdsr, Pitpnm1, Vwf, 1600012H06Rik, Lamc1, Pdgfrb, Rab34, Samd4, Chchd6, Cpeb1, Dbp, Elk3, Gnai1, Golt1a, Slc44a3, Synpo, Tox, Wdr73, B930041F14Rik, Smpdl3a |
| DEGs downregulated in both GSE52748 and GSE57425 (n = 46) | C8a, Dpy19l3, Hacl1, Arhgef10l, Plxnb1, B3galt1, Cyp2c44, Ugt2b1, Kdm5b, Adck5, Cml1, Slco1a1, Igfals, Cyp1a2, Agxt, Cyp2c54, Slc43a1, Ankrd33b, Cxcl13, Cyp2c70, Slc3a1, Nudt7, Pdia5, Keg1, Apom, Slc41a2, Chic1, Snord104, Osgin1, Igfbp2, Nnmt, Serpine2, Atp11a, Cdh1, Egfr, Slc22a7, Lifr, Cadm4, Cyp7b1, Susd4, Avpr1a, Gm16551, C8b, Obp2a, Ces2a, Hsd3b5 |
| DEGs upregulated in GSE52748 but downregulated in GSE57425 (n = 16) | Lcn2, Mt2, Btg2, Myc, Saa2, Gadd45g, Mt1, E2f8, Pfkfb3, Hmox1, Saa3, Slc25a30, Rnf186, Saa1, Steap4, Syt12 |
| DEGs downregulated in GSE52748 but upregulated in GSE57425 (n = 24) | Camk1d, Cryl1, Slc6a6, Rmdn2, Pde9a, Nr0b2, Hmgcs1, Mid1ip1, Csad, Dhcr7, Adck3, Acss2, Kcnk5, Ddc, Sucnr1, Lss, Fdps, Cyp26a1, Mmd2, Car14, Hmgcr, Angptl8, Rdh11, Etnppl |
The Molecular Function Enrichment Analysis of DEGs in NAFLD Mice Models
To identify the biological functions associated with identified and selected DEGs, GO analysis was conducted using the web-based tool g:Profiler,23 and 146 GO terms with upregulated DEGs and 10 GO terms with downregulated DEGs were found. According to the number of genes, the top 10 enriched GO terms of the upregulated and downregulated DEGs were selected. The results of the GO analysis show that the DEGs were enriched in a variety of biological processes (BPs), cellular components (CCs), and molecular functions (MFs). As shown in Figure 2A, the main enriched GO terms of BP in upregulated DEGs were associated with response to stimulus, positive regulation of BP, and developmental process. In addition, the main GO term of BP in the downregulated DEGs were related to response to glucocorticoid, monocarboxylic acid metabolic process, and organic anion transport (Fig. 2B). The upregulated DEGs were located in the organelle, cytoplasm, and vesicles, while the enriched CCs were not found in downregulated DEGs. The MF of the upregulated DEGs was related to protein, integrin, and growth factor binding. However, the MF of the downregulated DEGs was associated with oxidoreductase activity and steroid hydroxylase activity. We next compared the enriched GO terms between upregulated and downregulated DEGs, and as shown in Figure 2C, only one common GO term named response to organic cyclic compound was found.
Figure 2.

Gene Ontology (GO) analysis of identified DEGs in NAFLD mice models. (A) The top 10 GO terms of upregulated DEGs. (B) The identified GO terms of downregulated DEGs. (C) The comparison of GO terms between upregulated DEGs and downregulated DEGs in NAFLD mice models.
The Pathway and Crosstalk Analysis of DEGs in NAFLD Mice Models
We next performed KEGG pathway analysis based on the ConsensusPathDB database24, and 40 KEGG pathways with upregulated DEGs and 9 KEGG pathways related to downregulated DEGs were identified. According to the number of genes, the top 10 enriched KEGG pathways of upregulated and downregulated DEGs were selected and shown in Figure 3. As shown in Figure 3A, the results of the KEGG pathways indicated that upregulated DEGs were associated with phagosome, focal adhesion, and PI3K–AKT signaling pathway. The downregulated DEGs were related to linoleic acid metabolism, steroid hormone biosynthesis, and cytokine–cytokine receptor interaction (Fig. 3B). The enriched KEGG pathways between upregulated and downregulated DEGs were further compared, but no common pathway was found (Fig. 3C). Furthermore, according to the overlap of identified DEGs between different pathways, the pathway crosstalk was constructed using the online tool in the ConsensusPathDB database24. We observed that one complex crosstalk network of KEGG pathways was associated with upregulated DEGs (Fig. 3D). In addition, a crosstalk network of KEGG pathways related to downregulated DEGs was found (Fig. 3E).
Figure 3.

Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of identified DEGs in NAFLD mice models. (A) The top 10 KEGG pathways of upregulated DEGs. (B) The identified KEGG pathways of downregulated DEGs. (C) The comparison of KEGG pathways between upregulated DEGs and downregulated DEGs in NAFLD mice models. (D) The crosstalk of pathways associated with upregulated DEGs. (E) The crosstalk of pathways associated with downregulated DEGs.
The Interaction Network and Hub Genes Analysis of DEGs in NAFLD Mice Models
In order to better understand the interaction of identified DEGs, we constructed the interaction networks, and PPI information was from STRING databases25. As shown in Figure 4A, the upregulated DEGs constituted a large and complex network. Based on the degree of connection with other DEGs, the top 10 hub DEGs, including Itgb2, Hck, Rac2, CD48, Ptprc, Jun, Cd68, Tyrobp, Ctss, and Itgax, were identified in the interaction network. In addition, the downregulated DEGs also formed a complex network, and Hsd3b5, Cyp2c44, Cyp2c54, Cyp1a2, Cyp2c70, Ugt2b1, C8b, Egfr, Gyp7b1, and Slco1a1 were identified as the top 10 hub downregulated DEGs (Fig. 4B).
Figure 4.

The interaction networks of identified DEGs. (A) The interaction network of upregulated DEGs. (B) The interaction network of downregulated DEGs.
The Module Analysis of DEGs in NAFLD Mice Models
Next, module analysis of the PPI networks was performed with MCODE plugin in Cytoscape to discover the functionally homogenous cluster within the constructed interaction networks. Based on the MCODE score >3 and node numbers >5, three significant modules in the interaction network of upregulated DEGs and one significant module of downregulated DEGs were found (Fig. 5). In addition, we found that, among the identified hub upregulated DEGs, Hck, Rac2, Ptprc, Cd68, and Tyrobp were located in module 1, and Jun was located in module 3 (Fig. 5A). Besides, Hsd3b5, Cyp2c54, Cyp1a2, Cyp2c70, Cyp2c44, and Ugt2b1 were found in the identified module 1 of downregulated DEGs (Fig. 5B). We next performed the pathway enrichment analysis for DEGs in the identified four modules (Table 3). Enrichment analysis showed that the upregulated DEGs in module 1 were enriched in natural killer cell-mediated cytotoxicity, Fc γ R-mediated phagocytosis, and osteoclast differentiation. The upregulated DEGs in module 2 were associated with microRNAs in cancer and cytokine–cytokine receptor interaction. The upregulated DEGs in module 3 were mainly related to leishmaniasis and phagosome. Besides, the downregulated DEGs in module 1 were associated with retinol metabolism, chemical carcinogenesis, and metabolism of xenobiotics by cytochrome P450.
Figure 5.
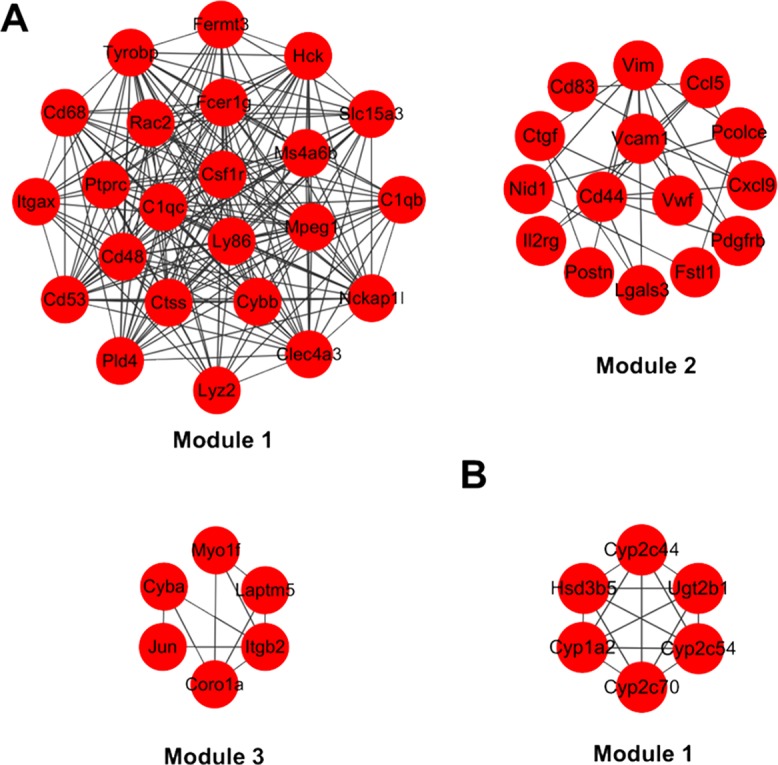
The module analysis in interaction networks of identified DEGs. (A) The identified three modules in the interaction network of upregulated DEGs. (B) The identified one module in interaction network of downregulated DEGs.
Table 3.
The Identified Significant KEGG Pathways in Different Modules
| DEGs/Modules | Pathways | Genes | p Value |
|---|---|---|---|
| Upregulated DEGs | |||
| Module 1 | Natural killer cell-mediated cytotoxicity | Tyrobp; Fcer1g; Rac2; Cd48 | 6.05E-05 |
| Fc γ R-mediated phagocytosis | Hck; Rac2; Ptprc | 0.0006 | |
| Osteoclast differentiation | Cybb; Tyrobp; Csf1r | 0.0017 | |
| Tuberculosis | Itgax; Fcer1g; Ctss | 0.0045 | |
| Regulation of actin cytoskeleton | Itgax; Nckap1l; Rac2 | 0.0080 | |
| Module 2 | MicroRNAs in cancer | Cd44;Pdgfrb;Vim | 0.0050 |
| Cytokine–cytokine receptor interaction | Cxcl9; Pdgfrb; Ccl5; Il2rg | 0.0003 | |
| HTLV-I infection | Pdgfrb; Vcam1; Il2rg | 0.0053 | |
| PI3K–Akt signaling pathway | Pdgfrb; Vwf; Il2rg | 0.0105 | |
| Module 3 | Leishmaniasis | Itgb2; Cyba; Jun | 2.39E-06 |
| Phagosome | Coro1a; Itgb2; Cyba | 4.41E-05 | |
| Downregulated DEGs | |||
| Module 1 | Retinol metabolism | Cyp1a2; Cyp2c70; Cyp2c54; Cyp2c44; Ugt2b1 | 2.30E-10 |
| Chemical carcinogenesis | Cyp1a2; Cyp2c70; Cyp2c54; Cyp2c44; Ugt2b1 | 3.60E-09 | |
| Metabolism of xenobiotics by cytochrome P450 | Cyp1a2; Cyp2c70; Cyp2c54; Cyp2c44; Ugt2b1 | 3.80E-09 | |
| Drug metabolism – cytochrome P450 | Cyp1a2; Cyp2c70; Cyp2c54; Cyp2c44; Ugt2b1 | 4.01E-09 | |
| Linoleic acid metabolism | Cyp2c54; Cyp1a2; Cyp2c44; Cyp2c70 | 7.36E-08 | |
| Arachidonic acid metabolism | Cyp2c44; Cyp2c54; Cyp2c70 | 1.59E-05 | |
| Serotonergic synapse | Cyp2c44; Cyp2c54; Cyp2c70 | 3.67E-05 | |
DISCUSSION
NAFLD is a chronic hepatic disease characterized by excessive accumulation of fat in hepatocytes. However, the exact mechanisms associated with NAFLD are not clear. It has been demonstrated that gene factors play important roles in the development of NAFLD. However, owing to lack of validated genetic targets, no available drugs have been reported for the effective and safe treatment of the disease, so our goal was to identify the key DEGs, associated pathways, and models that may be used as potential biomarkers or therapeutic targets for NAFLD. In this study, we identified and selected several common key DEGs that contribute to NAFLD via the microarray data from two NAFLD C57BL/6(N) mice models. Based on bioinformatics analysis, we found that the DEGs could exert distinct MFs, were associated with many significant pathways, and could form complex protein interaction networks. Furthermore, in the interaction network of DEGs, different modules were further identified, and the DEGs in each module were associated with distinct pathways.
Current studies show that a variety of cellular factors are responsible for the development of NAFLD14,15. In the study, based on the analysis of microarray data from liver tissues of mice fed with HFD, we identified that 379 common DEGs were associated with NAFLD that ranged from simple steatosis to NASH in two mice models. Among these identified common DEGs, a great number of DEGs were increased or decreased in both steatosis and NASH mice models. However, we also found that the expression patterns of some DEGs were different between steatosis and NASH in two mice models of NAFLD. Several factors may explain the differences. On the one hand, different experimental methods were used to obtain the gene expression data of microarrays in the two mice models. Generally, the probes used in the different microarray platforms were varied, and it may cause differences in the sensitivity and accuracy in the detection of gene expression and lead to false-positive or false-negative results. On the other hand, inflammation or fibrosis in NASH might induce a change in gene expression patterns that is different from simple steatosis. In the study, we were not sure whether the discrepancy in gene expression of identified common DEGs in two mice models was caused by different experimental methods or distinct pathological conditions in the different development stages of NAFLD. Therefore, for the accuracy of the bioinformatics analysis, the DEGs in both steatosis and NASH with similar expression patterns were selected for further investigation. Nevertheless, the exact reasons that caused the difference in gene expression in the identified common DEGs between steatosis and NASH in two mice models should be explored in future studies.
In order to better explore the molecular characteristics and associated biological functions related to key DEGs that are responsible for NAFLD, GO analysis was performed, and the results (Fig. 2) show the DEGs that contribute to NAFLD were associated with a variety of BPs, CCs, and MFs. As shown in Figure 2A, the BPs of upregulated DEGs were associated with response to stimulus, with the regulation of biological and development processes, and were different from those related to downregulated DEGs, which were mainly associated with the monocarboxylic acid metabolic process and organic anion transport. The upregulated DEGs were found to locate in various cellular areas, such as organelle, cytoplasm, membrane, and vesicle. These results indicated that the upregulated DEGs in different cellular areas might have diverse biological roles. As expected, we found that the MFs of upregulated DEGs were related to binding with protein, integrin, and growth factor (Fig. 2A and B). Besides, both upregulated and downregulated DEGs were found to be involved in one common BP related to response to organic cyclic compound, implying that the abnormal DEGs played vital roles in the development of NAFLD via mediating the disorders of organic cyclic compound.
The development of NAFLD is considered to be a complex multistep process, and several distinct molecular pathways are implicated16,17,27. Based on KEGG pathways, we discovered that many significant pathways were related to NAFLD. Current studies have reported that the pathways involved in lipid metabolism participated in the development of NAFLD16,17. Consistent with the published studies, we found that both upregulated and downregulated DEGs were involved in pathways that are associated with lipid metabolism. For example, upregulated DEGs were relevant to ether lipid metabolism. In addition, the downregulated DEGs were associated with linoleic acid metabolism and steroid hormone biosynthesis (Fig. 3E).
Moreover, in accordance with reported studies16, the pathway associated with phagosome was identified in upregulated DEGs. Phagosome pathway is related to phagocytosis, which is an important mechanism of innate immune response mediated by host cells to defense against infectious agents28. Our results implied that the abnormality of the phagosome pathway in NAFLD might disrupt the innate immune response mediated by phagocytosis. In addition, the results from our study showed that the PI3K–AKT signaling pathway was associated with the development of NAFLD, and the results were consistent with current research, which showed that the PI3K–AKT signaling pathway participated in lipid metabolic processes and regarded as a key treatment target for NAFLD29. The results of epidemiological studies show that NAFLD is a risk factor for HCC30. In the study, we found that the identified upregulated DEGs were associated with colorectal cancer (Fig. 3D), and downregulated DEGs were involved in the pathway of chemical carcinogenesis (Fig. 3E). The results implied that these DEGs associated with cancers might play important roles in the development of HCC induced by NAFLD.
Previous studies mainly focused on a single dysregulated pathway that contributes to the NAFLD16,17; the interactions among different pathways that facilitate the development of NAFLD are not well explored. Via pathway crosstalk analysis based on overlapping host factors in different signal pathways, we found that the identified pathways could form one complex interaction network in upregulated DEGs, and a crosstalk network was also observed in downregulated DEG. These results suggest that the identified pathways play important roles in NAFLD with a coordinated manner. Further understanding of the significant dysfunction crosstalk between identified pathways will help us to provide intense insights into the molecular mechanisms of NAFLD.
We next investigated the interaction of identified DEGs, and large and complex protein interaction networks were constructed. The results indicate that these DEGs could promote NAFLD via the interaction with each other. In addition, many hub DEGs with a high degree of connectivity were identified in the interaction networks. Among these identified upregulated hub DEGs, Cd68 is a molecular marker of macrophage31. In addition, Ctss is also known to be highly expressed in macrophages31. The upregulation of these molecules indicated that the number of macrophages increased in the liver of mice with NAFLD. These results are consistent with the study from Stanton et al.31, which has shown that the expressions of Cd68 and Ctss are increased in NAFLD. In addition, Ptprc (CD45) is a marker of leukocytes32. The upregulated expression of Ptprc indicated the increased infiltration of leukocytes in the liver of NAFLD mice. Itgb2 and Itgax belong to the integrin family that participates in cell adhesion33,34. Moreover, Itgax has been shown to be highly expressed in livers with NAFLD35. These results implied that the molecules associated with cell adhesion played vital roles in the development of NAFLD. Besides, Jun is an important transcription factor and has been reported to be correlated with inflammation and hepatic steatosis and contributes to the development of NASH21. The downregulated hub DEGs, including cyp2c54, cyp1a2, and cyp2c70, are members of cytochrome P450 enzyme superfamily that regulates xenobiotic metabolism, steroid transformation, and drug metabolism36–38. In addition, Cyp2c5437 and Cyp1a238 have been reported to be associated with NAFLD. The abnormal expressions of these members of cytochrome P450 enzyme superfamily may be associated with the dysregulation of multiple substance metabolism and transformation in NAFLD. Hsd3b5 is a member of 3-β-hydroxysteroid dehydrogenase family that regulates the BP of steroid hormones39. The dysregulation of Hsd3b5 might be related to the abnormality of steroid hormones in NAFLD.
Furthermore, based on the MOCDE analysis, three significant modules in the upregulated DEGs and one significant module in the downregulated DEGs were identified based on interaction networks. Furthermore, many of the hub DEGs were found to be located in these identified modules. Besides, we observed that the DEGs in these modules were enriched with distinct pathways. Given that hub DEGs and modules are considered to play important roles in maintaining the network system that is associated with distinct functions in specific physiological and pathological conditions40,41, targeting these hub DEGs or modules may offer new therapeutic opportunities for NAFLD.
In summary, via bioinformatics analysis of two microarray data from NAFLD mice models, we identified and selected several key DEGs that are involved in the development of NAFLD. Furthermore, our results indicate that the identified DEGs not only are associated with distinct functions and crosstalk pathways but also could construct complex networks. In addition, a lot of DEGs with distinct pathways could form distinct modules. Our study provides new insight on NAFLD development not only in the gene level but also in the pathway and module levels. The identified DEGs, associated pathways, and modules could serve as potentially diagnostic and therapeutic targets for NAFLD. In addition, our research has some limitations. For example, the identified DEGs, associated pathways, and modules were only generated from bioinformatics analysis. These data still need to be further confirmed in NAFLD mice and clinical specimens in future studies. Despite these limitations, the results of our study lay a foundation for further evaluating the roles and associated mechanisms mediated by identified key genes, associated pathways, and models in NAFLD.
ACKNOWLEDGMENTS
This study was supported by the Innovation and Entrepreneurship Training Programs for College Students (201610313063X and 201710313026Z), the research funding of the Qing Lan Project of Jiangsu province, a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and the Natural Science Foundation of the Jiangsu Higher Education Institutions (16KJB310017).
Footnotes
The authors declare no conflicts of interests.
REFERENCES
- 1. Farrell GC, Wong VW, Chitturi S. NAFLD in Asia—As common and important as in the West. Nat Rev Gastroenterol Hepatol. 2013;10:307–18. [DOI] [PubMed] [Google Scholar]
- 2. Rinella ME. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015;313:2263–73. [DOI] [PubMed] [Google Scholar]
- 3. Ahmed MH, Husain NE, Almobarak AO. Nonalcoholic fatty liver disease and risk of diabetes and cardiovascular disease: What is important for primary care physicians? J Family Med Prim Care 2015;4:45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Byrne CD, Targher G. NAFLD: A multisystem disease. J Hepatol. 2015;62:S47–64. [DOI] [PubMed] [Google Scholar]
- 5. Dongiovanni P, Romeo S, Valenti L. Genetic factors in the pathogenesis of nonalcoholic fatty liver and steatohepatitis. Biomed Res Int. 2015;2015:460190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arciello M, Gori M, Maggio R, Barbaro B, Tarocchi M, Galli A, Balsano C. Environmental pollution: A tangible risk for NAFLD pathogenesis. Int J Mol Sci. 2013;14:22052–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aqel B, DiBaise JK. Role of the gut microbiome in nonalcoholic fatty liver disease. Nutr Clin Pract. 2015;30:780–6. [DOI] [PubMed] [Google Scholar]
- 8. Qi S, Wang C, Li C, Wang P, Liu M. Candidate genes investigation for severe nonalcoholic fatty liver disease based on bioinformatics analysis. Medicine 2017;2017:96: e7743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takahashi Y, Soejima Y, Fukusato T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol. 2012;18:2300–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kanuri G, Bergheim I. In vitro and in vivo models of non-alcoholic fatty liver disease (NAFLD). Int J Mol Sci. 2013;14:11963–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2011;8:35–44. [DOI] [PubMed] [Google Scholar]
- 12. Ibrahim SH, Hirsova P, Malhi H, Gores GJ. Animal models of nonalcoholic steatohepatitis: Eat, delete, and inflame. Dig Dis Sci. 2016;61(5):1325–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Anstee QM, Goldin RD. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int J Exp Pathol. 2006;87:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakamura A, Terauchi Y. Lessons from mouse models of high-fat diet-induced NAFLD. Int J Mol Sci. 2013;14:21240–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Riordan JD, Nadeau JH. Modeling progressive non-alcoholic fatty liver disease in the laboratory mouse. Mamm Genome 2014;25:473–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guo XY, He CX, Wang YQ, Sun C, Li GM, Su Q, Pan Q, Fan JG. Circular RNA profiling and bioinformatic modeling identify its regulatory role in hepatic steatosis. Biomed Res Int. 2017;2017:5936171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jin X, Feng CY, Xiang Z, Chen YP, Li YM. CircRNA expression pattern and circRNA-miRNA-mRNA network in the pathogenesis of nonalcoholic steatohepatitis. Oncotarget 2016;7:66455–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li HY, Jin N, Han YP, Jin XF. Pathway crosstalk analysis in prostate cancer based on protein-protein network data. Neoplasma 2017;64:22–31. [DOI] [PubMed] [Google Scholar]
- 19. Xing C, Zhang R, Cui J, Li Y, Li G, Yang Y, Pang L, Ruan X, Li J. Pathway crosstalk analysis of non-small cell lung cancer based on microarray gene expression profiling. Tumori 2015;101(1):111–6. [DOI] [PubMed] [Google Scholar]
- 20. Ryall KA, Tan AC. Systems biology approaches for advancing the discovery of effective drug combinations. J Cheminform. 2015;7:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dorn C, Engelmann JC, Saugspier M, Koch A, Hartmann A, Müller M, Spang R, Bosserhoff A, Hellerbrand C. Increased expression of c-Jun in nonalcoholic fatty liver disease. Lab Invest. 2014;94:394–408. [DOI] [PubMed] [Google Scholar]
- 22. Lu Y, Liu X, Jiao Y, Xiong X, Wang E, Wang X, Zhang Z, Zhang H, Pan L, Guan Y, Cai D, Ning G, Li X. Periostin promotes liver steatosis and hypertriglyceridemia through downregulation of PPARalpha. J Clin Invest. 2014;124:3501–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Reimand J, Arak T, Adler P, Kolberg L, Reisberg S, Peterson H, Vilo J. g:Profiler-a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 2016;44:W83–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Herwig R, Hardt C, Lienhard M, Kamburov A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat Protoc. 2016;11:1889–907. [DOI] [PubMed] [Google Scholar]
- 25. Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang W, He Y, Liu S, Gan L, Zhang Z, Wang J, Liang J, Dong Y, Wang Q, Hou Z, Yang L. Integrative transcriptomic analysis of NAFLD animal model reveals dysregulated genes and pathways in metabolism. Gene 2016;595:99–108. [DOI] [PubMed] [Google Scholar]
- 28. Zhao L, Tu J, Zhang Y, Wang J, Yang L, Wang W, Wu Z, Meng Q, Lin L. Transcriptomic analysis of the head kidney of Topmouth culter (Culter alburnus) infected with Flavobacterium columnare with an emphasis on phagosome pathway. Fish Shellfish Immunol. 2016;57:413–8. [DOI] [PubMed] [Google Scholar]
- 29. Pisonero-Vaquero S, Martínez-Ferreras Á, García-Mediavilla MV, Martínez-Flórez S, Fernández A, Benet M, Olcoz JL, Jover R, González-Gallego J, Sánchez-Campos S. Quercetin ameliorates dysregulation of lipid metabolism genes via the PI3K/AKT pathway in a diet-induced mouse model of nonalcoholic fatty liver disease. Mol Nutr Food Res. 2015;59(5):879–93. [DOI] [PubMed] [Google Scholar]
- 30. De Minicis S, Day C, Svegliati-Baroni G. From NAFLD to NASH and HCC: Pathogenetic mechanisms and therapeutic insights. Curr Pharm Des. 2013;19(29):5239–49. [PubMed] [Google Scholar]
- 31. Stanton MC, Chen SC, Jackson JV, Rojas-Triana A, Kinsley D, Cui L, Fine JS, Greenfeder S, Bober LA, Jenh CH. Inflammatory signals shift from adipose to liver during high fat feeding and influence the development of steatohepatitis in mice. J Inflamm. (Lond) 2011;8:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Woudstra L, Biesbroek PS, Emmens RW, Heymans S, Juffermans LJ, van der Wal AC, van Rossum AC, Niessen HWM, Krijnen PAJ. CD45 is a more sensitive marker than CD3 to diagnose lymphocytic myocarditis in the endomyocardium. Hum Pathol. 2017;62:83–90. [DOI] [PubMed] [Google Scholar]
- 33. Dashti N, Mahmoudi M, Gharibdoost F, Kavosi H, Rezaei R, Imeni V, Jamshidi A, Aslani S, Mostafaei S, Vodjgani M. Evaluation of ITGB2 (CD18) and SELL (CD62L) genes expression and methylation of ITGB2 promoter region in patients with systemic sclerosis. Rheumatol Int. 2018;38(3):489–98. [DOI] [PubMed] [Google Scholar]
- 34. Bradford BM, Sester DP, Hume DA, Mabbott NA. Defining the anatomical localisation of subsets of the murine mononuclear phagocyte system using integrin alpha X (Itgax, CD11c) and colony stimulating factor 1 receptor (Csf1r, CD115) expression fails to discriminate dendritic cells from macrophages. Immunobiology 2011;216(11):1228–37. [DOI] [PubMed] [Google Scholar]
- 35. Latorre J, Moreno-Navarrete JM, Mercader JM, Sabater M, Rovira Ò, Gironès J, Ricart W, Fernández-Real JM, Ortega FJ. Decreased lipid metabolism but increased FA biosynthesis are coupled with changes in liver microRNAs in obese subjects with NAFLD. Int J Obes. (Lond) 2017;41(4):620–30. [DOI] [PubMed] [Google Scholar]
- 36. Munro AW, McLean KJ, Grant JL, Makris TM. Structure and function of the cytochrome P450 peroxygenase enzymes. Biochem Soc Trans. 2018;46(1):183–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang C, Tao Q, Wang X, Zhang X. Impact of high-fat diet on liver genes expression profiles in mice model of nonalcoholic fatty liver disease. Environ Toxicol Pharmacol. 2016;45:52–62. [DOI] [PubMed] [Google Scholar]
- 38. Merrell MD, Cherrington NJ. Drug metabolism alterations in nonalcoholic fatty liver disease. Drug Metab Rev. 2011;43:317–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Simard J, Ricketts ML, Gingras S, Soucy P, Feltus FA, Melner MH. Molecular biology of the 3 beta-hydroxysteroid dehydrogenase/delta5-delta4 isomerase gene family. Endocr Rev. 2005;26(4):525–82. [DOI] [PubMed] [Google Scholar]
- 40. He X, Zhang J. Why do hubs tend to be essential in protein networks? PLoS Genet. 2006;2:e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rhrissorrakrai K, Gunsalus KC. MINE: Module Identification in Networks. BMC Bioinformatics 2011;12:192. [DOI] [PMC free article] [PubMed] [Google Scholar]