

Abstract
Background
Friend leukemia virus integration 1 (FLI1), an ETS transcription factor family member, acts as an oncogenic driver in hematological malignancies and promotes tumor growth in solid tumors. However, little is known about the mechanisms underlying the activation of this proto-oncogene in tumors.
Results
Immunohistochemical staining showed that FLI1 is aberrantly overexpressed in advanced stage and metastatic breast cancers. Using a CRISPR Cas9-guided immunoprecipitation assay, we identify a circular RNA in the FLI1 promoter chromatin complex, consisting of FLI1 exons 4-2-3, referred to as FECR1.Overexpression of FECR1 enhances invasiveness of MDA-MB231 breast cancer cells. Notably, FECR1 utilizes a positive feedback mechanism to activate FLI1 by inducing DNA hypomethylation in CpG islands of the promoter. FECR1 binds to the FLI1 promoter in cis and recruits TET1, a demethylase that is actively involved in DNA demethylation. FECR1 also binds to and downregulates in trans DNMT1, a methyltransferase that is essential for the maintenance of DNA methylation.
Conclusions
These data suggest that FECR1 circular RNA acts as an upstream regulator to control breast cancer tumor growth by coordinating the regulation of DNA methylating and demethylating enzymes. Thus, FLI1 drives tumor metastasis not only through the canonical oncoprotein pathway, but also by using epigenetic mechanisms mediated by its exonic circular RNA.
Electronic supplementary material
The online version of this article (10.1186/s13059-018-1594-y) contains supplementary material, which is available to authorized users.
Keywords: Circular RNA, FLI1, Breast cancer, DNA methylation, TET1, DNMT1, Tumor
Introduction
The ETS gene family of transcription factors share a common ETS DNA binding domain responsible for sequence-specific DNA recognition on target promoters, and they play critical roles in controlling cell proliferation, transformation, apoptosis, and tumorigenesis [1–3]. Among the ETS family members, Friend leukemia virus integration 1 (FLI1) was first identified as a proto-oncogene activated by proviral integration in F-MuLV-induced erythroleukemias [4]. FLI1 is preferentially expressed in hematopoietic cells and tissues, where it regulates hematopoietic stem cell self-renewal and differentiation [5, 6]. Aberrant expression of FLI1 may act as a critical driver in the development of hematological malignancies [7–10]. Overexpression of FLI1 in erythroblasts causes inhibition of differentiation and ultimately the development of pre-T cell lymphoblastic leukemia/lymphoma [11]. In addition to erythroleukemia, the aberrantly deregulated FLI1 is also associated with other hematological malignancies, including pre-T cell lymphoblastic lymphoma, acute myeloid leukemia, and B cell lymphomas [1].
Recent studies have shown that FLI1 is also aberrantly expressed in some solid tumors, including Ewing sarcoma [12, 13], metastatic melanomas [14], nasopharyngeal carcinoma [13], and non-Ewing soft tissue tumors [15]. A series of clinical studies from our lab showed that FLI1 is overexpressed in breast cancers [16] and lung cancers [17]. In patients with breast cancer, expression of FLI1 is strongly correlated with advanced stage, poor differentiation, and lymph node metastasis. In highly metastatic human breast cancer cells, knockdown of FLI1 significantly attenuated tumor metastasis through the Rho GTPase pathway [16]. In non-small cell lung cancer, the FLI1 expression score is associated with the stage of SCLC. In these cells, FLI1 promoted tumor metastasis by activating the miR-17-92 cluster family [17].
However, little is known about the molecular mechanisms underlying the aberrant activation of FLI1 in these solid tumors. In this study, we harnessed a novel CRISPR Cas9-guided promoter immunoprecipitation (CasIP) assay to identify the molecular components that interact with the FLI1 promoter. We hypothesized that these components might actively participate in the control of FLI1 in the development of breast cancers. Using this CasIP assay, we identified FECR1, a FLI1 exonic circular RNA that binds to the FLI1 promoter and epigenetically activates FLI1 in breast cancer cells.
Results
CasIP identifies a novel FLI1 exonic circular RNA
To determine the role of FLI1 in breast cancer, we first examined its expression in tumor samples collected from patients with breast cancer. Using immunohistochemical staining, we found that FLI1 was significantly activated in breast tumor tissues as compared with adjacent normal tissues (Fig. 1a, b; red arrows). There was also a relatively high abundance of FLI1 in breast cancer tissues associated with metastases (Additional file 1: Figure S1A-S1B) and in advanced stages (Additional file 1: Figure S1C-S1D).
Fig. 1.

Identification of FLI1 circular RNA by Cas9IP. a Overexpression of FLI1 in breast cancer tissues. FLI1 expression was quantitated by immunohistochemical staining and was evaluated as the expression score. **p < 0.01 in breast carcinoma tissues as compared with their adjacent tissues. b High expression of FLI1 in carcinoma as compared with adjacent tissues. Red arrow: dark brown immunohistochemical staining of FLI1 oncoprotein. c CRISPR Cas9-guided chromatin immunoprecipitation (CasIP). Cas9, CRISPR Cas9; FLI1 gRNA, Cas9 guiding RNAs that target the FLI1 promoter. Cas9 binds to the FLI1 promoter through a mechanism of base pairing between the gRNA and target DNA. After fixation, the Cas9-FLI1 promoter chromatin complex was immunoprecipitated by an anti-Cas9 antibody. The CasIP-captured RNAs were sequenced to identify the RNA components that regulate the gene activity in breast cancers. d Identification of FLI1 circular RNA by CasIP. In the FLI1 Cas9-gRNA cassette vector, two Cas9 gRNAs are transcribed by U6 and H1 promoters, respectively, and guide Cas9 to the FLI1 promoter. The CasIP sequencing identifies a novel FLI1 exonic circular RNA that interacts with the gene promoter. e Enrichment of FECR1 in the FLI1 promoter. After CasIP, the captured RNAs were reverse transcribed to quantitate the abundance of FECR1 in the Cas9-captured promoter complex. M, 100 bp marker; IgG, ChIP with antibody control; Cas9, ChIP with anti-Cas9 antibody; nAb, the ChIP negative control, in which the anti dCas9-FLAG antibody was replaced by the equal amount of albumin protein
To identify the molecular components that aberrantly regulate FLI1 in breast cancer, we utilized a novel CasIP approach to immunoprecipitate the FLI1 promoter chromatin complex (Fig. 1c). We hypothesized that identification of the components that participate in the formation of the chromatin interaction network in the gene promoter might suggest mechanisms underlying the aberrant FLI1 activation in tumors.
We constructed a lentiviral vector containing the catalytically inactive CRISPR Cas9 (dCas9) and two FLI1 promoter gRNAs (Fig. 1d and Additional file 1: Figure S2) and transfected it into MDA-MB231 breast cancer cells. After puromycin selection, stable cells were treated with 1% formaldehyde to fix the dCas9-gRNA-FLI1 promoter chromatin complex. To avoid RNA degradation, we performed an in situ reverse transcription to convert chromatin RNAs into cDNA with biotin-dCTP. The FLI1 chromatin complex was immunoprecipitated with an anti Cas9-FLAG antibody. After reversing the crosslinks, the FLI1 promoter-associated cDNAs were separated from genomic DNAs with streptavidin beads and were cloned into pJet vector for sequencing. Through this Cas9 immunoprecipitation sequencing, we identified a novel FLI1 circular RNA that interacted with the FLI1 promoter that we refer to as FECR1 (FLI1 exonic circular RNA).
The interaction of FECR1 with the FLI1 promoter was confirmed using CasIP PCR. We found that FECR1 was enriched in breast cancer cells that were transfected with Cas9-FLI1 gRNAs (Fig. 1e, top panel, lane 2). No CasIP signals were detected in cells that were transfected with Cas9 control gRNAs (gCT, middle panel). Neither was the FECR1 signal detected in the IgG control (lane 1) and the non-antibody control (nAb, lane 3). Thus, FECR1 specifically interacted with the FLI1 promoter in breast cancer cells.
FLI1 circular RNA is derived from back splicing between exon 4 and exon 2
To delineate the structure of this newly identified circular RNA, we used a pair of circular RNA-specific PCR primers to amplify the cDNA fragment that covers the back-splicing site (Fig. 2a); with these primers, the linear FLI1 mRNA would not be amplified.
Fig. 2.

FECR1 is associated with the development of breast cancer. a Formation of FLI1 exonic circular RNA. FECR1 is formed by back splicing of FLI1 exon 4 to exon 2 and is composed of exons 4-2-3. b Detection of FECR1 circular RNA after RNase R digestion. RNA samples were treated with RNase R to remove linear RNAs. For qPCR normalization, the abundance of FECR1 was calculated by standardizing over the spike DNA control and setting the PBS control as 1. **p < 0.01 as compared with PBS and vector controls. c Sequencing of FECR1. Two FECR1 variants were detected by RT-PCR (left panel). V2 was expressed at very low level. We thus focused on the major form V1. The circular RNA (V1) was amplified with a forward primer (JH2532F) that is located at the end of exon 4 and a reverse primer (JH3273R) at the 5′-region of exon 2. Red arrow: back-splicing site. d Expression of FECR1 in breast cancer cell lines. e Expression of FECR1 in breast cancer tissues. Beta-actin was used as the PCR control
To confirm the circularity of FECR1, we treated the total RNA from MDA-MB231 tumor cells with RNase R, which specifically digests linear RNA. After RNase R digestion, the expression of the circular RNA was detected by FECR1-specific PCR primers. After removal of linear RNAs, we still detected the presence of FECR1 in MDA-MB231 cells and FECR1-overexpressing MDA-MB231 cells (Fig. 2b). The FLI1 linear mRNA was digested by RNase R, and only very weak singles were detected by PCR. These data further confirmed that FECR1 is a circular RNA.
To further characterize FECR1, we utilized PCR to amplify the major FECR1 isoform consisting of FLI1 exons 4-2-3 (Additional file 1: V1, Figure S3A-S3B), and the minor FECR1 isoform contained FLI1 exons 5-2-3-4 (Additional file 1: V2, Figure S3C-S3D). In support of this finding, the circular RNA sequencing database also showed the presence of FECR1 circular RNA (Additional file 1: Figure S4). Sequencing analysis revealed that FECR1 V1 was formed by back splicing between FLI1 exon 4 and exon 2, and the V2 variant was jointed between exon 5 and exon 2 (Fig. 2c and Additional file 1: Figure S5). Since V2 was found in such low abundance, we focused on the isoform V1.
We also used RT-PCR to examine the expression of FECR1 in other breast cancer cell lines (Fig. 2d). FECR1 was also expressed in breast cancer tissue samples collected from patients at surgery (Fig. 2e).
FECR1 promotes tumor cell invasion
To characterize its physiological function, we overexpressed FECR1 using a lentiviral FECR1 expression vector. The expression cassette was synthesized by including part of the intron 1 sequence carrying the splicing acceptor sequence, exon 2, exon 3, exon 4, and part of intron 4 containing the splicing donor sequence. The synthetic expression cassette was placed under the control of the CMV promoter. The expression of FECR1 was tracked by the DsRed fluorescence marker in the vector (Fig. 3a).
Fig. 3.

FLI1 circular RNA promotes invasion of breast cancer cells. a Ectopic expression of FECR1 in MDA-MB231 breast cancer cells. The FECR1 expression cassette is composed of FLI1 exons 4-2-3 and the intron fragments containing the back-splicing elements. pCMV, CMV promoter; pEF1a, EF1a promoter. DsRed fluorescent marker was used to track the transfection. b RT-PCR of FECR1. After stable transfection, cells were collected and FECR1 was amplified by PCR. PBS and vector, control groups. c Quantitation of FECR1 by qPCR. **p < 0.01 as compared with PBS and vector controls. d FECR1 promotes cell invasion. Cells that crossed through the collagen-coated membrane of the transwell were stained and photographed. e Quantitation of invading cells. All data shown are mean ± SEM from three independent experiments. **p < 0.01 as compared with PBS and vector control groups
After puromycin selection, stable clones were selected. RT-PCR revealed the overexpression of FECR1 in transfected cells (Fig. 3b, lane 2). Quantitative PCR also validated the overexpression of this circular RNA as compared with vector controls (Fig. 3c).
Recently, we showed that knockdown of FLI1 with small interfering RNAs significantly attenuated the potential for invasion of highly metastatic human breast cancer cells [16]. We thus examined if increased amounts of FECR1 enhances cell invasion. As seen in Fig. 3d, e, ectopic expression of FECR1 enhanced cell invasion as compared with the vector control. These data suggest that like FLI1, FECR1 circular RNA also has the ability to promote metastasis of breast cancer cells.
We also used shRNA to knockdown FECR1 in these tumor cells. As a control, cells were transfected with a random shRNA control. We found that knockdown of FECR1 with shRNA significantly inhibited the invasion of tumor cells (Additional file 1: Figure S6).
RNA reverse transcription-associated trap identifies the FECR1 target genes
As FECR1 binds to the FLI1 promoter, we examined the cellular distribution of FECR1 in MDA-MB231 tumor cells. Cytoplasmic RNA and nuclear RNA were separated and were reverse transcribed. Using quantitative PCR, we found that FECR1 was present in both the cytoplasm and nucleus (Additional file 1: Figure S7).
We then used an RNA reverse transcription-associated trap (RAT) assay [18, 19] to identify a target gene network that interacts with FECR1 in addition to the FLI1 promoter (Fig. 4a). After crosslinking, FECR1 was reverse transcribed in situ using a FECR1 circular RNA-specific complementary primer located at the exon 4-exon 2 splicing site (Additional file 2: Table S1 and Additional file 1: Figure S8). To reduce background from non-specific transcription, the reverse transcription reaction was performed at 65 °C using Maxima Reverse Transcriptase in the presence of biotin-dCTP. The FECR1 biotin-cDNA chromatin complex was pulled down with streptavidin beads. The FECR1-interacting target DNAs were eluted for library sequencing. The RAT-Seq bioinformatic analysis revealed that FECR1 circular RNA bound to multiple genes involved in pathways related to cell growth and proliferation (Additional file 1: Figure S9). Additional file 1: Figure S10 illustrates the partial FECR1 interactome of target genes.
Fig. 4.
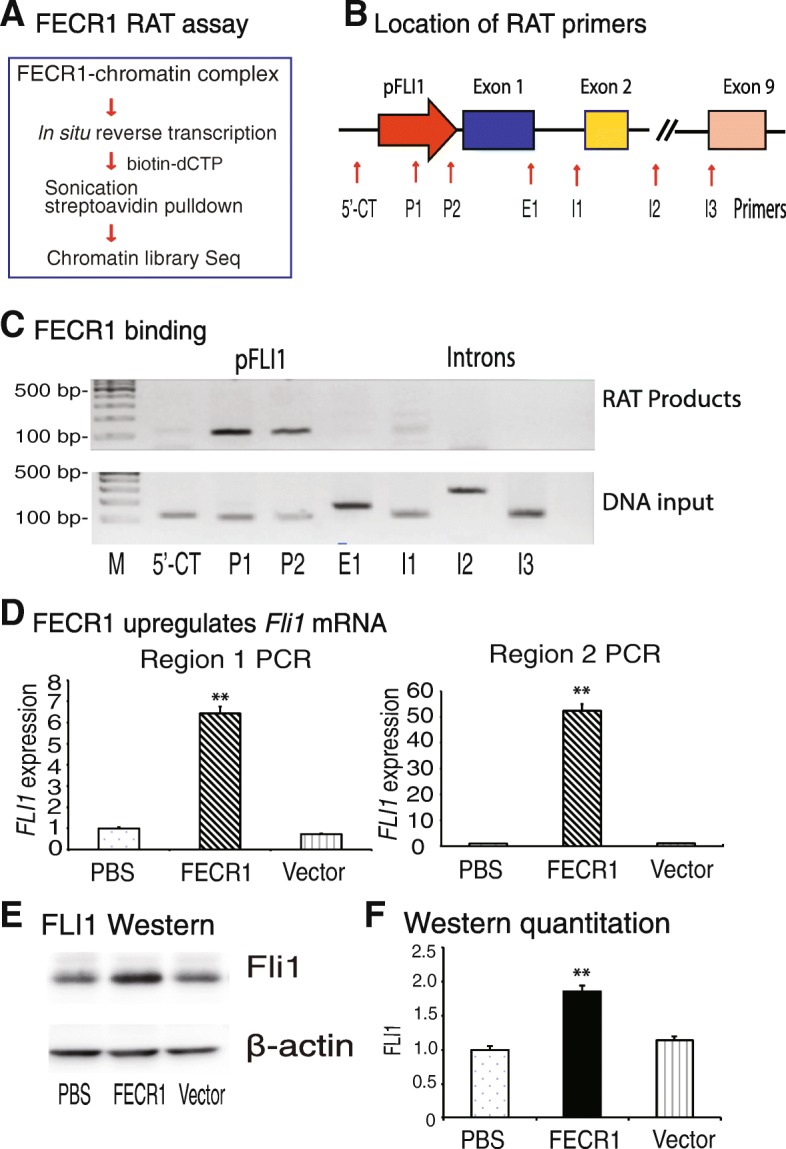
FECR1 upregulates FLI1. a Diagram of the RNA reverse transcription-associated trap (RAT) assay. FECR1 was in situ reverse transcribed using circular RNA-specific primers in the presence of biotin-dCTP. The FECR1-interacting chromatin DNAs were isolated for library sequencing. b Location of PCR primers to detect the interaction of FECR1 at the FLI1 locus. 5′-CT, 5′-upstream control site. c Binding of FECR1 in the FLI1 locus. The FECR1 RAT-captured chromatin DNAs were amplified by PCR using primers covering the FLI1 locus. Note the binding of FECR1 in the FLI1 promoter (P1, P2). d Activation of FLI1 by FECR1. Expression of FLI1 was quantitated by qPCR using two pairs of primers that cover different regions of FLI1. Region 1, the PCR product covers exon 4 to exon 6; region 2, the PCR product covers exon 3 to exon 4. **p < 0.01 as compared with PBS and vector control groups. Both qPCR data show that the overexpressed FECR1 upregulates the linear FLI1 mRNA. e Western blot of FLI1 protein. Cells that were stably transfected with FECR1-overexpression vector, vector control, and PBS were collected for Western blotting. f Quantitation of FLI1 oncoprotein Western blot. **p < 0.01 as compared with PBS and vector control groups
We then designed a series of PCR primers to cover the FLI1 gene locus (Fig. 4b) and examined the binding sites of FECR1. We found that FECR1 bound primarily to the promoter of FLI1 (Fig. 4c, pFLI1, P1–P2). There was no binding of the circular RNA to remote intron regions (I1–I3) and the 5′-upstream control region (5′-CT).
FECR1 binds to the FLI1 promoter in cis, and it is estimated that it may regulate its activity. To determine if FECR1 affects the activity of FLI1, we used two pairs of PCR primers (exons 4–6 and exons 3–4) to quantitate the expression of FLI1 in the FECR1-expressing cell clones. As seen in Fig. 4d, quantitative PCR showed that ectopic expression of the circular RNA significantly upregulated FLI1 as compared with the PBS and vector controls. Using Western blot, we found that overexpression of FECR1 significantly upregulated FLI1 in the treated cells (Fig. 4e, f). These data suggest that FECR1 binds to the promoter of FLI1, where it activates the gene transcription.
FECR1 induces extensive CpG DNA demethylation in the FLI1 promoter
We then examined the epigenetic mechanisms underlying the upregulation of FLI1 by this circular RNA. The binding sequences of FECR1 to the FLI1 promoter are very CpG-rich (Additional file 1: Figure S11). In order to learn if the FLI1 promoter is epigenetically regulated by FECR1, we focused on the methylation status of the CpG islands in the proximal promoter of FLI1 (Fig. 5a).
Fig. 5.

FECR1 induces DNA demethylation in the FLI1 promote. a CpG islands in the FLI1 promoter. In order to detect the status of DNA methylation in FECR1-expressing cells, we designed five pairs of methylation-specific primers located at each CpG island. Three restriction enzymes were used to separate methylated and unmethylated DNAs. b FECR1 induces DNA demethylation. DNA methylation was measured by combined bisulfite restriction analysis (COBRA). PCR products from FECR1-expressing cells and vector control cells were digested by TaiI, Bsh1236I, and BstB1 to separate the unmethylated and methylated DNAs. They recognize and digest the methylated ACGT, CGCG, and TTCGAA sites, respectively. After treatment with sodium bisulfate, unmethylated cytosines were converted to uracils, and the ATGT, TGTG, and TTTGAA sites are not digested by these enzymes. After digestion, unmethylated and methylated DNAs were separated on 3% agarose gels. Note the uncut demethylated bands in FECR1-expressing cells
We used sodium bisulfite sequencing to assess DNA methylation in the FLI1 promoter. After bisulfite treatment, three restriction enzymes were used to distinguish the methylated and unmethylated CpGs, including TaiI, Bsh1236I, and BstB1 (Fig. 5b). Bisulfite treatment converted the unmethylated CpGs into TpGs that will be not cut by these enzymes. Remarkably, we found that FECR1 induced extensive DNA demethylation when compared with the vector controls. Extensive DNA demethylation in the promoter correlates with the upregulation of FLI1 in FECR1-expressing tumor cells.
FECR1 promotes tumor metastasis by coordinately regulating DNMT1 and TET1
We then focused on the molecular mechanisms by which FECR1 epigenetically activates FLI1. DNMT1 is a key methyltransferase required for the maintenance of DNA methylation in mammals. It predominantly catalyzes methylation at hemimethylated CpG di-nucleotides [20]. Given the fact that FECR1 induces DNA demethylation in FLI1 promoter, we examined whether DNMT1 was involved in this epigenetic control process.
First, we analyzed FECR1 RAT sequencing data and found that FECR1 was enriched in the promoter area of DNMT1, a methyltransferase that is essential for the maintenance of DNA methylation (Fig. 6a), where histone 3 lysine 27 (H3K27) is highly acetylated. We then quantitated the abundance of DNMT1 in treated cells. Ectopic expression of FECR1 significantly reduced the abundance of DNMT1 mRNA as compared with the controls (Fig. 6b). These data thus suggest that FECR1 binds to the DNMT1 promoter, where it downregulates DNMT1 transcription.
Fig. 6.
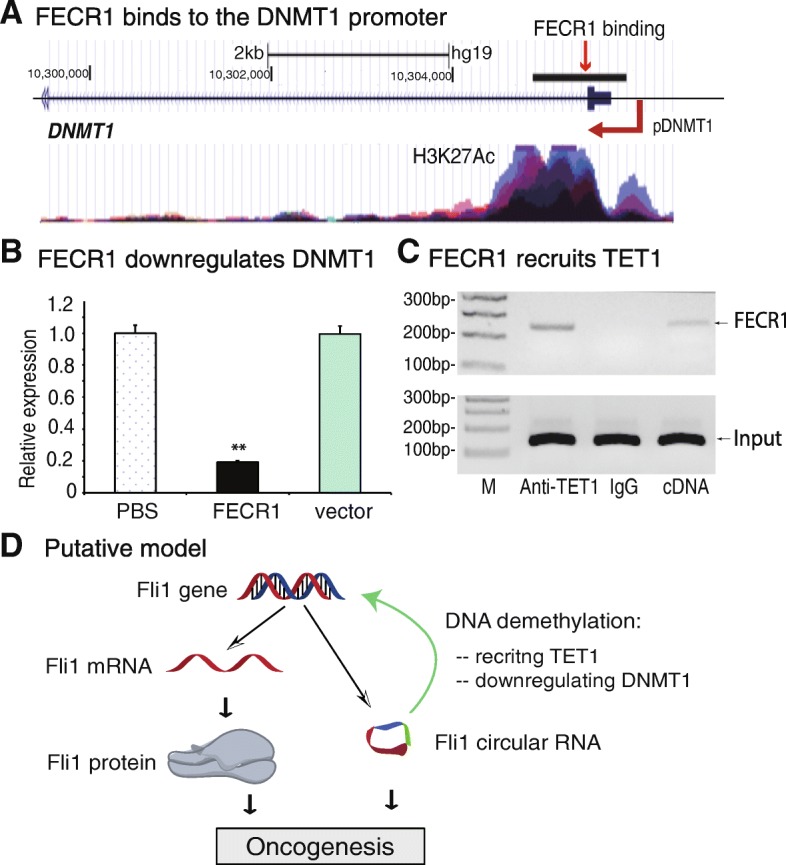
FECR1 coordinately regulates DNMT1 and TET1. a Binding of FECR1 to the DNMT1 promoter. After RAT sequencing, the FECR1 binding sequences were blasted to the human genome at the UCSC website. Histone 3 lysine 27 (H3K27) acetylation signal was also shown correspondingly. b FECR1 downregulates DNMT1. Expression of DNMT1 was measured by qPCR. **p < 0.01 as compared with PBS and vector control groups. c FECR1 recruits TET1 enzyme. RNA-chromatin immunoprecipitation (RIP) was performed to identify FECR1-TET1 binding. The TET-FECR1 chromatin complex was immunoprecipitated with an antibody against TET1. After removal of crosslinking, the immunoprecipitated RNAs were reverse transcribed, and the TET-interacting FECR1 was measured by PCR. IgG was use as the antibody control, and cDNA was used as the positive control. d Putative model of FECR1 in breast cancer. In addition to the conventional FLI1 mRNA-oncoprotein model, FLI1 also produces circular RNA FECR1. Through the interaction with FLI1 promoter, FECR1 recruits TET1 demethylase and induces extensive DNA demethylation in the CpG islands. In addition, FECR1 also inhibits DNMT1, the critical enzyme that maintains DNA demethylation during DNA replication. Working together, FECR1 activates FLI1, which in turn promotes tumor cell invasion in breast cancers
The status of DNA methylation in a given gene promoter is also determined by TET1, a Fe(II)/2-oxoglutarate-dependent dioxygenase. TET1 is a critical factor that induces the oxidation-deamination mechanism underlying active DNA demethylation [21, 22]. To determine if the binding of FECR1 to the FLI1 promoter would regulate this demethylation process, we used an RNA-chromatin immunoprecipitation (RIP) method to pull down TET1-interacting complexes. The pulled-down RNAs were reverse transcribed and quantitated by PCR using primers specific for FECR1. Using this assay, we detected an enrichment of FECR1 in the TET1 antibody-precipitated complex (Fig. 6c).
To further confirm the interaction between FECR1 and TET1, we collected FECR1-overexpressing and vector control MDA-MB231 cells and performed chromatin immunoprecipitation with an antibody against TET1 (Additional file 1: Figure S12). Using PCR primers from the FLI1 locus, we observed an enhanced binding of TET1 to the FLI1 promoter (P1, P2) in FECR1-overexpressing cells as compared with that in the vector control group. Taken together, these data suggest that FECR1 circular RNA may regulate its target genes, like FLI1, by binding to the gene promoter and recruiting TET1 DNA demethylase to induce DNA demethylation.
We also used quantitative PCR to measure the expression of several other FECR1 target genes. We found that in addition to the upregulated FLI1 and the downregulated DNMT1, FECR1 also significantly enhanced the expression of the SERTED2 gene in FECR1-expressing cells (P < 0.01) (Additional file 1: Figure S13).
Discussion
The present study demonstrates a non-canonical function for the FLI1 in the development of breast cancer. We uncovered a novel pathway by which FLI1 controls tumor metastasis in conjunction with its exonic circular RNA FECR1. To explore the molecular components that activate FLI1 in breast cancer, we utilized a CasIP assay to identify a FECR1 circular RNA that interacts with the FLI1 promoter. Increased expression of FECR1 enhanced tumor invasion in MDA-MB231 breast cancer cells. After binding to the FLI1 promoter, the circular RNA recruited TET1 demethylase which demethylated the promoter CpG islands. FECR1 also bound to the DNMT1 promoter and downregulated its transcription although the specific mechanisms were unknown. Together, FECR1 induced extensive DNA demethylation in the FLI1, thereby enhancing its expression to promote metastasis (Fig. 6d). In a related study, we recently found that knockdown of FECR1 with shRNA significantly inhibited tumor metastasis and prolonged tumor survival in a small cell lung cancer xenograft model in nude mice [23].
FLI1 is a member of the ETS family of transcription factors, sharing an ETS domain that binds specifically to their target promoters. Increased expression of FLI1 is associated with poor survival in cancer patients [24]. It is now clear that FLI1 promotes tumor growth by regulating a variety of target genes, including Bcl-2 in the regulation of apoptosis, gata-1 and RB1 in erythroid differentiation, MDM2 in the destabilization of the anti-apoptotic protein P53, VEGF-A in angiogenesis, and the Notch1 signaling pathway [1, 11, 25–28]. Additionally, the oncoprotein also negatively regulates tumor suppressors, including TP53 and RB1. Overall, like other members of the ETS family of transcription factors, FLI1 regulates the expression of oncogenes, tumor suppressor genes, and other genes involved in blood vessel formation, invasion, and metastasis. It is conventionally believed that the proto-oncogene functions primarily through the expression of its oncoprotein product.
In this study, however, we have identified a new player in this malignant cascade. FECR1 circular RNA, a transcriptional byproduct of the proto-oncogene, promotes tumor metastasis at the level of gene transcription. FECR1 is a 571-bp circular RNA, consisting of FLI1 exons 4-2-3. Using RNA reverse transcription-associated trap (RAT) sequencing, we demonstrate that this circular RNA binds to a variety of target genes that are involved in cell proliferation, transformation, apoptosis, and tumorigenesis. Notably, FECR1 binds to its own gene, FLI1, in cis and recruits TET1 demethylase. TET1 (ten-eleven translocation 1) is a member of a DNA hydroxylase family that possesses enzymatic activity toward converting 5mC into 5-hydroxymethylcytosine, 5-formylcytosine, and 5-carboxylcytosine through three consecutive oxidation reactions [29–32]. By inducing DNA hypomethylation in the gene promoter, FECR1 activates FLI1. Thus, FECR1 activates a positive feedback mechanism which promotes oncogene transcription. Unlike the conventional FLI1 oncoprotein that regulates its downstream target genes, FECR1 functions by targeting the FLI1 gene transcriptionally via a cis mechanism.
We also demonstrate that FECR1 binds to the DNMT1 promoter in trans. After promoter binding, FECR1 silences DNMT1 gene activity. In mammals, the genomic methylation process is executed by DNA methyltransferases DNMT1, DNMT2, DNMT3a, and DNMT3b [21, 33, 34]. Among these three methyltransferases, DNMT3A and DNMT3B are referred to as de novo methyltransferases that are responsible for establishing DNA methylation patterns and genomic imprints during embryogenesis. DNMT1, on the other hand, is essential for the maintenance of DNA methylation in the genome. During DNA replication, DNMT1 recognizes semi-methylated CpG as substrate and restores the specific methylation pattern on the daughter strand in a full copy of the parental DNA [20, 35, 36]. In this study, we show that FECR1 significantly downregulates DNMT1. We predict that the downregulation of DNMT1 by FECR1 may cause activation of many target genes, including FLI1, that are associated with tumor growth. However, it is unclear how the binding of FECR1 induces downregulation of DNMT1. Future studies are needed to address the specific mechanisms by which FECR1 downregulates its downstream target genes, like DNMT1.
Circular RNAs belong to the category of long noncoding RNAs. However, recent studies have demonstrated that circular RNAs are also translated into functional peptides. With a combination of bioinformatic tools, Legnini et al. found that zinc finger protein circular RNA cir-ZNF609 is associated with heavy polysomes and is translated into a protein that controls myoblast proliferation [37]. Using ribosome footprinting, Pamudurti et al. demonstrated that a group of circRNAs was associated with translating ribosomes [38]. Sequence analysis shows that FECR1 contains the translation initiation codon ATG for FLI1. If actively translated, it is predicted that FECR1 may produce a short truncated FLI1 (53 aa). Alternatively, it may use its internal ATGs as the initiation site and produces several short peptides. Future studies should validate the presence of these circular RNA-derived peptides and evaluate their functions in breast cancer.
It should be noted that the EWS/FLI1 t(11;22) translocation, although occurring in more than 80% of the cases with Ewing sarcoma/primitive neuroectodermal tumor (EWS/PNET), has not been reported in solid tumors. In the latter case, FLI1 is activated primarily by gene upregulation. In breast cancer, aberrant expression of FLI1 correlates with advanced stage, poor differentiation, lymph node metastasis, and disease-free survival [16, 39]. Knockdown of FLI1 attenuates tumor metastasis through multiple pathways, including the Rho GTPase pathway [16] and the epithelial-mesenchymal transition pathway [39]. Thus, FLI1 may function as a tumor enhancer or promoter in solid tumors. Future studies are needed to address whether the FLI1 gene may undergo subtle genetic alterations in solid tumors, including breast cancer.
Conclusion
We have discovered a novel epigenetic pathway by which FLI1 contributes to tumor metastasis. We identified a novel FLI1 circular RNA FECR1 that interacts with the FLI1 promoter. FECR1 binds to the FLI1 promoter in cis and recruits TET1 demethylase. FECR1 also downregulates DNMT1 in trans. Together, this circular RNA may regulate metastasis of breast cancer cells by coordinating DNA methylation and demethylation in target genes that are involved in tumor growth. These data thus suggest that FLI1 circular RNA may serve as a potential therapeutic target in the development of therapeutic interventions for metastatic breast cancer.
Materials and methods
Cell culture
Human breast cancer cell lines (MDA-MB231, MCF7, SKBR3, T47D, BT474, ZR751) and viral packaging 293 T cells were purchased from ATCC (Manassas, VA). Cells were routinely maintained in DMEM medium (Sigma, MO) containing 10% (v/v) fetal bovine serum (Sigma, MO), 100 U/ml of penicillin sodium, and 100 μg/ml of streptomycin sulfate (Invitrogen, CA), at 37 °C in 5% CO2.
Breast cancer tissue samples
The study protocol was approved by the Research Ethics Board of the First Hospital of Jilin University [16, 40]. Informed consent was obtained from patients with breast cancer. Formalin-fixed and paraffin-embedded tissues of breast cancer were obtained from the biological sample library of the First Hospital of Jilin University. The pathological diagnosis was made in accordance with the histological classification of tumors developed by the World Health Organization. Tumor stage was defined according to the American Joint Committee on Cancer/International Union Against Cancer classification system. Tumors were histologically graded according to the Elston and Ellis method.
Immunohistochemical staining of FLI1 oncoprotein
Expression of FLI1 in breast cancer tissues was evaluated by immunohistochemical staining as previously described [16]. Briefly, tissue slides were de-paraffinized, rehydrated, and incubated in 3% hydrogen peroxide for 15 min to block endogenous peroxidase activity. The slides were incubated with primary anti-FLI1 polyclonal antibodies (Neomarker) at 4 °C overnight. After washing with PBS, the secondary antibodies (biotinylated goat anti-rabbit immunoglobulin) and streptavidin peroxidase complex reagent were added. The FLI1 signal was visualized using Polink-2 HRP DAB Detection kit.
The abundance of FLI1 was quantitated by immunoreactivity scoring as evaluated by two independent investigators. The intensity of FLI1 staining was scored as 0 (negative), 1 (weak), 2 (moderate), and 3 (intense). The immunoreactivity score was calculated as the percentage of positive cells per field multiplied by the intensity of staining.
Construction of plasmids and viral transfection
A modified Cas9-guided chromatin immunoprecipitation assay [41, 42] was used to identify components that bind to the promoter of FLI1. The U6-gRNA1-T5-H1-gRNA2 cassette was synthesized by linking two Cas9-FLI1 gRNAs with the U6 promoter and H1 promoter. FLI1-gRNAs included FLI1-gRNA1 5′-GGGGTTGAGGACACGTGCTG-3′ and FLI1-gRNA2 5′-GAGCCAATATTCCGTAGCAT-3′. The expression cassette was ligated at the Pme I and Not I sites in the lenti CRISPR-EGFP sgRNA 2 vector (Addgene Plasmid #51761), in which the wild-type Cas9 was replaced with the catalytically inactive dCas9 (Additional file 1: Figure S2).
Lentiviruses were packaged in 293 T cells using lipofectamine 3000 (Invitrogen, USA) with 2 μg of lentiviral expression and packaging plasmids. Viral supernatants were collected at 24 and 48 h post transfection. After addition of polybrene (8 μg ml−1), the supernatants were added in MDA-MB231 cells for transfection.
Matrigel invasion assay
Invasion assays were carried out using 6-well BD Biocoat Matrigel Invasion Chambers (BD Biosciences, San Jose, CA) according to the manufacturer’s recommendations [43]. Briefly, 1.25 × 105 cells in 0.5 ml DMEM medium were added to the inner chambers of Matrigel-coated wells with DMEM medium containing 10% serum in the bottom chamber. The cells were incubated for 24 h at 37 °C, and cells that did not invade through the pores were removed by a cotton swab. Cells on the lower surface of the membrane were stained with crystal violet and counted.
RNA preparation and real-time PCR
Total RNA was extracted by TRIzol reagent (Sigma, MO) from cells and stored at − 80 °C. RT-PCR reaction was performed with an Eppendorf Thermol Cycler. The target amplification was performed by PCR of 1 cycle at 95 °C for 5 min; 33 cycles at 95 °C for 20 s, 60 °C for 15 s, and 72 °C for 20 s; and 1 cycle at 72 °C for 5 min. Quantitative real-time PCR was performed using 2× HotSybr Real-time PCR Kit (Mclab, CA). The threshold cycle (Ct) values of target genes were assessed by quantitative PCR in triplicate using a sequence detector (ABI Prism 7900HT; Applied Biosystems) and were normalized over the Ct of the β-ACTIN control.
Cas9-gRNA-guided chromatin immunoprecipitation
A Cas9-guided chromatin immunoprecipitation assay [41, 42] was used to identify components that bind to a target gene DNA fragment. In this study, we constructed the dCas9-FLI1 promoter-gRNA vector by cloning two FLI1 promoter gRNAs into the dCas9-2x gRNA vector that contains a mutated Cas9 (dCas9) and the tandem U6 and H1 promoters. Specifically, two oligonucleotides covering guiding RNA (gRNA) from the Fli1 promoter were synthesized and inserted immediately downstream of the U6 and H1 promoters, respectively, followed by the Cas9-hairpin RNA-(T)5 sequence.
The dCas9-Fli1 promoter lentivirus was produced in 293 T cells as previously described. The viral supernatants were filtered through a 0.45-μm filter, concentrated by a PEG-IT kit (SBI, CA), aliquoted, and stored in a − 80 °C freezer. An aliquot of the Cas9-Fli1 promoter-gRNA lentivirus was used to transfect MDA 231 cells. The stable cells were selected in 1 μg/ml puromycin and collected for immunoprecipitation following the method as described previously. In brief, cells were fixed with 1% formaldehyde and sonicated for 180 s (10 s on and 10 s off) on ice with a Branson sonicator with a 2-mm microtip at 40% output control and 90% duty cycle settings. The sonicated chromatin TNAs containing dCas9-gRNA-Fli1 promoter complex were immunoprecipitated with Cas9 antibody (#ab191468, Abcam, MA). After reversal of cross-linking and proteinase K treatment, the Cas9-bound chromatin DNA and RNA were released and subjected to DNA/RNA sequencing and analyses.
Identification of the FECR1 target genes by RNA reverse transcription-associated trap (RAT)
A RAT assay [18, 44] was modified to identify the interacting target genes of FECR1. Specifically, cells were cross-linked with 2% formaldehyde and lysed with hypotonic buffer (10 mM Hepes, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.4% NP-40, RNase inhibitor 100 U/ml, 1× protease inhibitors). Nuclei were suspended in 1× reverse transcription buffer. Reverse transcription was performed using FECR1-specific antisense primers and biotin-dCTP. To reduce non-specific reaction, the transcription was performed with Maxima Reverse Transcriptase (Thermo Fisher Scientific, CA) at 65 °C for 50 min. The reaction was stopped by adding 4 μl 0.5 M EDTA. After nuclear lysis, the chromatin complex was subjected to sonication for 180 s (10 s on and 10 s off) on ice with a Branson sonicator. The biotin-FECR1 cDNA/chromatin DNA complex was pulled down with biotin-streptavidin magic beads (Invitrogen, CA). After reversing the cross-links and washing with 10 mg/ml proteinase K at 65 °C for 2 h and treatment with 0.4 μg/ml RNase A for 30 min at 37 °C, the genomic DNA that interacts with the circular RNA was extracted and digested by Mbo I and ligated with the NEBNext adaptors (NEBNext® ChIP-Seq Library Prep Master Mix) to construct the library for Illumina sequencing (Shanghai Biotechnology, Shanghai). For the control group, a random RAT oligo (Additional file 2: Table S1) was used to generate RAT control library for Illumina sequencing.
Confirmation of FECR1 as a circRNA by RNase R treatment
To confirm FECR1 as a circular RNA, the RNA samples were treated with RNase R to remove the linear RNA [37, 45]. The untreated RNAs were used as the control. For RNase R treatment, 1 μg of total RNA was treated with 2 U/μg of RNase R (Epicentre Technologies, Madison, WI), at 37 °C for 30 min. For qPCR normalization, 1 pg spike-in DNA was added to each reaction [37]. Following phenol/chloroform/ethanol extraction, RNAs were converted into cDNAs by reverse transcription and quantitated by qPCR using FECR-specific primers. The abundance of FECR1 was calculated by standardizing over the spike-in DNA control [37].
RAT-seq data analysis
Raw data were filtered with Fastx (version: 0.0.13), and approximately 12 million single-end reads of 50 bp length from RAT-seq were mapped to hg38 reference genome using bowtie2 (version 2.2.5) with default parameters [46]. Only reads with at most one mismatch and a Phred quality score greater than or equal to 30 were kept for further analysis. Multiple reads that map to the exact same coordinates were further filtered out as PCR duplicates. The remaining high-quality mapping reads were extended to 500 bp fragments, which were then used to compute genome-wide coverage at 5 kb resolution with bedtools genomecov function [47]. The resulting coverage tracks (bedgraph file) were visualized in the UCSC genome browser.
DNA methylation analysis
Total cellular DNAs were extracted by Genomic DNA Purification Kit (Thermo Scientific, CA) and treated with sodium bisulfite using EZ DNA Methylation™ Kit (Zymo, CA) [48]. Bisulfite-treated DNA was amplified by polymerase chain reaction (PCR) under liquid wax in a 6-μl reaction containing 2 μl of 3× Klen-Taq I Mix, 2 μl template DNA, and 1 μl of each 2.5 μM primer. After incubation at 95 °C for 5 min, DNA was amplified by 38 cycles of 95 °C for 20 s, 60 °C for 20 s of annealing and 72 °C for 20 s of extension, and finally with extension at 72 °C for 5 min. Methylation PCR primer sequences are listed in Additional file 2: Table S1.
The status of DNA methylation was determined by restriction enzyme digestion [49]. Genomic DNA was treated with sodium bisulfite to convert the unmethylated cytosines into uracils. The treated genomic DNA was purified and amplified by PCR. After bisulfite treatment and PCR reaction, unmethylated CpGs will be converted into TpGs, while methylated CpGs remain unchanged. Three restriction enzymes (TaiI/Bsh1236I/BstBI) were used to separate the methylated and unmethylated CpG DNAs. Briefly, PCR DNAs were digested by TaiI/Bsh1236I/BstBI at 37 °C for 30 min and separated on 2% agarose gel. TaiI recognizes the ACGT site, Bsh1236I recognizes the CGCG site, and BstBI digests the TTCGAA site. The unmethylated CpGs were not to be digested by these restriction enzymes.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was used to examine the binding of TET1 to the FLI1 promoter and was performed using the method as described previously [50]. In brief, FECR1-overexpressing and vector control cells were fixed with 1% formaldehyde, sonicated on ice for 180 s (10 s on and 10 s off), and immunoprecipitated with an anti-TET1 antibody (Invitrogen, CA). The isotype-matched was used as the ChIP control. An aliquot of cell lysates was served as the input DNA control. Precipitated DNA was subjected to quantitative PCR analysis. The qPCR data were first adjusted over the input and were further standardized over the IgG control [43].
RNA-chromatin immunoprecipitations (RNA ChIP)
RNA-CHIP was performed based on the protocol by Zhang et al. with minor modifications [51]. MDA-MB231 cells were transfected with FECR1 vector and control vector; RNA immunoprecipitation was performed utilizing reversible chemical crosslinking of RNA-protein interactions by formaldehyde followed by immunoprecipitation using a TET1 antibody (Invitrogen, CA). After immunoprecipitation, extracts were reverse crosslinked; total RNA was extracted using TRIzol reagent (Sigma, MO) and treated with RNase-free DNase I (Invitrogen, CA). RT-PCR was conducted using either random-hexamer as the manufacturer’s instructions (Invitrogen, CA). PCR was performed using a specific set of primers.
Western blot analysis
Cells were lysed in RIPA buffer in the presence of the protease inhibitor cocktail and 1 mM phenylmethylsulfonyl fluoride. Equal amounts of protein were resolved by SDS-PAGE and subjected to Western blot analysis using enhanced chemiluminescence (Pierce). Antibodies to FLI1 and β-actin were obtained from Abcam (MA, USA).
Statistical analysis
Comparisons between groups were analyzed by t test. Comparison was made of groups with high FLI1 expression (score > median score) and low FLI1 expression (score ≤ median score). We assessed score comparisons between groups by one-way ANOVA test. Overall survival (OS) was calculated by using the Kaplan-Meier method, and the differences were assessed by using the log-rank test. p value of less than 0.05 was considered significant. Statistical calculations were performed using SPSS 13.0.
Additional files
Figure S1. FLI1 overexpression in metastatic breast cancer tissues. Figure S2. Location of Cas9 gRNAs in the FLI1 promoter. Figure S3. Sequences of FECR1 variants. Figure S4. FECR1 from circular RNA database websites. Figure S5. Sequencing of Fli1 circle RNAs. Figure S6. FECR1 knockdown inhibited cell invasion in MDA-MB231 cells. Figure S7. Cellular distribution of FECR1 circRNA. Figure S8. Location of the FECR1-specific RAT primer. Figure S9. FECR1 binding by RAT-Seq. Figure S10. The chromatin RAT interactome of the FECR1 circRNA. Figure S11. FECR1 binding sequences in the CpG-rich FLI1 promoter. Figure S12. FECR1 enhances the binding of TET1 to the Fli1 promoter. Figure S13. Expression of target genes in FECR1-overexpressing cells. (PDF 2053 kb)
Table S1. Oligonucleotide sequences of PCR primers. (PDF 119 kb)
Acknowledgments
Funding
This work was supported by Research on Chronic Noncommunicable Diseases Prevention and Control of National Ministry of Science and Technology (2016YFC1303804), Nation Key Research and Development Program of China grant (2016YFC13038000), Key Project of Chinese Ministry of Education grant (311015), National Natural Science Foundation of China grant (81672275), Jilin Provincial Key Laboratory of Biological Therapy (20170622011JC), Provincial Science Fund of Jilin Province Development and Reform Commission (2014 N147 and 2017C022), Natural Science Foundation of Jilin Province grant (20150101176JC), and National Health Development Planning Commission Major Disease Prevention and Control of Science and Technology Plan of Action, Cancer Prevention and Control (ZX-07-C2016004) to C.J.; the National Natural Science Foundation of China (31430021), the National Key R&D Program of China (2018YFA0106902), the National Basic Research Program of China (973 Program)(2015CB943303), Natural Science Foundation of Jilin Science and Technique (20180101117JC), and California Institute of Regenerative Medicine (CIRM) grant (RT2-01942) to J.F.H.; the National Natural Science Foundation of China grant (81372835, 81670143) and Jilin Science and Technique International Collaboration grant (20130413010GH) to W.L; and the Department of Veterans Affairs (BX002905) to A.R.H.
Availability of data and materials
The FECR1 RAT-seq data generated in this study have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO accession number GSE117727 [52].
Authors’ contributions
JFH, CJ, and NC conceived and designed the study. NC, XY, GZ, ZL, HY, SZ, WS, XL, LL, ZD, LJ, and LZ performed the experiments. JFH and JC wrote the paper. ARH, SG, and WL reviewed and edited the manuscript. All authors read and approved the manuscript.
Ethics approval and consent to participate
The study was conducted in compliance with the Helsinki Declaration. The study protocol was approved by the Research Ethics Board of the First Hospital of Jilin University (IRB 2015-260). Informed consent was obtained from each participant in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Ji-Fan Hu, Phone: +1 650-493-5000, Email: jifan@stanford.edu.
Jiuwei Cui, Phone: +86 431-8878-2178, Email: cuijw@jlu.edu.cn.
References
- 1.Li Y, Luo H, Liu T, Zacksenhaus E, Ben-David Y. The ets transcription factor Fli-1 in development, cancer and disease. Oncogene. 2015;34:2022–2031. doi: 10.1038/onc.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maroulakou IG, Bowe DB. Expression and function of Ets transcription factors in mammalian development: a regulatory network. Oncogene. 2000;19:6432–6442. doi: 10.1038/sj.onc.1204039. [DOI] [PubMed] [Google Scholar]
- 3.Oikawa T. ETS transcription factors: possible targets for cancer therapy. Cancer Sci. 2004;95:626–633. doi: 10.1111/j.1349-7006.2004.tb03320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ben-David Y, Giddens EB, Bernstein A. Identification and mapping of a common proviral integration site Fli-1 in erythroleukemia cells induced by Friend murine leukemia virus. Proc Natl Acad Sci U S A. 1990;87:1332–1336. doi: 10.1073/pnas.87.4.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hart A, Melet F, Grossfeld P, Chien K, Jones C, Tunnacliffe A, Favier R, Bernstein A. Fli-1 is required for murine vascular and megakaryocytic development and is hemizygously deleted in patients with thrombocytopenia. Immunity. 2000;13:167–177. doi: 10.1016/S1074-7613(00)00017-0. [DOI] [PubMed] [Google Scholar]
- 6.Spyropoulos DD, Pharr PN, Lavenburg KR, Jackers P, Papas TS, Ogawa M, Watson DK. Hemorrhage, impaired hematopoiesis, and lethality in mouse embryos carrying a targeted disruption of the Fli1 transcription factor. Mol Cell Biol. 2000;20:5643–5652. doi: 10.1128/MCB.20.15.5643-5652.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cui JW, Li YJ, Sarkar A, Brown J, Tan YH, Premyslova M, Michaud C, Iscove N, Wang GJ, Ben-David Y. Retroviral insertional activation of the Fli-3 locus in erythroleukemias encoding a cluster of microRNAs that convert Epo-induced differentiation to proliferation. Blood. 2007;110:2631–2640. doi: 10.1182/blood-2006-10-053850. [DOI] [PubMed] [Google Scholar]
- 8.Tyybakinoja A, Saarinen-Pihkala U, Elonen E, Knuutila S. Amplified, lost, and fused genes in 11q23-25 amplicon in acute myeloid leukemia, an array-CGH study. Genes Chromosomes Cancer. 2006;45:257–264. doi: 10.1002/gcc.20288. [DOI] [PubMed] [Google Scholar]
- 9.Kwiatkowski BA, Bastian LS, Bauer TR, Jr, Tsai S, Zielinska-Kwiatkowska AG, Hickstein DD. The ets family member Tel binds to the Fli-1 oncoprotein and inhibits its transcriptional activity. J Biol Chem. 1998;273:17525–17530. doi: 10.1074/jbc.273.28.17525. [DOI] [PubMed] [Google Scholar]
- 10.Kwiatkowski BA, Zielinska-Kwiatkowska AG, Bauer TR, Jr, Hickstein DD. The ETS family member Tel antagonizes the Fli-1 phenotype in hematopoietic cells. Blood Cells Mol Dis. 2000;26:84–90. doi: 10.1006/bcmd.2000.0282. [DOI] [PubMed] [Google Scholar]
- 11.Smeets MF, Chan AC, Dagger S, Bradley CK, Wei A, Izon DJ. Fli-1 overexpression in hematopoietic progenitors deregulates T cell development and induces pre-T cell lymphoblastic leukaemia/lymphoma. PLoS One. 2013;8:e62346. doi: 10.1371/journal.pone.0062346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mhawech-Fauceglia P, Herrmann FR, Bshara W, Odunsi K, Terracciano L, Sauter G, Cheney RT, Groth J, Penetrante R, Mhawech-Fauceglia P. Friend leukaemia integration-1 expression in malignant and benign tumours: a multiple tumour tissue microarray analysis using polyclonal antibody. J Clin Pathol. 2007;60:694–700. doi: 10.1136/jcp.2006.039230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson AD, Pambuccian SE, Andrade RS, Dolan MM, Aslan DL. Ewing sarcoma and primitive neuroectodermal tumor of the esophagus: report of a case and review of literature. Int J Surg Pathol. 2010;18:388–393. doi: 10.1177/1066896908316903. [DOI] [PubMed] [Google Scholar]
- 14.Torlakovic EE, Slipicevic A, Florenes VA, Chibbar R, DeCoteau JF, Bilalovic N. Fli-1 expression in malignant melanoma. Histol Histopathol. 2008;23:1309–1314. doi: 10.14670/HH-23.1309. [DOI] [PubMed] [Google Scholar]
- 15.Trancau IO, Huica R, Surcel M, Munteanu A, Ursaciuc C. Detection of EWS/FLI-1 fusion in non-Ewing soft tissue tumors. J Med Life. 2015;8:85–89. [PMC free article] [PubMed] [Google Scholar]
- 16.Song W, Li W, Li L, Zhang S, Yan X, Wen X, Zhang X, Tian H, Li A, Hu JF, Cui J. Friend leukemia virus integration 1 activates the Rho GTPase pathway and is associated with metastasis in breast cancer. Oncotarget. 2015;6:23764–23775. doi: 10.18632/oncotarget.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lingyu L, Song W, Yan X, Li A, Zhang X, Li W, Zhou XWL, Yu D, Hu JF, Cui J: Friend leukemia virus integration 1 promotes tumorigenesis of small cell lung cancer cells by activating the miR-17-92 pathway. Oncotarget 2017, doi: 10.18632/oncotarget.16715:[Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 18.Sun J, Li W, Sun Y, Yu D, Wen X, Wang H, Cui J, Wang G, Hoffman AR, Hu JF. A novel antisense long noncoding RNA within the IGF1R gene locus is imprinted in hematopoietic malignancies. Nucleic Acids Res. 2014;42:9588–9601. doi: 10.1093/nar/gku549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang H, Ge S, Qian G, Li W, Cui J, Wang G, Hoffman AR, Hu JF. Restoration of IGF2 imprinting by polycomb repressive complex 2 docking factor SUZ12 in colon cancer cells. Exp Cell Res. 2015;338:214–221. doi: 10.1016/j.yexcr.2015.09.016. [DOI] [PubMed] [Google Scholar]
- 20.Hamidi T, Singh AK, Chen T. Genetic alterations of DNA methylation machinery in human diseases. Epigenomics. 2015;7:247–265. doi: 10.2217/epi.14.80. [DOI] [PubMed] [Google Scholar]
- 21.Wu H, Zhang Y. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell. 2014;156:45–68. doi: 10.1016/j.cell.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hill PW, Amouroux R, Hajkova P. DNA demethylation, Tet proteins and 5-hydroxymethylcytosine in epigenetic reprogramming: an emerging complex story. Genomics. 2014;104:324–333. doi: 10.1016/j.ygeno.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 23.Li L, Li W, Chen N, Zhao H, Xu G, Zhao Y, Pan X, Zhang X, Zhou L, Yu D, et al. FLI1 exonic circular RNAs as a novel oncogenic driver to promote tumor metastasis in small cell lung cancer. Clin Cancer Res; 2018. 10.1158/1078-0432.CCR-18-1447. [Epub ahead of print]. [DOI] [PubMed]
- 24.Tsai HP, Tsai TH, Hsieh YJ, Chen YT, Lee CL, Tsai YC, She TC, Lin CL, Chai CY, Kwan AL. Overexpression of Fli-1 in astrocytoma is associated with poor prognosis. Oncotarget. 2017;8:29174–29186. doi: 10.18632/oncotarget.16303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yi H, Fujimura Y, Ouchida M, Prasad DD, Rao VN, Reddy ES. Inhibition of apoptosis by normal and aberrant Fli-1 and erg proteins involved in human solid tumors and leukemias. Oncogene. 1997;14:1259–1268. doi: 10.1038/sj.onc.1201099. [DOI] [PubMed] [Google Scholar]
- 26.Pereira R, Quang CT, Lesault I, Dolznig H, Beug H, Ghysdael J. FLI-1 inhibits differentiation and induces proliferation of primary erythroblasts. Oncogene. 1999;18:1597–1608. doi: 10.1038/sj.onc.1202534. [DOI] [PubMed] [Google Scholar]
- 27.Tamir A, Howard J, Higgins RR, Li YJ, Berger L, Zacksenhaus E, Reis M, Ben-David Y. Fli-1, an Ets-related transcription factor, regulates erythropoietin-induced erythroid proliferation and differentiation: evidence for direct transcriptional repression of the Rb gene during differentiation. Mol Cell Biol. 1999;19:4452–4464. doi: 10.1128/MCB.19.6.4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Athanasiou M, Mavrothalassitis G, Sun-Hoffman L, Blair DG. FLI-1 is a suppressor of erythroid differentiation in human hematopoietic cells. Leukemia. 2000;14:439–445. doi: 10.1038/sj.leu.2401689. [DOI] [PubMed] [Google Scholar]
- 29.Wu H, Zhang Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 2011;25:2436–2452. doi: 10.1101/gad.179184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016;30:733–750. doi: 10.1101/gad.276568.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scourzic L, Mouly E, Bernard OA. TET proteins and the control of cytosine demethylation in cancer. Genome Med. 2015;7:9. doi: 10.1186/s13073-015-0134-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu GL, Walsh CP. Enzymatic DNA oxidation: mechanisms and biological significance. BMB Rep. 2014;47:609–618. doi: 10.5483/BMBRep.2014.47.11.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uysal F, Akkoyunlu G, Ozturk S. Dynamic expression of DNA methyltransferases (DNMTs) in oocytes and early embryos. Biochimie. 2015;116:103–113. doi: 10.1016/j.biochi.2015.06.019. [DOI] [PubMed] [Google Scholar]
- 34.Subramaniam D, Thombre R, Dhar A, Anant S. DNA methyltransferases: a novel target for prevention and therapy. Front Oncol. 2014;4:80. doi: 10.3389/fonc.2014.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li E, Zhang Y. DNA methylation in mammals. Cold Spring Harb Perspect Biol. 2014;6:a019133. doi: 10.1101/cshperspect.a019133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin B, Robertson KD. DNA methyltransferases, DNA damage repair, and cancer. Adv Exp Med Biol. 2013;754:3–29. doi: 10.1007/978-1-4419-9967-2_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Legnini I, Di Timoteo G, Rossi F, Morlando M, Briganti F, Sthandier O, Fatica A, Santini T, Andronache A, Wade M, et al. Circ-ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol Cell. 2017;66:22–37. doi: 10.1016/j.molcel.2017.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pamudurti NR, Bartok O, Jens M, Ashwal-Fluss R, Stottmeister C, Ruhe L, Hanan M, Wyler E, Perez-Hernandez D, Ramberger E, et al. Translation of CircRNAs. Mol Cell. 2017;66:9–21. doi: 10.1016/j.molcel.2017.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahmad S, Mu X, Yang F, Greenwald E, Park JW, Jacob E, Zhang CZ, Hur S. Breaching self-tolerance to Alu duplex RNA underlies MDA5-mediated inflammation. Cell. 2018;172:797–810. doi: 10.1016/j.cell.2017.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kang L, Sun J, Wen X, Cui J, Wang G, Hoffman AR, Hu JF, Li W. Aberrant allele-switch imprinting of a novel IGF1R intragenic antisense non-coding RNA in breast cancers. Eur J Cancer. 2015;51:260–270. doi: 10.1016/j.ejca.2014.10.031. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y, Hu JF, Wang H, Cui J, Gao S, Hoffman AR, Li W. CRISPR Cas9-guided chromatin immunoprecipitation identifies miR483 as an epigenetic modulator of IGF2 imprinting in tumors. Oncotarget. 2017;8:34177–34190. doi: 10.18632/oncotarget.10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fujita T, Fujii H. Efficient isolation of specific genomic regions and identification of associated proteins by engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP) using CRISPR. Biochem Biophys Res Commun. 2013;439:132–136. doi: 10.1016/j.bbrc.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 43.Zhao X, Liu X, Wang G, Wen X, Zhang X, Hoffman AR, Li W, Hu JF, Cui J. Loss of insulin-like growth factor II imprinting is a hallmark associated with enhanced chemo/radiotherapy resistance in cancer stem cells. Oncotarget. 2016;7:51349–51364. doi: 10.18632/oncotarget.9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang H, Li W, Guo R, Sun J, Cui J, Wang G, Hoffman AR, Hu JF. An intragenic long noncoding RNA interacts epigenetically with the RUNX1 promoter and enhancer chromatin DNA in hematopoietic malignancies. Int J Cancer. 2014;135:2783–2794. doi: 10.1002/ijc.28922. [DOI] [PubMed] [Google Scholar]
- 45.Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 46.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi X, Li M, Cui M, Niu C, Xu J, Zhou L, Li W, Gao Y, Kong W, Cui J, et al. Epigenetic suppression of the antitumor cytotoxicity of NK cells by histone deacetylase inhibitor valproic acid. Am J Cancer Res. 2016;6:600–614. [PMC free article] [PubMed] [Google Scholar]
- 49.Yu D, Du Z, Pian L, Li T, Wen X, Li W, Kim SJ, Xiao J, Cohen P, Cui J, et al. Mitochondrial DNA hypomethylation is a biomarker associated with induced senescence in human fetal heart mesenchymal stem cells. Stem Cells Int. 2017;2017:1764549. doi: 10.1155/2017/1764549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang H, Guo R, Du Z, Bai L, Li L, Cui J, Li W, Hoffman AR, Hu JF. Epigenetic targeting of granulin in hepatoma cells by synthetic CRISPR dCas9 epi-suppressors. Mol Ther Nucleic Acids. 2018;11:23–33. doi: 10.1016/j.omtn.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang H, Jiao W, Sun L, Fan J, Chen M, Wang H, Xu X, Shen A, Li T, Niu B, et al. Intrachromosomal looping is required for activation of endogenous pluripotency genes during reprogramming. Cell Stem Cell. 2013;13:30–35. doi: 10.1016/j.stem.2013.05.012. [DOI] [PubMed] [Google Scholar]
- 52.Chen N, Cui J, Hu JF: RAT-seq mapping of the FLI1 exonic circular RNA FECR1 target gene network in MDA-MB231 breast cancer cells. Gene Expression Omnibus; 2018:GSE117727. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?&acc=GSE117727.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. FLI1 overexpression in metastatic breast cancer tissues. Figure S2. Location of Cas9 gRNAs in the FLI1 promoter. Figure S3. Sequences of FECR1 variants. Figure S4. FECR1 from circular RNA database websites. Figure S5. Sequencing of Fli1 circle RNAs. Figure S6. FECR1 knockdown inhibited cell invasion in MDA-MB231 cells. Figure S7. Cellular distribution of FECR1 circRNA. Figure S8. Location of the FECR1-specific RAT primer. Figure S9. FECR1 binding by RAT-Seq. Figure S10. The chromatin RAT interactome of the FECR1 circRNA. Figure S11. FECR1 binding sequences in the CpG-rich FLI1 promoter. Figure S12. FECR1 enhances the binding of TET1 to the Fli1 promoter. Figure S13. Expression of target genes in FECR1-overexpressing cells. (PDF 2053 kb)
Table S1. Oligonucleotide sequences of PCR primers. (PDF 119 kb)
Data Availability Statement
The FECR1 RAT-seq data generated in this study have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO accession number GSE117727 [52].