

Recent studies have suggested that DDX3 functions in antiviral innate immunity, but the underlying mechanism remains elusive. We previously identified target mRNAs whose translation is controlled by DDX3.
KEYWORDS: DEAD-box RNA helicase, inflammation, innate immunity, phagocytosis, translational control
ABSTRACT
Recent studies have suggested that DDX3 functions in antiviral innate immunity, but the underlying mechanism remains elusive. We previously identified target mRNAs whose translation is controlled by DDX3. Pathway enrichment analysis of these targets indicated that DDX3 is involved in various infections and inflammation. Using immunoblotting, we confirmed that PACT, STAT1, GNB2, Rac1, TAK1, and p38 mitogen-activated protein kinase (MAPK) proteins are downregulated by DDX3 knockdown in human monocytic THP-1 cells and epithelial HeLa cells. Polysome profiling revealed that DDX3 knockdown reduces the translational efficiency of target mRNAs. We further demonstrated DDX3-mediated translational control of target mRNAs by luciferase reporter assays. To examine the effects of DDX3 knockdown on macrophage migration and phagocytosis, we performed in vitro cell migration assay and flow cytometry analysis of the uptake of green fluorescent protein-expressing Escherichia coli in THP-1 cells. The DDX3 knockdown cells exhibited impaired macrophage migration and phagocytosis. Moreover, we used a human cytokine antibody array to identify the cytokines affected by DDX3 knockdown. Several chemokines were decreased considerably in DDX3 knockdown THP-1 cells after lipopolysaccharide or poly(I·C) stimulation. Lastly, we demonstrated that DDX3 is crucial for the recruitment of phagocytes to the site of inflammation in transgenic zebrafish.
INTRODUCTION
Asp-Glu-Ala-Asp (DEAD)-box RNA helicases play crucial roles in almost all aspects of cellular RNA metabolism (1, 2). Human DDX3 is a member of the DEAD box RNA helicase family, which are generally involved in ATP-dependent unwinding of double-stranded RNAs and remodeling of ribonucleoprotein complexes (1, 2). DDX3 and its yeast homolog Ded1 have been implicated in various cellular processes (3), including transcription (4, 5), precursor mRNA splicing (6–8), mRNA transport (9, 10), and translation initiation (11–19). DDX3 has been reported to be primarily distributed throughout the cytoplasm in HeLa cells (15, 17). Under stress conditions, DDX3 is recruited to cytoplasmic stress granules (15, 20), suggesting its potential role in translational control. The role of DDX3 in translation initiation is evolutionarily conserved from yeasts to humans (21). We have demonstrated that DDX3 is required for the translation of some mRNAs that contain long or structured 5′ untranslated regions (UTRs) in human cells (14–16). Given that the RNA helicase activity of DDX3 is required for its function in translation initiation, DDX3 may facilitate ribosome scanning by resolving secondary structures in the 5′ UTRs of certain mRNAs (14–16).
DDX3 has been shown to participate in various cellular processes, including the cell cycle (16, 22, 23), cancer progression (4, 24–26), and antiviral innate immunity (27–31). DDX3 knockdown impedes cell proliferation by inhibiting the G1/S phase transition during the cell cycle (16, 23, 32). We demonstrated that DDX3 regulates the cell cycle through translational control of cyclin E1 (16), which plays a regulatory role at the G1/S phase transition. In a hamster temperature-sensitive DDX3 mutant cell line, tsET24, the G1/S phase transition of the cell cycle was impeded at the nonpermissive temperature (32). The helicase activity of DDX3 can be repressed by RK-33, a small-molecule inhibitor of DDX3. RK-33 treatment has been shown to cause cell cycle arrest at the G1 phase in various cancer cells, thus indicating that DDX3 may have potential as a therapeutic target for cancer treatment (33–35).
It has recently emerged that DDX3 plays a key role in antiviral innate immune signaling pathways (27–31). DDX3 was reported to be a transcriptional regulator of the beta interferon (IFN-β) promoter (30), a signal transducer downstream of TANK-binding kinase 1 (TBK1) and IκB kinase ε (IKKε) (27, 29), and a viral RNA sensor through its interactions with retinoic acid-inducible gene I (RIG-I) and IFN-β promoter stimulator 1 (IPS-1) (36). The RIG-I/IPS-1 complex is required for the recognition of viral RNA and the induction of IFN-β. However, the mechanism by which DDX3 functions in antiviral innate immunity has not been elucidated. We recently reported that DDX3 is required for translation of the mRNA of protein activator of the interferon-induced protein kinase (PACT) in human cells (19). PACT is a double-stranded RNA-binding protein which functions as an activator of RIG-I to facilitate the detection of viral RNAs (37). Some viruses evade immune detection by directly targeting PACT (38–42). The perturbation of RIG-I activation by various viral proteins suggests that PACT plays a critical role in antiviral host defense. Viruses have various kinds of defensive strategies to resist antiviral innate immunity, either by avoiding pattern recognition receptor (PRR) recognition or by interfering with IFN production. The finding that DDX3 is a target of viral immune evasion proteins highlights the importance of the role of DDX3 in antiviral innate immunity.
In this study, we obtained compelling evidence that DDX3 regulates the translation of several genes involved in viral and bacterial infections, inflammation, and phagocytosis. DDX3 knockdown results in the inhibition of phagocyte migration and phagocytosis in macrophages. The expression of several proinflammatory cytokines was markedly downregulated in DDX3 knockdown cells after either lipopolysaccharide (LPS) or poly(I·C) stimulation. Morpholino oligonucleotide-mediated knockdown of zebrafish DDX3 proteins resulted in the attenuation of phagocyte recruitment to the sites of inflammation caused by tail fin injury. In summary, our results suggest that DDX3 plays multiple roles in the inflammatory response caused by viral and bacterial infections and injuries.
RESULTS
DDX3-regulated genes are mainly involved in infections and inflammation.
We previously performed sucrose gradient fractionation coupled with a whole human genome expression microarray to screen cellular mRNAs whose translation is regulated by DDX3 (16), and we identified 375 candidate target genes of DDX3. To gain insight into the biological functions of DDX3, we performed pathway enrichment analysis by using DAVID Bioinformatics Resources 6.8 software to map genes to biological pathways defined by the Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway database. As expected, DDX3 was found to control the translation of genes involved in viral infections, such as those caused by herpes simplex virus, Epstein-Barr virus, human T-cell lymphotropic virus 1, influenza A virus, and hepatitis C virus (Table 1). Notably, DDX3 also controls the translation of genes involved in bacterial infections, such as infections by Helicobacter pylori and Vibrio cholerae as well as shigellosis and tuberculosis (Table 1). Pathogen-associated molecular patterns are unique microbial molecules that are recognized by PRRs in host cells during innate immunity. DDX3-regulated genes are also involved in PRR signaling pathways, such as the Toll-like receptor signaling pathway, RIG-I-like receptor signaling pathway, and NOD-like receptor signaling pathway (Table 1). Inflammation is triggered when innate immune cells detect infection or tissue injury. DDX3 also controls the translation of genes involved in inflammatory signaling pathways, such as the tumor necrosis factor (TNF) signaling pathway, rheumatoid arthritis, and the chemokine signaling pathway (Table 1). Therefore, we speculated that DDX3 may play multiple roles in viral and bacterial infections and inflammation.
TABLE 1.
Functional classification of DDX3 target genes
| KEGG pathway | P value | Genes |
|---|---|---|
| Epithelial cell signaling in Helicobacter pylori infection | 3.66E−4 | P38γ, RAC1, MAP2K4, P38β, CCL5, ATP6V0A2, ATP6V0B |
| Toll-like receptor signaling pathway | 7.66E−4 | TAK1, P38γ, IRF7, RAC1, MAP2K4, P38β, CCL5, STAT1, PACT |
| MAPK signaling pathway | 9.88E−4 | TAK1, FGFR4, P38γ, ELK4, RRAS2, RAC1, MAP2K4, PRKACA, CACNB3, P38β, DUSP9, TGFB1 |
| American trypanosomiasis | 0.0036 | P38γ, PPP2CB, MAP2K4, P38β, GNAS, CCL5, TGFB1 |
| Oocyte meiosis | 0.0045 | ANAPC1, CCNE1, ANAPC5, PPP2R5B, PPP2CB, PRKACA, FBXW11 |
| Sphingolipid signaling pathway | 0.0072 | PRKCZ, PPP2R5B, P38γ, PPP2CB, RAC1, SPHK1, P38β |
| Dopaminergic synapse | 0.0098 | GNB2, PPP2R5B, P38γ, PPP2CB, PRKACA, P38β, GNAS |
| Shigellosis | 0.0130 | ATG5, P38γ, RAC1, P38β, FBXW11 |
| Herpes simplex virus infection | 0.0156 | TAK1, CSNK2A2, IFIT1, IRF7, IL15, CDC34, CCL5, STAT1 |
| RIG-I-like receptor signaling pathway | 0.0176 | TAK1, ATG5, P38γ, IRF7, P38β, PACT |
| TNF signaling pathway | 0.0177 | TAK1, P38γ, MAP2K4, P38β, IL15, CCL5 |
| Adherens junction | 0.0185 | TAK1, CSNK2A2, PARD3, RAC1, SSX2IP |
| Leishmaniasis | 0.0185 | TAK1, P38γ, P38β, STAT1, TGFB1 |
| Epstein-Barr virus infection | 0.0188 | TAK1, CSNK2A2, GTF2E2, P38γ, MAP2K4, PRKACA, P38β, POLR3E |
| Hippo signaling pathway | 0.0209 | PRKCZ, PARD3, NF2, PPP2CB, YAP1, FBXW11, TGFB1 |
| Pathways in cancer | 0.0243 | CCNE1, ARHGEF1, GNB2, RAC1, RALA, PRKACA, EGLN1, GNAS, STAT1, TGFB1, TRAF4, FH |
| Human T-cell lymphotropic virus 1 infection | 0.0299 | ANAPC1, ANAPC5, ELK4, RRAS2, MAP2K4, IL15RA, PRKACA, IL15, TGFB1 |
| Rap1 signaling pathway | 0.0305 | PRKCZ, FGFR4, PARD3, P38γ, RAC1, RALA, P38β, GNAS |
| Amyotrophic lateral sclerosis | 0.0344 | P38γ, RAC1, TOMM40, P38β |
| Progesterone-mediated oocyte maturation | 0.0357 | ANAPC1, ANAPC5, P38γ, PRKACA, P38β |
| Rheumatoid arthritis | 0.0370 | IL15, CCL5, TGFB1, ATP6V0A2, ATP6V0B |
| Influenza A | 0.0382 | P38γ, IRF7, MAP2K4, P38β, CPSF4, CCL5, STAT1 |
| Platelet activation | 0.0383 | PRKCZ, ARHGEF1, P38γ, PRKACA, P38β, GNAS |
| Osteoclast differentiation | 0.0394 | TAK1, P38γ, RAC1, P38β, STAT1, TGFB1 |
| Vibrio cholerae infection | 0.0399 | PRKACA, GNAS, ATP6V0A2, ATP6V0B |
| Tuberculosis | 0.0410 | P38γ, SPHK1, P38β, STAT1, TGFB1, ATP6V0A2, ATP6V0B |
| GnRH signaling pathway | 0.0411 | P38γ, MAP2K4, PRKACA, P38β, GNAS |
| mRNA surveillance pathway | 0.0411 | PPP2R5B, PPP2CB, CPSF6, CPSF4, PABPC1L |
| Hepatitis C | 0.0416 | IFIT1, P38γ, IRF7, PPP2CB, P38β, STAT1 |
| NOD-like receptor signaling pathway | 0.0437 | TAK1, P38γ, P38β, CCL5 |
| Ubiquitin-mediated proteolysis | 0.0463 | ANAPC1, ANAPC5, CDC34, SMURF1, FBXW11, STUB1 |
| Chemokine signaling pathway | 0.0501 | PRKCZ, PARD3, GNB2, RAC1, PRKACA, CCL5, STAT1 |
DDX3 regulates the expression of PACT, STAT1, GNB2, Rac1, TAK1, and p38 MAPK proteins in THP-1 and HeLa cells.
We have identified numerous DDX3-regulated genes involved in infections and inflammation. To verify whether DDX3 can regulate their expression, we used a lentivirus expressing short hairpin RNA (shRNA) to specifically knock down DDX3 expression in human monocytic THP-1 cells and HeLa (human cervical epithelioid carcinoma) cells. The results of immunoblotting analysis revealed that the level of DDX3 protein was significantly reduced in the THP-1 and HeLa cells transduced with either of the DDX3-targeting shRNAs (Fig. 1, shDDX3-1 and shDDX3-2); however, shDDX3-2 was more effective than shDDX3-1 in inhibiting DDX3 expression. PACT has been identified as a target gene of DDX3; thus, it served as a positive control (19). The immunoblotting results showed that the STAT1, GNB2, Rac1, TAK1, and p38 mitogen-activated protein kinase (MAPK) (α, β, and γ) proteins were downregulated because of DDX3 knockdown in the THP-1 and HeLa cells (Fig. 1). STAT1 is a transcription factor that can be activated by interferons, and its activation results in the increased expression of interferon-stimulated genes (43). The heterotrimeric G protein subunit GNB2 plays a regulatory role in neutrophil function (44). The small G protein Rac1 is required for phagocytosis in macrophages during inflammation (45–47). TAK1 is known to be a crucial regulator in proinflammatory cytokine signaling (48). p38 MAPK participates in a signaling cascade that controls many cellular processes, including inflammation (49). Therefore, DDX3 knockdown may not only reduce the production of interferons and inflammatory cytokines but also inhibit the inflammatory response and phagocytosis.
FIG 1.
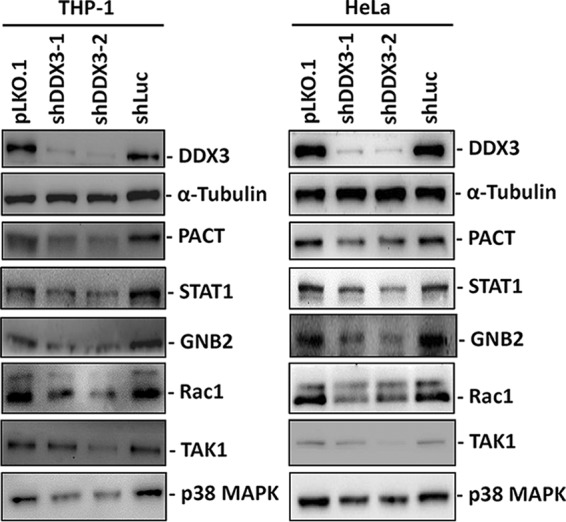
DDX3 regulates the expression of PACT, STAT1, GNB2, Rac1, TAK1, and p38 MAPK proteins in THP-1 and HeLa cells. THP-1 and HeLa cells were transduced with either the empty lentiviral vector (pLKO.1) or the pLKO.1 vector expressing the indicated shRNAs. After 24 h, puromycin was added to the culture medium for selection. The cells were harvested for analysis at 3 days posttransduction. Immunoblotting was performed using antibodies against DDX3, α-tubulin, PACT, STAT1, GNB2, Rac1, TAK1, and p38 MAPK. The detection of α-tubulin served as a loading control.
Translation of STAT1, GNB2, Rac1, TAK1, and p38 MAPK mRNAs is repressed by DDX3 knockdown in HEK293 cells.
To determine whether DDX3 is involved in the translation of STAT1, GNB2, Rac1, TAK1, and p38 MAPK (α, β, and γ) mRNAs, we performed sucrose gradient sedimentation and polysome profile analysis to evaluate the translational status in the DDX3 knockdown and mock-treated HEK293 cells. HEK293 is embryonic kidney immortalized cell line which represents epithelium and is suitable for polysome profile analysis (15). The results of polysome profiling showed that the knockdown of DDX3 caused by shDDX3-2 did not result in a significant change in global translation (Fig. 2A), indicating that DDX3 is not required for general translation (15, 16). Consistently, the fractional distribution of 18S and 28S rRNAs was not significantly affected by DDX3 knockdown (Fig. 2B). In contrast, the polysome associations of STAT1, GNB2, Rac1, TAK1, and p38 MAPK (α, β, and γ) mRNAs were decreased in the DDX3 knockdown HEK293 cells compared to the mock-treated controls (Fig. 2C). A decrease in polysome association was accompanied by an increase in 40S ribosome association. This indicated that the translation of the aforementioned proteins was inhibited because of DDX3 knockdown in the HEK293 cells. We also assessed the translational efficiencies of STAT1, GNB2, Rac1, TAK1, and p38 MAPK (α, β, and γ) mRNAs by polysome profiling and quantitative real-time reverse transcription-PCR (RT-PCR). The translational efficiency of a transcript was defined as the ratio of the mRNA abundance in the polysome fractions to the mRNA abundance in the total fractions (50). After calculation, the translational efficiencies of STAT1, GNB2, Rac1, TAK1, and p38 MAPK (α, β, and γ) mRNAs were significantly decreased in the DDX3 knockdown cells compared to the mock-treated controls (Fig. 2D). Therefore, DDX3 may regulate the translation of STAT1, GNB2, Rac1, TAK1, and p38 MAPK (α, β, and γ) mRNAs in HEK293 cells.
FIG 2.

Polysome profile analysis of STAT1, GNB2, Rac1, TAK1, and p38 MAPK translation in the DDX3 knockdown and mock-treated HEK293 cells. HEK293 cells were transduced with the empty lentiviral vector (pLKO.1) or the pLKO.1 vector expressing DDX3 shRNA (shDDX3-2). The cells were harvested for analysis at 3 days posttransduction. The cytoplasmic extracts of the cells were loaded on a linear 15% to 40% sucrose gradient and subjected to ultracentrifugation. (A) After sucrose gradient centrifugation, the polysome profiles were plotted using A254 values. (B) After sucrose gradient centrifugation, the mRNA/ribosome complexes were separated into 12 fractions. Total RNA was extracted from each fraction for analysis. The purified RNA was resolved by 1% denaturing agarose gel electrophoresis. 18S and 28S rRNAs were visualized using ethidium bromide for staining. (C) Total RNA was extracted from each fraction for analysis. The amount of mRNA was detected by quantitative real-time RT-PCR with specific primers for STAT1, GNB2, Rac1, TAK1, MAPK14 (p38α), MAPK11 (p38β), and MAPK12 (p38γ) mRNAs. The sum of a specific mRNA from the total fractions (fractions 1 to 11) was assumed to be 1 (or 100%), and the relative mRNA level in each fraction was expressed as a percentage of the amount of total specific mRNA. (D) The translational efficiencies of STAT1, GNB2, Rac1, TAK1, MAPK14 (p38α), MAPK11 (p38β), MAPK12 (p38γ), and GAPDH mRNAs in the mock-treated (pLKO.1) and DDX3 knockdown (shDDX3-2) HEK293 cells were calculated and are represented as percentages. The detection of GAPDH mRNA served as a negative control. The bar graph shows the changes in translational efficiencies as means ± SDs from three independent experiments.*, P < 0.05; **, P < 0.01.
DDX3 may facilitate the translation of PACT, STAT1, GNB2, Rac1, TAK1, and p38 MAPK (α, β, and γ) mRNAs by resolving their 5′ UTRs.
We have previously proposed that DDX3 is required for the efficient translation of mRNAs containing long or structured 5′ UTRs (14–16). The average length of 5′ UTRs in PACT, STAT1, GNB2, Rac1, TAK1, and p38 MAPK (α, β, and γ) mRNAs is ∼300 nucleotides (nt), with a GC content of 75.6% (Fig. 3A). In contrast, the average length of 5′ UTRs in randomly selected mRNAs is only about 246 nt, with a GC content of 63% (16). The results of computational analysis showed that PACT, STAT1, GNB2, Rac1, TAK1, and p38 MAPK (α, β, and γ) mRNAs contain long or GC-rich 5′ UTRs that are likely to form stable secondary structures. The RNALfold program was used to calculate the minimum free energy (MFE) of RNA secondary structures (Fig. 3A). Secondary structures were predicted using the RNAfold web server in ViennaRNA Web Services; the centroid secondary structures are shown in Fig. 3B. To determine how DDX3 functions in viral and bacterial infections and inflammation, firefly luciferase (Fluc) reporter genes containing the 5′ UTRs of PACT, STAT1, GNB2, Rac1, TAK1, and p38β MAPK mRNAs were constructed (Fig. 3C), and the dual-luciferase reporter assay was performed. A Renilla luciferase (Rluc) reporter with an unstructured 5′ UTR was cotransfected with the Fluc reporter as an internal control. Translation of the reporter mRNAs was assessed using the ratio of firefly to Renilla luciferase (Fluc/Rluc) activities. The relative Fluc/Rluc activity of reporters with the 5′ UTRs of PACT, STAT1, GNB2, Rac1, TAK1, and p38β MAPK mRNAs was significantly decreased in the DDX3 knockdown cells compared to the mock-treated controls (Fig. 3C). The Fluc reporter with the 5′ UTR of β-actin mRNA that is not a target of DDX3 was not affected by DDX3 knockdown. Therefore, DDX3 may facilitate the translation of PACT, STAT1, GNB2, Rac1, TAK1, and p38 MAPK mRNAs by resolving secondary structures within the complex 5′ UTRs.
FIG 3.

DDX3 controls the translation of PACT, STAT1, GNB2, Rac1, TAK1, and p38 MAPK mRNAs that contain complex 5′ UTRs. (A) The 5′ UTR sequences of PACT, STAT1, GNB2, Rac1, TAK1, and p38 MAPK (α, β, and γ) mRNAs obtained from the NCBI Reference Sequence (RefSeq) database were analyzed. The length, GC content, and minimum free energy of the 5′ UTR sequences are indicated. (B) Secondary structures within the 5′ UTRs were predicted using the RNAfold web server (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). The nucleotides are colored according to their probabilities in the structure. (C) HEK293 cells were transduced with the empty lentiviral vector (pLKO.1) or the pLKO.1 vector expressing DDX3 shRNA (shDDX3-2). After 48 h, the HEK293 cells were cotransfected with firefly luciferase (Fluc) reporters containing the 5′ UTRs of PACT, STAT1, GNB2, Rac1, TAK1, p38β MAPK, and β-actin mRNAs and the control pRL-SV40 vector encoding the Renilla luciferase (Rluc). The cells were lysed for analysis at 24 h posttransfection. For each transfectant, the Fluc activity was normalized to that of the Rluc control. The bar graph shows the relative Fluc/Rluc activities in DDX3 knockdown cells compared with those in the mock-treated controls. Data are shown as means ± SDs from three independent experiments. **, P < 0.01; ***, P < 0.001.
DDX3 participates in macrophage migration and phagocytosis.
Because STAT1, GNB2, and Rac1—DDX3 targets—are involved in phagocyte migration and phagocytosis (44–47, 51, 52), we investigated the effects of DDX3 knockdown on cell migration and phagocytosis in macrophages. Human monocytic THP-1 cells can be induced to differentiate into macrophages by treatment with phorbol 12-myristate 13-acetate (PMA). To detect phagocyte migration, in vitro Transwell cell migration assay was performed using PMA-induced THP-1 cells. Cell migration was quantified by counting the cells that passed through the filter membrane. As expected, the DDX3 knockdown cells showed markedly lower cell migration than the mock-treated cells did. A significant reduction (approximately 60% and 80% by shDDX3-1 and shDDX3-2, respectively) of cell migration was observed in the DDX3 knockdown PMA-induced THP-1 cells (Fig. 4A), suggesting that DDX3 plays a critical role in macrophage migration. In addition, we also quantified macrophage phagocytosis using green fluorescent protein (GFP)-expressing Escherichia coli in combination with flow cytometry (53, 54). The kinetics of phagocytosis in the PMA-induced THP-1 and U937 monocytic cells was analyzed by flow cytometry. Our results showed that the DDX3 knockdown cells exhibited reduced phagocytic activity, which was quantified using the uptake of GFP-expressing E. coli in the two monocytic cell lines (Fig. 4B). We also treated the THP-1 cells with a DDX3-specific inhibitor, RK‐33, which can bind to DDX3 and abrogate its helicase activity (33). Consistently, RK-33 treatment of THP-1 cells reduced the phagocytosis of E. coli in a dose-dependent manner (Fig. 4C). Therefore, we suggest that DDX3 should be required for macrophage migration and phagocytosis.
FIG 4.

DDX3 is involved in macrophage migration and phagocytosis. (A) THP-1 cells were transduced with the empty lentiviral vector (pLKO.1) or the pLKO.1 vector expressing the indicated shRNAs. After 24 h, puromycin was added to the culture medium for selection. At 2 days posttransduction, phorbol 12-myristate 13-acetate (PMA) was added to induce the differentiation of monocytes to macrophages. The PMA-induced THP-1 cells were loaded into the upper well of 8-μm BD Falcon cell culture inserts. After incubation at 37°C for 24 h, migrated cells were stained purple with 0.5% crystal violet. Representative images of the staining are shown in left graphs. The migrated cells were counted using a microscope at a magnification of ×100 in six randomly selected fields. The bar graph shows the relative number of migrated cells as means ± SDs from three independent experiments. *, P < 0.05; **, P < 0.01. (B) The THP-1 and U937 cells were transduced with the empty lentiviral vector (pLKO.1) or the pLKO.1 vector expressing DDX3 shRNA (shDDX3-2). After 24 h, puromycin was added to the culture medium for selection. At 2 days posttransduction, PMA was added to induce differentiation of monocytes to macrophages. At 3 days posttransduction, phagocytosis was quantified using the uptake of GFP-expressing E. coli at a multiplicity of infection (MOI) of 20. The mean fluorescent intensity of the GFP signal was measured by flow cytometry. Data are shown as means ± SDs from three independent experiments. *, P < 0.05. (C) The THP-1 cells were treated with different doses of RK-33 for 24 h. Phagocytosis was quantified using the uptake of GFP-expressing E. coli at an MOI of 20. The mean fluorescent intensity of the GFP signal was measured by flow cytometry. Data are shown as means ± SDs from three independent experiments. *, P < 0.05.
DDX3 regulates the expression of proinflammatory cytokines during viral and bacterial infections.
PACT, TAK1, and p38 MAPK are known to be involved in the induction of proinflammatory cytokines (37, 48, 55). PACT, a double-stranded RNA-binding protein, functions as a cellular activator of RIG-I to facilitate the detection of viral infection and IFN production (19, 37). TAK1 is a key mediator of the MAPK and nuclear factor κB (NF-κB), a major transcription factor that controls the expression of many proinflammatory cytokines (56). The p38 MAPK pathway is activated by a wide range of cellular stresses, including the regulation of proinflammatory cytokine expression (57). Therefore, we analyzed the effects of DDX3 knockdown on proinflammatory cytokine expression. A human cytokine antibody array (Fig. 5A) which detects the relative levels of 36 cytokine proteins was used to analyze changes in cytokine expression in THP-1 cells after either LPS or poly(I·C) stimulation. Under LPS treatment, the levels of CCL1, CCL2, CCL5, CXCL10, and TNF-α were decreased in the DDX3 knockdown THP-1 cells compared to the mock-treated controls (Fig. 5B). TNF-α is a key regulator of the inflammatory response (58), and TNF-α was released extensively in response to LPS. Similarly, the expression of CCL2, CCL5, TNF-α, and interleukin-1β (IL-1β) was impaired in the DDX3 knockdown THP-1 cells after poly(I·C) treatment (Fig. 5B). IL-1β is also a crucial mediator of the inflammatory response to injury and infections (59). Quantitative real-time RT-PCR was simultaneously performed to detect the mRNA levels of the cytokines affected by DDX3 knockdown. The mRNA expression patterns of cytokines are relatively similar to those of proteins in LPS-treated THP-1 cells (Fig. 5C), indicating that knockdown of DDX3 downregulates cytokine expression mainly at the transcriptional level. In the poly(I·C)-treated THP-1 cells, the knockdown of DDX3 also inhibited transcription of the CCL2, CCL5, TNF-α, and IL-1β genes (Fig. 5D). Consistent with previous findings (27–30), DDX3 knockdown resulted in the downregulation of IFN-β production induced by poly(I·C) at the mRNA level (Fig. 5D). We therefore propose that DDX3 may function in proinflammatory cytokine expression, at least in part, through translational control of PACT, TAK1, and p38 MAPK. To test this hypothesis, we ectopically expressed p38 MAPK in the DDX3 knockdown cells. Quantitative real-time RT-PCR showed that ectopic expression of p38 MAPK rescued the LPS-stimulated elevation of CCL2 mRNA expression in DDX3 knockdown THP-1 cells (Fig. 5E), supporting our hypothesis. However, we cannot rule out the possibility that DDX3 is also involved in regulating transcription or RNA stability of cytokine mRNAs (60).
FIG 5.

Expression of proinflammatory cytokines is impaired in DDX3 knockdown THP-1 cells. (A) The locations of various cytokine capture antibodies on the human cytokine array (R&D Systems) are indicated. (B) THP-1 cells were transduced with the empty lentiviral vector (pLKO.1) or the pLKO.1 vector expressing DDX3 shRNA (shDDX3-2). At 3 days posttransduction, the cells were stimulated with 80 ng/ml of LPS or 1 μg/ml of poly(I·C) for 6 h to mimic bacterial or viral infection, respectively. After 6 h of treatment, cell culture supernatants were collected and analyzed according to the manufacturer’s instructions. (C) The bar graph shows the relative protein level of LPS-induced cytokines in DDX3 knockdown THP-1 cells compared to those in the mock-treated controls (top). The relative mRNA levels of the cytokines normalized to GAPDH were analyzed using quantitative real-time RT-PCR (bottom). (D) The bar graph shows the relative protein level of poly(I·C)-induced cytokines in DDX3 knockdown THP-1 cells compared to mock-treated controls (top). The relative mRNA level of cytokines normalized to GAPDH was analyzed using quantitative real-time RT-PCR (bottom). Data are shown as means ± SDs from two independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (E) The experiment was essentially analogous to that for panel B, except that cells were transiently transfected with the mock or p38α-expressing vector for 24 h. At 3 days posttransduction, total RNA was extracted for analysis after 6 h of LPS stimulation. The relative mRNA level of CCL2 normalized to GAPDH was analyzed using quantitative real-time RT-PCR. Data are shown as means ± SDs from three independent experiments. *, P < 0.05; ***, P < 0.001.
DDX3 is crucial for the recruitment of phagocytes to the sites of inflammation in zebrafish.
As mentioned previously, the expression of chemokines CCL1, CCL2, CCL5, and CXCL10 was impaired in the DDX3 knockdown THP-1 cells (Fig. 5). CCL1 and CCL2, also called monocyte chemoattractant protein 1 (MCP1), are crucial for the recruitment of monocytes/macrophages to sites of inflammation (61). CCL5 and CXCL10 also participate in the recruitment of monocytes/macrophages in human adipose tissue and kidney (62, 63). Therefore, we analyzed the effects of DDX3 knockdown on the recruitment of phagocytic cells. Because of its evolutionarily conserved immune system, the zebrafish (Danio rerio) is a suitable model organism with which to investigate phagocyte recruitment and inflammation (64–66). Live imaging of transgenic zebrafish is used to track the migration of neutrophils and macrophages to a site of inflammation (64–66). Two homologous DDX3 genes (the DDX3a and DDX3b genes) are present in zebrafish. Antisense morpholino oligonucleotides (MOs) have been used successfully in zebrafish to knock down the expression of specific genes in vivo (67). In addition to inhibiting mRNA translation, MOs can efficiently block pre-mRNA splicing in zebrafish embryos (68). We designed two 25-mer MOs complementary to the exon 2/intron 2 junction of zebrafish DDX3a and DDX3b pre-mRNAs to interfere with normal splicing. Both MOs span the exon/intron junction, which contains the most conserved residues of the splice donor consensus sequence. To knock down zebrafish DDX3a and DDX3b simultaneously, we mixed the DDX3a and DDX3b MOs in a ratio of 1:1 and then microinjected them into the developing zebrafish embryos. At 2 days after microinjection, we observed that knockdown of zebrafish DDX3a/b reduced the larval size and caused the development of the bent-tail phenotype (Fig. 6A). Consistent with this finding, a recent study showed that zebrafish DDX3b is involved in embryonic development (69). RT-PCR analysis revealed that the DDX3a/b MOs efficiently blocked the correct splicing of DDX3a and DDX3b pre-mRNAs; consequently, several aberrant transcripts were detected (Fig. 6B). Immunoblotting analysis further confirmed MO-mediated knockdown of DDX3 in zebrafish. We used a human DDX3 antibody to detect the expression of zebrafish DDX3a/b proteins (Fig. 6C), and a weak band was observed in the region corresponding to the predicted molecular weight of DDX3 (approximately 80 kDa).
FIG 6.
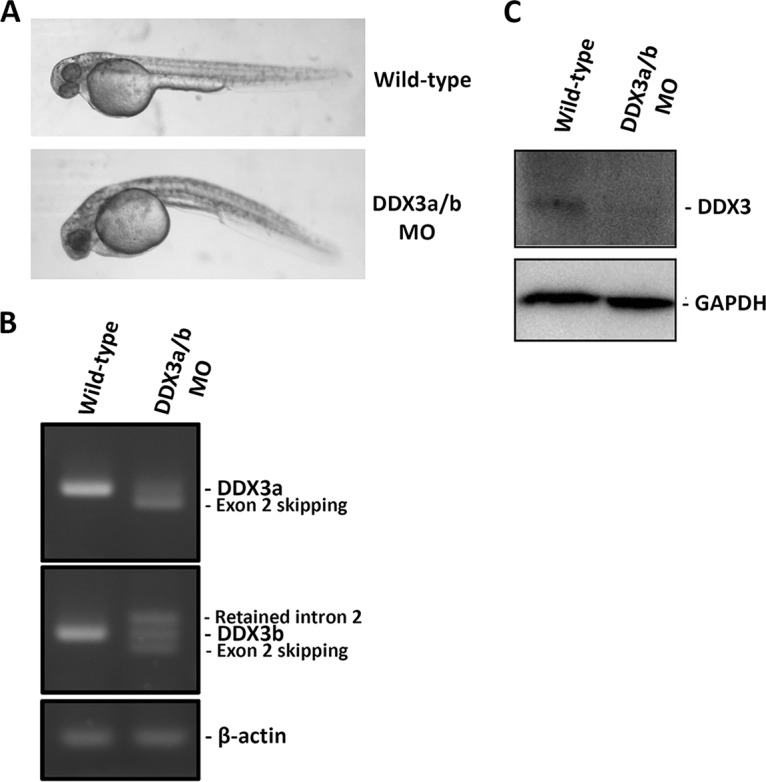
Confirmation of morpholino oligonucleotide (MO)-mediated DDX3a/b knockdown in zebrafish. Zebrafish embryos were injected with a mixture of DDX3a and DDX3b splice-blocking MOs (0.25 mM). (A) Phenotypic differences between wild-type and DDX3a/b MO-injected zebrafish at 2 days postfertilization. (B) RT-PCR analysis of DDX3a and DDX3b splicing patterns in the wild-type and DDX3a/b MO-injected zebrafish. Specific primers were designed for DDX3a, DDX3b, and β-actin. The PCR products were resolved on a 2% agarose gel. The detection of β-actin mRNA served as a loading control. (C) Immunoblot analysis showing the expression of DDX3 in wild-type and DDX3a/b MO-injected zebrafish. GAPDH protein served as a loading control.
Phagocyte migration is a fundamental step in the inflammatory response. We used the transgenic Tg(mpx:EGFP) zebrafish line, in which neutrophils are labeled with GFP, to determine the role of DDX3 in phagocyte recruitment to the site of inflammation. An inflammatory response was induced by tail transection, and the kinetics of neutrophil recruitment to the site of tail fin injury was recorded for 18 h. Live imaging of transgenic zebrafish larvae revealed that neutrophil recruitment to the site of inflammation in DDX3a/b knockdown zebrafish was less than that in the control zebrafish (Fig. 7). At 8 h post-tail fin transection, the recruitment of neutrophils was 50% lower in the DDX3a/b knockdown zebrafish than in the controls (Fig. 7). Therefore, DDX3 may play a crucial role in phagocyte migration and the recruitment of phagocytes to sites of inflammation.
FIG 7.

Recruitment of phagocytes to inflammation sites is impaired in DDX3a/b knockdown zebrafish. Control MO- and DDX3a/b MO-injected transgenic zebrafish (mpx:EGFP) larvae at the long-pec stage (2 days postfertilization) were anesthetized using 4.2% tricaine, and tail fin injury was inflicted using a sterile scalpel to induce inflammation (top). Each zebrafish larva was placed in a layer of 3% (wt/vol) methylcellulose in a 35-mm glass-bottom dish. Live imaging showed the recruitment of neutrophils at the indicated time points after tail fin transection (middle). The number of recruited neutrophils was determined by sampling 4 or 5 zebrafish larvae per group (bottom). Data are shown as means ± SDs from four independent experiments. *, P < 0.05; **, P < 0.01.
DISCUSSION
Recent reports have suggested that DDX3 has functions in antiviral innate immunity (19, 27, 29, 30, 36). However, the exact mechanism by which DDX3 participates in antiviral innate immunity is incompletely understood. We here provide, for the first time, compelling evidence that DDX3 has functions in inflammation induced by bacterial infections and injuries. Pathway enrichment analysis showed that DDX3 may be involved in infections caused by viruses, bacteria, and even parasites (Table 1). DDX3 also participates in inflammatory signaling pathways, including PRR signaling pathways, TNF signaling pathway, rheumatoid arthritis, and chemokine signaling pathway (Table 1). We further demonstrated that DDX3 is involved in phagocyte migration and phagocytosis process. It is therefore not surprising that DDX3 is involved in two protozoan diseases, namely, American trypanosomiasis and leishmaniasis (Table 1).
The results of immunoblotting showed that the expression levels of PACT, STAT1, GNB2, Rac1, TAK1, and p38 MAPK proteins were lower in the DDX3 knockdown cells than in the mock-treated control cells (Fig. 1). Like PACT (19), DDX3-mediated translational control of STAT1, GNB2, Rac1, TAK1, and p38 MAPK mRNAs was further confirmed by polysome profile analysis (Fig. 2) and luciferase reporter assay (Fig. 3). Notably, all these target mRNAs of DDX3 contain a long and/or structured 5′ UTR (Fig. 3). This suggests that DDX3 may unwind secondary structures in the 5′ UTR of target mRNAs to facilitate ribosome scanning during translation initiation. More recently, it was reported that repressing the activity of Ded1p, the yeast homolog of DDX3, leads to ribosome stalling at structural 5′ UTR and activates the usage of near-cognate translation initiation codons that are proximal to mRNA structure in 5′ UTRs (70). This may provide a mechanistic explanation of the role of DDX3 in translation initiation.
Previous studies have reported that STAT1, GNB2, and Rac1 are involved in phagocyte migration and phagocytosis (44–47, 51, 52). According to the KEGG pathway database, STAT1, GNB2, and Rac1 are included in chemokine signaling pathway (Table 1). This pathway is required for phagocyte migration and the production of inflammatory cytokines. Therefore, we speculated that DDX3 plays a role in the regulation of phagocytosis in macrophages. The phagocytic process can be divided into the following three stages: phagocyte migration, engulfment of pathogens, and intracellular killing and digestion. We used an in vitro Transwell cell migration assay and flow cytometry to confirm whether DDX3 participates in macrophage migration and phagocytosis (Fig. 4). The human cytokine antibody array was employed to identify cytokines and chemokines that are affected by DDX3 knockdown. It is notable that the expression levels of the chemokines CCL1, CCL2, CCL5, and CXCL10 were downregulated primarily at the transcriptional level (Fig. 5). CCL1, CCL2, CCL5, and CXCL10 are crucial chemokines that attract phagocytes to sites of inflammation. Impaired transcription of CCL1, CCL2, CCL5, and CXCL10 was most likely due to low levels of TAK-1 and p38 MAPK kinases in the DDX3 knockdown cells. More recently, a study demonstrated that DDX3 regulates the NF-κB signal pathway (71). Wang et al. reported that DDX3 knockdown reduced the phosphorylation of NF-κB p65 and IKK-β, thus attenuating the production of inflammatory cytokines induced by poly(I·C) stimulation. Notably, TAK1 is known to play an essential role in the phosphorylation of IKK-β (72), and IKK-β is required for the phosphorylation of NF-κB p65 (73). It seems likely that knockdown of DDX3 results in a decrease in the TAK1 protein level; thus, it reduces the phosphorylation of NF-κB p65 and IKK-β.
In summary, DDX3 may directly regulate phagocyte migration and phagocytosis through translational control of STAT1, GNB2, and Rac1 and affect the levels of chemokines indirectly. Consistent with this conclusion, the recruitment of neutrophils to sites of inflammation induced by tail fin injury was impaired in DDX3a/b knockdown zebrafish (Fig. 7). Therefore, we suggest that DDX3 has a function in the inflammatory response triggered by pathogen infections and tissue injury.
Here we propose a model of DDX3-mediated translational control in inflammation induced by viral and bacterial infections (Fig. 8). In epithelial (HeLa and HEK293) and phagocytic (THP-1 and U937) cells, DDX3 functions in the detection of viral infections through translational control of PACT; thus, it affects the expression of IFN-β (19). DDX3 also regulates the translation of TAK1 and p38 MAPK kinases, which play central roles in the production of proinflammatory cytokines (e.g., CCL2, CCL5, CXCL10, and TNF-α) during bacterial or viral infection. TNF-α is a major proinflammatory cytokine involved in early inflammatory events. CCL2, CCL5, and CXCL10 are key chemokines that attract monocytes/macrophages to sites of inflammation. In phagocytes, DDX3 also participates in phagocyte migration and phagocytosis through translational control of STAT1, GNB2, and Rac1 (Fig. 8). STAT1 and Rac1 are involved in phagocyte migration to inflamed tissues. Rac1 is also required for phagocytosis (45). GNB2 controls neutrophil recruitment through a Rac1-dependent pathway (44). Therefore, DDX3-mediated translational control plays multiple roles in viral and bacterial infections and the inflammatory response.
FIG 8.

Model of DDX3-mediated translational control in virus- and bacterium-induced inflammation and phagocytosis. In epithelial and phagocytic cells, DDX3 has functions in the signaling pathways of proinflammatory cytokines and IFN-β through translational control of PACT, TAK1, and p38 MAPK. In phagocytes, DDX3 also participates in phagocyte migration and phagocytosis through translational control of STAT1, GNB2, and Rac1.
Several recent studies suggest that DDX3 functions in cancer progression (4, 24–26). We also observed that DDX3 controls the translation of several genes involved in cancer pathways (Table 1). Overexpression of DDX3 represses E-cadherin expression and induces an epithelial-mesenchymal-like transformation (EMT) in breast cancer cells (4). DDX3 may promote cancer progression by increasing the transcription factor Snail (24), which represses the expression of cellular adhesion proteins and thus induces cell migration and metastasis. It has been shown that DDX3 promotes cancer cell migration/metastasis via Rac1 and the Wnt/β-catenin signaling pathway (25, 26), which modulates the expression of genes involved in the processes of EMT, invasion, and cell migration. However, whether DDX3 regulates phagocyte migration and phagocytosis by a similar mechanism remains to be determined.
More recently, a study showed upregulation of DDX3 in inflammatory bowel diseases (74), which are characterized by chronic production of proinflammatory cytokines and high levels of matrix metalloproteinases, suggesting that DDX3 is one of the inflammatory markers. This study further showed that RK-33 treatment of human colonic epithelial cells could reduce the expression of matrix metalloproteinases (74). Matrix metalloproteinases play a crucial role in the development of chronic inflammation. Therefore, DDX3 inhibitors have a high potential of being used as anti-inflammatory drugs. A thorough understanding of DDX3-mediated translational control in inflammation may open new avenues for the development of anti-inflammatory therapeutics.
MATERIALS AND METHODS
Cell culture and transfection.
HeLa and HEK293 cells were grown at 37°C in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml of penicillin, and 100 µg/ml of streptomycin. THP-1 and U937 cells were grown at 37°C in RPMI 1640 supplemented with 10% fetal bovine serum, 100 U/ml of penicillin, and 100 µg/ml of streptomycin. Phorbol 12-myristate 13-acetate (PMA; 200 nM) was added to the culture medium for 24 h to induce macrophage differentiation. Cell transfection was performed using Lipofectamine 2000 (Thermo Fisher Scientific), essentially according to the manufacturer’s instructions.
Plasmid constructs.
For in vivo translation assay, the 5′ UTR fragments of STAT1, GNB2, Rac1, and TAK1 mRNAs were obtained by RT-PCR using total RNA from HEK293 cells as the template and inserted into an unique HindIII site upstream of the firefly luciferase coding region in the pFL-SV40 vector (15). The 5′ UTR fragment of p38β was synthetic oligonucleotides. All plasmid constructs were confirmed by autosequencing analysis. The control reporter pRL-SV40 (Promega) was described previously (15). The p38α-expressing vector pMT3 p38 was a gift from John Kyriakis (Addgene plasmid 12658).
Lentivirus-mediated RNA interference knockdown.
All of the plasmids required for lentivirus production were provided by the National RNAi Core Facility (Academia Sinica, Taiwan). The two pLKO.1-shRNA vectors used to knock down DDX3 were as follows: TRCN0000000002 (shDDX3-1) and TRCN0000000004 (shDDX3-2). The control plasmid was TRCN0000231693 (shLuc). The transfection reagent Lipofectamine 2000 (Thermo Fisher Scientific) was used for lentivirus production in 293T cells with a packaging construct (pCMV ΔR8.91), an envelope construct (pMD.G), and pLKO.1-shRNA. To knock down endogenous DDX3, cells were transduced with shRNA-expressing lentivirus at a multiplicity of infection (MOI) of 10 virus particles/cell in growth medium containing 8 µg/ml of Polybrene at 37°C and 5% CO2. After 24 h, puromycin (2 μg/ml) was added to the medium for selecting infected cells. Cells were harvested 3 days posttransduction for analysis.
Immunoblotting.
For immunoblot analysis, proteins were transferred onto a polyvinylidene difluoride (PVDF) transfer membrane (Perkin Elmer). Protein blots were blocked with 3% skim milk in TBST buffer (100 mM Tris-HCl [pH 7.6], 150 mM NaCl, and 0.1% Tween 20) at room temperature for 1 h. The primary antibodies used included affinity-purified rabbit anti-DDX3 (0.1 µg/ml) (15), mouse anti-α-tubulin (0.2 µg/ml; Santa Cruz Biotechnology, Inc.), rabbit anti-PACT (1:1,000 dilution; Cell Signaling, Beverly, MA), rabbit anti-STAT1 (1:1,000 dilution; Cell Signaling), rabbit anti-GNB2 (1:1,000 dilution; GeneTex, Inc.), mouse anti-Rac1 (1:1,000 dilution; Millipore, Billerica, MA), rabbit anti-TAK1 (1:1,000 dilution; Cell Signaling), rabbit anti-p38 MAPK (1:1,000 dilution; Cell Signaling), rabbit anti-DDX3 (1:1,000 dilution; GeneTex, Inc.), and rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH; 1:5,000 dilution; GeneTex, Inc.). Zebrafish DDX3 was detected by rabbit anti-DDX3 (1:1,000 dilution; GeneTex, Inc.). Blots were incubated with primary antibodies in blocking buffer at room temperature for 2 h, followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for 2 h. Signals were detected using Immobilon Western chemiluminescent HRP substrate (Millipore), and images from autoradiograms were captured with the ChemiDoc touch imaging system (Bio-Rad).
Sucrose gradient sedimentation and polysome profiling.
Cells were collected in cold phosphate-buffered saline (PBS) containing 100 µg/ml of cycloheximide. All subsequent steps were performed at 4°C. Cell pellets were resuspended in RSB-150 (10 mM Tris-HCl [pH 7.4], 3 mM MgCl2, and 150 mM NaCl) containing 100 µg/ml of cycloheximide, 40 µg/ml of digitonin (Calbiochem), 20 U/ml of RNasin (Promega), and 1× protease inhibitor cocktail (Thermo Fisher Scientific). After incubation on ice for 5 min, cells were disrupted by passage through a 26-gauge needle five times. Cytoplasmic extracts were collected by centrifugation at 3,000 × g for 2 min and clarified by further centrifugation at 11,000 × g for 15 min. The samples were loaded on a linear 15 to 40% sucrose gradient and centrifuged at 178,000 × g for 3 h in a Beckman SW41 rotor. For polysome profile analysis, the gradients were monitored at 254 nm using an ISCO fractionation system (Lincoln, NE). After centrifugation, total RNA was extracted from each fraction using phenol-chloroform extraction in the presence of 1% SDS and 0.25 M NaCl, followed by ethanol precipitation.
RT-PCR and quantitative real-time PCR.
RT-PCR was used to detect the mRNA expression level. Extracted total RNA was reverse transcribed into cDNA using high-capacity cDNA reverse transcription kits (Thermo Fisher Scientific) according to the manufacturer’s instructions. The resulting cDNA was subjected to conventional PCR or quantitative real-time PCR analysis. Conventional PCR was performed using GoTaq DNA polymerase (Promega) and the forward and reverse primers as follows: for zebrafish DDX3a, forward primer (FP) 5′GTCATTACAAGGTCTCAAGAAGG3′ and reverse primer (RP) 5′CAAATGAGGAGGAATGTAACGCC3′); for zebrafish DDX3b, FP 5′AGTTTCATCGAGCACCGTCGACC3′ and RP 5′GCTT CTTTGTTCCTTAAATGTGGTGG3′); and for β-actin, FP 5′GCCCTGAGGCACTCTT CCA3′ and RP 5′CGGATGTCCACGTCACACTT3′.
Quantitative real-time PCR was performed using StepOne real-time PCR systems (Thermo Fisher Scientific) according to the supplier’s recommendations. Quantitative real-time PCR was performed using fast SYBR green master mix (Thermo Fisher Scientific) and the forward and reverse primers (Table 2). Quantitative analysis was performed by the measurement of threshold cycle (CT) values during the exponential phase of amplification. Relative quantitation values are calculated using the 2−ΔΔCT method.
TABLE 2.
List of primers used for quantitative RT-PCR
| Transcript | Primer sequencesa |
|---|---|
| STAT1 | FP, 5′GTGGAAAGACAGCCCTGCAT3′; RP, 5′GCAATTTCACCAACAGTCTCAACT3′ |
| GNB2 | FP, 5′CGCCTGTGATGCCTCTATCA3′; RP, 5′GGAAGAAAGCCACTGCATTGA3′ |
| Rac1 | FP, 5′ATGGGATACAGCTGGACAAGAAG3′; RP, 5′GCCCCGGGAGGTTATATCCT3′ |
| TAK1 | FP, 5′GGTGCTGAACCATTGCCATA3′; RP, 5′CTTTGGGTTGCATGCTGTGA3′ |
| MAPK14 (p38α) | FP, 5′TGGCTCTGTGTGTGCTGCTT3′; RP, 5′GGTTCTTTTCGCATGAATGATG3′ |
| MAPK11 (p38β) | FP, 5′GACCTGAACAACATCGTCAAGTG3′; RP, 5′CGGCCGAGTGGATGTACTTC3′ |
| MAPK12 (p38γ) | FP, 5′AGGCAGGCAGACAGTGAGATG3′; RP, 5′TGTCCACCGTCTGCGTGTAG3′ |
| CCL1 | FP, 5′GCAGGTACCCTTCTCCAGATGT3′; RP, 5′ATTGGAGCAGATGGAGCTGGTAT3′ |
| CCL2 | FP, 5′GCAATCAATGCCCCAGTCA3′; RP, 5′GACACTTGCTGCTGGTGATTCT3′ |
| CCL5 | FP, 5′CAGCCCTCGCTGTCATCCT3′; RP, 5′GGGCAATGTAGGCAAAGCA3′ |
| CXCL10 | FP, 5′ACGCTGTACCTGCATCAGCAT3′; RP, 5′CTCAACACGTGGACAAAATTGG3′ |
| TNF-α | FP, 5′TGAGG CCAAGCCCTGGTAT3′; RP, 5′GAGATAGTCGGGCCGATTGA3′ |
| IL-1β | FP, 5′AAATACCTGTGGCCTTGGGC3′; RP, 5′TTTGGGATCTACACTCTCCAGCT3′ |
| IFN-β1 | FP, 5′CAGAAGCTCCTGTGGCAATTG3′; RP, 5′GGAACTGCT GCAGCTGCTTA3′ |
| GAPDH | FP, 5′CCATCTTCCAGGAGCGAGATC3′; RP, 5′GCCTTCTCCATGGTGGTGAA3′ |
FP, forward primer; RP, reverse primer.
In vivo translation assay.
DDX3 knockdown and mock-treated HEK293 cells were transfected with a pFL-SV40-derived reporter (0.25 µg) and the control pRL-SV40 vector (0.25 µg). At 24 h posttransfection, cells were lysed in 1× passive lysis buffer (Promega). The activities of firefly luciferase and Renilla luciferase were determined using a Dual-Luciferase reporter assay system (Promega) and a GloMax 20/20 luminometer (Promega).
In vitro Transwell cell migration assay.
Migration of PMA-induced THP-1 cells was measured in a modified Boyden chamber migration assay (75). In brief, 2 × 105 THP-1 cells in RPMI 1640 supplemented with 0.1% FBS were loaded into 8-μm BD Falcon cell culture inserts. Chambers were inserted into 24-well culture dishes containing RPMI 1640 supplemented with 10% FBS. After cell migration for 24 h, cells were fixed and stained with 0.5% crystal violet in 25% methanol for 20 min. Migrated cells were quantified by the number of cells that migrated to the underside of the Transwell inserts. The images were captured at a magnification of ×100 with a digital camera in an Olympus CKX41 inverted microscope.
In vitro macrophage phagocytosis assay.
THP-1 and U937 cells were transduced with the empty lentiviral vector (pLKO.1) or the pLKO.1 vector expressing shDDX3-2. After 24 h, puromycin (2 μg/ml) was added to the culture medium for selection. At 54 h posttransduction, PMA was added to the culture medium to induce macrophage differentiation. Macrophages were infected with GFP-expressing E. coli at an MOI of 20 bacteria per cell at 3 days posttransduction. Macrophages were harvested for analysis via a BD Accuri C6 flow cytometer. To inhibit the helicase activity of DDX3, PMA-induced THP-1 cells were treated with RK-33 for 24 h and then infected with GFP-expressing E. coli at an MOI of 20.
Human cytokine antibody array.
THP-1 cells were transduced with the empty lentiviral vector (pLKO.1) or the pLKO.1 vector expressing shDDX3-2. After 3 days, THP-1 cells were treated with 80 ng/ml of LPS or transfected with 1 μg/ml of poly(I·C) to mimic bacterial or viral infection, respectively. After 6 h of treatment, cell culture supernatants were collected by centrifugation at 12,000 rpm for 5 min to remove the cell debris. The supernatant were analyzed using a human cytokine array (R&D Systems) according to the manufacturer’s instructions.
Morpholino oligonucleotide-mediated DDX3a/b knockdown in zebrafish.
Antisense morpholino oligonucleotides (MOs) were purchased from Gene Tools, LLC (Philomath, OR). Two MOs against zebrafish DDX3a and DDX3b were as follows: DDX3a MO (5′ATAAATGAGACTTACGGCCAGTGCC3′) and DDX3b MO (5′TGAGGAACAGAAACTCACTTCCAGT3′). Both MOs were designed to bind the exon2/intron2 splice site (E2I2) to interfere with the normal splicing process. A nontargeting MO (5′CCTCTTACCTCAGTTACAATTTATA3′) served as the control. Zebrafish embryos were injected with MOs at the one- to two-cell stage blastomeres. An injection volume of 500 pl was commonly used, and an MO concentration of 0.25 mM was used to achieve effective results.
Tail transection in transgenic zebrafish.
At 2 days postfertilization (dpf), transgenic zebrafish larvae injected with MOs were placed into the center of a glass-bottom 35-mm dish. Zebrafish larvae were anesthetized by immersion in E3 medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, and 0.33 mM MgSO4) with 4.2% tricaine. Each embryo was placed into a layer of 3% methylcellulose (wt/vol) on a glass-bottom 35-mm dish. Transection of the tail fin was performed with a sterile scalpel to induce inflammation. Images were acquired with the EVOS FL cell imaging system (Thermo Fisher Scientific).
Pathway enrichment analysis.
Functional annotation of DDX3 targets was performed using the publicly available DAVID Bioinformatics Resources 6.8 software (https://david.ncifcrf.gov/) with the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database. The statistical significance was assessed by a modified Fisher exact test. A P value of 0 represents perfect enrichment. A P value of <0.05 is considered to be strongly enriched in the annotation category.
Statistical analysis.
All data are presented as means ± standard deviations and were analyzed using Prism 4.0 software (GraphPad Software Inc., La Jolla, CA). Statistical analysis was performed using an unpaired t test. A P value of <0.05 was considered statistically significant.
ACKNOWLEDGMENTS
We thank Ching-Yu Lin (Graduate Institute of Biomedical Sciences, Chang Gung University) and Chuan-Sheng Lin (Department of Medical Biotechnology and Laboratory Science, Chang Gung University) for technical assistance in microinjection of zebrafish embryos and flow cytometry analysis, respectively. The manuscript was edited by Wallace Academic Editing.
This work was supported by grants from Ministry of Science and Technology, Taiwan (MOST 107-2320-B-182-031), and Chang Gung Memorial Hospital, Taiwan (CMRPD3E0013, CMRPD1H0051, and CMRPG3F0181).
REFERENCES
- 1.Cordin O, Banroques J, Tanner NK, Linder P. 2006. The DEAD-box protein family of RNA helicases. Gene 367:17–37. doi: 10.1016/j.gene.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 2.Rocak S, Linder P. 2004. DEAD-box proteins: the driving forces behind RNA metabolism. Nat Rev Mol Cell Biol 5:232–241. doi: 10.1038/nrm1335. [DOI] [PubMed] [Google Scholar]
- 3.Sharma D, Jankowsky E. 2014. The Ded1/DDX3 subfamily of DEAD-box RNA helicases. Crit Rev Biochem Mol Biol 49:343–360. doi: 10.3109/10409238.2014.931339. [DOI] [PubMed] [Google Scholar]
- 4.Botlagunta M, Vesuna F, Mironchik Y, Raman A, Lisok A, Winnard P Jr, Mukadam S, Van Diest P, Chen JH, Farabaugh P, Patel AH, Raman V. 2008. Oncogenic role of DDX3 in breast cancer biogenesis. Oncogene 27:3912–3922. doi: 10.1038/onc.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chao CH, Chen CM, Cheng PL, Shih JW, Tsou AP, Lee YH. 2006. DDX3, a DEAD box RNA helicase with tumor growth-suppressive property and transcriptional regulation activity of the p21waf1/cip1 promoter, is a candidate tumor suppressor. Cancer Res 66:6579–6588. doi: 10.1158/0008-5472.CAN-05-2415. [DOI] [PubMed] [Google Scholar]
- 6.Jamieson DJ, Rahe B, Pringle J, Beggs JD. 1991. A suppressor of a yeast splicing mutation (prp8-1) encodes a putative ATP-dependent RNA helicase. Nature 349:715–717. doi: 10.1038/349715a0. [DOI] [PubMed] [Google Scholar]
- 7.Stevens SW, Ryan DE, Ge HY, Moore RE, Young MK, Lee TD, Abelson J. 2002. Composition and functional characterization of the yeast spliceosomal penta-snRNP. Mol Cell 9:31–44. doi: 10.1016/S1097-2765(02)00436-7. [DOI] [PubMed] [Google Scholar]
- 8.Merz C, Urlaub H, Will CL, Luhrmann R. 2007. Protein composition of human mRNPs spliced in vitro and differential requirements for mRNP protein recruitment. RNA 13:116–128. doi: 10.1261/rna.336807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yedavalli VS, Neuveut C, Chi YH, Kleiman L, Jeang KT. 2004. Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell 119:381–392. doi: 10.1016/j.cell.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 10.Kanai Y, Dohmae N, Hirokawa N. 2004. Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron 43:513–525. doi: 10.1016/j.neuron.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 11.Chuang RY, Weaver PL, Liu Z, Chang TH. 1997. Requirement of the DEAD-box protein ded1p for messenger RNA translation. Science 275:1468–1471. doi: 10.1126/science.275.5305.1468. [DOI] [PubMed] [Google Scholar]
- 12.Soto-Rifo R, Rubilar PS, Limousin T, de Breyne S, Decimo D, Ohlmann T. 2012. DEAD-box protein DDX3 associates with eIF4F to promote translation of selected mRNAs. EMBO J 31:3745–3756. doi: 10.1038/emboj.2012.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hilliker A, Gao Z, Jankowsky E, Parker R. 2011. The DEAD-box protein Ded1 modulates translation by the formation and resolution of an eIF4F-mRNA complex. Mol Cell 43:962–972. doi: 10.1016/j.molcel.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lai MC, Wang SW, Cheng L, Tarn WY, Tsai SJ, Sun HS. 2013. Human DDX3 interacts with the HIV-1 Tat protein to facilitate viral mRNA translation. PLoS One 8:e68665. doi: 10.1371/journal.pone.0068665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lai MC, Lee YH, Tarn WY. 2008. The DEAD-box RNA helicase DDX3 associates with export messenger ribonucleoproteins as well as tip-associated protein and participates in translational control. Mol Biol Cell 19:3847–3858. doi: 10.1091/mbc.e07-12-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lai MC, Chang WC, Shieh SY, Tarn WY. 2010. DDX3 regulates cell growth through translational control of cyclin E1. Mol Cell Biol 30:5444–5453. doi: 10.1128/MCB.00560-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee CS, Dias AP, Jedrychowski M, Patel AH, Hsu JL, Reed R. 2008. Human DDX3 functions in translation and interacts with the translation initiation factor eIF3. Nucleic Acids Res 36:4708–4718. doi: 10.1093/nar/gkn454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soto-Rifo R, Rubilar PS, Ohlmann T. 2013. The DEAD-box helicase DDX3 substitutes for the cap-binding protein eIF4E to promote compartmentalized translation initiation of the HIV-1 genomic RNA. Nucleic Acids Res 41:6286–6299. doi: 10.1093/nar/gkt306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lai MC, Sun HS, Wang SW, Tarn WY. 2016. DDX3 functions in antiviral innate immunity through translational control of PACT. FEBS J 283:88–101. doi: 10.1111/febs.13553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shih JW, Wang WT, Tsai TY, Kuo CY, Li HK, Wu Lee YH. 2012. Critical roles of RNA helicase DDX3 and its interactions with eIF4E/PABP1 in stress granule assembly and stress response. Biochem J 441:119–129. doi: 10.1042/BJ20110739. [DOI] [PubMed] [Google Scholar]
- 21.Tarn WY, Chang TH. 2009. The current understanding of Ded1p/DDX3 homologs from yeast to human. RNA Biol 6:17–20. doi: 10.4161/rna.6.1.7440. [DOI] [PubMed] [Google Scholar]
- 22.Chang PC, Chi CW, Chau GY, Li FY, Tsai YH, Wu JC, Wu Lee YH. 2006. DDX3, a DEAD box RNA helicase, is deregulated in hepatitis virus-associated hepatocellular carcinoma and is involved in cell growth control. Oncogene 25:1991–2003. doi: 10.1038/sj.onc.1209239. [DOI] [PubMed] [Google Scholar]
- 23.Li Q, Zhang P, Zhang C, Wang Y, Wan R, Yang Y, Guo X, Huo R, Lin M, Zhou Z, Sha J. 2014. DDX3X regulates cell survival and cell cycle during mouse early embryonic development. J Biomed Res 28:282–291. doi: 10.7555/JBR.27.20130047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun M, Song L, Zhou T, Gillespie GY, Jope RS. 2011. The role of DDX3 in regulating Snail. Biochim Biophys Acta 1813:438–447. doi: 10.1016/j.bbamcr.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen HH, Yu HI, Cho WC, Tarn WY. 2015. DDX3 modulates cell adhesion and motility and cancer cell metastasis via Rac1-mediated signaling pathway. Oncogene 34:2790–2800. doi: 10.1038/onc.2014.190. [DOI] [PubMed] [Google Scholar]
- 26.Wu DW, Lin PL, Cheng YW, Huang CC, Wang L, Lee H. 2016. DDX3 enhances oncogenic KRAS-induced tumor invasion in colorectal cancer via the betacatenin/ZEB1 axis. Oncotarget 7:22687–22699. doi: 10.18632/oncotarget.8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu L, Fullam A, Brennan R, Schroder M. 2013. Human DEAD box helicase 3 couples IkappaB kinase epsilon to interferon regulatory factor 3 activation. Mol Cell Biol 33:2004–2015. doi: 10.1128/MCB.01603-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oshiumi H, Ikeda M, Matsumoto M, Watanabe A, Takeuchi O, Akira S, Kato N, Shimotohno K, Seya T. 2010. Hepatitis C virus core protein abrogates the DDX3 function that enhances IPS-1-mediated IFN-beta induction. PLoS One 5:e14258. doi: 10.1371/journal.pone.0014258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schroder M, Baran M, Bowie AG. 2008. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. EMBO J 27:2147–2157. doi: 10.1038/emboj.2008.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soulat D, Burckstummer T, Westermayer S, Goncalves A, Bauch A, Stefanovic A, Hantschel O, Bennett KL, Decker T, Superti-Furga G. 2008. The DEAD-box helicase DDX3X is a critical component of the TANK-binding kinase 1-dependent innate immune response. EMBO J 27:2135–2146. doi: 10.1038/emboj.2008.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fullam A, Schroder M. 2013. DExD/H-box RNA helicases as mediators of anti-viral innate immunity and essential host factors for viral replication. Biochim Biophys Acta 1829:854–865. doi: 10.1016/j.bbagrm.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fukumura J, Noguchi E, Sekiguchi T, Nishimoto T. 2003. A temperature-sensitive mutant of the mammalian RNA helicase, DEAD-BOX X isoform, DBX, defective in the transition from G1 to S phase. J Biochem 134:71–82. doi: 10.1093/jb/mvg126. [DOI] [PubMed] [Google Scholar]
- 33.Bol GM, Vesuna F, Xie M, Zeng J, Aziz K, Gandhi N, Levine A, Irving A, Korz D, Tantravedi S, Heerma van Voss MR, Gabrielson K, Bordt EA, Polster BM, Cope L, van der Groep P, Kondaskar A, Rudek MA, Hosmane RS, van der Wall E, van Diest PJ, Tran PT, Raman V. 2015. Targeting DDX3 with a small molecule inhibitor for lung cancer therapy. EMBO Mol Med 7:648–669. doi: 10.15252/emmm.201404368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heerma van Voss MR, Vesuna F, Trumpi K, Brilliant J, Berlinicke C, de Leng W, Kranenburg O, Offerhaus GJ, Burger H, van der Wall E, van Diest PJ, Raman V. 2015. Identification of the DEAD box RNA helicase DDX3 as a therapeutic target in colorectal cancer. Oncotarget 6:28312–28326. doi: 10.18632/oncotarget.4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xie M, Vesuna F, Tantravedi S, Bol GM, Heerma van Voss MR, Nugent K, Malek R, Gabrielson K, van Diest PJ, Tran PT, Raman V. 2016. RK-33 radiosensitizes prostate cancer cells by blocking the RNA helicase DDX3. Cancer Res 76:6340–6350. doi: 10.1158/0008-5472.CAN-16-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oshiumi H, Sakai K, Matsumoto M, Seya T. 2010. DEAD/H BOX 3 (DDX3) helicase binds the RIG-I adaptor IPS-1 to up-regulate IFN-beta-inducing potential. Eur J Immunol 40:940–948. doi: 10.1002/eji.200940203. [DOI] [PubMed] [Google Scholar]
- 37.Kok KH, Lui PY, Ng MH, Siu KL, Au SW, Jin DY. 2011. The double-stranded RNA-binding protein PACT functions as a cellular activator of RIG-I to facilitate innate antiviral response. Cell Host Microbe 9:299–309. doi: 10.1016/j.chom.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 38.Cardenas WB, Loo YM, Gale M Jr, Hartman AL, Kimberlin CR, Martinez-Sobrido L, Saphire EO, Basler CF. 2006. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J Virol 80:5168–5178. doi: 10.1128/JVI.02199-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li S, Min JY, Krug RM, Sen GC. 2006. Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology 349:13–21. doi: 10.1016/j.virol.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 40.Kew C, Lui PY, Chan CP, Liu X, Au SW, Mohr I, Jin DY, Kok KH. 2013. Suppression of PACT-induced type I interferon production by herpes simplex virus 1 Us11 protein. J Virol 87:13141–13149. doi: 10.1128/JVI.02564-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luthra P, Ramanan P, Mire CE, Weisend C, Tsuda Y, Yen B, Liu G, Leung DW, Geisbert TW, Ebihara H, Amarasinghe GK, Basler CF. 2013. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe 14:74–84. doi: 10.1016/j.chom.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siu KL, Yeung ML, Kok KH, Yuen KS, Kew C, Lui PY, Chan CP, Tse H, Woo PC, Yuen KY, Jin DY. 2014. Middle East respiratory syndrome coronavirus 4a protein is a double-stranded RNA-binding protein that suppresses PACT-induced activation of RIG-I and MDA5 in the innate antiviral response. J Virol 88:4866–4876. doi: 10.1128/JVI.03649-13. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Lehtonen A, Matikainen S, Julkunen I. 1997. Interferons up-regulate STAT1, STAT2, and IRF family transcription factor gene expression in human peripheral blood mononuclear cells and macrophages. J Immunol 159:794–803. [PubMed] [Google Scholar]
- 44.Block H, Stadtmann A, Riad D, Rossaint J, Sohlbach C, Germena G, Wu D, Simon SI, Ley K, Zarbock A. 2016. Gnb isoforms control a signaling pathway comprising Rac1, Plcbeta2, and Plcbeta3 leading to LFA-1 activation and neutrophil arrest in vivo. Blood 127:314–324. doi: 10.1182/blood-2015-06-651034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee DJ, Cox D, Li J, Greenberg S. 2000. Rac1 and Cdc42 are required for phagocytosis, but not NF-kappaB-dependent gene expression, in macrophages challenged with Pseudomonas aeruginosa. J Biol Chem 275:141–146. doi: 10.1074/jbc.275.1.141. [DOI] [PubMed] [Google Scholar]
- 46.Massol P, Montcourrier P, Guillemot JC, Chavrier P. 1998. Fc receptor-mediated phagocytosis requires CDC42 and Rac1. EMBO J 17:6219–6229. doi: 10.1093/emboj/17.21.6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoppe AD, Swanson JA. 2004. Cdc42, Rac1, and Rac2 display distinct patterns of activation during phagocytosis. Mol Biol Cell 15:3509–3519. doi: 10.1091/mbc.e03-11-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. 1999. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature 398:252–256. doi: 10.1038/18465. [DOI] [PubMed] [Google Scholar]
- 49.Schieven GL. 2005. The biology of p38 kinase: a central role in inflammation. Curr Top Med Chem 5:921–928. doi: 10.2174/1568026054985902. [DOI] [PubMed] [Google Scholar]
- 50.Thomas JD, Johannes GJ. 2007. Identification of mRNAs that continue to associate with polysomes during hypoxia. RNA 13:1116–1131. doi: 10.1261/rna.534807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dominici S, Schiavano GF, Magnani M, Buondelmonte C, Celeste AG, Brandi G. 2012. Involvement of Stat1 in the phagocytosis of M. avium. Clin Dev Immunol 2012:652683. doi: 10.1155/2012/652683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xie B, Zhao J, Kitagawa M, Durbin J, Madri JA, Guan JL, Fu XY. 2001. Focal adhesion kinase activates Stat1 in integrin-mediated cell migration and adhesion. J Biol Chem 276:19512–19523. doi: 10.1074/jbc.M009063200. [DOI] [PubMed] [Google Scholar]
- 53.Lehmann AK, Sornes S, Halstensen A. 2000. Phagocytosis: measurement by flow cytometry. J Immunol Methods 243:229–242. doi: 10.1016/S0022-1759(00)00237-4. [DOI] [PubMed] [Google Scholar]
- 54.Gille C, Spring B, Tewes L, Poets CF, Orlikowsky T. 2006. A new method to quantify phagocytosis and intracellular degradation using green fluorescent protein-labeled Escherichia coli: comparison of cord blood macrophages and peripheral blood macrophages of healthy adults. Cytometry A 69:152–154. doi: 10.1002/cyto.a.20222. [DOI] [PubMed] [Google Scholar]
- 55.Neuder LE, Keener JM, Eckert RE, Trujillo JC, Jones SL. 2009. Role of p38 MAPK in LPS induced pro-inflammatory cytokine and chemokine gene expression in equine leukocytes. Vet Immunol Immunopathol 129:192–199. doi: 10.1016/j.vetimm.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 56.Taniguchi F, Harada T, Miyakoda H, Iwabe T, Deura I, Tagashira Y, Miyamoto A, Watanabe A, Suou K, Uegaki T, Terakawa N. 2009. TAK1 activation for cytokine synthesis and proliferation of endometriotic cells. Mol Cell Endocrinol 307:196–204. doi: 10.1016/j.mce.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 57.Cuenda A, Rousseau S. 2007. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta 1773:1358–1375. doi: 10.1016/j.bbamcr.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 58.Bradley JR. 2008. TNF-mediated inflammatory disease. J Pathol 214:149–160. doi: 10.1002/path.2287. [DOI] [PubMed] [Google Scholar]
- 59.Lopez-Castejon G, Brough D. 2011. Understanding the mechanism of IL-1beta secretion. Cytokine Growth Factor Rev 22:189–195. doi: 10.1016/j.cytogfr.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shah A, Rashid F, Awan HM, Hu S, Wang X, Chen L, Shan G. 2017. The DEAD-box RNA helicase DDX3 interacts with m(6)A RNA demethylase ALKBH5. Stem Cells Int 2017:8596135. doi: 10.1155/2017/8596135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ajuebor MN, Flower RJ, Hannon R, Christie M, Bowers K, Verity A, Perretti M. 1998. Endogenous monocyte chemoattractant protein-1 recruits monocytes in the zymosan peritonitis model. J Leukoc Biol 63:108–116. doi: 10.1002/jlb.63.1.108. [DOI] [PubMed] [Google Scholar]
- 62.Keophiphath M, Rouault C, Divoux A, Clement K, Lacasa D. 2010. CCL5 promotes macrophage recruitment and survival in human adipose tissue. Arterioscler Thromb Vasc Biol 30:39–45. doi: 10.1161/ATVBAHA.109.197442. [DOI] [PubMed] [Google Scholar]
- 63.Petrovic-Djergovic D, Popovic M, Chittiprol S, Cortado H, Ransom RF, Partida SS. 2015. CXCL10 induces the recruitment of monocyte-derived macrophages into kidney, which aggravate puromycin aminonucleoside nephrosis. Clin Exp Immunol 180:305–315. doi: 10.1111/cei.12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wittamer V, Bertrand JY, Gutschow PW, Traver D. 2011. Characterization of the mononuclear phagocyte system in zebrafish. Blood 117:7126–7135. doi: 10.1182/blood-2010-11-321448. [DOI] [PubMed] [Google Scholar]
- 65.Mathias JR, Dodd ME, Walters KB, Yoo SK, Ranheim EA, Huttenlocher A. 2009. Characterization of zebrafish larval inflammatory macrophages. Dev Comp Immunol 33:1212–1217. doi: 10.1016/j.dci.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gray C, Loynes CA, Whyte MK, Crossman DC, Renshaw SA, Chico TJ. 2011. Simultaneous intravital imaging of macrophage and neutrophil behaviour during inflammation using a novel transgenic zebrafish. Thromb Haemost 105:811–819. doi: 10.1160/TH10-08-0525. [DOI] [PubMed] [Google Scholar]
- 67.Corey DR, Abrams JM. 2001. Morpholino antisense oligonucleotides: tools for investigating vertebrate development. Genome Biol 2:REVIEWS1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Draper BW, Morcos PA, Kimmel CB. 2001. Inhibition of zebrafish fgf8 pre-mRNA splicing with morpholino oligos: a quantifiable method for gene knockdown. Genesis 30:154–156. doi: 10.1002/gene.1053. [DOI] [PubMed] [Google Scholar]
- 69.Deciphering Developmental Disorders Study. 2015. Large-scale discovery of novel genetic causes of developmental disorders. Nature 519:223–228. doi: 10.1038/nature14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guenther UP, Weinberg DE, Zubradt MM, Tedeschi FA, Stawicki BN, Zagore LL, Brar GA, Licatalosi DD, Bartel DP, Weissman JS, Jankowsky E. 2018. The helicase Ded1p controls use of near-cognate translation initiation codons in 5′ UTRs. Nature 559:130–134. doi: 10.1038/s41586-018-0258-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang X, Wang R, Luo M, Li C, Wang HX, Huan CC, Qu YR, Liao Y, Mao X. 2017. (DEAD)-box RNA helicase 3 modulates NF-kappaB signal pathway by controlling the phosphorylation of PP2A-C subunit. Oncotarget 8:33197–33213. doi: 10.18632/oncotarget.16593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen ZJ, Bhoj V, Seth RB. 2006. Ubiquitin, TAK1 and IKK: is there a connection? Cell Death Differ 13:687–692. doi: 10.1038/sj.cdd.4401869. [DOI] [PubMed] [Google Scholar]
- 73.Yang F, Tang E, Guan K, Wang CY. 2003. IKK beta plays an essential role in the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. J Immunol 170:5630–5635. doi: 10.4049/jimmunol.170.11.5630. [DOI] [PubMed] [Google Scholar]
- 74.Tantravedi S, Vesuna F, Winnard PT Jr, Van Voss MRH, Van Diest PJ, Raman V. 2017. Role of DDX3 in the pathogenesis of inflammatory bowel disease. Oncotarget 8:115280–115289. doi: 10.18632/oncotarget.23323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Park YM, Febbraio M, Silverstein RL. 2009. CD36 modulates migration of mouse and human macrophages in response to oxidized LDL and may contribute to macrophage trapping in the arterial intima. J Clin Invest 119:136–145. doi: 10.1172/JCI35535. [DOI] [PMC free article] [PubMed] [Google Scholar]