

Welander distal myopathy (WDM) is a muscle dystrophy characterized by adult-onset distal muscle weakness, prevalently impacting the distal long extensors of the hands and feet. WDM is an autosomal dominant disorder caused by a missense mutation (c.1362G>A; p.E384K) in the TIA1 (T-cell intracellular antigen 1) gene, which encodes an RNA-binding protein basically required for the posttranscriptional regulation of RNAs.
KEYWORDS: TIA1, Welander distal myopathy, autophagy, gene expression, stress granules
ABSTRACT
Welander distal myopathy (WDM) is a muscle dystrophy characterized by adult-onset distal muscle weakness, prevalently impacting the distal long extensors of the hands and feet. WDM is an autosomal dominant disorder caused by a missense mutation (c.1362G>A; p.E384K) in the TIA1 (T-cell intracellular antigen 1) gene, which encodes an RNA-binding protein basically required for the posttranscriptional regulation of RNAs. We have developed a heterologous cell model of WDM to study the molecular and cellular events associated with mutated TIA1 expression. Specifically, we analyzed how this mutation affects three regulatory functions mediated by TIA1: (i) control of alternative SMN2 (survival motor neuron 2) splicing; (ii) formation, assembly, and disassembly of stress granules; and (iii) mitochondrial dynamics and its consequences for mitophagy, autophagy, and apoptosis. Our results show that whereas WDM-associated TIA1 expression had only a mild effect on SMN2 splicing, it led to suboptimal adaptation to environmental stress, with exacerbated stress granule formation that was accompanied by mitochondrial dysfunction and autophagy. Overall, our observations indicate that some aspects of the cell phenotype seen in muscle of patients with WDM can be recapitulated by ectopic expression of WDM-TIA1 in embryonic kidney cells, highlighting the potential of this model to investigate the pathogenesis of this degenerative disease and possible therapeutics.
INTRODUCTION
Welander distal myopathy (WDM) is an autosomal dominant muscle dystrophy with late-adult onset (40 to 60 years of age), which starts by initial weakness of index finger extensors that progresses to extension weakness in the other fingers, ultimately involving all small hand muscles and those of the lower legs (1, 2). Skeletal muscle biopsy specimens commonly show myopathic alterations and rimmed vacuoles; however, cardiac muscle involvement has not been observed (3, 4). WDM is mainly located in a geographical area around the Baltic Sea. The estimated incidence in Sweden and Finland is 1/10,000 (1–4). The clinical progression of WDM is benign, and life expectancy is normal, although fine motor hand skills are usually lost (4). Rare homozygous individuals show an earlier onset and proximal muscle involvement, with faster progression, and they become wheelchair bound by the age of 50 years (5, 6).
WDM is caused by a missense nucleotide change (c.1362G>A; p.E384K) in T-cell intracellular antigen 1 (TIA1) (2p13) (5–8), which encodes an RNA-binding protein that regulates/modulates many regulatory aspects of gene expression (9–11). Human TIA1 has 13 exons and is transcribed into two major mRNA isoforms (identified as isoforms a and b) (12), which are generated by the inclusion and skipping, respectively, of exon 5 (13). TIA1 is ubiquitously expressed in human cells and tissues, suggesting that its role is cell context dependent (12–14). Both protein isoforms consist of three RNA recognition motifs (RRMs) and a C-terminal glutamine/asparagine-rich, low-complexity domain (Q/N-rich domain), which contains the mutation (12, 13). TIA1 plays a key role in regulating the metabolism and fate of cellular RNAs in both the nucleus and cytoplasm (11, 15–20), involving transcription, alternative splicing, localization, stability/turnover, and translation (11, 15–20). Indeed, together with TIA1-like/TIA1-related protein (TIAL1/TIAR) (21, 22), TIA1 has direct regulatory control of over 10 to 20% of the human transcriptome (19, 20). Consequently, TIA1 targets the cellular and molecular biology of RNAs and proteins, determining their fate in ribonucleoprotein (RNP) complexes (15–20). TIA1 impacts genes involved in main biological programs such as embryonic development, cell survival and death, differentiation, stress responses, inflammation, virus infection, and oncogenesis, all of which are important in physiopathology (11, 15–20, 23–25). Targeted ablation of TIA1 results in embryonic lethality (26, 27), demonstrating its importance in early development, but the penetrance varies between TIA proteins: around 50% in mice lacking TIA1 (26, 27) and close to 100% in TIAR knockout mice (28). Moreover, nullizygotes of TIA1 are fully fertile, whereas TIAR knockout mice are sterile (26, 27), indicating that weighted TIA1/TIAR expression is required for early embryogenesis. Mice lacking TIA1 and TIAR die around embryonic days 7 to 8 (26, 29).
Two established regulatory processes directly mediated by TIA1 have been linked to WDM: the control of alternative splicing of survival motor neuron 2 (SMN2) exon 7 (8) and the formation and dynamics of TIA1-dependent stress granules (SGs) (7). TIA1 functions as a splicing auxiliary factor by improving the splicing of pre-mRNAs through facilitating the selection of constitutive and atypical 5′ splice sites (SS) on introns (16, 17). Mechanistically, TIA1 binds to U-rich sequences close to 5′ SS, enhancing the recruitment of U1 snRNP by the protein-protein interaction between the Q/N-rich C-terminal domain of TIA1 and the U1-C component of U1 snRNP (18). Furthermore, TIA1 is a canonical protein in SGs under conditions of environmental stress (15, 30–34). SGs are cytoplasmic foci that assemble subsets of messenger RNP (mRNP) complexes in a translationally repressed state (15, 30–34). SGs have been described as highly dynamic liquid-like droplets (35–37). They are enriched in prion-like proteins containing disordered regions, which aggregate spontaneously in vitro (35–37). Accordingly, SGs can modify the cell information flow, as they influence the adaptive reprogramming of the transcriptome and proteome. Thus, SGs have a dual function in adaption and protection in eukaryotic cells by responding to environmental challenges (38–42).
The pathological events associated with WDM were originally identified from a small number of muscle biopsy specimens of patients, and an in-depth study of these events has been limited by the difficulty in accessing patient samples. For this reason, the development of cellular and/or animal models expressing the mutated version of WDM-associated TIA1 (WDM-TIA1) would allow a better characterization of the cellular and molecular dysfunctions associated with this pathology; however, no cellular and/or animal models have been reported. Thus, we sought to develop a cell model to address this issue using novel genetically engineered isogenic human cell lines. This would allow us to investigate and characterize key regulatory nodes in WDM, which might open new horizons for therapeutic interventions.
We describe here a cell model overexpressing the pathogenic missense variant (c.1362G>A; p.E384K [GenBank accession number NM_022173.2]) of TIA1 (a and b isoforms). Analysis of the heterologous cell lines reveals a strong link between dysfunctions in SG dynamics under environmental stress and suboptimal responses in mitochondrial dynamics and autophagy.
RESULTS
Human TIA1 gene isoforms and the single mutation associated with Welander distal myopathy.
The human TIA1 gene is located on chromosome 2p13 (12) and comprises 13 exons encoding the two major mRNA/protein isoforms, TIA1a and TIA1b (Fig. 1A). Exons 1 to 4, 5 to 8, and 9 to 11 encode the RRM1, -2, and -3 domains, respectively, whereas exons 12 and 13 encode the Q/N-rich domain (Fig. 1A). The inclusion or skipping of exon 5 generates TIA1a or TIA1b, respectively (12, 14) (Fig. 1A). The production of TIA1 isoforms is controlled in a tissue/cell-specific manner (14) and involves posttranscriptional regulatory events such as alternative splicing and/or translational control (14, 43).
FIG 1.

Welander distal myopathy is associated with a single mutation in the human TIA1 gene. (A) Schematic representation of human TIA1, major mRNA variants, and protein isoforms. Also shown is the organization of exons and introns and major mRNA and protein isoforms generated by alternative splicing of exon 5. TIA1 protein isoforms include three RNA recognition motifs (RRMs) and a low-complexity domain rich in asparagine and glutamine residues. The distinct peptide between protein isoforms a and b of human TIA1 as well as the WDM-related peptide located at the C terminus in exon 13 are shown. (B) The WDM-associated mutation is located in TIA1 exon 13. Artificial chromatograms illustrate the WDM mutation: normal control (top), heterozygous (middle), and homozygous (bottom) c.1362G>A (green); p.E384K (green) (7, 8). (C) Multialignment analysis of TIA1 and TIAL1/TIAR orthologs. The blue bar shows the highly conserved p.E384 residue of TIA1, which is absent in TIAL1/TIAR orthologs (7, 8). (D) Schematic representation of plasmid constructs used to generate FT293 cell lines expressing wild-type (WT) and mutated (WDM) TIA1 isoforms (and TIAR and HuR proteins of interest) using the Flp-In T-Rex system. FRT, Flp recombination target.
The WDM-associated TIA1 mutation was identified independently by two research groups through screening of muscle biopsy specimens (7, 8), finding a guanine-to-adenine substitution in exon 13, resulting in the exchange of glutamic acid for lysine (Fig. 1B). Whereas the glutamic acid residue is highly conserved in tetrapods, it is not conserved in TIAL1/TIAR (Fig. 1C).
We constructed wild-type (WT) and mutated (WDM) versions of both TIA1a and TIA1b by directed mutagenesis and used the Flp-In T-Rex system to generate the corresponding inducible and isogenic HEK293 cell lines coexpressing a green fluorescent protein (GFP) fusion at the 5′ terminus: TIA1aWT, TIA1bWT, TIA1a(E384K)WDM (here called TIA1aWDM), and TIA1b(E373K)WDM (here called TIA1bWDM). An empty vector (GFP) was used as a negative control, and a vector coexpressing GFP and the RNA-binding protein HuR (human R antigen) was used in some experiments as an additional control (Fig. 1D) (24, 25). We used these cell lines to investigate the three specific regulatory aspects of TIA1 in WDM, namely, regulation of exon 7 splicing in SMN2; the formation, assembly, and/or disassembly of SGs; and effects on mitochondrial dynamics and function (e.g., autophagy, mitophagy, and apoptosis).
Expression of WDM-associated TIA1 has a modest effect on SMN2 exon 7 skipping.
A previous work reported that the TIA1 mutation in WDM had an effect on alternative exon 7 splicing of SMN2 in patients’ muscle (i.e., the levels of SMN2 transcripts without exon 7 were moderately increased in WDM patients versus controls) (8). We thus tested this splicing phenomenon in the different cell models using SMN1- and SMN2-derived minigenes (44). As an additional control for these experiments, we used a neurofibromin 1 (NF1) minigene, which is targeted by TIA1 at exon 23a (Fig. 2A) (45). Transfection of SMN2 into TIA1aWT, TIA1bWT, TIA1aWDM, and TIA1bWDM FT293 cells (Fig. 2B) showed that both WDM- and WT-TIA1 (a and b) isoforms promoted the inclusion of exon 7, with only a slight difference (5 to 10%) in this capacity between mutated and control proteins, as evaluated by reverse transcription-elongation PCR (RT-PCR) (Fig. 2B). To further document the function of WDM-TIA1, we performed transient cotransfection of WT-TIA1b and WDM-TIA1b isoforms with the SMN1 and SMN2 minigenes into HEK293, SH-SY5Y (neurons), and C2C12 (myoblasts/myotubes) cells. The observations were similar to those in FT293 cells, showing only small differences (5 to 10%) in SMN2 exon 7 inclusion between WDM- and WT-TIA1b. In contrast, these modest effects were not observed with the NF1 minigene, perhaps because exon 23a skipping was very limited in the cellular types analyzed (data not shown). As a control for transfection, the relative expression levels of each GFP-tagged fusion protein were evaluated by Western blotting (Fig. 2B to E) using the appropriate antibodies.
FIG 2.

WDM-TIA1 mutation moderately promotes SMN2 exon 7 skipping. (A) Schematic representation of human SMN2 and NF1 minigenes and location of exons (colored boxes), introns (solid lines), and splice variants (dashed lines). The TIA1-dependent splicing event is illustrated, indicating the U-rich sequence where TIA is bound to improve U1 snRNP recruitment on 5′ splice sites by facilitating the inclusion of SMN2 exon 7 and NF1 exon 23a. (B) Analysis of alternative splicing of SMN1/SMN2 and NF1 in FT293 cells. GFP (control)- and TIA1-expressing FT293 cells were transiently transfected with SMN1/SMN2 and NF1 minigenes for 24 h. Western blots of GFP-tagged fusion proteins upon tetracycline treatment for 24 h are shown. Analysis of alternative splicing was carried out using RT-PCR assays as described previously (25, 44, 45). (C to E) Analysis of ectopic SMN1/SMN2 and NF1 alternative splicing in HEK293 (C), SH-SY5Y (D), and C2C12 (E) cells cotransfected with the corresponding minigenes and expression plasmids. The relative levels of GFP-tagged proteins and endogenous α-tubulin (TUBA) levels were analyzed by Western blotting, and the splicing patterns from reporter chimeric minigenes were determined as described above for panel B. In all cases, molecular weight markers for protein and DNA and the identities of protein bands and amplified DNA fragments are shown. (F) GST recombinant fusion proteins used for GST pulldown analysis were expressed in and purified from E. coli, fractionated on 12% SDS-PAGE gels, and stained with Coomassie brilliant blue reagent. The positions of molecular weight (MW) markers for protein and GST, GST-TIA1bWT, and GST-TIA1bWDM recombinant proteins are indicated. (G) Precipitation of the U1-C component from U1 snRNP by WT and WDM GST-TIA1b proteins. GST pulldown assays were carried out after the addition of either GST alone, GST-TIA1bWT, or GST-TIA1WDM (top) to either the U1-C(6×His) fusion protein or [35S]methionine-cysteine-labeled U1-C. Aliquots of recombinant fusion/[35S]methionine-cysteine-labeled U1-C proteins (25%) prior to pulldown assays, the corresponding supernatants (SN) (10%), and pulled-down proteins (P) were analyzed by Western blotting and autoradiography. The positions of U1-C protein versions and molecular weight markers are indicated.
TIA1 functions as an auxiliary splicing factor because it directly interacts with the U1-C component of U1 snRNP to promote its recruitment at atypical 5′ SS (18). To reinforce our findings, we performed pulldown assays using glutathione S-transferase (GST)-tagged versions of the proteins to evaluate whether WT- and WDM-TIA1 had different capacities to interact with U1-C (Fig. 2F). As shown in Fig. 2G, both WT and mutant TIA1b proteins had similar abilities to interact with 35S-labeled and also His-tagged U1-C recombinant proteins (Fig. 2G). Overall, these results strongly suggest that the WDM mutation has a moderate/limited effect on SMN2 exon 7 skipping/splicing.
Expression of the WDM-TIA1 mutation alters the dynamics of stress granule assembly and disassembly.
It has been reported that WDM caused by mutated TIA1 occurs through a dominant pathomechanism likely involving altered SG dynamics (7). We thus tested the capacity of the FT293 cell lines to produce spontaneous and triggered SGs in the absence and presence, respectively, of environmental stress. Results showed that TIA1aWDM and TIA1bWDM cells had a moderate capacity to generate spontaneous granules (aggregates with GFP-TIA1 expression) compared to the equivalent TIA1aWT and TIA1bWT cells (data not shown). This was particularly striking for TIA1bWDM, which showed a time-dependent increase in SG formation (data not shown). We extended this analysis by subjecting cell lines to sodium arsenite treatment for 0.5 to 1 h and monitoring SG formation for a further 1 to 3 h. Results showed an alteration in the dynamics of SG formation, assembly, and/or disassembly (i.e., number and size of SGs) between WT- and WDM-expressing cells during arsenite treatment. The size/number of SGs was larger in TIA1aWDM and TIA1bWDM cells than in TIA1aWT and TIA1bWT cells, with considerably more granules being formed and with a larger size (≤1 μm for S [small] granules versus ≥2 μm for M/L [medium/large] granules) in WDM-TIA1-expressing cells (Fig. 3A to C). Furthermore, the dynamics of assembly and/or disassembly was altered, since WDM-TIA1-expressing SGs were more stable over 3 h after removal of sodium arsenite (Fig. 3B and C). However, no major differences were observed in SG formation between isoforms a and b. A similar, albeit less striking, result was found when the FT293 cell lines were subjected to heat shock for 1 h at 45°C and allowed to recover for an additional 3 h (data not shown). Collectively, these results demonstrate the dynamics of SG assembly and/or disassembly during environmental stress by the expression of the WDM-TIA1 mutation.
FIG 3.

Altered dynamics of stress granule formation and assembly/disassembly in TIA1WDM cells under oxidative stress. (A) Time course of stress granule (SG) formation in NaAsO2 (arsenite)-treated FT293 cells. Shown are fluorescence images of GFP-TIA1a/bWT (top panels) or GFP-TIA1a/bWDM (bottom panels) FT293 cells (green). (B) Fluorescence images of GFP-TIA1a/bWT (left panels) or GFP-TIA1a/bWDM (right panels) cells showing the differential dynamics of TIA1WT and TIA1WDM SGs in arsenite-treated FT293 cells for 60 min followed by recovery for 1 to 3 h. Nuclei were stained with To-Pro3 (blue). Bars, 10 μm. (C) Estimation of the number and size of SGs in TIA1a/bWT- and TIA1a/bWDM-expressing FT293 cells. S and M/L indicate relative sizes (S, ≤1 μm; M/L, ≥2 μm) of SGs, expressed as a percentage. Data represent means ± SEM (n = 60 to 180 cells under each condition) (*, P < 0.05; **, P < 0.01). h.p.a., hours post-arsenite addition.
Zinc enhances the formation and maintenance of stress granules in WDM-TIA1-expressing cells.
Because divalent zinc has been reported to be important for TIA1 self-multimerization and recruitment into SGs, and also for phase separation (40), we questioned whether it was also necessary for the genesis and maintenance of SGs in WDM-TIA1-expressing FT293 cells upon environmental stress induced by arsenite. Sodium arsenite treatment induced an increase in SG frequency in TIA1aWDM- versus TIA1aWT-expressing cells, as indicated by the greater presence of green cytoplasmic foci 30 and 60 min after stimulation (Fig. 4, top and middle panels). Moreover, in TIA1aWT and TIA1aWDM cells pretreated with the zinc chelator TPEN [N,N,N′,N′-tetrakis(2-pyridylmethyl)ethane-1,2-diamine] before arsenite treatment, the number of SGs was notably lower in TIA1aWT cells than in TIA1aWDM cells. In contrast, TIAR-expressing FT293 cells (used as an additional control) were relatively insensitive to zinc chelation under the same conditions (Fig. 4, compare top and bottom panels). Accordingly, the effects of zinc chelation on SG formation in FT293 cells appear to be specific to TIA1aWT and TIA1aWDM cells, with a lesser effect on TIAR-expressing FT293 cells, in agreement with previous observations (40). In addition, whereas the sole addition of exogenous zinc (or calcium chloride and dimethyl sulfoxide [DMSO], used as selective controls) was insufficient to promote the formation of SGs in TIA1aWT and TIA1aWDM cells, it enhanced the effects of arsenite for SG formation in TIA1aWDM cells (Fig. 4, middle panels). Interestingly, sequestration/chelation of zinc by TPEN before arsenite treatment inhibited the recruitment of WT- and WDM-TIA1a into stress granules (Fig. 4A). Taken together, our observations support a specific role for the preferential interaction of WDM-TIA1a with zinc during SG assembly.
FIG 4.

Dynamics of stress granules in TIA1WT and TIA1WDM cells during arsenite treatment is regulated by divalent zinc. Shown are fluorescence images of GFP-TIA1aWT (top panels), GFP-TIA1aWDM (middle panels), and GFP-TIARb (bottom panels) FT293 cells (green) that were untreated or treated with DMSO (vehicle), NaAsO2 (0.5 mM), ZnCl2 (1 μM), TPEN (20 μM), and CaCl2 (1 μM), or different combinations of these reagents, as indicated. Nuclei were stained with To-Pro3 (blue). Bars, 10 μm.
Enhanced stress granule formation is prevalent with the E384K mutation.
To better characterize the functional relevance of the p.E384/373K mutated residue, we created mutants with mimetic, antagonist, and neutral potentials by introducing amino acids with positive (Arg), negative (Asp), and no (Gly) charge at the same position of the WDM-TIA1 mutation, p.E373R, p.E373D, and p.E373G, respectively (Fig. 5A). From the corresponding plasmid constructs, we generated the inducible cell lines GFP-TIA1b-E373R, GFP-TIA1b-E373D, and GFP-TIA1b-E373G FT293 (Fig. 5B), and their capacity to produce SGs was compared with that of TIA1bWT and TIA1bWDM cells in the absence and presence of sodium arsenite (Fig. 5C). Results showed that the capacity of the p.E373R, p.E373D, and p.E373G TIA1b mutations to generate SGs, as well as assembly/disassembly dynamics, was progressively reproduced compared with the observations in TIA1bE373KWDM cells (Fig. 5C and D). The observations suggest that the expression of p.E373K>p.E373R>p.E373G>p.E373D=p.E373E leads to higher average numbers and sizes of SGs under oxidative stress. Thus, we conclude that the functional relevance of the change of the amino acid charge from negative to positive seems to be important per se for the development of SGs, which suggests a priori an altered function associated with the mutant lysine residue at the C terminus of TIA1 for the deleterious effects and cellular consequences linked to WDM. These observations are in agreement with recent findings reported while our paper was under revision (46).
FIG 5.

The dynamics of WDM-TIA1-dependent stress granules is prevalently linked to the p.E373K mutation. (A) Schematic representation of plasmid constructs expressing additional mutated TIA1 proteins by replacement of glutamic acid with glycine (p.E373G), aspartic acid (p.E373D), and arginine (p.E373R). (B) Western blotting of GFP-TIA1bE373WT, GFP-TIA1bE373KWDM, GFP-TIA1bE373G, GFP-TIA1bE373D, and GFP-TIA1bE373R FT293 cells using anti-GFP and anti-α-tubulin (TUBA) antibodies. Molecular weight markers for proteins and the identities of protein bands are shown. (C) Fluorescence images from the FT293 cells described in panel B (green) expressing the indicated fusion proteins treated with sodium arsenite (0.5 mM) for 1 h and allowed to recover for 1 to 3 h. Nuclei were stained with To-Pro3 (blue). Bars, 10 μm. (D) Estimation of the number and size of SGs was carried out as described in the legend of Fig. 3C.
Mitochondrial dynamics is altered in WDM-TIA1-expressing FT293 cells.
Our results showing an increase in SG formation in TIA1WDM cells in the presence of zinc are particularly relevant, as zinc is associated with a number of redox signaling pathways and acts as a sensor of the cellular redox state (40, 47). Major alterations in the cellular redox state must be quickly interpreted by signaling systems/pathways to execute adaptive cellular responses and maintain homeostasis (40, 47). Mitochondria are essential for these functions and strongly contribute to the cellular redox state (47). We recently demonstrated that short-term TIA1b overexpression in FT293 cells drives mitochondrial remodeling, involving mitochondrial clustering, fission, and, ultimately, dysfunction (25). We thus evaluated mitochondrial dynamics in uninduced FT293 cells and WDM- and WT-TIA1-expressing FT293 cells. Results showed that mitochondrial populations in TIA1WDM cells formed clusters close to the nucleus (Fig. 6A), which was reminiscent of the mitochondrial distribution that we described previously in WT TIA1b-expressing FT293 cells (25). A detailed analysis using transmission electron microscopy confirmed mitochondrial clustering in WT-TIA1b cells and also revealed mitochondrial fission and mitochondrial swelling, with an abnormal distribution of cristae (Fig. 6B). These findings led us to evaluate mitochondrial membrane potential (MMP) and redox levels using specific probes and flow cytometry analysis (Fig. 7). Results indicated that both TIA1bWT and TIA1bWDM cells presented a significant reduction in MMP compared with control (GFP) cells and also an increase in redox levels (Fig. 7A and B). These results suggest that the expression per se of the WT- and WDM-TIA1 forms could favor mitochondrial dysfunction and elevated redox signaling, possibly enhancing its ability to regulate SG dynamics by modulating/regulating mitochondrial physiology.
FIG 6.

Mitochondrial spatial distribution and morphology in TIA1WT and TIA1WDM cells. (A) Mitochondrial spatial distribution in uninduced and TIA1a/bWT- and TIA1a/bWDM-expressing FT293 cells visualized by confocal microscopy. Shown are GFP fluorescence (green) and immunolabeling for mitochondrial Tom20 (red). Nuclei were stained with To-Pro3 (blue). The outlined box in the merged image is enlarged in the zoom panel. The detail is a 2× zoom image. Bars, 10 μm. (B) Details of mitochondrial morphology and crista architecture in the indicated cells visualized by transmission electron microscopy (25). Arrowheads on images indicate mitochondrial fragmentation and swelling. Bars, 2 μm (left images) and 500 nm (right images).
FIG 7.
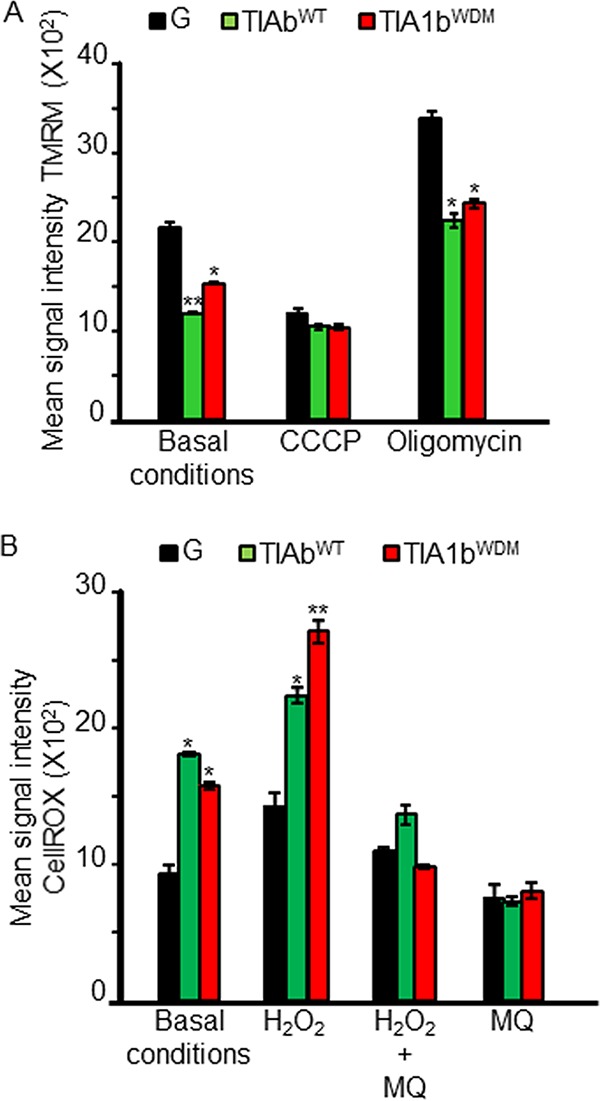
Low levels of mitochondrial membrane potential and high rates of reactive oxygen species production in TIA1bWT and TIA1bWDM FT293 cells. (A) Estimation of mitochondrial membrane potential with 100 nM tetramethylrhodamine methylester (TMRM) and flow cytometry. Values represent means ± SEM (n = 3) (*, P < 0.05; **, P < 0.01). (B) Estimation of reactive oxygen species (ROS) production with 5 μM CellROX deep red and flow cytometry. Values represent means ± SEM (n = 3) (*, P < 0.05; **, P < 0.01). In panels A and B, the final concentrations of oligomycin and CCCP were 10 μM, and the final concentrations of H2O2 and MitoQ (MQ) were 1 mM.
Expression of WDM-TIA1 increases autophagy.
Autophagy is a complex cellular program to be activated in situations of compromised cell survival (25). Next, we decided to investigate whether the ectopic expression of WT- or WDM-TIA1(a/b) enhances autophagy. Autophagic flux can be monitored by evaluating the fusion between autophagosomes and lysosomes through conversion of microtubule-associated protein light chain 3-I (LC3-I) to phosphatidylethanolamine (PE)-conjugated LC3-II, a specific marker of autophagic vesicles, using the dual-fluorescence probe GFP-LC3B-red fluorescent protein (RFP) (25). As shown in Fig. 8A, the improved expression of LC3B-II was concomitant with the formation of autophagosomes (yellow dots), as indicated by the colocalization of autophagosomes (green dots) and lysosomes (red dots) (Fig. 8A). Autophagy flux is illustrated by the fusion between autophagosomes and lysosomes to produce autolysosomes (free red dots). Therefore, the formation of autophagosomes and/or autolysosomes was greater in TIA1WDM cells than in TIA1WT cells (Fig. 8B). We confirmed these results by Western blotting for the autophagic markers LC3-I/LC3-II, p62/SQSTM1, and EIF2AK2 (Fig. 8C). We additionally analyzed mitophagy and apoptosis connected to WDM-TIA1b expression, finding similar increases in mitophagy (Fig. 9A) and apoptosis (Fig. 9B) in TIA1bWT and TIA1bWDM cells. Finally, we performed immunocytochemistry to evaluate the presence of vacuoles containing LC3B and protein aggregates of the DNA/RNA-binding protein TDP-43. We found that the expression of TIA1WDM enhanced the cytoplasmic localization of these molecular markers of autophagy (Fig. 10A) and protein aggregates (Fig. 10B). Taken together, these data reveal that WDM-TIA1 drives the progressive development of autophagy, mitophagy, and apoptosis-linked phenotypes.
FIG 8.

Expression of WDM-TIA1 triggers autophagy. (A) Autophagic flux analysis. Cells were transiently transfected with the GFP-LC3B-RFP plasmid and visualized by confocal microscopy. Nuclei were stained with To-Pro3 (blue). Bars, 10 μm. (B) Histograms showing the ratio of autophagosomes to autolysosomes estimated, as yellow dots per cell and free red dots per cell, respectively (n = 3) (*, P < 0.05; **, P < 0.01) as previously described (25). (C) Analysis of molecular markers of autophagy by Western blotting using specific antibodies against the indicated proteins. G, GFP-expressing FT293 cells.
FIG 9.

Expression of WDM-TIA1 triggers mitophagy and apoptosis. (A) Mitophagic flux analysis using the mitochondrial Keima probe. Cells were transiently transfected with the mt-Keima-expressing plasmid and visualized at 3 days postinduction. The histogram shows normalized red/green mt-Keima signals (n = 3) (**, P < 0.01). Bars, 10 μm. a.u., arbitrary units. (B) Estimation of apoptosis rates in cells for 24 to 72 h measured by flow cytometry. Values represent means ± SEM (n = 3) (**, P < 0.01).
FIG 10.

Subcellular localization of LC3B and TDP-43 markers associated with vacuoles, and protein aggregates during autophagy. Fluorescence images of GFP-TIA1a/bWT and GFP-TIA1a/bWDM FT293 cells (green) were immunolabeled (red) for LC3B (A) and TDP-43 (B). Nuclei were stained with To-Pro3 (blue in merged image). Bars, 10 μm.
DISCUSSION
We generated cell models of WDM to investigate the impact of TIA1 protein variants on two known regulatory processes: exon 7 splicing of SMN2 and the dynamics of the formation and assembly/disassembly of TIA1-dependent SGs (7, 8). Whereas our results showed a moderate impact on the regulation of alternative SMN2 splicing by WDM-TIA1, in agreement with previous findings (8), a stronger phenotype was noted for SG formation after oxidative stress, which was, in part, dependent on divalent zinc.
TIA1 is a multifunctional protein with a well-established role as a splicing regulator (16, 17). TIA1 regulates 5 to 10% of total splicing events in the human genome (20) and selectively functions by facilitating U1 snRNP recruitment to typical and atypical 5′ SS located on introns (18). Our results suggest that WDM-TIA1, despite having a specific mutation in the Q/N-rich domain, functions similarly to WT-TIA1 to regulate SMN2 splicing in different cell types. However, we believe that the small change in SMN2 regulation and its interpretation should be carefully interpreted. Since WDM is a late-onset disease, the disease phenotype may reflect relatively subtle cellular changes. Thus, the results obtained from muscle biopsy specimens of WDM patients could potentially be more physiologically relevant than HEK293, SH-SY5Y, and C2C12 cells. In fact, cell type itself could play a relevant role in sensitivity to the TIA1 E384K mutation.
TIA1 is a key component of SGs (15, 30–34). Indeed, the expression of TIA1 promotes the formation of small SGs in the absence of stress cues (15, 30–34). Furthermore, overexpression of the C-terminal Q/N-rich domain of TIA1 alone inhibits SG formation in a dominant negative manner (15, 33). Our results show that inducible expression of WT- and WDM-TIA1 variants generates SGs in the absence and presence of stress stimuli, although this process was more rapid and efficient in cells expressing WDM-TIA1 variants under oxidative stress. In a similar vein, the dynamics of SG clearance after removal of the stress stimulus was slower in WDM-TIA1-expressing cells. A recent study proposed that divalent zinc is a physiological ligand of TIA1 (40), which is consistent with our results showing the dependency of TIA1-positive SGs on the ectopic and/or endogenous levels of zinc.
The zinc ion has many roles within the cellular environment, including catalytic (with phosphoproteases), structural (coordinates zinc fingers [ZFs]), regulatory (coenzyme in metalloproteases), and/or antioxidant (regulates redox levels) roles (47). Indeed, a recent report suggested that both zinc release and redox changes are necessary to trigger TIA1 multimerization into SGs (40), and oxidative stress triggers zinc release (40). Cells of aerobic organisms are persistently exposed to reactive oxygen species (ROS) as part of normal metabolism (47). Oxidative stress is caused by imbalanced redox states, owing to either excessive production of ROS or disturbances in antioxidant pathways to detoxify or scavenge them. Accordingly, perturbations of intracellular levels of redox have a major effect on cell functions because many cellular signaling pathways regulating cell division and stress response systems are highly sensitive to a redox variation (47). Maintained oxidative stress can lead to damage of cell membranes and other functional components, such as proteins, lipids, and DNA, and is associated in humans with age-related switches affecting cells, tissues, and organs (47). Mitochondria are the most relevant source of oxidative stress in cells, and afunctional mitochondria are major drivers of cellular aging and age-related disorders (48). Furthermore, mitochondrial dynamics change during cellular activities and can result in the overproduction of ROS, which drives oxidative DNA damage and contributes to genomic instability and a wide range of pathologies such as inflammation, neurodegeneration, atherosclerosis, cancer, and premature aging (48). We and others have recently reported that TIA1 plays a significant role in mitochondrial homeostasis by regulating mitochondrial dynamics (mitochondrial distribution and fission/fusion) through the regulation of gene expression of mitochondrion-associated genes, including OPA1, OMA1, MFN1/2, and MFF (25, 49). In this regard, our observations suggest that the expression of WDM-TIA1 has a negative impact on mitochondrial dynamics, in particular mitochondrial crista density and organization, mitochondrial membrane potential, and ROS production.
Paradoxically, oxidative stress might also suppress SG formation (50). For example, low concentrations of hydrogen peroxide oxidize TIA1 at Cys36 and subsequently suppress SG formation by inhibiting the interaction of TIA1 and its target mRNAs (50). Moreover, hydrogen peroxide attenuates arsenite/endoplasmic reticulum stress-induced SG formation, although a high concentration of hydrogen peroxide (1 mM) moderately induces SG formation. The Cys36 residue is situated inside the N-terminal RRMs of TIA1 (Fig. 1A), suggesting that oxidation and posterior disulfide bond formation at this site induce conformational changes that are essential for mRNA binding and/or SG assembly. This inhibition of SG nucleation by oxidative stress may underlie the cell death in neurons observed in some neurodegenerative diseases (50). These findings may be relevant for understanding why zinc improves TIA1 self-multimerization. Zinc ions interact with proteins through electrostatic interactions in a coordination-dependent manner, known as ZFs (51–53). This structural element has been described in several transcription factors. The C2H2 (Cys-Cys-His-His motif and variants thereof) zinc finger proteins are members of a superfamily of nucleic acid-binding proteins in eukaryotes (51–53). There are data revealing large diversities in both the structures of ZF domains and the mechanisms through which they can interact with molecular partners. Thus, there are both classical and nonclassical ZF domains that retain RNA-binding activity (51–53). Accordingly, it is possible that the positive charge of the lysine residue introduced by the WDM mutation in TIA1 somewhat reinforces the pool of positive charges of TIA1, improving and stabilizing the interaction between zinc, TIA1, and/or RNA-binding proteins and RNAs. This agrees with recent findings (which our observations confirm) showing that there is a molecular syntax based on specific contexts of amino acids (for example, tyrosine, arginine, and lysine) governing the driving forces for phase separation of prion-like RNA-binding proteins such as FUS (46) and TIA1 (our study).
Considering our present findings, and those of others, we propose a working model to explain WDM as a complex mitochondriopathy, linking the TIA1 mutation to aging-dependent mitochondrial dysfunction and the generation of transient and pathological SGs (Fig. 11). Human TIA1 isoforms are highly expressed in human skeletal muscle (14). Also, TIA1 and TIAR are downregulated in an age-dependent manner (54), and aging accelerates the loss of mitochondrial function and quality since mitochondrial biogenesis and turnover (mitophagy) are impaired during aging, which favors the development of oxidative stress (48). The quality of mitochondria in skeletal muscle is essential for maintaining metabolic homeostasis during adaptive stress responses; however, how this is controlled remains unclear. It was recently demonstrated that FUNDC1, a referee of mitophagy, plays a key role in regulating muscle mitochondrial quality and metabolic homeostasis (55). Furthermore, there is evidence showing a nonlinear dose-response characteristic of stressing agents promoting adaptive responses in a process known as hormesis, with transient increases in ROS levels acting as transducers of mitochondrion-induced hormesis, known as mitohormesis (56). These adaptive responses along with exposure to redox stressors can produce an increase in the amount of free zinc, which may be released from redox-sensitive metallothioneins (40, 56). Free zinc might more efficiently bind to WDM-TIA1, promoting aggregation and phase separation. The increase of the size of this membraneless particle would lead to the accumulation of untranslated mRNPs, RNPs with long noncoding RNAs, and other proteins containing intrinsically disordered regions, in addition to canonical and noncanonical RNA-binding proteins (57). These tangles establish a network of noncovalent protein-protein and protein-RNA interactions that constitute heterogeneous SGs, which can sequester additional proteins and RNAs and evolve toward pathological SGs (57) (Fig. 11). Posttranslational modification (PTM) of RNP components is a straightforward mechanism to modulate/regulate mRNA function during the stress response, because rapid and reversible PTMs allow adaptation to stress without de novo protein synthesis. Identification of the critical physiological targets of PTMs, and the mechanisms underlying their effects, will therefore be an important future goal (57). Thus, WDM-TIA1 variants could be a source of additional PTMs linked to mutated lysine, such as ubiquitination, methylation, and/or acetylation (an excellent molecular context with two aromatic [Y] amino acids), which have been shown to improve polymerization by fibrillation (46, 58–63). These PTMs could further allow dynamic cellular responses to different cell environmental conditions to trigger signaling, RNA decay, and/or autophagy/apoptosis (58–63). This scenario can be even more complicated with WDM patients harboring additional mutations involving MYH7 (c.5459G>A; p.Arg1820Gln) or SQSTM1/p62 (c.1175C>T; p.P392L together with c.1070A>G; p.N357S in the TIA1 gene), which could accentuate the phenotypes and pathophysiological contexts (58–63). In this regard, WDM-TIA1 may display altered biophysical properties with enhanced liquid-liquid-phase separation, promoting TDP-43 recruitment in TIA1-positive SGs (58–62).
FIG 11.

Working model to explain the pathophysiological consequences of the dynamics of TIA1-dependent stress granules associated with WDM. Shown is a summary of cellular, aging-associated mitochondrial, redox, and molecular events linked to the expression of TIA1WDM. lncRNAs, long noncoding RNAs; RBPs, RNA-binding proteins; MTs, metallothioneins.
Finally, whereas oxidative stress is a well-known SG inducer, its effects are controversial, and additional investigations are required to establish the functional heterogeneity of SG subtypes and the molecular mechanisms underlying granule formation driven by TIA1. This will imply knowledge of the molecular components involving specific transcriptomes and proteomes as well as the contribution of amino acid sequences and the PTMs in intrinsically disordered regions. These future goals may reveal processes that govern both physiological and pathological aggregation of normal and WDM-mutated TIA1, respectively. Clarification of these issues will provide clues to implement possible therapeutic strategies for TIA1-related myopathies. The muscles, but also brain and heart, are especially susceptible to these damaging effects because of their high demand for oxygen, their abundance of highly oxidizable substrates, and their low antioxidant activity. In this way, excessive ROS are believed to be a cause of neurodegenerative diseases such as Alzheimer disease, Parkinson disease, Huntington disease, and amyotrophic lateral sclerosis (ALS) (58–64).
MATERIALS AND METHODS
Cell culture.
Isogenic and inducible GFP-tagged fusion-expressing FT293 cell lines were generated using the Flp-In T-Rex system (Invitrogen) and cultured as described previously (24, 25). HEK293 (human kidney embryonic cells; ATCC CRL-1573), SH-SY5Y (human neuroblastoma; ATCC CRL-2266), and C2C12 (mouse myoblasts; ATCC CRL-1772) cells were grown in Dulbecco’s modified Eagle’s medium with 10% fetal calf serum (Sigma) (decomplemented for 30 min at 56°C) supplemented with 2 mM l-glutamine, 0.4 mM a mixture of nonessential amino acids, penicillin (100 U/ml), and streptomycin (100 μg/ml). Cells were cultured in an incubator (Thermo Electron Corporation) at 37°C, with 95% humidity and 5% CO2. Cell stress analysis was performed as described previously (40).
Minigenes, transfections, RNA isolation, and RT-PCR analysis.
The SMN1/SMN2 and NF1 reporter minigenes used for transient transfections were previously described (44, 45). TurboFect and Lipofectin reagents (Thermo Scientific) were used for transfection of DNA plasmids according to the manufacturer’s instructions. After incubation for 24 h, protein and RNA samples were prepared and analyzed as reported previously (25, 44, 45). RNAs were purified using the RNeasy kit (Qiagen), treated, and amplified by reverse transcription-elongation PCR (RT-PCR) as described previously (25). DNA oligonucleotides used to analyze the splicing patterns of human SMN1/SMN2 and NF1 were previously reported (44, 45).
Protein purification and Western blot analysis.
Cells were cultured and processed for protein isolation and Western blot analysis using the appropriate antibodies (24, 25).
GST- and His-tagged recombinant proteins and GST pulldown analysis.
GST fusion proteins and recombinant histidine-tagged U1-C were expressed in and purified from Escherichia coli as described previously (18). 35S-labeled U1-C was generated in rat reticulocyte lysates using the T7 Quick transcription/translation system (Promega). GST pulldown and Western blot analysis were performed as reported previously (18).
Immunofluorescence and electron microscopy analysis.
Cells were processed for immunofluorescence and transmission electron microscopy as described previously (24, 25).
Mitophagy and autophagy analysis.
To visualize and quantify mitophagy and autophagy rates, we used the fluorescence probes mt-Keima (mitochondrial Keima) (25, 65) and GFP-LC3-RFP (25, 66, 67), respectively.
Fluorescence-activated cell sorter analysis.
Cell death/apoptosis rates were quantified using the annexin V-PE apoptosis detection kit (BD Pharmingen). Mitochondrial membrane potential and ROS levels were measured with 100 nM tetramethylrhodamine methylester and 5 μM CellROX deep red, respectively, by flow cytometry. In both cases, the estimations were carried out in the absence and presence of oligomycin and CCCP (carbonyl cyanide m-chlorophenyl hydrazine) (10 μM) as well as H2O2 and MitoQ (MQ) (a derivative of coenzyme Q10 that is conjugated to a triphenylphosphonium cation) (1 mM), respectively (24, 25).
Statistical analysis.
The data are represented as means ± standard errors of the means (SEM). Student’s t test (paired and two tailed) was applied to determine statistical significance between 2 groups. P values of <0.05 were considered statistically significant.
ACKNOWLEDGMENTS
We are indebted to the generosity and invaluable support with reagents and tips from the following researchers and facilities: F. E. Baralle, T. Finkel, M. Guerra, T. Johansen, H. Lou, A. Miyawaki, N. Mizushima, M. Murphy, G. Pruijn, M. Rejas, J. M. Sierra, R. N. Singh, J. Valcárcel, and confocal and electron microscopy and flow cytometry facilities at the CBMSO.
This work was supported by a grant from the Spanish Ministry of Economic Affairs and Competitiveness to J.M.I. (BFU2014-57735R). The CBMSO receives an institutional grant from the Fundación Ramón Areces and Banco Santander.
We declare that we have no competing interests.
J.M.I. conceived the research. I.C. and J.M.I. designed all the experiments. I.C., C.S.-J., E.S., J.A., and J.M.I. performed the experiments. J.M.I. wrote the paper. All authors provided feedback and approved the final manuscript.
REFERENCES
- 1.Welander L. 1951. Myopathia distalis tarda hereditaria; 249 examined cases in 72 pedigrees. Acta Med Scand Suppl 265:1–124. [PubMed] [Google Scholar]
- 2.von Tell D, Somer H, Udd B, Edström L, Borg K, Åhlberg G. 2002. Welander distal myopathy outside the Swedish population: phenotype and genotype. Neuromuscul Disord 12:544–547. doi: 10.1016/S0960-8966(01)00338-8. [DOI] [PubMed] [Google Scholar]
- 3.Ählberg G, Borg K, Edström L, Anvret M. 1998. Welander hereditary distal myopathy, a molecular genetic comparison to hereditary myopathies with inclusion bodies. Neuromuscul Disord 8:111–114. doi: 10.1016/S0960-8966(98)00007-8. [DOI] [PubMed] [Google Scholar]
- 4.Borg K, Ählberg G, Anvret M, Edström L. 1998. Welander distal myopathy: an overview. Neuromuscul Disord 8:115–118. doi: 10.1016/S0960-8966(98)00008-X. [DOI] [PubMed] [Google Scholar]
- 5.Edström L. 1975. Histochemical and histopathological changes in skeletal muscle in late-onset hereditary distal myopathy (Welander). J Neurol Sci 26:147–157. doi: 10.1016/0022-510X(75)90027-1. [DOI] [PubMed] [Google Scholar]
- 6.Ählberg G, von Tell D, Borg K, Edström L, Anvret M. 1999. Genetic linkage of Welander distal myopathy to chromosome 2p13. Ann Neurol 46:399–404. doi:. [DOI] [PubMed] [Google Scholar]
- 7.Hackman P, Sarparanta J, Lehtinen S, Vihola A, Evilä A, Jonson PH, Luque H, Kere J, Screen M, Chinnery PF, Ählberg G, Edström L, Udd B. 2013. Welander distal myopathy is caused by a mutation in the RNA-binding protein TIA1. Ann Neurol 73:500–509. doi: 10.1002/ana.23831. [DOI] [PubMed] [Google Scholar]
- 8.Klar J, Sobol M, Melberg A, Mäbert K, Ameur A, Johansson AC, Feuk L, Entesarian M, Orlén H, Casar-Borota O, Dahl N. 2013. Welander distal myopathy caused by an ancient founder mutation in TIA1 associated with perturbed splicing. Hum Mutat 34:572–577. doi: 10.1002/humu.22282. [DOI] [PubMed] [Google Scholar]
- 9.Tian Q, Streuli M, Saito H, Schlossman SF, Anderson P. 1991. A polyadenylate binding protein localized to the granules of cytolytic lymphocytes induces DNA fragmentation in target cells. Cell 67:629–639. doi: 10.1016/0092-8674(91)90536-8. [DOI] [PubMed] [Google Scholar]
- 10.Kawakami A, Tian Q, Duan X, Streuli M, Schlossman SF, Anderson P. 1992. Identification and functional characterization of a TIA-1-related nucleolysin. Proc Natl Acad Sci U S A 89:8681–8685. doi: 10.1073/pnas.89.18.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sánchez-Jiménez C, Izquierdo JM. 2015. T-cell intracellular antigens in health and disease. Cell Cycle 14:2033–2043. doi: 10.1080/15384101.2015.1053668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawakami A, Tian Q, Streuli M, Poe M, Edelhoff S, Disteche CM, Anderson P. 1994. Intron-exon organization and chromosomal localization of the human TIA-1 gene. J Immunol 152:4937–4945. [PubMed] [Google Scholar]
- 13.Beck AR, Medley QG, O’Brien S, Anderson P, Streuli M. 1996. Structure, tissue distribution and genomic organization of the murine RRM-type RNA binding proteins TIA-1 and TIAR. Nucleic Acids Res 24:3829–3835. doi: 10.1093/nar/24.19.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Izquierdo JM, Valcárcel J. 2007. Two isoforms of the T-cell intracellular antigen 1 (TIA-1) splicing factors display distinct splicing regulation activities. Control of TIA-1 isoform ratio by TIA-1 related protein. J Biol Chem 282:19410–19417. doi: 10.1074/jbc.M700688200. [DOI] [PubMed] [Google Scholar]
- 15.Kedersha NL, Gupta M, Li W, Miller I, Anderson P. 1999. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2 alpha to the assembly of mammalian stress granules. J Cell Physiol 147:1431–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Del Gatto-Konczak F, Bourgeois CF, Le Guiner C, Kister L, Gesnel MC, Stévenin J, Breathnach R. 2000. The RNA-binding protein TIA-1 is a novel mammalian splicing regulator acting through intron sequences adjacent to a 5′ splice site. Mol Cell Biol 20:6287–6299. doi: 10.1128/MCB.20.17.6287-6299.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Förch P, Puig O, Kedersha N, Martinez C, Granneman S, Seraphin B, Anderson P, Valcarcel J. 2000. The apoptosis-promoting factor TIA-1 is a regulator of alternative pre-mRNA splicing. Mol Cell 6:1089–1098. doi: 10.1016/S1097-2765(00)00107-6. [DOI] [PubMed] [Google Scholar]
- 18.Förch P, Puig O, Martínez C, Seraphin B, Valcárcel J. 2002. The splicing regulator TIA-1 interacts with U1-C to promote U1 snRNP recruitment to 5′ splice sites. EMBO J 21:6882–6892. doi: 10.1093/emboj/cdf668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.López de Silanes I, Galbán S, Martindale JL, Yang X, Mazan-Mamczarz K, Indig FE, Falco G, Zhan M, Gorospe M. 2005. Identification and functional outcome of mRNAs associated with RNA-binding protein TIA-1. Mol Cell Biol 25:9520–9531. doi: 10.1128/MCB.25.21.9520-9531.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Z, Kayikci M, Briese M, Zarnack K, Luscombe NM, Rot G, Zupan B, Curk T, Ule J. 2010. iCLIP predicts the dual splicing effects of TIA-RNA interactions. PLoS Biol 8:e1000530. doi: 10.1371/journal.pbio.1000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mazan-Mamczarz K, Lal A, Martindale JL, Kawai T, Gorospe M. 2006. Translational repression by RNA-binding protein TIAR. Mol Cell Biol 26:2716–2727. doi: 10.1128/MCB.26.7.2716-2727.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim HS, Kuwano Y, Zhan M, Pullmann R Jr, Mazan-Mamczarz K, Li H, Kedersha N, Anderson P, Wilce MC, Gorospe M, Wilce JA. 2007. Elucidation of a C-rich signature motif in target mRNAs of RNA-binding protein TIAR. Mol Cell Biol 27:6806–6817. doi: 10.1128/MCB.01036-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reyes R, Alcalde J, Izquierdo JM. 2009. Depletion of T-cell intracellular antigen proteins promotes cell proliferation. Genome Biol 10:R87. doi: 10.1186/gb-2009-10-8-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sánchez-Jiménez C, Ludeña MD, Izquierdo JM. 2015. T-cell intracellular antigens function as tumor suppressor genes. Cell Death Dis 6:e1669. doi: 10.1038/cddis.2015.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carrascoso I, Alcalde J, Sánchez-Jiménez C, González-Sánchez P, Izquierdo JM. 2017. T-cell intracellular antigens and Hu antigen R antagonistically modulate mitochondrial activity and dynamics by regulating optic atrophy 1 gene expression. Mol Cell Biol 37:e00174-17. doi: 10.1128/MCB.00174-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Piecyk M, Wax S, Beck AR, Kedersha N, Gupta M, Maritim B, Chen S, Gueydan C, Kruys V, Streuli M, Anderson P. 2000. TIA-1 is a translational silencer that selectively regulates the expression of TNF-alpha. EMBO J 19:4154–4163. doi: 10.1093/emboj/19.15.4154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Howell MD, Ottesen EW, Singh NN, Anderson RL, Seo J, Sivanesan S, Whitley EM, Singh RN. 2017. TIA1 is a gender-specific disease modifier of a mild mouse model of spinal muscular atrophy. Sci Rep 7:7183. doi: 10.1038/s41598-017-07468-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beck AR, Miller IJ, Anderson P, Streuli M. 1998. RNA-binding protein TIAR is essential for primordial germ cell development. Proc Natl Acad Sci U S A 95:2331–2336. doi: 10.1073/pnas.95.5.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meyer C, Garzia A, Mazzola M, Gerstberger S, Molina H, Tuschl T. 2018. The TIA1 RNA-binding protein family regulates EIF2AK2-mediated stress response and cell cycle progression. Mol Cell 69:622–635. doi: 10.1016/j.molcel.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kedersha N, Chen S, Gilks N, Li W, Miller IJ, Stahl J, Anderson P. 2002. Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol Biol Cell 13:195–210. doi: 10.1091/mbc.01-05-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anderson P, Kedersha N. 2002. Visibly stressed: the role of eIF2, TIA-1, and stress granules in protein translation. Cell Stress Chaperones 7:213–221. doi:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson P, Kedersha N. 2002. Stressful initiations. J Cell Sci 115:3227–3234. [DOI] [PubMed] [Google Scholar]
- 33.Gilks N, Kedersha N, Ayodele M, Shen L, Stoecklin G, Dember LM, Anderson P. 2004. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol Biol Cell 15:5383–5398. doi: 10.1091/mbc.e04-08-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waris S, Wilce MC, Wilce JA. 2014. RNA recognition and stress granule formation by TIA proteins. Int J Mol Sci 15:23377–23388. doi: 10.3390/ijms151223377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Protter DSW, Rao BS, Van Treeck B, Lin Y, Mizoue L, Rosen MK, Parker R. 2018. Intrinsically disordered regions can contribute promiscuous interactions to RNP granule assembly. Cell Rep 22:1401–1412. doi: 10.1016/j.celrep.2018.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khong A, Matheny T, Jain S, Mitchell SF, Wheeler JR, Parker R. 2017. The stress granule transcriptome reveals principles of mRNA accumulation in stress granules. Mol Cell 68:808–820. doi: 10.1016/j.molcel.2017.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boeynaems S, Alberti S, Fawzi NL, Mittag T, Polymenidou M, Rousseau F, Schymkowitz J, Shorter J, Wolozin B, Van Den Bosch L, Tompa P, Fuxreiter M. 2018. Protein phase separation: a new phase in cell biology. Trends Cell Biol 28:420–435. doi: 10.1016/j.tcb.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Namkoong S, Ho A, Woo YM, Kwak H, Lee JH. 2018. Systematic characterization of stress-induced RNA granulation. Mol Cell 70:175–187. doi: 10.1016/j.molcel.2018.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Markmiller S, Soltanieh S, Server KL, Mak R, Jin W, Fang MY, Luo EC, Krach F, Yang D, Sen A, Fulzele A, Wozniak JM, Gonzalez DJ, Kankel MW, Gao FB, Bennett EJ, Lécuyer E, Yeo GW. 2018. Context-dependent and disease-specific diversity in protein interactions within stress granules. Cell 172:590–604. doi: 10.1016/j.cell.2017.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rayman JB, Karl KA, Kandel ER. 2018. TIA-1 self-multimerization, phase separation, and recruitment into stress granules are dynamically regulated by Zn2+. Cell Rep 22:59–71. doi: 10.1016/j.celrep.2017.12.036. [DOI] [PubMed] [Google Scholar]
- 41.Van Treeck B, Protter DSW, Matheny T, Khong A, Link CD, Parker R. 2018. RNA self-assembly contributes to stress granule formation and defining the stress granule transcriptome. Proc Natl Acad Sci U S A 115:2734–2739. doi: 10.1073/pnas.1800038115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang K, Daigle JG, Cunningham KM, Coyne AN, Ruan K, Grima JC, Bowen KE, Wadhwa H, Yang P, Rigo F, Taylor JP, Gitler AD, Rothstein JD, Lloyd TE. 2018. Stress granule assembly disrupts nucleocytoplasmic transport. Cell 173:958–971. doi: 10.1016/j.cell.2018.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pullmann R Jr, Kim HH, Abdelmohsen K, Lal A, Martindale JL, Yang X, Gorospe M. 2007. Analysis of turnover and translation regulatory RNA-binding protein expression through binding to cognate mRNAs. Mol Cell Biol 27:6265–6278. doi: 10.1128/MCB.00500-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh NN, Seo J, Ottesen EW, Shishimorova M, Bhattacharya D, Singh RN. 2011. TIA1 prevents skipping of a critical exon associated with spinal muscular atrophy. Mol Cell Biol 31:935–954. doi: 10.1128/MCB.00945-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu H, Hinman MN, Hasman RA, Mehta P, Lou H. 2008. Regulation of neuron-specific alternative splicing of neurofibromatosis type 1 pre-mRNA. Mol Cell Biol 28:1240–1251. doi: 10.1128/MCB.01509-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang J, Choi JM, Holehouse AS, Lee HO, Zhang X, Jahnel M, Maharana S, Lemaitre R, Pozniakovsky A, Drechsel D, Poser I, Pappu RV, Alberti S, Hyman AA. 2018. A molecular grammar governing the driving forces for phase separation of prion-like RNA binding proteins. Cell 174:688–699. doi: 10.1016/j.cell.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bratic A, Larsson NG. 2013. The role of mitochondria in aging. J Clin Invest 123:951–957. doi: 10.1172/JCI64125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim Y, Zheng X, Ansari Z, Bunnell MC, Herdy JR, Traxler L, Lee H, Paquola ACM, Blithikioti C, Ku M, Schlachetzki JCM, Winkler J, Edenhofer F, Glass CK, Paucar AA, Jaeger BN, Pham S, Boyer L, Campbell BC, Hunter T, Mertens J, Gage FH. 2018. Mitochondrial aging defects emerge in directly reprogrammed human neurons due to their metabolic profile. Cell Rep 23:2550–2558. doi: 10.1016/j.celrep.2018.04.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tak H, Eun JW, Kim J, Park SJ, Kim C, Ji E, Lee H, Kang H, Cho DH, Lee K, Kim W, Nam SW, Lee EK. 2017. T-cell-restricted intracellular antigen 1 facilitates mitochondrial fragmentation by enhancing the expression of mitochondrial fission factor. Cell Death Differ 24:49–58. doi: 10.1038/cdd.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arimoto-Matsuzaki K, Saito H, Takekawa M. 2016. TIA1 oxidation inhibits stress granule assembly and sensitizes cells to stress-induced apoptosis. Nat Commun 7:10252. doi: 10.1038/ncomms10252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brown RS. 2005. Zinc finger proteins: getting a grip on RNA. Curr Opin Struct Biol 15:94–98. doi: 10.1016/j.sbi.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 52.Collins KM, Kainov YA, Christodolou E, Ray D, Morris Q, Hughes T, Taylor IA, Makeyev EV, Ramos A. 2017. An RRM-ZnF RNA recognition module targets RBM10 to exonic sequences to promote exon exclusion. Nucleic Acids Res 45:6761–6774. doi: 10.1093/nar/gkx225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Font J, Mackay JP. 2010. Beyond DNA: zinc finger domains as RNA-binding modules. Methods Mol Biol 649:479–491. doi: 10.1007/978-1-60761-753-2_29. [DOI] [PubMed] [Google Scholar]
- 54.Masuda K, Marasa B, Martindale JL, Halushka MK, Gorospe M. 2009. Tissue- and age-dependent expression of RNA-binding proteins that influence mRNA turnover and translation. Aging (Albany NY) 1:681–698. doi: 10.18632/aging.100073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fu T, Xu Z, Liu L, Guo Q, Wu H, Liang X, Zhou D, Xiao L, Liu L, Liu Y, Zhu MS, Chen Q, Gan Z. 2018. Mitophagy directs muscle-adipose crosstalk to alleviate dietary obesity. Cell Rep 23:1357–1372. doi: 10.1016/j.celrep.2018.03.127. [DOI] [PubMed] [Google Scholar]
- 56.Yun J, Finkel T. 2014. Mitohormesis. Cell Metab 19:757–766. doi: 10.1016/j.cmet.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Buchan JR, Roy PR. 2009. Eukaryotic stress granules: the ins and out of translation. Mol Cell 36:932–941. doi: 10.1016/j.molcel.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vanderweyde T, Apicco DJ, Youmans-Kidder K, Ash PEA, Cook C, Lummertz da Rocha E, Jansen-West K, Frame AA, Citro A, Leszyk JD, Ivanov P, Abisambra JF, Steffen M, Li H, Petrucelli L, Wolozin B. 2016. Interaction of tau with the RNA-binding protein TIA1 regulates tau pathophysiology and toxicity. Cell Rep 15:1455–1466. doi: 10.1016/j.celrep.2016.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mackenzie IR, Nicholson AM, Sarkar M, Messing J, Purice MD, Pottier C, Annu K, Baker M, Perkerson RB, Kurti A, Matchett BJ, Mittag T, Temirov J, Hsiung GR, Krieger C, Murray ME, Kato M, Fryer JD, Petrucelli L, Zinman L, Weintraub S, Mesulam M, Keith J, Zivkovic SA, Hirsch-Reinshagen V, Roos RP, Züchner S, Graff-Radford NR, Petersen RC, Caselli RJ, Wszolek ZK, Finger E, Lippa C, Lacomis D, Stewart H, Dickson DW, Kim HJ, Rogaeva E, Bigio E, Boylan KB, Taylor JP, Rademakers R. 2017. TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron 95:808–816. doi: 10.1016/j.neuron.2017.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Niu Z, Pontifex CS, Berini S, Hamilton LE, Naddaf E, Wieben E, Aleff RA, Martens K, Gruber A, Engel AG, Pfeffer G, Milone M. 2018. Myopathy with SQSTM1 and TIA1 variants: clinical and pathological features. Front Neurol 9:147. doi: 10.3389/fneur.2018.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee Y, Jonson PH, Sarparanta J, Palmio J, Sarkar M, Vihola A, Evilä A, Suominen T, Penttilä S, Savarese M, Johari M, Minot MC, Hilton-Jones D, Maddison P, Chinnery P, Reimann J, Kornblum C, Kraya T, Zierz S, Sue C, Goebel H, Azfer A, Ralston SH, Hackman P, Bucelli RC, Taylor JP, Weihl CC, Udd B. 2018. TIA1 variant drives myodegeneration in multisystem proteinopathy with SQSTM1 mutations. J Clin Invest 128:1164–1177. doi: 10.1172/JCI97103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Apicco DJ, Ash PEA, Maziuk B, LeBlang C, Medalla M, Al Abdullatif A, Ferragud A, Botelho E, Ballance HI, Dhawan U, Boudeau S, Cruz AL, Kashy D, Wong A, Goldberg LR, Yazdani N, Zhang C, Ung CY, Tripodis Y, Kanaan NM, Ikezu T, Cottone P, Leszyk J, Li H, Luebke J, Bryant CD, Wolozin B. 2018. Reducing the RNA binding protein TIA1 protects against tau-mediated neurodegeneration in vivo. Nat Neurosci 21:72–80. doi: 10.1038/s41593-017-0022-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Peskett TR, Rau F, O’Driscoll J, Patani R, Lowe AR, Saibil HR. 2018. A liquid to solid phase transition underlying pathological Huntingtin exon1 aggregation. Mol Cell 70:588–601. doi: 10.1016/j.molcel.2018.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ramdzan YM, Trubetskov MM, Ormsby AR, Newcombe EA, Sui X, Tobin MJ, Bongiovanni MN, Gras SL, Dewson G, Miller JML, Finkbeiner S, Moily NS, Niclis J, Parish CL, Purcell AW, Baker MJ, Wilce JA, Waris S, Stojanovski D, Böcking T, Ang CS, Ascher DB, Reid GE, Hatters DM. 2017. Huntingtin inclusions trigger cellular quiescence, deactivate apoptosis, and lead to delayed necrosis. Cell Rep 19:919–927. doi: 10.1016/j.celrep.2017.04.029. [DOI] [PubMed] [Google Scholar]
- 65.Sun N, Yun J, Liu J, Malide D, Liu C, Rovira II, Holmström KM, Fergusson MM, Yoo YH, Combs CA, Finkel T. 2015. Measuring in vivo mitophagy. Mol Cell 60:685–696. doi: 10.1016/j.molcel.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kimura S, Noda T, Yoshimori T. 2007. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 67.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. 2007. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]