

Abstract
Tyrosinase, an enzyme involved in melanin synthesis, is expressed in nearly all primary and metastatic melanoma lesions and thus is an attractive target for TCR-based gene therapy using adoptive cell transfer. The TCR α- and β-chain genes from a tumor-infiltrating lymphocyte, which recognized the tyrosinase 368–376 peptide in the context of HLA-A2, were cloned into a γ-retroviral vector. Following transduction of PBL, specific reactivity was confirmed by cytokine production following coculture with tumor targets. Experiments using Ab blockade and CD4/CD8 sorting of the transduced PBLs demonstrated that this antityrosinase TCR was CD4/CD8 independent. The introduction of a second disulfide bond between the TCR constant regions and/or creation of a chimeric protein in which the human constant regions were replaced by murine homologs resulted in enhanced TCR expression as demonstrated by tetramer staining and improved tumor reactivity that was comparable to PBL transduced with either antimelanoma Ag recognized by T cells-1 or anti-gp100 TCR vectors currently used in clinical trials. The chimeric TCR also allowed us to test antitumor function of in HLA-A2/Kb–transgenic mice. Transfer of the antityrosinase TCR into mouse splenocytes conferred CD4/CD8-independent, HLA-A2–restricted Ag reactivity against B16/A2Kb murine melanoma in vitro. Furthermore, adoptive transfer of transduced splenocytes mediated B16/A2Kb melanoma tumor regression in lymphodepleted mice, and, surprisingly, both CD8 and CD4 T cells were equally effective in mediating tumor regression. These results suggest that this highly active tyrosinase-specific TCR could be of value in adoptive cell transfer for melanoma.
Immunotherapy with adoptive cell transfer (ACT) has proven a useful strategy for the treatment of metastatic melanoma (1–4). Objective response rates of 72% have been achieved with 16% of patients rendered completely free of disease (2). These studies involved the extraction and ex vivo expansion of tumor-infiltrating lymphocytes (TILs). Only one-half of patients are able to receive this therapy due to lack of harvestable tumors, inability to successfully grow TIL, or a lack of cellular reactivity. Recent strategies aimed at broadening the applicability of this therapy involve the cloning of TCR genes specific for melanoma-associated Ags (MAAs) from reactive TILs and retrovirally transducing these genes into PBLs (5). This has been applied with some success in two recent trials targeting melanoma Ag recognized by T cells-1 (MART-1) and gp100 (5, 6). Efforts aimed at improving the response rate have focused on targeting different Ags with higher avidity receptors (7, 8) and modifications to TCR genes to increase their function (9–11).
Tyrosinase, an enzyme involved in melanin synthesis, is found in nearly all primary and metastatic melanomas (12–14), and may be expressed in the absence of other MAA such as gp100 and MART-1 (15, 16). Although multiple MHC-expressed epitopes have been identified (17–20), the HLA-A2–presented 368–376 peptide is best characterized (21–23) and has successfully generated reactive lymphocytes via vaccination both in vitro (24) and in vivo (25). TIL-1383i, previously described by Nishimura et al. (26), reacts with the tyrosinase 368–376 epitope in an HLA-A2–restricted manner. This TIL is unique in that it is MHC class I-restricted despite being a CD4+/CD8− T cell, and thus, the TCR appears to recognize Ag in a CD8-independent manner. The use of TIL-1383i in ACT did not result in objective cancer regression, and initial attempts to clone and transfer this TCR gene generated relatively poor overall reactivity (27). In this study, we isolated the TCR α and β genes from TIL-1383i and employed improvements in gene transfer as well as amino acid modifications to increase cell surface expression and reactivity following gene transfer. This new reagent was demonstrated to have high-avidity CD8-independent properties and mediated equal tumor regression in a murine melanoma model using either CD8- or CD4-engineered T cells.
Materials and Methods
Mice and cell lines
TIL-1383i was obtained from a melanoma metastasis resected from a patient treated at the Surgery Branch, National Cancer Institute (Bethesda, MD), as previously described (26). All PBLs were collected via leuka-pheresis from patients enrolled in Institutional Review Board-approved studies. Lymphocytes were cultured as described (5) using AIM-V media (Invitrogen, Rockville, MD) with IL-2 (Chiron, Emmerville, CA) at a concentration of 300 IU/ml for PBL and 6000 IU/ml for TIL.
T2 cells (28) from American Type Culture Collection (Rockville, MD) were pulsed with peptides for tyrosinase 368–376 (YMDGTMSQV) (23) or MART-1 27–35 (AAGIGILTV) (29) as previously described (30). Melanoma lines 526mel (tyrosinase+, HLA-A2+), 624mel (tyrosinase+, HLA-A2+), 1359mel (tyrosinase+, HLA-A2−), 836mel (tyrosinase−, HLA-A2+), A375 (tyrosinase−, HLA-A2+), and 888mel (tyrosinase+, HLA-A2−) were generated in the Surgery Branch from resected tumors as previously described (31). H1299-A2 (tyrosinase−, HLA-A2+) is a small cell lung cancer line transduced to stably express HLA-A2. All lines were maintained in RPMI 1640 (Invitrogen) supplemented with 10% FCS (Biofluid, Gaithersburg, MD) and penicillin/streptomycin (50 U/ml).
HLA-A*0201/Kb–transgenic mice were acquired from L. Sherman (The Scripps Research Institute, La Jolla, CA) and have been previously described (32). These mice express the human HLA-A*201 Ag modified to include the α3, transmembrane, and cytoplasmic regions of the murine H-2Kb. Similarly, a B16/A2/Kb line was created by transfecting wild-type B16 melanoma cells with a plasmid containing A2Kb and a puromycin selection gene (33). Transfected cells were selected and maintained in RPMI 1640 with 1 μg/ml puromycin.
TCR gene isolation
RNA was isolated from TIL-1383i and reverse transcribed using the Thermoscript RT-PCR system (Invitrogen). The product was subjected to 5′ RACE using a 5′ universal primer and 3′ gene-specific primers for the α 5′-CACTGTTGCTCTTGAAGTCC-3′ and β 5′-CAGGCAGTATCTGGAGTCATTGAG-3′ constant regions. The purified products were cloned into a TOPO2.1 vector (Invitrogen) for sequencing and the variable regions identified using published international ImMunoGeneTics information system sequences (http://imgt.cines.fr/). In accordance with ImMunoGeneTics nomenclature, the TIL-1383i TCR was α-chain TRAV4/TRAJ42/TRAC and β-chain TRBV10-3/TRBJ1-3/TRBD1/TRBC1. Gene-specific primers were used to clone the full-length α-chain (5′-gacatgtcgcaa-3′ and 5′-aattctcagctggacca-3′) and β-chain (5′-gaaatgggcacaag-3′ and 5′-gcttatcagaaatcct-3′).
In vitro RNA transcription and expression in PBLs
α- and β-chain cDNAs were cloned into distinct pGEM RNA expression vectors (Promega, Madison, WI) modified to contain a 64 polyA tail. RNA synthesis and electroporation was performed as previously described (34). Briefly, donor PBLs were stimulated with OKT3 (Ortho Biotech, Raritan, NJ) (50 ng/ml) and cultured for 7–10 d. PBLs were washed, resuspended in serum-free media, and transferred to electroporation cuvettes (Harvard Apparatus, Holliston, MA) on ice. Each curette, containing 2 μg α- and β-chain RNA per 1 × 106 cells was electroporated at 500 V for 500 μsec. Cells were then placed into 37°C AIM-V media for 2 h prior to use.
Construction of retroviral vectors
The γ-retroviral backbone used in this study was pMSGV1 (30). An insert containing the cDNA of the α- and β-chains linked by a P2A ribosomal skip element (35) was created using overhang PCR (36). Primers used to create the wild-type insert included α-chain primers 5′-cccagcccatctccatggactcat-3′ and 5′-ccggcctgcttcagcaggctgaagttggtggctccggatccggaccgcttggcccggctggaccacagccgcag-3′ and β-chain primers 5′-agcctgctgaagcaggccggcgacgtggaggagaaccccggcccgatgggcacaaggttgttcttc-3′ and 5′-tttttttgaattctcagaaatcctttct-3′. The 3′ region of the PCR-amplified α-chain and the 5′ region of the PCR-amplified β-chain contain overlapping sequences that fused in a second PCR reaction that used the α forward (5′-cccagcccatctccatggactcat-3′) and β reverse (5′-tttttttgaattctcagaaatcctttct-3′) primers to generate the full-length insert.
A second construct (cysteine replacement) was created by replacing an amino acid from the α- and β-chains with a cysteine as previously described (9). Briefly, an initial overhang PCR was performed for both the α- and β-chains using primers containing a α-Cys48 and a β-Cys57. α primers included the wild-type α forward primer (5′-cccagcccatctccatggactcat-3′) with the reverse primer 5′-agacctcatgtctagcacgcatttgtctgtgatatacacatc-3′, and the forward primer 5′-gatgtgtatatcacagacaaatgcgtgctagacatgaggtct-3′ with the wild-type α reverse primer (5′-ccggcctgcttcagcaggctgaagttggtggctccggatccggaccgcttggcccggctggaccacagccgcag-3′). A second PCR using the wild-type forward (5′-cccagcccatctccatggactcat-3′) and reverse (5′-ccggcctgcttcagcaggctgaagttggtggctccggatccggaccgcttggcccggctggaccacagccgcag-3′) primers completed the α-chain. The β-chain was created in a similar manner using the wild-type β forward primer (5′-agcctgctgaagcaggccggcgacgtggaggagaaccccggcccgatgggcacaaggttgttcttc-3′) with the reverse primer 5′-gaggggctgcgggtccgtgcagaccccactgtgcacctcctt-3′ and the forward primer 5′-aaggaggtgcacagtggggtctgcacggacccgcagcccctc-3′ with the wild-type β reverse primer (5′-tttttttgaattctcagaaatcctttct-3′). The full-length insert was formed in a third PCR using the α and β products with the wild-type α forward (5′-cccagcccatctccatggactcat-3′) and β reverse primers (5′-tttttttgaattctcagaaatcctttct-3′).
A third construct (mouse C region) replaced the α- and β-chain constant regions with murine homologs creating a human-murine chimera (10). Primers were created for use in overhang PCR as described above. α primers included the wild-type α forward primer (5′-cccagcccatctccatggactcat-3′) with the reverse primer 5′-ggttctgggttctggatatttggtttaacagagagtttagtgcc-3′ and the forward primer 5′-ggcactaaactctctgttaaaccaaatatccagaacccagaacc-3′ with the reverse primer 5′-ccggcctgcttcagcaggctgaagttggtggctccggatccggaccgcttggcccgactggaccacagcctcagcgt-3′. A murine TCR vector (37) was used as the template for the latter two amplifications. A second PCR using the first (5′-cccagcccatctccatggactcat-3′) and last (5′-ccggcctgcttcagcaggctgaagttggtggctccggatccggaccgcttggcccgactggaccacagcctcagcgt-3′) primers above completed formation of the α-chain. The β-chain was made in a similar using the wild-type β forward primer (5′-agcctgctgaagcaggccggcgacgtggaggagaaccccggcccgatgggcacaaggttgttcttc-3′) with the reverse primer 5′-tggagtcacatttctcagatcctctacaacagtgagccaacttccctc-3′ and the forward primer 5′-gagggaagttggctcactgttgtagaggatctgagaaatgtgactcca-3′ with the reverse primer 5′-gactacgtagcggccgctcatgaattc-3′. The full-length insert was formed in a third PCR using the α and β products with the wild-type α forward primer (5′-cccagcccatctccatggactcat-3′) and β reverse primer (5′-gactacgtagcggccgctcatgaattc-3′). A fourth construct (mouse C region with cysteine replacement), which contains both the cysteine replacement and murine constant regions, was made using a combination of the above primers.
Production of retroviral supernatant
Retroviral supernatant was produced using 293GP cells as previously described (35). Briefly, 9 μg plasmid DNA and 4 μg RD-114 envelope plasmid were mixed with Lipofectamine 2000 (Invitrogen) in serum-free media and incubated at room temperature for 30 min. This mixture was then applied to 293GP cells, which had been plated the prior day on polylysine-coated plates (Becton Dickinson, San Jose, CA). After 7 h of incubation, the media was replaced with DMEM (Invitrogen) with 10% FBS and the viral supernatant harvested 48 h later. Supernatants were centrifuged at 2000 × g for 10 min to ensure no carryover of packaging cells. All supernatants were used fresh and made simultaneously for vector comparison. Retroviral supernatant for murine transductions was made in a similar fashion using the packaging line Platinum-E (Plat-E), which stably expresses the gag-pol-env genes, therefore requiring only transfection of the vector plasmid DNA (38).
PBLs and splenocyte transduction
Lymphocytes were stimulated with OKT3 Ab (50 ng/ml) in AIM-V with 300 IU/ml IL-2 for 2 d prior to transduction. Retroviral supernatant was spin-loaded onto RetroNectin (Takara Bio, Otsu, Japan) coated plates by centrifugation at 2000 × g for 2 h at 32°C. PBLs were then loaded onto plates by centrifugation at 1000 × g for 10 min and incubated overnight. The transduction was thena repeated the next day with a new virus-loaded plate. Cells were cultured for 5–7 d prior to FACS and coculture.
For splenocyte transduction, whole C57BL/6 HLA-A*0201/Kb spleens were harvested and passed through a 40-μm filter to achieve a single-cell suspension and placed into ACK lysing buffer (Biosource, Rockville, MD). CD3+ cells were separated by negative selection over a column (Miltenyi Biotec, Auburn, CA) and stimulated with plate-bound OKT3 and soluble CD28 in AIM-V with 60 IU/ml IL-2 for 48 h prior to transduction.
FACS analysis
Cell-surface expression of TCR was assessed by anti-TCR Vβ12 (Immunotech, Westbrook, ME) or PE-conjugated tetramer specific for the antityrosinase, anti-gp100, or anti–MART-1 TCRs (Beckman Coulter, Fullerton, CA). T cell marker proteins were evaluated using PE- or FITC-conjugated Abs against CD8, CD4, and CD3 (Becton Dickinson). Immunofluorescence was measured as relative log fluorescence of live cells using an FACScan flow cytometer (Becton Dickinson).
Measurement of lymphocyte and splenocyte reactivity
Peptide-specific reactivity was assessed by incubation of varying concentrations (0.0001–1 μM) of peptide with T2 cells at 37°C for 2 h followed by washing and coculturing with various effectors for 20 h. Supernatants were harvested, and IFN-γ was measured by colorimetric ELISA (Endogen, Cambridge, MA). Cocultures of lymphocytes or splenocytes with melanoma tumor lines were carried out in a similar fashion. Lysis was assessed by radioactivity of supernatant following coculture of 51Cr-labeled tumor targets with effectors for 4 h as previously described (31). Patient-derived tumor cells used as targets were acquired from surgically resected lesions, which were digested overnight in enzymes until a single-cell suspension was obtained.
Ab blockade and cell sorting
For blocking of costimulatory molecules CD4 and CD8, 5 × 104 TCR-transduced PBLs were incubated with 20 μg/ml appropriate Ab (Becton Dickinson) for 1 h at 37°C then cocultured for 20 h with 1 × 105 targets. Values recorded as percent activity compared with isotype (mouse IgG2) control Ab. CD4 and CD8 cells were purified from the bulk population by negative selection using Ab cocktails and Fc Ab-conjugated beads (Miltenyi Biotec) per the manufacturer’s protocol.
Adoptive cell transfer
All animal experiments were approved by the National Institutes of Health animal care and use committee. HLA-A*0201/Kbαtransgenic mice at 6–12 wk were s.c. injected in the flank region with 5 × 105 B16/A2/Kb tumor cells in PBS 2 wk prior to ACT. Mice were irradiated with 500 cGy and then separated into four groups of five mice per group. Cells were administered by tail vein injection (10 × 106) in 200 μl PBS. For mice receiving vaccine, 2 × 107 PFU of recombinant fowlpox virus expressing human tyrosinase was given by tail vein injection. All mice except those in the untreated group were given 180,000 IU recombinant human IL-2 i.p. twice per day for 3 d. Measurements were done using a caliper by a blinded investigator. Mice were sacrificed when tumors reached 300 m2 or at the discretion of the veterinary staff at National Institutes of Health.
Results
Molecular cloning of antityrosinase TCR genes
TIL-1383i was grown from a metastatic melanoma deposit in the inguinal lymph node of a patient who progressed after receiving systemic IL-2. It was previously demonstrated to be reactive to autologous tumor cells and a variety of HLA-A*0201–restricted melanoma cell lines (27). TIL-1383i was cocultured with T2 cells pulsed with decreasing concentrations of tyrosinase 368–376 peptide and demonstrated to recognize T2 cells pulsed with peptide as low as 0.001 μM (data not shown). IFN-γ was also released following coculture of TIL-1383i with tyrosinase and HLA-A2–expressing melanoma cells (526mel and 624mel), but not non–HLA-A2 (938mel), or tyrosinase-negative (H1299-A2) cells (data not shown). FACS analysis revealed TIL-1383i to be CD8 negative, and 50% of cells stained positive using tyrosinase 368–376-specific tetramer and 34% were Vβ12 positive (data not shown).
To identify and isolate the genes encoding the α and β TCR chains, TIL-1383i was lysed and RNA extracted. cDNA was made via reverse transcription and 5′ RACE reactions performed using TCR α- and β-specific primers. An appropriate-sized cDNA for each chain was identified (~700–800 bp), isolated, molecularly cloned, and sequenced. A single α-chain TRAV 4.1 (10 out of 10 clones) and a dominant β-chain TRBV 10.3 (11 of 13 clones) were identified and the full-length genes cloned using gene-specific primers. Postinsertion into an RNA expression vector, in vitro transcription was performed and the RNA products electroporated into stimulated PBLs. TCR surface expression was confirmed using tyrosinase 368–376-specific tetramer (17.2%), and reactivity against HLA-A2 and tyrosinase-expressing cells, as well as peptide-pulsed T2 cells, was demonstrated by IFN-γ release afterovernight coculture (data not shown).
Construction of a retroviral vector
To allow stable expression of the antityrosinase TCR in PBL and their progeny, a γ-retroviral vector was constructed by inserting the genes for the α- and β-chains linked with a P2A ribosomal skip element (35), which allows for two proteins to be translated from a single mRNA transcript (Fig. 1A). Retroviral transduction was performed and receptor expression determined at 7 d posttransduction by FACS and coculture (Fig. 1B–D). Fifteen percent of the TCR-transduced cells now bound tyrosinase tetramer compared with 0.05% of the untransduced controls. PBL transduced with the antityrosinase TCR and not untransduced PBLs released IFN-α when cocultured with varying concentrations of tyrosinase 368–376 peptide (Fig. 1C) and were reactive against HLA-A2 and tyrosinase-positive (624mel and 526mel), but not tyrosinase-negative (A375) or HLA-A2–negative (888mel) cells (Fig. 1D). TCR expression and reactivity persisted for over 60 d in culture (data not shown).
FIGURE 1.
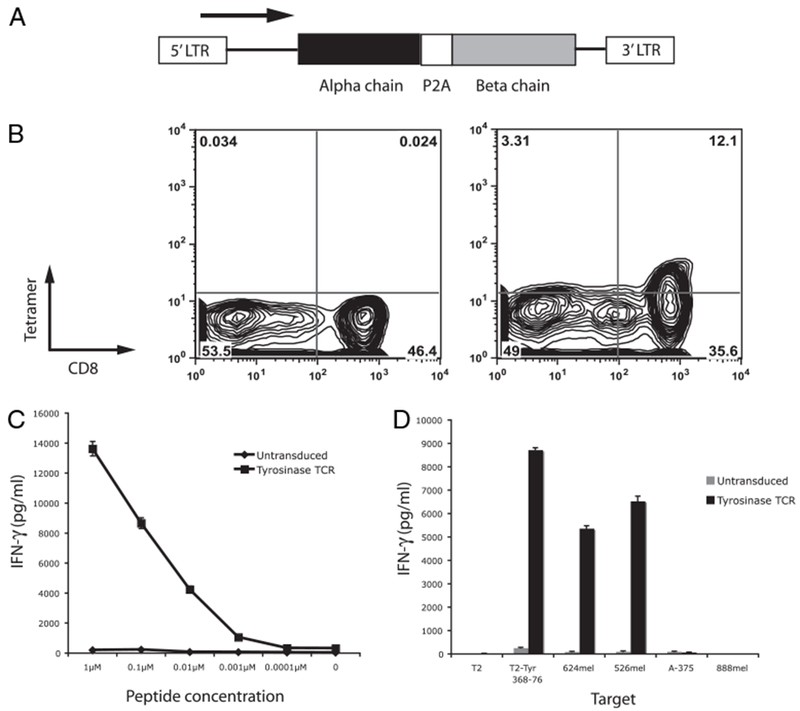
Transduction of PBL with a γ-retrovirus encoding the α and β TCR chains from TIL-1383i. A, Schematic representation of the γ-retrovirus composed of the murine stem cell virus long terminal repeat and cDNA encoding the α and β TCR chains linked with a P2A ribosomal skip element. B, Tyrosinase 368–376-specific tetramer binding of retrovirally transduced PBLs. IFN-γ release following coculture of PBLs transduced with the antityrosinase TCR and decreasing concentrations of tyrosinase 368–376 peptide (C) and tumor targets 624mel, 526mel, A-375 (HLA-A2+, tyrosinase−), and 888mel (HLA-A2−, tyrosinase+) (D).
Antityrosinase TCR is CD4 and CD8 independent and functions in both cell types
Ab blockade was used to assess the contribution of the CD4 and CD8 coreceptors to the reactivity of PBL transduced with the antityrosinase TCR. Controls included PBL transduced with an anti–MART-1 TCR previously demonstrated to be CD8 dependent (8) and a CD4-dependent TIL clone 1749-E11. CD4- and CD8-blocking Abs were able to suppress the reactivity of the TIL clone and anti–MART-1 TCR transduced PBLs, respectively, but had no effect on antityrosinase TCR-transduced PBLs (Fig. 2A). To further test coreceptor independence, PBLs were transduced with an anti–MART-1 TCR, an anti-gp100 154–162 TCR previously shown to be CD8 independent (37), and the antityrosinase TCR followed by enrichment into CD8+ and CD4+ populations (purity was assessed by FACS) (Fig. 2B). Both the CD8+ and CD4+ populations of the gp100 154–162 and tyrosinase TCR-transduced PBLs secreted IFN-γ when cultured with appropriate targets (Fig. 2C). The CD8-dependent anti–MART-1 TCR showed significantly reduced cytokine release in CD4+ cells compared with CD8+ cells. Lysis of the separated populations was assessed by [51Cr] release. In contrast to results using cytokine secretion, CD4+ cells engineered with the antityrosinase TCR showed decreased capability to lyse target cells. This was true for MART-1 and gp100-reactive CD4+ cells as well (Fig. 2D).
FIGURE 2.
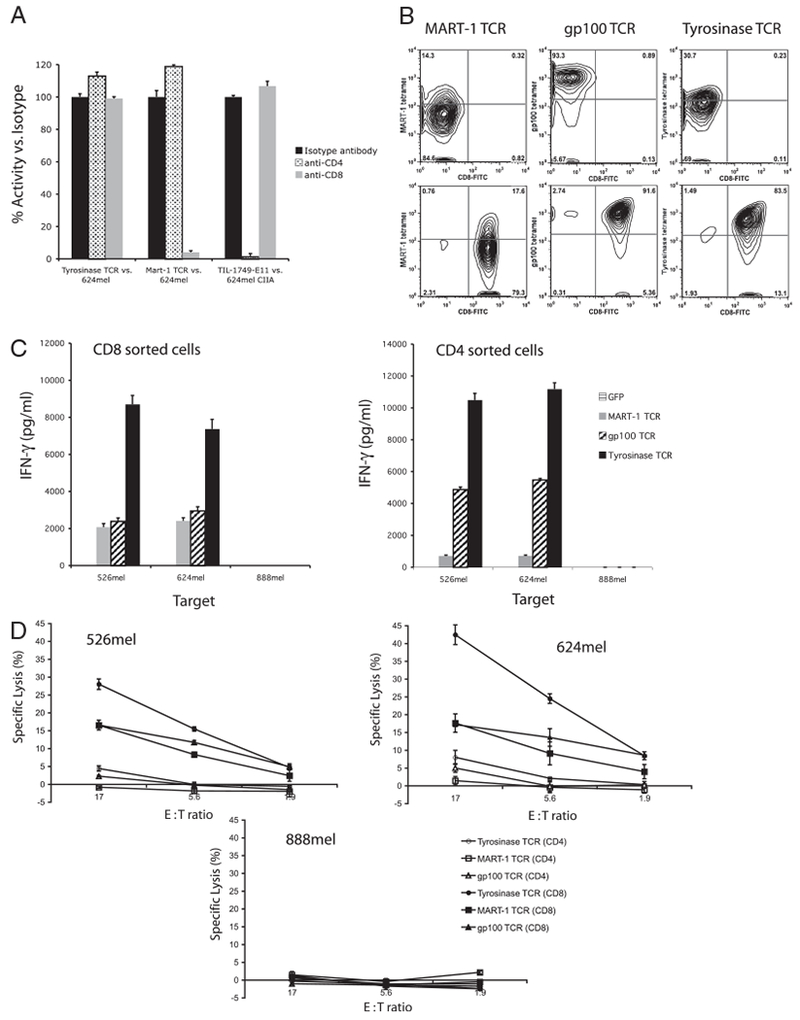
The high-avidity antityrosinase TCR functioned independently of the costimulatory molecules CD4 and CD8. A, Percent remaining activity following Ab blockade with anti-CD4 or anti-CD8. Controls include the CD8-dependent MART-1 TCR and the CD4-dependent TIL 1749-E11. B, FACS analysis using specific tetramer and CD8 Ab following CD4 (top panels) and CD8 (bottom panels) enrichment of antityrosinase TCR-transduced PBLs. C, IFN-γ release following coculture of the enriched cells with tumor targets 624mel, 526mel, and 888mel. D, Percent specific lysis of tumor lines 526mel, 624mel, and 888mel by CD4- or CD8-enriched transduced PBLs. These data are representative of four experiments.
Increasing TCR expression and reactivity via protein engineering
It has previously been reported that introduction of a second disulfide bond into the α and β constant regions improved TCR surface pairing and therefore reactivity (9, 11, 39). It has also been shown that replacement of the human TCR constant regions with the murine C region homologs resulted in improved TCR expression and function (10). To test whether these protein modifications would enhance the antityrosinase reactivity of the TIL1383i TCR, three constructs were generated employing these techniques (Fig. 3A) and used to transduce PBLs. Seven days posttransduction, TCR surface expression was evaluated by FACS using tyrosinase 368–376-specific tetramer and Vβ12 Ab (Fig. 3B). A small improvement in tetramer and Vβ12 staining was seen in the cysteine replacement construct compared with the wild-type vector (12.5% and 65.6% versus 6.4% and 62%, respectively). A more dramatic increase in surface expression was seen in the mouse C region construct, in which both tetramer- and Vβ12-specific staining increased (48% and 82.7%, respectively). There did not appear to be any improvement in expression with the addition of the second disulfide bond to the mouse C region. Differences in TCR gene expression were not due to differences in gene transfer efficiencies, as quantitative PCR of transduced cells demonstrated similar levels of gene transfer (data not shown). In coculture assays, there was no improvement in reactivity using the TCR engineered with a second disulfide bond, whereas the constructs with the mouse constant regions demonstrated increased reactivity against melanoma cell line 624mel (Fig. 3C). This was repeated using PBLs from additional donors with similar results (data not shown). The increased cytokine production was associated with improved lytic ability, as PBLs transduced with the mouse C region TCR lysed 60% more tyrosinase-expressing tumor cells than the wild-type TCR (49% versus 31% at a 17:1 E:T ratio) (Fig. 3D). PBLs transduced with each construct was subjected to a rapid expansion as previously described (1). Differences in TCR surface expression and reactivity were maintained through the rapid expansion after 300-fold expansion (data not shown).
FIGURE 3.
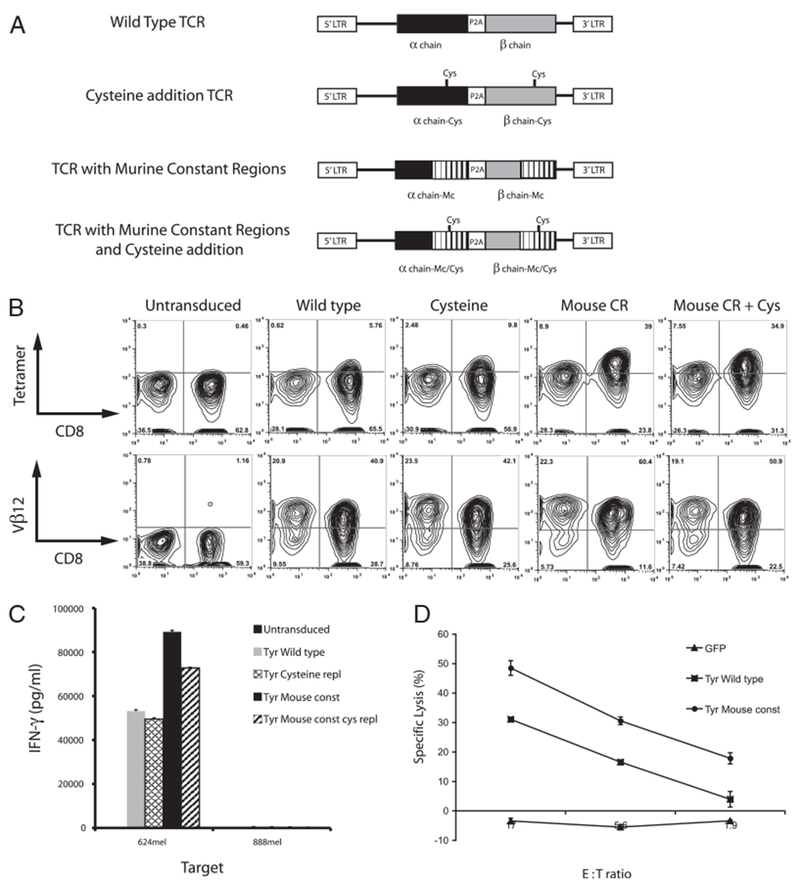
Protein engineering yielded improved TCR expression and reactivity. A, Schematic representation of the wild-type, cysteine replacement, mouse C region, and mouse C region with cysteine replacement vectors. B, FACS analysis using both tyrosinase 368–376 specific tetramer and anti-Vβ12 Ab. C, Comparative IFN-γ release following coculture of 624mel and 888mel with PBL transduced with the four vectors described above. D, Percent specific lysis of 624mel by PBL transduced with the wild-type versus the murine chimeric TCRs. These findings were repeated in two additional donor lymphocytes.
Comparison of antityrosinase TCR to vectors targeting gp100 and MART-1
To determine the potential clinical utility of the antityrosinase TCR modified to contain the murine constant regions, we first compared it to another CD8-independent TCR, the anti-gp100 154–162, which has demonstrated objective clinical responses in patients with melanoma (6). As target cells, tumor digests were prepared from freshly resected melanomas as previously described (40). Patient tumors were chosen for their tyrosinase, gp100, and HLA-A2 expression as determined by immunohistochemical analysis. The tumor from patient 1 had >75% of cells expressing both gp100 and tyrosinase and was HLA-A2+. Tumor from patient 2 had >75% tyrosinase-positive cells, <5% gp100-positive cells, and was HLA-A2+. Tumor from patient 3 had no tyrosinase or gp100 expression and was HLA-A2+. Finally, tumor from patient 4 had >75% of cells expressing tyrosinase and gp100, but was HLA-A2 negative. Overnight coculture with tumor digests revealed greater IFN-γ release with antityrosinase TCR compared with anti-gp100 154–162 TCR-transduced PBLs (Fig. 4A). Digests that failed to express the target Ag or HLA-A2 elicited no response. In a 4-h [51Cr]-release assay, the antityrosinase TCR-transduced PBLs demonstrated a higher percentage lysis of the tumor digests compared with the anti-gp100 154–162 TCR-transduced PBLs at the 17 and 5.6 E:T ratios (Fig. 4B).
FIGURE 4.
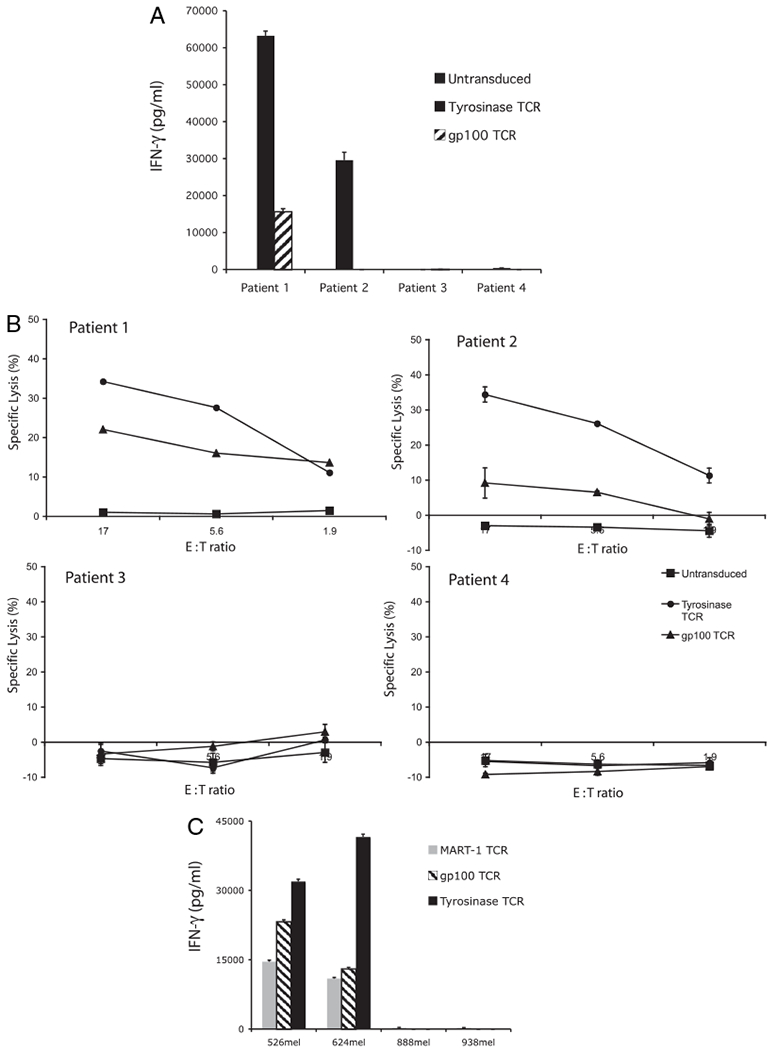
Antityrosinase TCR vector compared favorably to the anti–MART-1 and anti-gp100 154–162 TCR vectors. A and B, IFN-γ release and specific lysis following coculture of antityrosinase or anti-gp100 TCR-transduced PBLs with melanoma tumor digests from patient 1 (tyrosinase+, gp100+, HLA-A2+), patient 2 (tyrosinase+, gp100−, HLA-A2+), patient 3 (tyrosinase−, gp100−, HLA-A2+), and patient 4 (tyrosinase+, gp100+, HLA-A2−). C, Cytokine release following coculture of anti–MART-1, anti-gp100, and antityrosinase TCR-transduced PBLs with established melanoma lines 526mel, 624mel, 888mel, and 938mel.
Finally, the antityrosinase TCR was compared with the previously reported high-affinity anti–MART-1 (DMF5 clone) TCR vector (8). This particular vector was identified by screening 24 MART-1–reactive TIL clones and was chosen for its high avidity against peptide and tumor targets. Identical γ-retroviral vector preparations were produced for the three vectors (anti–MART-1, anti-gp100 154–162, and antityrosinase TCR) and used to transduce PBLs using the same protocol and subsequently cocultured with melanoma cell lines. Anti-tyrosinase TCR transduced cells displayed superior reactivity compared with the MART-1 and gp100 154–162 TCR vectors (Fig. 4C).
Expression and reactivity ofmouse splenocytes transduced with the antityrosinase TCR
Murine Ag-processing machinery cleaves the tyrosinase protein into peptides, one of which is homologous to the human 368–378 epitope. To determine the reactivity of the modified human antityrosinase TCR in murine cells, splenocytes were stimulated and transduced in a similar fashion to human lymphocytes. FACS analysis using Abs against the human Vβ12 Ag revealed a transduction efficiency of 36%, with 93% of cells staining positive for CD8 (Fig. 5A). To determine if the CD4/CD8 independence of the antityrosinase TCR was maintained in murine model, mouse splenocytes were sorted into CD4+ and CD8+ populations prior to transduction. Overnight coculture with tyrosinase 368–376 peptide-pulsed T2 cells and tumor targets revealed reactivity of both CD4+ and CD8+ cells as measured by IFN-γ release (Fig. 5B). Transduced cells were reactive against the transgenic murine melanoma B16/A2/Kb line but not the B16 wild-type line, confirming expression of both tyrosinase and HLA-A*0201 by this line as well as the ability of a receptor raised against the human tyrosinase 368–376 to recognize the naturally processed and presented murine epitope.
FIGURE 5.
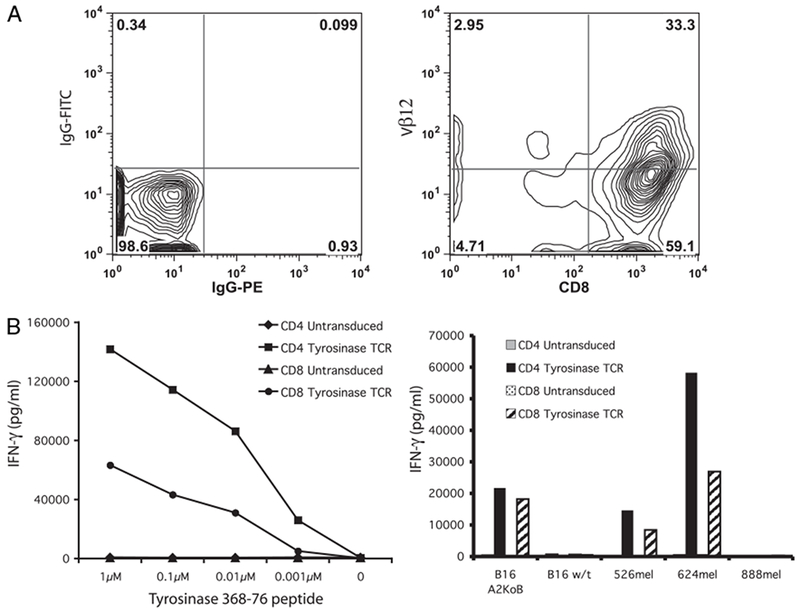
Transduction of mouse splenocytes with the antityrosinase TCR confers peptide reactivity and tumor recognition. A, Isotype, Vβ12, and CD8 staining of splenocytes following 48 h stimulation in OKT3 and CD28 and a single transduction with retrovirus encoding the antityrosinase TCR. B, IFN-γ release following coculture with T2 cells pulsed with decreasing concentrations of peptide (left panel) and tumor targets (right panel) B16/A2/Kb (HLA-A2+, tyrosinase+), B16 wild-type (HLA-A2−, tyrosinase+), 526mel, 624mel, and 888mel.
Tumor regression following ACT ofCD4 splenocytes transduced with the antityrosinase TCR
To test whether the immune cells transduced with the modified antityrosinase TCR could mediate in vivo tumor regression, HLA-A*0201/Kb–transgenic mice were implanted with B16/A2/Kb. Mice were separated into four groups receiving 1) irradiation alone; 2) irradiation, vaccine, and IL-2; 3) ACT with untransduced splenocytes, irradiation, vaccine, and IL-2; and 4) ACT with antityrosinase TCR-transduced splenocytes, irradiation, vaccine, and IL-2. Tumor size was measured on the day of treatment and every 3 d after until two groups had less than three mice alive. Tumor size and survival were plotted to compare growth and survival between the groups. Mice receiving splenocytes transduced with the antityrosinase TCR demonstrated greater tumor regression and delayed growth compared with mice treated with untransduced cells (p < 0.05) (Fig. 6A). There was no difference seen between ACT with untransduced cells and irradiation alone. The group receiving vaccine alone and IL-2 appeared to do slightly worse, although this was not statistically significant. Survival among mice treated with the antityrosinase TCR was 80% at the end of the experiment compared with 0% in the group receiving untransduced cells (Fig. 6B).
FIGURE 6.
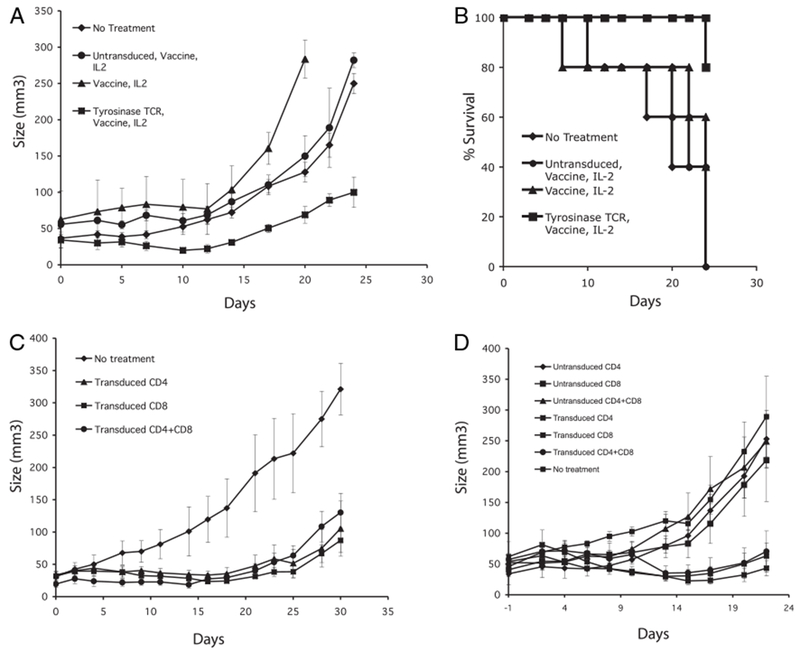
In vivo tumor treatment using antityrosinase TCR-engineered T cells. A and B, Tumor growth and survival following 1) no treatment, 2) vaccine and IL-2 alone, 3) ACT with untransduced splenocytes with IL-2 and vaccine, and 4) ACT with antityrosinase-transduced splenocytes with vaccine and IL-2. In all experiments, B16/A2Kb tumor-bearing mice were given 500 cGy irradiation on day 0 of cell transfer (five mice per group). AH mice except the no treatment group also received 2e7pfu of rVVhTYR vaccination and 180,000 IU/ml of recombinant human IL-2 twice a day three times. C and D, TYR-TCR transduced CD4, CD8, or CD4+CD8 cells were i.v. injected into mice and tumor growth followed.
To further analyze the in vivo antitumor activity of this TCR, transduced murine splenocytes were separated into CD8 and CD4 T cells and transduced with the modified antityrosinase TCR vector. Animals bearing established tumors were administered equal numbers of antityrosinase transduced CD8, CD4, or CD4 plus CD8 T cells, and tumor growth followed. All groups treated with antityrosinase TCR-engineered cells demonstrated statistically significant (p < 0.05) delay in tumor growth. In two independent experiments (Fig. 6C, 6D), we observed equal tumor treatment with all three groups of transduced cells compared with no treatment; there was no statistical difference between animals receiving both CD4 and CD8 T cells versus the animals that received CD8 or CD4 T cells alone.
Discussion
Cell-based immunotherapy for cancer involves the adoptive transfer of tumor-reactive T lymphocytes to the tumor-bearing host. TILs grown from surgically resected metastatic lesions, expanded ex vivo and infused into the autologous host, have mediated dramatic and durable clinical responses, rendering some patients with bulky metastatic lesions, free of disease (2, 41).
TIL therapy, however, has requirements that limit its applicability. First, to grow TIL, metastatic lesions need to be surgically harvested. Although this often requires only soft tissue or lymph node resection, occasionally laparotomy or thoracotomy is necessary, and this can be associated with morbidity and prolonged recovery. A second shortcoming is variability in ex vivo expansion of extracted lymphocytes, as TILs from some patients do not proliferate in sufficient quantities for use in ACT. Third, reactive TILs are difficult to obtain from histologies other than melanoma and renal cell carcinoma, thus limiting its utility for treatment of more common malignancies.
Cloning of TCR genes and insertion into peripheral lymphocytes can broaden the applicability of ACT (4). This technique involves the identification of Ag-specific reactive TIL clones (8) or the generation of high-avidity TCRs via immunization of HLA-transgenic mice using human Ags (37). When the cDNAs encoding the α- and β-chains of the TCR were extracted from these reactive cells, cloned into γ-retroviral vectors, and transferred into PBLs, the resulting gene-modified T lymphocytes were Ag specific. In the first trial administrating TCR gene-modified PBLs, Morgan et al. (5) reported an objective response rate of 12% in 15 patients with IL-2–resistant metastatic melanoma. Although these results were promising, the response rates were lower than prior TIL trials (1). Strategies aimed at increasing the efficacy of this treatment involve improvements in gene transfer efficiency, use of higher avidity TCRs (8), modifications to improve α- and β-chain surface pairing (9, 10), and induction of higher Ag expression in tumors in vivo (35).
TILs have been identifiedthat target the cancer testes Ags (35, 42), MAAs (8, 18, 27, 29, 30, 43), as well as mutated self-Ags (42). Recent interest has focused on MAAs, as they are present on most melanomas and are easily evaluable by immunohistochemistry. The three most commonly targeted Ags—MART-1, gp100, and tyrosinase—are highly immunogenic proteins and are capable of breaking immune tolerance in both tumor- and nontumor-bearing individuals (44). MAAs differ in abundance and distribution in melanomas. Comparative analysis of Ag expression in 30 metastatic lesions found that although tyrosinase was present in every specimen, 20% of lesions failed to express MART-1 or gp-100 (12). These findings have been echoed by others and indicate that tyrosinase may be an ideal candidate for immunotherapeutic targeting (15).
TIL-1383i was previously identified as a unique CD4+ T cell reactive against the tyrosinase 368–376 epitope in an MHC class I-restricted manner (27). This defies the traditional model of MHC class I/TCR interaction, which requires CD8 costimulation to stabilize the interaction. It appeared likely that the specific T cell responsible for TIL-1383i reactivity had a high-affinity TCR, which could overcome the need for costimulation, making it well suited for antityrosinase-based TCR therapy. Although previous attempts were made to clone and transfer the antityrosinase TCR from TIL-1383i (27), the resultant transduced PBLs had weak reactivity against tumor cells (100-fold less than reported in this study), suggesting the need to reisolate the TCR genes responsible for TIL-1383i’s high avidity.
In this report, we sought to isolate the high-avidity TCR gene from TIL1383i and further modify it to improve reactivity and cell surface expression. The 5′ RACE was used to identify the genes encoding the α- and β-chains and determined TIL1383i to be oli-goclonal with dominant TRAV4.1 and TRBV10.3 TCR chains. After confirming reactivity of the TCR via electroporation, we inserted the genes encoding the α- and β-chains into a γ-retroviral vector MSGV1, previously shown to allow for high gene transfer efficiency (5, 8, 30, 35). Initial transduction resulted in only modest surface expression as determined by FACS analysis, and thus, we employed techniques described to improve functional α-chain/β-chain pairing. One such method involves substituting a specific amino acid in the constant regions of both chains for a cysteine (9, 39), forming a second disulfide bond, which improves pairing, expression, and reactivity. Although this modification had little effect on β-chain surface expression (a 6% increase), it improved tetramer binding (2-fold), implying superior surface pairing of the introduced chains. Interestingly, this 2-fold increase in surface expression was insufficient to mediate improved reactivity against tumor targets.
A second method to improve surface pairing replaced the human constant regions with the murine homologs. Previous reports demonstrated this improved pairing of the introduced α- and β-chains, inhibited mismatching and increased CD3 binding (10). Using this strategy, we were able to enhance surface expression as exhibited by an 8-fold increase in tetramer binding and improved overall reactivity (Fig. 3B, 3C). There was a 26% increase in surface expression of the introduced TRBV12 chain, possibly due to greater CD3 binding, which we have previously demonstrated results from improved stability on the cell surface (10). The murine C region-transduced cells displayed greater cytokine release following stimulation with tyrosinase-expressing tumor cells and had superior lytic ability when compared with PBL transduced with the wild-type receptor. When the cysteine modification was applied to the TCR containing the murine constant regions, there was no additional change in expression or reactivity.
We next sought to determine if the T lymphocytes genetically modified to express the antityrosinase TCR would display the same costimulatory independence as TIL-1383i from which it was derived. The high-avidity TCR isolated in this report was subjected to Ab blockade as well as cell enrichment, which confirmed that the antityrosinase TCR-transduced PBLs could be stimulated in the absence of CD8. The ability of this class I-restricted TCR is not unique, and it has been previously reported that highly avid human TCRs and murine-derived TCRs, when introduced into human T cells, can also display this behavior (8,37). The utility of CD4 cells in tumor immunotherapy has not been extensively investigated, but in has been reported in murine models that, dependent on the specific CD4 T cell subset used, that these cells can either enhance the elimination of established tumors (using Th cells) or can actively inhibit antitumor activity (using regulatory T cells) (45).
A direct role for CD4+ T cell in tumor eradication was recently reported by two groups using the same B16 melanoma tumor model along with the transfer of a transgenic T cell directed to MAA TRP-1 (46, 47). It was reported that the transfer of naive CD4+ T cells into lymphopenic hosts resulted in the differentiation of these cells into Th type 1 cytotoxic T cells, which then mediated MHC class II-restricted tumor regression. In humans, a recent case report administering CD4+ cloned T cells reactive to the NY-ESO-1 Ag in an MHC class II manner demonstrated a complete response in one of nine patients with metastatic melanoma (41). These authors were able to identify anti–MART-1 and anti-MAGE–specific lymphocytes that were not present prior to ACT, suggesting that reactive CD4+ cells in the tumor microenvironment may recruit cytotoxic CD8+ cells that contributed to tumor eradication. Although it is unknown whether CD4+ cells engineered with an MHC class I-restricted receptor will behave similarly, it is possible that these cells may provide helper cytokines to both improve persistence of transferred cells and recruit endogenous tumor reactive lymphocytes.
One goal of this project was to create an antityrosinase vector that could be used clinically in ACT antimelanoma trials. We therefore compared this new receptor to the high-avidity antiMART-1 (DMF5) (8) and anti-gp100 154–162 vectors currently used in clinical trials (6). To limit confounding variables, transient retroviral supernatants were simultaneously made for the three vectors and transductions carried out using identical protocols. When cocultured with established melanoma lines, PBLs transduced with the antityrosinase TCR displayed superior reactivity compared with the other receptors. It should be noted, however, that the relative Ag expression of these lines is not known and could account for some of the observed differences. We next compared the antityrosinase and anti-gp100 TCR-transduced cells for their ability to secrete IFN-γ and lyse primary tumor digests. Lesions from HLA-A2+ and non–HLA-A2 patients expressing one or both Ags were surgically resected and digested to a single-cell suspension. In HLA-A2+ tumors containing >75% gp100 and tyrosinase Ag expression, the antityrosinase TCR-transduced PBL showed superior IFN-γ release and lytic ability.
Finally, the receptor modified with the murine constant regions enabled us a unique opportunity to test the in vivo reactivity of a human TCR in an immune-competent mouse. It has been previously demonstrated that although the murine and human epitopes for tyrosinase 368–376 differ by one amino acid (F for Y at the first position), this has no effect on recognition by TCR raised against either peptide (48). Colella et al. (48) determined that not only could a TCR targeting the human tyrosinase 368–376 recognize the homologous mouse peptide, but also this epitope was equally processed and presented by murine cells. We confirmed this by demonstrating reactivity of transduced splenocytes against a murine melanoma tumor line expressing both murine tyrosinase and human HLA-A*0201. There was no reactivity in cells lacking the human HLA, confirming specificity. It has been well established that when transferred into HLA-A*0201–transgenic mice, these tumors were not spontaneously rejected, making it an ideal tumor model for in-vivo testing of this receptor. Following ACT of antityrosinase TCR-transduced splenocytes, there was tumor regression, significantly delayed tumor growth, and prolonged animal survival (Fig. 6). This antitumor effect was not seen in mice receiving untransduced cells or the immune modulatory vaccine or IL-2 alone.
Most significantly, when we determined that relative reactivity of CD4 and CD8 T cells in this in vivo model, both cell types yielded equal tumor treatments. Although CD4 cells demonstrated a reduced ability to lyse tumor targets in vitro (Fig. 6B), in vivo they may have additional abilities not provided by CD8 cells, including the ability to interact with APCs to license APCs to activate endogenous CD8 cells, as well as having the potential to recruit macrophages and NK cells to mediate non–MHC-restricted tumor lysis (49, 50). This is the first report of successful in vivo antitumor reactivity of murine CD4 cells engineered to express a human class I-restricted TCR.
We have described the cloning and modification of a high-affinity TCR gene from a TIL reactive against the tyrosinase 368–376 epitope. Using an optimized retroviral vector and transduction techniques, we have demonstrated the ability to confer antityrosinase activity to peripheral PBLs. Substitution of mouse constant regions improved the pairing of the introduced a and β receptors and the stability of the TCR. By comparison with vectors currently used in human clinical trials, this vector has the potential to be an additional and valuable tool for antimelanoma-based ACT using genetically modified PBLs. ACT using antityrosinase TCR-transduced murine splenocytes mediated tumor regression and improved overall survival, further suggesting the potential clinical utility of this therapy.
Acknowledgments
This work was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
Abbreviations used in this paper:
- ACT
adoptive cell transfer
- MAA
melanoma-associated Ag
- MART
melanoma Ag recognized by T cells
- TIL
tumor-infiltrating lymphocyte
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, Royal RE, Kammula U, White DE, Mavroukakis SA, et al. 2005. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J. Clin. Oncol 23: 2346–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang J, Citrin DE, Leitman SF, et al. 2008. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J. Clin. Oncol 26: 5233–5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg SA, and Dudley ME. 2004. Cancer regression in patients with metastatic melanoma after the transfer of autologous antitumor lymphocytes. Proc. Natl. Acad. Sci. U.S.A 101(Suppl 2): 14639–14645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, and Dudley ME. 2008. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 8: 299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, et al. 2006. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314: 126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, et al. 2009. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 114: 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jorritsma A, Gomez-Eerland R, Dokter M, van de Kasteele W, Zoet YM, Doxiadis II, Rufer N, Romero P, Morgan RA, Schumacher TN, and Haanen JB. 2007. Selecting highly affine and well-expressed TCRs for gene therapy of melanoma. Blood 110: 3564–3572. [DOI] [PubMed] [Google Scholar]
- 8.Johnson LA, Heemskerk B, Powell DJ Jr., Cohen CJ, Morgan RA, Dudley ME, Robbins PF, and Rosenberg SA. 2006. Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. J. Immunol 177: 6548–6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, and Morgan RA. 2007. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res 67: 3898–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, and Morgan RA. 2006. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res 66: 8878–8886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuball J, Dossett ML, Wolfl M, Ho WY, Voss RH, Fowler C, and Greenberg PD. 2007. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood 109: 2331–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cormier JN, Abati A, Fetsch P, Hijazi YM, Rosenberg SA, Marincola FM, and Topalian SL. 1998. Comparative analysis of the in vivo expression of tyrosinase, MART-1/Melan-A, and gp100 in metastatic melanoma lesions: implications for immunotherapy. J. Immunother 21: 27–31. [DOI] [PubMed] [Google Scholar]
- 13.de Vries TJ, Fourkour A, Wobbes T, Verkroost G, Ruiter DJ, and van Muijen GN. 1997. Heterogeneous expression of immunotherapy candidate proteins gp100, MART-1, and tyrosinase in human melanoma cell lines and in human melanocytic lesions. Cancer Res 57: 3223–3229. [PubMed] [Google Scholar]
- 14.de Vries TJ, Smeets M, de Graaf R, Hou-Jensen K, Bro¨cker EB, Renard N, Eggermont AM, van Muijen GN, and Ruiter DJ. 2001. Expression of gp100, MART-1, tyrosinase, and S100 in paraffin-embedded primary melanomas and locoregional, lymph node, and visceral metastases: implications for diagnosis and immunotherapy. A study conducted by the EORTC Melanoma Cooperative Group. J. Pathol 193: 13–20. [DOI] [PubMed] [Google Scholar]
- 15.Chen YT, Stockert E, Tsang S, Coplan KA, and Old LJ. 1995. Immunophenotyping of melanomas for tyrosinase: implications for vaccine development. Proc. Natl. Acad. Sci. U.S.A 92: 8125–8129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cormier JN, Hijazi YM, Abati A, Fetsch P, Bettinotti M, Steinberg SM, Rosenberg SA, and Marincola FM. 1998. Heterogeneous expression of melanoma-associated antigens and HLA-A2 in metastatic melanoma in vivo. Int. J. Cancer 75: 517–524. [DOI] [PubMed] [Google Scholar]
- 17.Brichard VG, Herman J, Van Pel A, Wildmann C, Gaugler B, Wo¨lfel T, Boon T, and Lethe´ B. 1996. A tyrosinase nonapeptide presented by HLA-B44 is recognized on a human melanoma by autologous cytolytic T lymphocytes. Eur. J. Immunol 26: 224–230. [DOI] [PubMed] [Google Scholar]
- 18.Kang X, Kawakami Y, el-Gamil M, Wang R, Sakaguchi K, Yannelli JR, Appella E, Rosenberg SA, and Robbins PF. 1995. Identification of a tyrosinase epitope recognized by HLA-A24-restricted, tumor-infiltrating lymphocytes. J. Immunol 155: 1343–1348. [PubMed] [Google Scholar]
- 19.Kittlesen DJ, Thompson LW, Gulden PH, Skipper JC, Colella TA, Shabanowitz J, Hunt DF, Engelhard VH, Slingluff CL Jr., and Shabanowitz JA. 1998. Human melanoma patients recognize an HLA-A1-restricted CTL epitope from tyrosinase containing two cysteine residues: implications for tumor vaccine development. [Published erratum appears in 1999 J. Immunol. 162: 3106.] J. Immunol 160: 2099–2106. [PubMed] [Google Scholar]
- 20.Morel S, Ooms A, Van Pel A, Wo¨ lfel T, Brichard VG, van der Bruggen P, Van den Eynde BJ, and Degiovanni G. 1999. A tyrosinase peptide presented by HLA-B35 is recognized on a human melanoma by autologous cytotoxic T lymphocytes. Int. J. Cancer 83: 755–759. [DOI] [PubMed] [Google Scholar]
- 21.Wo¨lfel T, Van Pel A, Brichard V, Schneider J, Seliger B, Meyer zum Bu¨schenfelde KH, and Boon T. 1994. Two tyrosinase nonapeptides recognized on HLA-A2 melanomas by autologous cytolytic T lymphocytes. Eur. J. Immunol 24: 759–764. [DOI] [PubMed] [Google Scholar]
- 22.Kawakami Y, Robbins PF, and Rosenberg SA. 1996. Human melanoma antigens recognized by T lymphocytes. Keio J. Med 45: 100–108. [DOI] [PubMed] [Google Scholar]
- 23.Skipper JC, Hendrickson RC, Gulden PH, Brichard V, Van Pel A, Chen Y, Shabanowitz J, Wolfel T, Slingluff CL Jr., Boon T, et al. 1996. An HLA-A2-restricted tyrosinase antigen on melanoma cells results from post-translational modification and suggests a novel pathway for processing of membrane proteins. J. Exp. Med 183: 527–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tjandrawan T, Martin DM, Maeurer MJ, Castelli C, Lotze MT, and Storkus WJ. 1998. Autologous human dendriphages pulsed with synthetic or natural tumor peptides elicit tumor-specific CTLs in vitro. J. Immunother 21: 149–157. [DOI] [PubMed] [Google Scholar]
- 25.Lau R, Wang F, Jeffery G, Marty V, Kuniyoshi J, Bade E, Ryback ME, and Weber J. 2001. Phase I trial of intravenous peptide-pulsed dendritic cells in patients with metastatic melanoma. J. Immunother 24: 66–78. [DOI] [PubMed] [Google Scholar]
- 26.Nishimura MI, Avichezer D,Custer MC, Lee CS, Chen C,Parkhurst MR, Diamond RA, Robbins PF, Schwartzentruber DJ, and Rosenberg SA. 1999. MHC class I-restricted recognition of a melanoma antigen by a human CD4+ tumor infiltrating lymphocyte. Cancer Res 59: 6230–6238. [PubMed] [Google Scholar]
- 27.Roszkowski JJ, Lyons GE, Kast WM, Yee C, Van Besien K, and Nishimura MI. 2005. Simultaneous generation of CD8+ and CD4+ melanoma-reactive T cells by retroviral-mediated transfer of a single T-cell receptor. Cancer Res 65: 1570–1576. [DOI] [PubMed] [Google Scholar]
- 28.Salter RD, Howell DN, and Cresswell P. 1985. Genes regulating HLA class I antigen expression in T-B lymphoblast hybrids. Immunogenetics 21: 235–246. [DOI] [PubMed] [Google Scholar]
- 29.Kawakami Y, Eliyahu S, Sakaguchi K, Robbins PF, Rivoltini L, Yannelli JR, Appella E, and Rosenberg SA. 1994. Identification of the immunodominant peptides of the MART-1 human melanoma antigen recognized by the majority of HLA-A2-restricted tumor infiltrating lymphocytes. J. Exp. Med 180: 347–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hughes MS, Yu YY, Dudley ME, Zheng Z, Robbins PF, Li Y, Wunderlich J, Hawley RG, Moayeri M, Rosenberg SA, and Morgan RA. 2005. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum. Gene Ther 16: 457–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Topalian SL, Solomon D, and Rosenberg SA. 1989. Tumor-specific cytolysis by lymphocytes infiltrating human melanomas. J. Immunol 142: 3714–3725. [PubMed] [Google Scholar]
- 32.Vitiello A, Marchesini D, Furze J, Sherman LA, and Chesnut RW. 1991. Analysis of the HLA-restricted influenza-specific cytotoxic T lymphocyte response in transgenic mice carrying a chimeric human-mouse class I major histocompatibility complex. J. Exp. Med 173: 1007–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Irvine KR, Parkhurst MR, Shulman EP, Tupesis JP, Custer M, Touloukian CE, Robbins PF, Yafal AG, Greenhalgh P, Sutmuller RP, et al. 1999. Recombinant virus vaccination against “self” antigens using anchorfixed immunogens. Cancer Res 59: 2536–2540. [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao Y, Zheng Z, Cohen CJ, Gattinoni L, Palmer DC, Restifo NP, Rosenberg SA, and Morgan RA. 2006. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol. Ther 13: 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wargo JA, Robbins PF, Li Y, Zhao Y, El-Gamil M, Caragacianu D, Zheng Z, Hong JA, Downey S, Schrump DS, et al. 2009. Recognition of NY-ESO-1+ tumor cells by engineered lymphocytes is enhanced by improved vector design and epigenetic modulation of tumor antigen expression. Cancer Immunol. Immunother 58: 383–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heckman KL, and Pease LR. 2007. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat. Protoc 2: 924–932. [DOI] [PubMed] [Google Scholar]
- 37.Yang S, Cohen CJ, Peng PD, Zhao Y, Cassard L, Yu Z, Zheng Z, Jones S, Restifo NP, Rosenberg SA, and Morgan RA. 2008. Development of optimal bicistronic lentiviral vectors facilitates high-level TCR gene expression and robust tumor cell recognition. Gene Ther 15: 1411–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morita S, Kojima T, and Kitamura T. 2000. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther 7: 1063–1066. [DOI] [PubMed] [Google Scholar]
- 39.Boulter JM, Glick M, Todorov PT, Baston E, Sami M, Rizkallah P, and Jakobsen BK. 2003. Stable, soluble T-cell receptor molecules for crystallization and therapeutics. Protein Eng 16: 707–711. [DOI] [PubMed] [Google Scholar]
- 40.Romeo MJ, Wunderlich J, Ngo L, Rosenberg SA, Steinberg SM, and Berman DM. 2006. Measuring tissue-based biomarkers by immunochromatography coupled with reverse-phase lysate microarray. Clin. Cancer Res 12: 2463–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, Jungbluth A, Gnjatic S, Thompson JA, and Yee C. 2008. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N. Engl. J. Med 358: 2698–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khong HT, and Rosenberg SA. 2002. Pre-existing immunity to tyrosinaserelated protein (TRP)-2, a new TRP-2 isoform, and the NY-ESO-1 melanoma antigen in a patient with a dramatic response to immunotherapy. J. Immunol 168: 951–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robbins PF, el-Gamil M, Kawakami Y, Stevens E, Yannelli JR, and Rosenberg SA. 1994. Recognition of tyrosinase by tumor-infiltrating lymphocytes from a patient responding to immunotherapy. Cancer Res 54: 3124–3126. [PubMed] [Google Scholar]
- 44.Marincola FM, Rivoltini L, Salgaller ML, Player M, and Rosenberg SA. 1996. Differential anti–MART-1/MelanA CTL activity in peripheral blood of HLA-A2 melanoma patients in comparison to healthy donors: evidence of in vivo priming by tumor cells. J. Immunother. Emphasis Tumor Immunol 19: 266–277. [DOI] [PubMed] [Google Scholar]
- 45.Antony PA, Piccirillo CA, Akpinarli A, Finkelstein SE, Speiss PJ, Surman DR, Palmer DC, Chan CC, Klebanoff CA, Overwijk WW, et al. 2005. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J. Immunol 174: 2591–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon EK, Muranski P, Restifo NP, and Antony PA. 2010. Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. J. Exp. Med 207: 651–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, Blasberg R, Yagita H, Muranski P, Antony PA, et al. 2010. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J. Exp. Med 207: 637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Colella TA, Bullock TN, Russell LB, Mullins DW, Overwijk WW, Luckey CJ, Pierce RA, Restifo NP, and Engelhard VH. 2000. Self-tolerance to the murine homologue of a tyrosinase-derived melanoma antigen: implications for tumor immunotherapy. J. Exp. Med 191: 1221–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Corthay A, Skovseth DK, Lundin KU, Røsjø E, Omholt H, Hofgaard PO, Haraldsen G, and Bogen B. 2005. Primary antitumor immune response mediated by CD4+ T cells. Immunity 22: 371–383. [DOI] [PubMed] [Google Scholar]
- 50.Perez-Diez A, Joncker NT, Choi K, Chan WF, Anderson CC, Lantz O, and Matzinger P. 2007. CD4 cells can be more efficient at tumor rejection than CD8 cells. Blood 109: 5346–5354. [DOI] [PMC free article] [PubMed] [Google Scholar]