

Abstract
A series of metallosupramolecular [Fe2L3](BF4)4 “click” cylinders have been synthesized in excellent yields (90%–95%) from [Fe(H2O)6](BF4)2 and bis(bidentate) pyridyl-1,2,3-triazole ligands. All complexes were characterized by elemental analysis, IR, UV-vis, 1H-, 13C- and DOSY-NMR spectroscopies and, in four cases, the structures confirmed by X-ray crystallography. Molecular modeling indicated that some of these “click” complexes were of similar size and shape to related biologically active pyridylimine-based iron(II) helicates and suggested that the “click” complexes may bind both duplex and triplex DNA. Cell-based agarose diffusion assays showed that the metallosupramolecular [Fe2L3](BF4)4 “click” cylinders display no antifungal activity against S. cerevisiae. This observed lack of antifungal activity appears to be due to the poor stability of the “click” complexes in DMSO and biological media.
Keywords: iron(II), CuAAC, metallosupramolecular cylinders, biological activity
1. Introduction
Helicates [1,2,3,4,5,6] are one of the most studied types of discrete metallosupramolecular architectures. With judicious choice of the metal ion and linker ligand cyclic-, doubly-, triply- or quadruply-stranded helicates can be generated. These systems display interesting electronic [7,8,9], optical [10,11,12,13], catalytic [14] and molecular recognition [15,16,17,18,19,20] properties. They have also been used as building blocks to generate novel mechanically interlocked systems [21]. Pioneering work by Hannon and co-workers [22] showed that the triply stranded [Fe2L3]4+ helicate 1 (Figure 1) displays unique biological properties [23,24]. This tetracationic diiron(II) cylinder binds strongly and non-covalently to the major groove of duplex DNA [25,26,27] and, even more remarkably, can bind at the center of three-way (Y-shaped) DNA junctions [28,29]. The interaction of 1 with duplex DNA induces intramolecular DNA coiling [25,26,27]. Additionally, 1 has been shown to display both anti-cancer [30] and anti-bacterial [31] properties, but is not mutagenic or genotoxic [30]. Recently, Scott and co-workers reported that the related flexicates 2 (Figure 1) can bind DNA and act as anti-bacterial agents [32].
Figure 1.

Hannon’s (compound 1) and Scott’s (compound 2) biologically active [Fe2L3]4+ helicates and the CuAAC “click”-derived bis(bidentate) ligands 3.
Due to its reliability, mild reaction conditions and wide substrate scope the copper(I)-catalyzed 1,3-cycloaddition of organic azides with terminal alkynes (the CuAAC “click” reaction) [33,34,35,36,37,38] has become the reaction of choice in the last decade for functional molecule synthesis in a wide range of fields, including the biological [39,40,41,42,43] and materials sciences [44,45,46,47,48]. This synthetic versatility has attracted the attention of coordination chemists and a vast array of 1,4-disubstituted-1,2,3-triazole- containing ligand architectures and the corresponding “click” complexes have been developed in the past five years [49,50,51,52]. We [53,54,55,56,57] and others [58,59] have explored the use of “click” ligands in the development of functionalized metallosupramolecular architectures, and have shown that metallo-macrocycles, cages and coordination polymers can be generated with these 1,4-disubstituted-1,2,3-triazole-containing ligands. In that work we developed a small family of bis-2-(1-R-1H-1,2,3-triazol-4-yl)pyridine ligands 3 (Figure 1) and showed that they assemble into metallomacrocyclic species with Ag(I) ions [57]. We reasoned that these ligands should react with Fe(II) ions to generate metallosupramolecular helicates of a similar size and shape to 1 and 2. Given the wide substrate scope of the CuAAC “click” reaction used to construct the ligands, it should be possible to prepare a library of helicates that would enable convenient structure-activity studies. Herein we report the synthesis and biological properties [60,61,62,63,64,65,66,67,68,69] of a small family of Fe(II) “click” helicates.
2. Results and Discussion
2.1. Synthesis of Model Iron(II) Complexes
As tris(bidentate) Fe(II) complexes of the 2-(1-R-1H-1,2,3-triazol-4-yl)pyridine ligands 4a–b [70,71] had not been described when we began this work, we initially examined the formation of the model Fe(II) complexes 5a–b [72,73]. These complexes were prepared by reacting one of the ligands, either 4a or 4b (3 eq.), with iron(II) tetrafluoroborate hexahydrate (1 eq.) in acetonitrile at room temperature (Scheme 1). These compounds were isolated in excellent yields (90%–95%) as X-ray quality crystals by vapor diffusion of diethyl ether into the acetonitrile reaction mixtures. The presence of both the ligand (3000–2900 cm−1) and the BF4− counter-anions (1049 cm−1) in the isolated orange red solids was confirmed by IR spectroscopy, and elemental analyses were consistent with the expected [Fe(4a or 4b)3](BF4)2 formulation.
Scheme 1.

The synthesis of the model iron(II) complexes 5a–b.
The molecular structures of the complexes 5a–b were confirmed using X-ray crystallography (Figure 2 and Supporting Information). Both complexes were found to contain the expected [Fe(L)3]2+ cations with three of the 2-(1-R-1H-1,2,3-triazol-4-yl)pyridine ligands 4a–b coordinated to the iron atoms in a pseudo-octahedral arrangement. The Fe-Npyridine bond lengths vary between 1.987(4)–2.007(4) Å and Fe-Ntriazole bond lengths ranges from 1.919(4)–1.948(4) Å. These data are consistent with the formation of the low-spin Fe(II) complexes. Interestingly, the complex 5a crystallizes exclusively as the fac isomer, whereas 5b is found to exist only as the mer isomer in the solid state. This diastereoselectivity is presumably caused by crystal packing effects. In contrast, the analogous ruthenium(II) complexes of these ligands exist as a 1:1 mixture of the mer and fac isomers in solution [74,75]. 1H-NMR spectroscopy was employed to further examine this behavior.
Figure 2.

ORTEP diagrams of the solid state structures of fac-5a (a) and mer-5b (b). The thermal ellipsoids are shown at the 50% probability level. Solvent molecules and counteranions are omitted for clarity. Selected bond lengths (Å) and bond angles (°) for the complexes; 5a: Fe1-N1 2.006(3), Fe1-N2 1.941(3) N1-Fe-N2 81.5(1); 5b: Fe1-N1 1.990(5), Fe1-N21 2.007(4), Fe1-N41 1.987(4), Fe1-N2 1.918(4), Fe1-N22 1.947(4), Fe1-N42 1.940(4), N1-Fe-N2 80.6(2); N21-Fe-N22 80.8(2); N41-Fe-N42 81.7(2).
1H-NMR spectra of the iron(II) model complexes 5a–b recorded at room temperature in d3-acetonitrile and d6-acetone (Figure 3 and Supporting Information) show a simple pattern containing one set of proton signals, consistent with the quantitative formation of a single metal-containing species. Compared with the spectra of the ligands 4a–b, the downfield shift of the proton signals of the corresponding iron complexes indicates metal complexation in solution (Figure 3 and Supporting Information). Closer inspection of the spectra shows that most of the peaks are broad at room temperature, indicative of some type of fluxional process. Variable-temperature (VT) 1H-NMR studies in d6-acetone were used to investigate the possible interconversion of the two diasteriomeric (mer and fac) forms of the model iron(II) complexes. Figure 3 shows the results for compound 5a. At, and slightly above, room temperature (298 and 308 K) the spectra of 5a display a single set of broadened resonances. As the solution is cooled these resonances sharpen and then split into two sets of peaks, consistent with the presence of both the mer and fac isomers of 5a at around 208 K. These VT 1H-NMR data suggest that both the mer and fac isomers are present in solution, but at room temperature their interconversion on the NMR time scale leads to a single set of broad 1H-NMR resonances. This behaviour differs from that observed for structurally similar iron(II) [76] and analogous ruthenium(II) [74] complexes and suggests the Fe(II) complexes 5a-b are stereochemically more labile than these related compounds. However, this lability is important, as access to the fac isomer is required for the bis(bidentate) ligands 3 to assemble into iron(II) metallosupramolecular cylinders.
Figure 3.

Partial 1H-NMR (500 MHz, d6-acetone) spectra of the Fe(II) model complex, 5a at (a) 308 K, (b) 273 K, (c) 248 K, (d) 223 K, (e) 208 K.
2.2. Synthesis of Iron(II) Metallosupramolecular Cylinders
The iron(II) metallosupramolecular cylinders 6a–f were synthesized in an analogous fashion to the model complexes 5a–b. The complexes were prepared by reacting each one of the ligands 3a–f (3 eq.), with iron(II) tetrafluoroborate hexahydrate (2 eq.) in acetonitrile at room temperature (Scheme 2). The compounds were isolated in excellent yields (90%–95%) as either X-ray quality orange crystals (compounds 6a–d, 6f) or microcrystalline powders (compound 6e) by vapor diffusion of diethyl ether into the acetonitrile reaction mixtures. Hannon’s related iron(II) metallosupramolecular cylinders 1 and 8 (X = BF4−) were also synthesized to allow direct comparison of the structural and biological properties of these complexes with 6a–f. Complexes 1 and 8 were prepared by reacting the ligand 7 (3 eq.) either with iron(II) tetrafluoroborate hexahydrate (2 eq.) or iron(II) chloride, respectively, in acetonitrile at 328 K for one hour and isolated as purple solids in good yields (90%–94%, Scheme 2). All Fe(II) metallosupramolecular cylinders were characterized by elemental analysis, IR, UV-vis, 1H-, 13C- and DOSY-NMR spectroscopy and for 1 and 8, by ESI-MS. The presence of both the ligand (3000–2900 cm−1) and the BF4− counter-anions (1049 cm−1) in the isolated orange–red (compounds 6a–f) or purple (compound 8) solids was confirmed by IR spectroscopy and elemental analyses were consistent with the expected [Fe2(L)3](BF4)4 formulation.
Scheme 2.

The synthesis of the iron(II) metallosupramolecular cylinders, 1, 6a–f and 8.
2.2.1. Solid State Structures
X-ray quality single crystals were isolated for the complexes 6a–d, f and 8 but they all diffracted modestly and in certain cases synchrotron radiation was required to obtain satisfactory diffraction. Although the weak diffraction was, at least in part, due to the presence of multiple disordered solvent molecules within the crystal lattice (vide infra), the cationic cylinders were all readily identified. In all cases the crystals contained the expected iron(II) metallosupramolecular cylinders [Fe2L3]4+, along with some additional diffuse electron density peaks due to BF4− counter-anions and solvent (usually CH3CN) molecules (Figure 4 and Supporting Information). The cationic cylinders were all well resolved and modeled completely. Where possible, BF4− anions and solvent molecules were also modeled; the SQUEEZE routine within PLATON [77] was applied to the structural model before the final refinement due to the highly diffuse nature of some of the electron density associated with these species. Full details of the structural refinement and the crystallographic data are reported in the supporting information. Our X-ray data for the cylinders 6a and 6d showed these structures to be identical to those reported during the course of our study by Petitjean and co-workers [78].
Figure 4.

ORTEP diagrams of the solid state structures of the iron(II) metallosupramolecular cylinders, (a) 6b, (b) 6c, (c) 6f, and (d) 8. The thermal ellipsoids are shown at the 50% probability level. Solvent molecules, hydrogen atoms and counteranions are omitted for clarity. Selected bond lengths (Å) and bond angles (°) for the complexes; 6b: Fe-N1 1.998 (3), Fe-N2 1.923(3); N1-Fe-N2 81.19(13); 6c: Fe1-N1 2.001(4), Fe1-N2 1.944(3); N1-Fe-N2 81.05(15); 6f: Fe1-N1 1.938, Fe1-N6 2.009, N1-Fe1-N6 81.30; 8: Fe1-N1 1.994(4), Fe1-N3 2.010(4); N1- Fe- N3 80.65(15).
Each of the pyridyl-1,2,3-triazole complexes 6a–d, f contained the expected [Fe2L3]4+ cations and like the model complexes 5a–b, the bond lengths and angles were consistent with the presence of low-spin iron(II). For the most part these complexes obey the odd-even rule [6,79,80]. Compounds 6b–d [78], with an odd number of carbons in their spacer units, crystalize as the Λ∆-mesocate, whereas 6a [78], with an even number of carbons in its spacer unit, was a racemic mixture of the ∆∆- and ΛΛ-helicates. The exception is 6f, which despite having an odd number of atoms in its spacer, crystallizes as a racemic mixture of the ∆∆- and ΛΛ-helicates. This was not unexpected as the pyridyl imine complexes, 1 [81] and 8 (vide infra), with the analogous spacer unit, exist exclusively as helicates in both the solid and solution state. Ligand 3c has two different potential chelate pockets ‒ the bidentate “regular” pocket and a central “inverse” tridentate pocket. Although tridentate complexes of similar “inverse click” ligands [82,83] are known, complex 6c forms the desired [Fe2L3]4+ cylinder. This is presumably because coordination through the N3 nitrogen of the 1,2,3-triazole units, as observed for the bidentate coordination mode, is more thermodynamically favored [70,84]. Related Cu(II) complexes show similar behavior [85]. As the central pyridyl nitrogen atom of 6c is not involved in any coordination interactions, complexes 6b and 6c are essentially isostructural (Supporting Information). Complex 6f has a larger, more rigid spacer unit than the other cylinders and in the solid state this complex has an accessible central cavity filled with two acetonitrile “guest” molecules. None of the other cylinders have a cavity and the complexes with benzyl/pyridyl linkers display stabilizing π-π interactions between their central aromatic rings.
The solid state structure of 8 (the BF4− analogue of Hannon’s complex 1) was also determined using X-ray crystallography. The compound has been crystallographically characterized previously as the PF6− salt [81]. The cylinder 8 crystallizes as a racemic mixture of the ∆∆- and ΛΛ-helicates. As expected, changing the counter-anion from PF6− to BF4− has little effect on the structure of the cylinder (Supporting Information). The structure of 8 allows a direct (size and shape) comparison with the “click” cylinders 6a–d, f. The iron(II)-iron(II) distances (Table 1) and the overlays of the molecular structures (Supporting Information) for 8, 6a, 6b and 6c suggest that these complexes are structurally very similar. The same analysis for 8 with 6e and 6f showed that these pyridyl-triazole complexes are much smaller and significantly larger, respectively, than Hannon’s compounds. Their differing molecular size and shape might give rise to new biological properties.
Table 1.
Metal - Metal distances within the Fe(II) cylinders.
2.2.2. Solution Structures
The solution structures of the cylinders 6a–f and 8 were examined using 1H- and diffusion ordered (DOSY)-NMR spectroscopy (Figure 5, Table 2, and Supporting Information). The compounds 6a–d and 8 appear to maintain their observed solid state structures in solution. 1H-NMR spectra (CD3CN, 298 K) of 6a–d and 8 are generally sharp and display a set of peaks consistent with the quantitative formation of a single species in solution [Figures 5(a–h)]. 1H-DOSY-NMR spectroscopy of 6a–d and 8 provides further support for this idea (vide infra). The triazole (compounds 6a–f) or imine (compound 8) proton signals in the 1H-NMR spectra of the cylinders are shifted downfield relative to their position in the “free” ligands (compounds 3a–f and 7), indicating complexation to Fe(II) ions. Consistent with the observed solid state structures the upfield shifts of proton resonances due to the aromatic spacer units of 6a–c, f and 8 suggest the presence of π-π stacking interactions. The 1H-NMR spectra of 6e–f were different. While they indicate that complexation had occurred, the spectrum of 6e showed the formation of multiple species in solution, with a series of major peaks consistent with the formation of the [Fe2L3]4+ cylinder, along with smaller peaks attributed to the formation of larger oligomeric/polymeric species. This is probably due to the added flexibility of the hexyl linker, as similar behavior has been observed previously [6]. While the crystal structure of 6f indicates exclusive formation of the helicate [Figure 4(c)], 1H-NMR analysis of the complex suggests that it exists as an approximately 1:1 mixture of the rac-helicate and the mesocate in solution [Figure 5(g)]. This is in contrast to the pyridyl imine complexes, 1 and 8, which have the same spacer unit and are exclusively rac-helicates in solution and the solid state [Figure 4(d) and Figure 5(h)]. This difference in behavior appears related to the larger size of 6f compared to 1 and 8. In 1 and 8 the aryl rings of the spacer are in close contact and interdigitate, locking in the helical arrangement. The larger size of 6f lessens this steric interdigitation of the spacer aryl groups making the mesocate arrangement more energetically accessible, thus giving rise to the mixture observed in solution [86].
Figure 5.

Partial 1H-NMR spectra (500 MHz, CD3CN, 298 K) of (a) ligand, 3a, and the Fe(II) cylinders, (b) 6a (c) 6b, (d) 6c, (e), 6d, (f) 6e, (g) 6f, (h) 8. The assignments correspond to the lettering shown in Scheme 2.
Table 2.
Diffusion coefficients obtained from 1H DOSY spectra (CD3CN, 298 K).
| Ligand | Diffusion coefficient (×10−10 m2·s−1) | Fe(II) cylinder | Diffusion coefficient (×10−10 m2·s−1) |
|---|---|---|---|
| 7 | 12.9 | 8 | 6.9 |
| 3a | 12.9 | 6a | 4.7 |
| 3b | 12.8 | 6b | 4.7 |
| 3c | 11.9 | 6c | 4.0 |
| 3d | 14.4 | 6d | 3.9 |
| 3e | 13.3 | 6e | 4.9 |
| 3f | 9.6 | 6f | 3.0 |
1H-DOSY-NMR spectra (CD3CN, 298 K) were obtained for the ligands 3a–f and 7 and the iron(II) cylinder complexes 6a–f and 8 (Table 2, Supporting Information). All of the proton signals in the individual spectra of 6a–d and 8 displayed the same diffusion coefficients (D) indicating that only one species was present in solution. The ligands (compounds 3a–f and 7) showed similar diffusion coefficients, ranging between 9.6–14.4 × 10−10 m2·s−1, consistent with similar molecular sizes. The diffusion coefficients of the iron(II) cylinders (compounds 6a–f and 8) ranged between 3.0–6.9 × 10−10 m2·s−1 indicating that all the cages are similar in size. The Dcylinder/Dligand ratios are approximately 0.3–0.5:1 suggesting that the iron(II) complexes are much larger than the free ligands, providing strong evidence for the retention of the cylinder architectures in CD3CN solution. The DOSY spectrum of 6e was consistent with the existence of this complex as a mixture of the desired [Fe2L3]4+ cylinders, along with larger oligomeric/polymeric species. The major peaks in solution all displayed a diffusion coefficient of 4.9 × 10−10 m2·s−1. This value is similar to the other [Fe2L3]4+ cylinders, indicating that this major species is indeed the dimetallic metallosupramolecular architecture. The other less intense peaks had smaller diffusion coefficients, suggesting the presence of larger oligomeric/polymeric systems in solution. The DOSY spectrum of 6f showed two different sets of proton signals with the same diffusion coefficient of 3.0 × 10−10 m2·s−1 indicating that two species present in solution are indeed the rac-(∆∆- and ΛΛ)-helicates and the (∆Λ)-mesocate.
ESI-MS spectra (CH3CN) were also obtained for the cylinders 6a-f and 8. Despite extensive efforts, including the use of pseudo cold-spray conditions [87,88], ions due to the intact [Fe2L3]4+ cylinders for the “click” complexes 6a–f were not observed. Only peaks due to the “free” ligands and [FeL]n+ fragments could be detected for these complexes. In contrast, an isotopically resolved [Fe2L3]4+ ion was readily observed for the pyridyl imine complex 8 under standard ESI-MS conditions. The pyridylimine complexes appear much more stable than 6a–f under the conditions of the ESI-MS experiment.
2.2.3. Electronic Spectra
UV-vis spectroscopy (CH3CN) of compounds 5a–b, 6a–f and 8 was indicative of the formation of the iron(II) complexes in solution (Figure 6). Titration of [Fe(H2O)6](BF4)2 into acetonitrile solutions of all of the “click” ligands 3a–f or 4a–b gave rise to new absorption features in the visible region of the spectra (300–450 nm). All the complexes 5a–b, 6a–f display these absorption features with maxima for each compound observed at 420–430 nm (Figure 6 and Supporting Information). These absorption maxima are assigned as MLCT bands and are consistent with the formation of low spin tris-(diimine)Fe(II) complexes [22,32,76]. The MLCT bands of the “click” complexes 5a–b, 6a–f are blue-shifted relative to related the pyridylimine- 1, 2 and 8 (λmax = 570–600 nm) [22,32,76] and bipyridine (λmax = 500–540 nm) [20,79]-based iron(II) complexes but occur in a similar region to complexes containing pyridylpyrazole chelators (λmax = 423 nm) [89]. This shift to lower wavelength for the MLCT band of the “click” complexes compared to these other systems is consistent with the reduced π-acceptor nature of the triazole unit [74,75,89,90]. The combined solution data indicate that the [Fe2L3]4+ “click” cylinders are stable in acetonitrile solution and adopt similar structures to those observed in the solid state.
Figure 6.

UV-vis. spectra of Fe(II) cylinders 6a–f and 8 (10−5 M, CH3CN).
2.3. Molecular Modeling (Docking) and Biological Activity Studies
With the molecules in hand we examined the biological properties of the [Fe2L3]4+ cylinders, 6a–f and 8. Hannon and Scott have shown that a key biological target for these metallosupramolecular cylinders is DNA. We therefore examined the interaction between complexes 6a–f and DNA via computational techniques. As the related [Fe2L3]4+ cylinders, 1 and 2, have been shown to interact with both duplex and triplex DNA architectures, docking studies with both forms were carried out.
2.3.1. Duplex Modeling
Molecular docking studies of metal complexes 1(8), 6a, 6b, 6d, and 6f were carried out using generic B-DNA duplex structure (pdb3bse). The docking revealed that all Fe(II) cylinders, including 1(8), fit into the major groove in a similar fashion to that reported previously by Erlend et al. [25] and Hannon et al. [27].
2.3.2. Triplex Modeling
Molecular docking studies of metal complexes 6a, 6b, 6d and 6f were conducted using the triplex DNA structure (pdb2et0). Initially the Fe(II) complex 1(8) was docked as a control. This procedure effectively replicated the reported crystal structure. Of the present complexes, 6d ranked the highest, followed by 6b, 6a and 6f. Complex 6d binds in a fashion similar to 1(8), with possible π interactions via the triazole ring, even though the ring is not planar with the DNA (Figure 7). Complex 6b is shifted almost centrally within the cavity by approximately 6 Å. No clear π-stacking interactions are seen. Some close contacts exist between the non-aromatic spacer carbon and the thymine residue lining the cavity. Complex 6a is shifted in the opposite direction (~4 Å; Figure 7). The largest “click” cylinder complex 6f failed to dock within the central cavity of the DNA Y-junction. This complex binds on the surface of the DNA perpendicular to the other compounds. The docking studies support the postulate that some of the smaller “click” cylinders, 6b–d, are of suitable size and shape to interact with both duplex and triplex DNA in a similar fashion to the known biologically active complexes, 1(8) and 2. These results led us to test the biological activity of the “click” cylinders using the yeast S. cerevisiae strain ADΔ/pABC3 [91].
Figure 7.

Docking of metal complexes into DNA triplex. The crystal structure of the triplex DNA (pdb2et0) is shown as a transparent surface representation with the Fe(II) complex, 1(8), shown with white carbons. The smallest “click” complex, 6d, best matches the crystal structure and is shown with cyan carbons (left). Complex 6b is shown centrally located within the cavity (magenta carbons; center). The right panel shows the lowest ranking compounds 6a and 6f (yellow and brown respectively) adjacent to the cavity.
2.3.3. Biological Testing on Yeast Cells
Cell-based agarose diffusion assays were used to test the antifungal activity of both ligands, 3a–f, 4a–b and 7, and the Fe(II) complexes, 1, 5a–b, 6a–f and 8. As the ligands and complexes were not soluble at high concentrations in aqueous media, stock solutions of each of the molecules were prepared in DMSO. Interestingly, none of the “click” ligands (compounds 3a–f and 4a–b) or complexes (compounds 5a–b and 6a–f) displayed any antifungal activity at pH 6.8 when 100 nmol was added per disk (Supporting Information). In contrast, the Fe(II) cylinders 1 and 8 displayed antifungal activity under identical conditions (100 nmol per disk). The cylinder 8 had a zone of inhibition of 12 mm and the related chloride salt 1 showed a zone of inhibition of 11 mm diameter (Figure 8). The control compound at 7 nmol per disk (amphotericin B) gave an inhibition zone of 9 mm. These results indicate that Hannon’s metallosupramolecular cylinders, 1 and 8 have modest antifungal activity compared with amphotericin B under the conditions of the experiment. These results suggest that suitably designed metallosupramolecular cylinders have potential as a novel class of antifungal agents. Despite the strong structural similarity between 1(8) and 6a–f, the lack of biological activity of the Fe(II) “click” cylinders may indicate that the molecular size and shape of the cationic cylinders are not the only factors important in the design of biologically active metallosupramolecular complexes.
Figure 8.
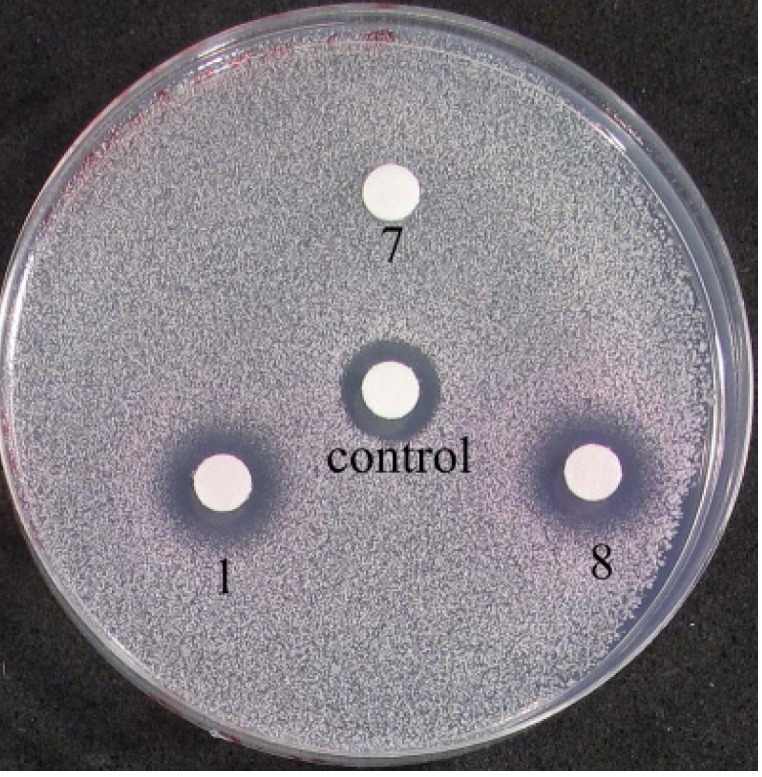
Agarose disc diffusion assay showing the effect of ligand, 7, and Fe(II) complexes, 1 and 8 on the growth of Saccharomyces cerevisiae strain ADΔ/pABC3. Amphotericin B is used as a control.
2.3.4. Solution Stability Studies of the Iron(II) Metallosupramolecular Architectures
Since the biological activities of the iron(II) metallosupramolecular architectures may depend on their stability, this property was tested for the Fe(II) cylinders 6a and 8 in the presence of a biologically relevant nucleophile (histidine) [92,93] and in DMSO solution using 1H-NMR and UV-vis spectroscopy and ESI-MS.
Competition experiments were carried out in CD3CN/D2O (97:3) at 40 °C using histidine as a surrogate for other biological nucleophiles. Both the Fe(II) cylinders, 6a and 8, were stable in the solvent mixture for a period of 24 h as judged by 1H-NMR spectroscopy and ESI-MS (for 8). Addition of D,L-histidine monohydrochloride monohydrate (6 eq.) to a CD3CN/D2O (97:3) solution of the “click” cylinder 6a led to instantaneous decomposition of the complex as shown by 1H-NMR and UV-vis spectroscopy (Supporting Information and Figure 9). The color of the solution changed immediately from orange/yellow to colorless and the MLCT (λmax = 423 nm) in the UV-vis spectrum was completely extinguished, indicating that the cylinder was destroyed (Figure 9). This was confirmed by 1H-NMR spectroscopy as the 6a/D,L-histidine containing mixture displayed only peaks due to the “free” ligand 3a. The ESI-MS (positive mode) of this mixture also only displayed peaks due to the ligand 3a. Presumably the iron(II) ions form neutral [Fe(his)2] [94] complexes which are not detected by ESI-MS under these conditions. Carrying out the same experiment with the pyridyl imine cylinder 8 gave a different result. The CD3CN/D2O (97:3) solutions of 8 were deep purple in color and lightened only slightly upon addition of D,L-histidine monohydrochloride monohydrate (Figure 9 inset, Supporting Information). The intense purple color remained even after 24 h. ESI-MS of the reaction mixture after 24 h showed the presence of a mixture of compounds, including free ligand, 7, and the ligand precursors and the aldehyde precursor to the ligand. Isotopically resolved peaks due to the intact cylinder complex were also present. 1H-NMR spectroscopy of the mixture of 8 and histidine showed the presence of additional species in solution which corresponded to the free ligand, 7, and the aldehyde precursor to the ligand. However, there were also peaks consistent with the intact cylinder complex and these remain even after 24 h.
Figure 9.

UV-vis spectra of the cylinder 6a and a mixture of 6a and histidine (6 eq.) in CD3CN/D2O (97:3). Inset photographs of 6a, 6a + histidine (6 eq.), 8, and 8 + histidine (6 eq.) in CD3CN/D2O (97:3).
The stability of the iron(II) metallosupramolecular cylinders in DMSO solution was examined using the same techniques. While acetonitrile solutions of 6a were orange/yellow in color, dissolution of 6a in DMSO gave a colorless solution that had no MLCT band (Supporting Information). 1H-NMR spectroscopy of 6a in d6-DMSO was consistent with the UV-vis spectroscopy data. The position of the single set of broad resonances indicated that the Fe(II) cylinder had decomposed in the coordinating DMSO solvent (Supporting Information). Analogous experiments with 8 in DMSO indicated that pyridyl imine complexes were also somewhat unstable in the coordinating solvent. However, when 8 was dissolved in DMSO solution the deep purple color of the complex persists, and the intensity of the MLCT band is reduced. This indicates that the amount of the intact complex in DMSO was reduced compared to acetonitrile but some of the Fe(II) cylinder remains present in solution.
These results suggest that the pyridylimine helicates, 1 and 8, are far more stable than the “click” cylinders 6a–f under the conditions used to carry out the biological assays. This difference explains why the pyridyl imine cylinders display antifungal activity, whereas the “click” cylinders do not. The high lability of these Fe(II) “click” complexes is due to two factors: 1) the weaker σ-donor ability of the ligands’ 1,2,3-triazole units, and 2) lone pair-lone pair repulsion between the uncoordinated N2 nitrogen atoms of the “click” ligands when they are in the fac-arrangement found in the cylinder structures. The weaker donor ability of the 2-(1-R-1H-1,2,3-triazol-4-yl)pyridine ligands compared to other diimines has been observed experimentally [95] and presumably results from the presence of the extra electronegative nitrogens in the 1,2,3-triazole units of the ligand. The enforced fac-arrangement of the 2-(1-R-1H-1,2,3-triazol-4-yl)pyridine chelate units may additionally destabilize the “click” cylinders relative to the pyridyl imine, i.e. the lone pair-lone pair repulsion between the uncoordinated N2 nitrogen atoms of the “click” ligands when they are in the fac-arrangement are unlikely to be favorable and further weaken the metal ligand interaction in comparison with systems that do not contain this motif.
3. Experimental
3.1. General Information
Unless otherwise stated, all reagents were purchased from commercial sources and used without further purification. Solvents were laboratory reagent grade with the following exceptions: dry acetonitrile was obtained by passing the solvent through an activated alumina column on a PureSolv TM solvent purification system (Innovative Technologies, Inc., Amesbury, MA, USA). 1H and 13C-NMR spectra were recorded on either a 400 MHz Varian 400 MR or Varian 500 MHz VNMRS spectrometers. Chemical shifts are reported in parts per million and referenced to residual solvent peaks (CD3CN: 1H δ 1.94, 13C δ 1.32, 118.26 ppm, d6-DMSO: 1H δ 2.50 ppm; 13C δ 39.52 ppm). Coupling constants (J) are reported in Hertz (Hz). Standard abbreviations indicating multiplicity were used as follows: m = multiplet, q = quartet, t = triplet, dt = double triplet, d = doublet, dd = double doublet, s = singlet. IR spectra were recorded on a Bruker ALPHA FT-IR spectrometer with an attached ALPHA-P measurement module. Microanalyses were performed at the Campbell Microanalytical Laboratory at the University of Otago. Electrospray mass spectra (ESI-MS) were collected on a Bruker micro-TOF-Q spectrometer. UV-visible absorption spectra were acquired with a Perkin Elmer Lambda-950 spectrophotometer in acetonitrile (10−5 M concentrations). The ligands 3a–f, [57] 4a–b, [70,71] and 7 [22] and the complex 1 [22] were prepared according to the literature procedures.
3.2. General Method for the Synthesis of the Fe(II) Model Complexes 5a–b
An acetonitrile solution (3 mL) of iron(II) tetrafluoroborate hexahydrate (0.048 g, 0.14 mmol, 1 eq.) was added to an acetonitrile solution (3 mL) of one of the ligands 4a–b (0.100 g, 0.43 mmol, 3 eq.). The resulting orange solution was stirred for one hour, filtered through cotton wool and vapor diffused with diethyl ether resulting in light red colored X-ray quality crystals of 5a or 5b. The crystals were isolated by filtration, washed with Et2O and dried in vacuo.
5a. Red crystals (0.110 g, 90%). MP: 140–142 °C. 1H-NMR (500 MHz, CD3CN): δ 8.78 (s, 3H, He), 8.19 (d, J = 7.9 Hz, 3H, Hd), 7.99–7.96 (m, 3H, Hc), 7.78–7.76 (m, 3H, Ha), 7.43–7.28 (m, 12H, Hb, Hg, Hi), 7.21–7.16 (m, 6H, Hh), 5.63 (s, 6H, Hf). 13C-NMR (125 MHz, CD3CN): δ 155.9, 152.1, 150.8, 139.8, 134.1, 130.3, 129.1, 128.1, 127.3, 126.4, 123.7, 56.9. ATR-IR: υ (cm−1) 3123, 1645, 1623, 1584, 1497, 1452, 1428, 1356, 1328, 1277, 1245, 1216, 1156, 1126, 1049. Anal. calcd. for C42H36B2F8FeN12•(0.5H2O): C 53.67, H 4.19, N 17.47%; found: C 53.30, H 4.23, N 17.67%. UV-Vis (CH3CN) λmax/nm (ε/L mol−1cm−1) = 241 (4.4 × 104), 280 (5.2 × 104), 425 (9.5 × 103).
5b. Red crystals (0.111 g, 91%). MP: 91–92 °C. 1H-NMR (500 MHz, CD3CN): δ 9.32 (s, 3H, He), 8.27 (d, J = 7.8 Hz, 3H, Hd), 8.12 (t, J = 7.8 Hz, 3H, Hc), 7.86–7.83 (m, 3H, Ha), 7.75 (d, J = 7.6 Hz, 6H, Hf), 7.63–7.56 (m, J = 8.3 Hz, 9H, Hg & Hh), 7.44–7.41 (m, 3H, Hb). 13C-NMR (125 MHz, CD3CN): δ 156.3, 153.3, 151.6, 140.2, 137.4, 131.3, 131.1, 126.8, 124.4, 123.4, 121.7. ATR-IR: υ (cm−1) 3548, 3129, 1619, 1595, 1502, 1464, 1419, 1359, 1281, 1260, 1212, 1164, 1142, 1049, 1031. Anal. calcd. for C39H30B2F8FeN12•(0.5H2O): C 51.75, H 3.45, N 18.57%; found: C 52.00, H 3.60, N 18.12%. UV-Vis (CH3CN) λmax/nm (ε/L mol−1cm−1) = 240 (7.4 × 104), 280 (9.0 × 104), 425 (1.8 × 104).
3.3. General Method for the Synthesis of the Fe(II) Cylinders 6a–f
An acetonitrile solution (3 mL) of iron(II) tetrafluoroborate hexahydrate (0.048 g, 0.14 mmol, 2 eq.) was added to an acetonitrile solution (3 mL) of one of the ligands 3a–f (0.21 mmol, 3 eq.). The resulting orange solution was stirred at room temperature for 1 h then filtered through cotton wool and vapor diffused with diethyl ether resulting in orange/red X-ray quality crystals. The crystals were isolated by filtration and washed with Et2O and then vacuum dried.
6a. Orange/red crystals (0.110 g, 94%). MP: > 230 °C. 1H-NMR (500 MHz, CD3CN): δ 8.83 (s, 6H, He), 8.20–8.06 (m, 12H, Hd and Hc), 7.83 (dt, J = 5.6, 1.2 Hz, 6H, Ha), 7.44 (ddd, J = 7.4, 5.6, 1.5 Hz, 6H, Hb), 7.27 (s, 12H, Hg), 5.53 (s, 6H, Hf), 5.33 (s, 6H, Hf). 13C-NMR (125 MHz, CD3CN): δ 155.9, 153.9, 151.1, 140.1, 136.8, 129.5, 126.9, 125.6, 123.3, 55.9. ATR-IR: υ (cm−1) 3417, 3298, 3130, 1622, 1583, 1518, 1450, 1426, 1360, 1329, 1275, 1246, 1214, 1159, 1126, 1049, 1031. Anal. calcd for C66H54B4F16Fe2N24•(15H2O): C 41.45, H 4.43, N 17.58%; found: C 41.20, H 4.33, N 17.71%. UV/Vis (CH3CN) λmax/nm (ε/L mol−1cm−1) = 240 (2.6 × 104), 279 (5.2 × 104), 425 (1.0 × 104).
6b. Orange/red crystals (0.110 g, 94%). MP >230 °C. 1H-NMR (500 MHz, CD3CN): δ 8.85 (s, 6H, He), 8.24 (d, J = 7.9 Hz, 6H, Hd), 8.16 (td, J = 7.8, 1.4 Hz, 6H, Hc), 8.01 (d, J = 5.7 Hz, 6H, Ha), 7.50 (t, J = 6.7 Hz, 6H, Hb), 7.33 (s, 3H, Hg), 5.84–5.81 (m, 6H, Hh), 5.78 (s, 6H, Hf), 5.75 (s, 6H, Hf), 5.20 (t, J = 7.9 Hz, 3H, Hi). 13C-NMR (125 MHz, CD3CN): δ 156.0, 154.0, 150.7, 140.1, 136.9, 130.6, 127.3, 126.8, 123.4, 123.3, 123.1, 54.8. ATR-IR: υ (cm−1) 3114, 1623, 1584, 1499, 1471, 1451, 1361, 1331, 1280, 1245, 1229, 1165, 1129, 1050. Anal. calcd for C66H54B4F16Fe2N24•(3H2O): C 46.73, H 3.57, N 19.82%; found: C 46.89, H 3.63, N 19.66%. UV/Vis (CH3CN) λmax/nm (ε/L mol−1cm−1) = 239 (3.0 × 104), 277 (3.5 × 104), 418 (5.6 × 103).
6c. Orange/red crystals (0.110 g, 90%). MP: >230 °C. 1H-NMR (500 MHz, CD3CN): δ 8.89 (s, 6H, He), 8.27 (d, J = 7.7 Hz, 6H, Hd), 8.18 (td, J = 7.8, 1.4 Hz, 6H, Hc), 8.02 (d, J = 5.5 Hz, 6H, Ha), 7.54–7.49 (m, 6H, Hb), 5.95–5.90 (m, 9H, Hg and Hh), 5.81 (s, 12H, Hf). 13C-NMR (125 MHz, CD3CN): δ 155.6, 155.4, 153.1, 150.4, 139.5, 138.2, 126.7, 126.3, 122.7, 55.8. ATR-IR: υ (cm−1) 3128, 1621, 1601, 1576, 1467, 1451, 1360, 1346, 1323, 1282, 1247, 1228, 1162, 1128, 1049. Anal. calcd for C63H51B4F16Fe2N27•(6H2O): C 43.16, H 3.62, N 21.57%; found: C 43.45, H 3.26, N 21.57%. UV/Vis (CH3CN) λmax/nm (ε/L mol−1cm−1): 239 (1.7 × 104), 278 (2.6 × 104), 424 (3.5 × 103).
6d. Orange/red crystals (0.105 g, 90%). MP: >230 °C. 1H-NMR (500 MHz, CD3CN): δ 8.71 (s, 6H, He), 8.18 (dd, J = 7.9, 1.1 Hz, 6H, Hd), 8.08 (td, J = 7.8, 1.4 Hz, 6H, Hc), 7.87 (d, J = 5.6 Hz, 6H, Ha), 7.43 (td, J = 5.8, 2.9 Hz, 6H, Hb), 4.46–4.34 (m, 12H, Hf), 2.53–2.35 (m, 6H, Hg). 13C-NMR (125 MHz, CD3CN): δ 155.9, 153.1, 149.9, 139.1, 126.7, 125.1, 122.1, 49.1, 31.1. ATR-IR: υ (cm‒1) 3132, 1621, 1581, 1563, 1472, 1454, 1438, 1393, 1361, 1336, 1279, 1248, 1204, 1166, 1129, 1102, 1051, 1029, 1015. Anal. calcd for C51H48B4F16Fe2N24•(6H2O): C 39.16, H 3.87, N 21.4%; found: C 38.92, H 3.54, N 21.39%. UV/Vis (CH3CN) λmax/nm (ε/L mol−1cm−1) = 238 (3.6 × 104), 278 (2.2 × 104), 425 (4.0 × 103).
6e. Orange/red crystals (0.097 g, 90%). MP: >230 °C. 1H-NMR (500 MHz, CD3CN): chemical shift values for the major component δ 8.71 (s, 6H, He), 8.15 (d, J = 8.0 Hz, 6H, Hd), 8.08 (t, J = 7.8 Hz, 6H, Hc), 7.86 (d, J = 5.6 Hz, 6H, Ha), 7.44 (t, J = 6.7 Hz, 6H, Hb), 4.42–4.33 (m, 12H, Hf), 1.90–1.68 (m, 12H, Hh), 1.20–1.14(m, 12H, Hg). 13C-NMR (125 MHz, CD3CN): δ 155.1, 154.1, 150.5, 139.1, 126.7, 125.6, 122.1, 53.0, 30.9, 26.6. ATR-IR: υ (cm−1) 3129, 2930, 2860, 1619, 1580, 1455, 1432, 1360, 1279, 1214, 1161, 1049. Anal. calcd for C60H66B4F16Fe2N24•(5.5H2O): C 42.86, H 4.62, N 19.99%; found: C 47.73, H 4.31, N 19.61%. UV/Vis (CH3CN) λmax/nm (ε/L mol−1cm−1) = 240 (3.5 × 104), 278 (3.5 × 104), 420 (5.4 × 103).
6f. Red crystals (0.115 g, 90%). MP: >230 °C. 1H-NMR (500 MHz, CD3CN): δ 9.26 (s, 6H, He), 8.27 (d, J = 7.9 Hz, 6H, Hd), 8.18 (t, J = 7.8 Hz, 6H, Hc), 7.86 (d, J = 5.7 Hz, 6H, Ha), 7.58 (dd, J = 8.1, 5.2 Hz, 12H, Hf), 7.50 (t, J = 6.8 Hz, 6H, Hb), 7.40 (d, J = 8.1 Hz, 6H, Hg), 7.33 (d, J = 8.1 Hz, 6H, Hg1), 4.13–4.05 (m, 6H, Hh). 13C-NMR (125 MHz, CD3CN): δ 155.9, 153.6, 151.2, 144.1, 143.9, 140.3, 135.4, 131.2, 131.1, 127.1, 123.4, 123.2, 121.3. ATR-IR: υ (cm−1) 3363, 3127, 1620, 1579, 1515, 1464, 1414, 1359, 1279, 1259, 1216, 1164, 1050. Anal. calcd for C81H60B4F16Fe2N24•(5H2O + 3.5CH3CN): C 51.25, H 3.93, N 18.68%; found: C 51.24, H 3.79, N 19.01%. UV/Vis (CH3CN) λmax/nm (ε/L mol−1cm−1) = 245 (3.0 × 104), 282 (5.3 × 104), 410 (1.9 × 104).
3.4. Synthesis of the Fe(II) Pyridylimine Cylinder 8
An acetonitrile solution (3 mL) of iron(II) tetrafluoroborate hexahydrate (0.026 g, 0.18 mmol, 2 eq.) was added to an acetonitrile solution (3 mL) of the ligand 7 (0.100 g, 0.27 mmol, 3 eq.) and heated at 55 °C for one hour which resulted in the formation of a purple colored solution. This purple solution was filtered through cotton wool and vapor diffused with diethyl ether resulting in deep purple X-ray quality crystals of 8. The crystals were isolated by filtration and washed with Et2O and dried in vacuo (0.125 g, 90%). MP: >230 °C. 1H-NMR (500 MHz, CD3CN): δ 8.96 (s, 6H, He), 8.60 (d, J = 7.7 Hz, 6H, Hd), 8.43 (t, J = 7.6 Hz, 6H, Hc), 7.81–7.75 (m, 6H, Hb), 7.38 (d, J = 5.5 Hz, 6H, Ha), 7.08–6.87 (m, 12H, Hf), 5.55–5.51 (m, 12H, Hg), 4.05 (s, 6H, Hh). 13C-NMR (125 MHz, CD3CN): δ 175.5, 159.1, 156.8, 150.2, 142.7, 140.7, 132.4, 131.1, 130.1, 122.5, 40.1. ATR-IR: υ (cm−1) 3747, 3605, 3035, 2908, 1613, 1583, 1556, 1500, 1471, 1441, 1414, 1358, 1302, 1258, 1238, 1049. Anal. calcd for C75H60B4F16Fe2N12•(3.5H2O): C 54.55, H 4.09, N 10.18%; found: C 54.41, H 4.00, N 10.20%. UV/Vis (CH3CN) λmax/nm (ε/L mol−1cm−1) = 237 (7.7 × 104), 324 (3.6 × 104), 571 (1.7 × 104).
3.5. General Method for the Molecular Docking Studies
Molecular docking studies were carried out using GOLD 5.1. [96]. The X-ray crystal structures of the Fe(II) complexes (6a, 6b, 6c and 6f) were used for the docking experiments with some modifications. The cif formatted files were converted to mol2 files using Mercury (CCDC) and edited to remove any counter ions and to add appropriate atom and bond types using SYBYLX 2.0. The Fe(II) cylinders were docked as rigid molecules into either the major groove of B-DNA (pdb3BSE) [97] or the three-way (triplex) DNA junction (pdb2et0) [29]. The docking cavity of B-DNA was identified as the solvent accessible surface centered on the position of water 209 and was adjacent to the sequence 5'(CAATGTTGC) and 5'(GCAACATTG). The cavity of the triplex DNA was identified as the solvent accessible surface (of the DNA) centered around the Fe(II) atom centered within the triplex.
3.6. Yeast Agarose Diffusion Assays
A base layer of Complete Synthetic Medium (CSM, 20 mL) was solidified with agarose (0.6%) in a Petri dish. A 5 mL overlay of the same medium containing S. cerevisiae cells (strain ADΔ/pABC3; 100 μL, OD600 = 0.4 in CSM, pH 4.8–5) at 45 °C was poured and allowed to solidify. Discs (5 mm diameter; BBL, Becton Dickinson Co., Sparks, MD, USA) containing Fe(II) cylinders (5 μL of 20 mM solution in DMSO; 100 nmol/disc) were placed on the agarose (strain ADΔ/pABC3). The plate was then incubated at 30 °C for 48 h. Amphotericin B (80% Sigma) was used as a positive control (7 μL of 1 mM solution in DMSO; 7 nmol/disc). DMSO was used as a negative control.
3.7. General X-ray Experimental Section
X-Ray data were recorded at 89 K using a Bruker APEX II CCD diffractometer using Mo-Kα radiation (λ = 0.71073 Å) except for 6f which was collected at 100 K on crystals mounted on a Hampton Scientific cryoloop at the MX2 beamline of the Australian synchrotron [98]. Absorption corrections were applied by semi-empirical methods (SADABS) [99].The structures were solved by direct methods using SIR97 [100], or SHELXS-97 [101] with the resulting Fourier maps revealing the location of most non-hydrogen atoms. Full-matrix least-squares refinement on F2 was carried out using SHELXL-97 with most non-hydrogen atoms being refined anisotropically. The non-hydrogen atoms that were isotropically refined are described in the Supporting Information for each structure. The hydrogen atoms were included in calculated positions and were refined as riding atoms with individual (or group, if appropriate) isotropic displacement parameters. The PLATON SQUEEZE [77] procedure was used to treat regions of diffuse solvent and counter anions which could not be sensibly modeled in terms of atomic sites, see Supporting Information. Some of the crystal structures contained disordered components. The ORTEP [102] diagrams have been drawn with 50% probability ellipsoids. Crystal data and collection parameters are given in the Supporting Information. The CIF files CCDC 931616–931621 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: (+44) 1223-336-033.
3.8.1H-NMR and UV-Vis Histidine Competition Experiments
1H-NMR study of 6a: Fe(II) cylinder 6a (0.010 g, 0.006 mmol, 1 eq.), D,L-histidine hydrochloride monohydrate (0.007 g, 0.03 mmol, 6 eq.), and sodium hydrogen carbonate (0.004 g, 0.05 mmol, 8 eq.), were dissolved in a mixture of CD3CN and D2O (97:3). The resulting suspension was sonicated for few seconds until the histidine and sodium hydrogen carbonate were completely dissolved and then the 1H NMR spectrum was recorded.
1H-NMR study of 8: Fe(II) cylinder 8 (0.010 g, 0.006 mmol, 1 eq.), D,L-histidine hydrochloride monohydrate (0.008 g, 0.04 mmol, 6 eq.), and sodium hydrogen carbonate (0.003 g, 0.04 mmol, 6 eq.), were dissolved in a mixture of CD3CN and D2O (97:3). The resulting suspension was sonicated for few seconds, until the histidine and sodium hydrogen carbonate were completely dissolved and then 1H NMR spectrum was immediately recorded.
UV-Vis study for 6a and 8: The Fe(II) cylinder, either 6a (0.0050 g, 1 eq.) or 8 (0.0050 g, 1 eq.), was dissolved in 10 mL of acetonitrile:water (97:3) giving a stock solution. D,L-Histidine hydrochloride monohydrate (0.0035 g, 6 eq.) and sodium hydrogen carbonate (0.0025 g, 6 eq.) were also dissolved in 10 mL of acetonitrile/water (97:3). The stock solution containing Fe(II) cylinders (5 mL), either 6a or 8, was added to the stock solution containing histidine and sodium hydrogen carbonate (5 mL), mixed thoroughly and UV-Vis spectra were recorded immediately.
4. Conclusions
A series of metallosupramolecular [Fe2L3]4+ “click” cylinders have been synthesized in excellent yields (90%–95%) from [Fe(H2O)6](BF4)2 and bis(bidentate) pyridyl-1,2,3-triazole ligands. All complexes were characterized by elemental analysis, IR, UV-vis, 1H-, 13C- and DOSY-NMR spectroscopies. In four cases the structures were confirmed by X-ray crystallography. Molecular modeling studies indicated that some of these complexes were of suitable size and shape to bind both duplex and triplex DNA in a similar fashion to some related biologically active iron(II) helicates. In contrast to the previously reported pyridylimine based iron(II) helicates, the “click” cylinders display no antifungal activity against the yeast S. cerevisiae. This is probably due to the poor stability of the “click” complexes in the presence of DMSO and the amino acid histidine found in the biological test media. The Fe(II) “click” cylinders rapidly decomposed in these media. The high lability of these Fe(II) “click” complexes is due to two factors; 1) the weaker σ-donor ability of the ligand’s 1,2,3-triazole units, and 2) lone pair-lone pair repulsion between the uncoordinated N2 nitrogen atoms of the “click” ligands when they are in the fac-arrangement found in the cylinder structures. Exchanging the labile Fe(II) for more kinetically inert metal ions may enable the development of more robust biologically active metallosupramolecular “click” cylinders. Efforts towards these types of architectures are underway.
Acknowledgments
We thank Dr Mervyn Thomas for his assistance collecting NMR data. The University of Otago, Department of Chemistry provided financial support for this work. SVK and JEML thank the University of Otago for PhD scholarships. Data for the structure of complex 6f was obtained on the MX2 beamline at the Australian Synchrotron, Victoria, Australia.
Supplementary Materials
Supplementary materials can be accessed at: http://www.mdpi.com/1420-3049/18/6/6383/s1.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 3a–f, 6a–f, and 8 are available from the authors.
References
- 1.Miyake H., Tsukube H. Coordination chemistry strategies for dynamic helicates: Time-programmable chirality switching with labile and inert metal helicates. Chem. Soc. Rev. 2012;41:6977–6991. doi: 10.1039/c2cs35192g. [DOI] [PubMed] [Google Scholar]
- 2.Howson S.E., Scott P. Approaches to the synthesis of optically pure helicates. Dalton Trans. 2011;40:10268–10277. doi: 10.1039/c1dt11423a. [DOI] [PubMed] [Google Scholar]
- 3.Albrecht M., Froehlich R. Symmetry driven self-assembly of metallo-supramolecular architectures. Bull. Chem. Soc. Jpn. 2007;80:797–808. [Google Scholar]
- 4.Piguet C., Borkovec M., Hamacek J., Zeckert K. Strict self-assembly of polymetallic helicates: the concepts behind the semantics. Coord. Chem. Rev. 2005;249:705–726. doi: 10.1016/j.ccr.2004.08.023. [DOI] [Google Scholar]
- 5.Hannon M.J., Childs L.J. Helices and Helicates. Beautiful supramolecular motifs with emerging applications. Supramol. Chem. 2004;16:7–22. doi: 10.1080/10610270310001632386. [DOI] [Google Scholar]
- 6.Albrecht M. “Let’s Twist Again”-double-stranded, triple-stranded, and circular helicates. Chem. Rev. 2001;101:3457–3497. doi: 10.1021/cr0103672. [DOI] [PubMed] [Google Scholar]
- 7.Archer R.J., Hawes C.S., Jameson G.N.L., McKee V., Moubaraki B., Chilton N.F., Murray K.S., Schmitt W., Kruger P.E. Partial spin crossover behaviour in a dinuclear iron(II) triple helicate. Dalton Trans. 2011;40:12368–12373. doi: 10.1039/c1dt11381j. [DOI] [PubMed] [Google Scholar]
- 8.Pelleteret D., Clerac R., Mathoniere C., Harte E., Schmitt W., Kruger P.E. Asymmetric spin crossover behavior and evidence of light-induced excited spin state trapping in a dinuclear iron(II) helicate. Chem. Commun. 2009:221–223. doi: 10.1039/b816196h. [DOI] [PubMed] [Google Scholar]
- 9.Telfer S.G., Bocquet B., Williams A.F. Thermal spin crossover in binuclear iron(II) helicates: Negative cooperativity and a mixed spin state in solution. Inorg. Chem. 2001;40:4818–4820. doi: 10.1021/ic015521h. [DOI] [PubMed] [Google Scholar]
- 10.Lincheneau C., Peacock R.D., Gunnlaugsson T. Europium directed synthesis of enantiomerically pure dimetallic luminescent “squeezed” triple-stranded helicates; solution studies. Chem. Asian J. 2010;5:500–504. doi: 10.1002/asia.200900515. [DOI] [PubMed] [Google Scholar]
- 11.Stomeo F., Lincheneau C., Leonard J.P., O'Brien J.E., Peacock R.D., McCoy C.P., Gunnlaugsson T. Metal-directed synthesis of enantiomerically pure dimetallic lanthanide luminescent triple-stranded helicates. J. Am. Chem. Soc. 2009;131:9636–9637. doi: 10.1021/ja9032204. [DOI] [PubMed] [Google Scholar]
- 12.Bünzli J.-C.G., Chauvin A.-S., Vandevyver C.D.B., Bo S., Comby S. Lanthanide bimetallic helicates forin vitroimaging and sensing. Ann. N.Y. Acad. Sci. 2008;1130:97–105. doi: 10.1196/annals.1430.010. [DOI] [PubMed] [Google Scholar]
- 13.Vandevyver C.D.B., Chauvin A.-S., Comby S., Bunzli J.-C.G. Luminescent lanthanide bimetallic triple-stranded helicates as potential cellular imaging probes. Chem. Commun. 2007:1716–1718. doi: 10.1039/b701482a. [DOI] [PubMed] [Google Scholar]
- 14.Sham K.-C., Yeung H.-L., Yiu S.-M., Lau T.-C., Kwong H.-L. New binuclear double-stranded manganese helicates as catalysts for alkene epoxidation. Dalton Trans. 2010;39:9469–9471. doi: 10.1039/c0dt00953a. [DOI] [PubMed] [Google Scholar]
- 15.Ayme J.-F., Beves J.E., Leigh D.A., McBurney R.T., Rissanen K., Schultz D. Pentameric circular iron(II) double helicates and a molecular pentafoil knot. J. Am. Chem. Soc. 2012;134:9488–9497. doi: 10.1021/ja303355v. [DOI] [PubMed] [Google Scholar]
- 16.Wenzel M., Knapp Q.W., Plieger P.G. A bis-salicylaldoximato-copper(II) receptor for selective sulfate uptake. Chem. Commun. 2011;47:499–501. doi: 10.1039/c0cc02230f. [DOI] [PubMed] [Google Scholar]
- 17.Stevens J.R., Plieger P.G. Anion-driven conformation control and enhanced sulfate binding utilising aryl linked salicylaldoxime dicopper helicates. Dalton Trans. 2011;40:12235–12241. doi: 10.1039/c1dt10808e. [DOI] [PubMed] [Google Scholar]
- 18.Wenzel M., Bruere S.R., Knapp Q.W., Tasker P.A., Plieger P.G. Zwitterionic dicopper helicates: anion encapsulation and binding studies. Dalton Trans. 2010;39:2936–2941. doi: 10.1039/b922998a. [DOI] [PubMed] [Google Scholar]
- 19.Wenzel M., Jameson G.B., Ferguson L.A., Knapp Q.W., Forgan R.S., White F.J., Parsons S., Tasker P.A., Plieger P.G. Anion-induced contraction of helical receptors. Chem. Commun. 2009:3606–3608. doi: 10.1039/b902772f. [DOI] [PubMed] [Google Scholar]
- 20.Goetz S., Kruger P.E. A new twist in anion binding: metallo-helicate hosts for anionic guests. Dalton Trans. 2006:1277–1284. doi: 10.1039/b514580e. [DOI] [PubMed] [Google Scholar]
- 21.Ayme J.-F., Beves J.E., Leigh D.A., McBurney R.T., Rissanen K., Schultz D. A synthetic molecular pentafoil knot. Nat. Chem. 2012;4:15–20. doi: 10.1038/nchem.1193. [DOI] [PubMed] [Google Scholar]
- 22.Hannon M.J., Painting C.L., Jackson A., Hamblin J., Errington W. An inexpensive approach to supramolecular architecture. Chem. Commun. 1997:1807–1808. [Google Scholar]
- 23.Hannon M.J. Supramolecular DNA recognition. Chem. Soc. Rev. 2007;36:280–295. doi: 10.1039/b606046n. [DOI] [PubMed] [Google Scholar]
- 24.Hannon M.J. Metal-based anticancer drugs: from a past anchored in platinum chemistry to a post-genomic future of diverse chemistry and biology. Pure Appl. Chem. 2007;79:2243–2261. doi: 10.1351/pac200779122243. [DOI] [Google Scholar]
- 25.Moldrheim E., Hannon M.J., Meistermann I., Rodger A., Sletten E. Interaction between a DNA oligonucleotide and a dinuclear iron(II) supramolecular cylinder; an NMR and molecular dynamics study. J. Biol. Inorg. Chem. 2002;7:770–780. doi: 10.1007/s00775-002-0354-2. [DOI] [PubMed] [Google Scholar]
- 26.Meistermann I., Moreno V., Prieto M.J., Moldrheim E., Sletten E., Khalid S., Rodger P.M., Peberdy J.C., Isaac C.J., Rodger A., et al. Intramolecular DNA coiling mediated by metallo-supramolecular cylinders: differential binding of P and M helical enantiomers. Proc. Natl. Acad. Sci. USA. 2002;99:5069–5074. doi: 10.1073/pnas.062634499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hannon M.J., Moreno V., Prieto M.J., Moldrheim E., Sletten E., Meistermann I., Isaac C.J., Sanders K.J., Rodger A. Intramolecular DNA coiling mediated by a metallo-supramolecular cylinder. Angew. Chem. Int. Ed. 2001;40:880–884. doi: 10.1002/1521-3773(20010302)40:5<879::AID-ANIE879>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 28.Cerasino L., Hannon M.J., Sletten E. DNA three-way junction with a dinuclear iron(II) supramolecular helicate at the center: A NMR structural study. Inorg. Chem. 2007;46:6245–6251. doi: 10.1021/ic062415c. [DOI] [PubMed] [Google Scholar]
- 29.Oleksi A., Blanco A.G., Boer R., Uson I., Aymami J., Rodger A., Hannon M.J., Coll M. Molecular recognition of a three-way DNA junction by a metallosupramolecular helicate. Angew. Chem. Int. Ed. 2006;45:1227–1231. doi: 10.1002/anie.200503822. [DOI] [PubMed] [Google Scholar]
- 30.Hotze A.C.G., Hodges N.J., Hayden R.E., Sanchez-Cano C., Paines C., Male N., Tse M.-K., Bunce C.M., Chipman J.K., Hannon M.J. Supramolecular iron cylinder with unprecedented DNA binding is a potent cytostatic and apoptotic agent without exhibiting genotoxicity. Chem. Biol. 2008;15:1258–1267. doi: 10.1016/j.chembiol.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 31.Richards A.D., Rodger A., Hannon M.J., Bolhuis A. Antimicrobial activity of an iron triple helicate. Int. J. Antimicrob. Agents. 2009;33:469–472. doi: 10.1016/j.ijantimicag.2008.10.031. [DOI] [PubMed] [Google Scholar]
- 32.Howson S.E., Bolhuis A., Brabec V., Clarkson G.J., Malina J., Rodger A., Scott P. Optically pure, water-stable metallo-helical ‘flexicate’ assemblies with antibiotic activity. Nat. Chem. 2012;4:31–36. doi: 10.1038/nchem.1206. [DOI] [PubMed] [Google Scholar]
- 33.Buckley B.R., Heaney H. Mechanistic investigations of copper(I)-catalysed alkyne-azide cycloaddition reactions. Top. Heterocycl. Chem. 2012;28:1–29. doi: 10.1007/7081_2011_71. [DOI] [Google Scholar]
- 34.Liang L., Astruc D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview. Coord. Chem. Rev. 2011;255:2933–2945. doi: 10.1016/j.ccr.2011.06.028. [DOI] [Google Scholar]
- 35.Tornoe C.W., Christensen C., Meldal M. Peptidotriazoles on solid phase: [1,2,3]-Triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 36.Rostovtsev V.V., Green L.G., Fokin V.V., Sharpless K.B. A stepwise Huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 37.Meldal M., Tornoe C.W. Cu-catalyzed azide-alkyne cycloaddition. Chem. Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- 38.Wu P., Fokin V.V. Catalytic azide-alkyne cycloaddition: reactivity and applications. Aldrichimica Acta. 2007;40:7–17. [Google Scholar]
- 39.Zheng T., Rouhanifard S.H., Jalloh A.S., Wu P. Click triazoles for bioconjugation. Top. Heterocycl. Chem. 2012;28:163–183. doi: 10.1007/7081_2011_72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Agalave S.G., Maujan S.R., Pore V.S. Click chemistry: 1,2,3-Triazoles as pharmacophores. Chem. Asian J. 2011;6:2696–2718. doi: 10.1002/asia.201100432. [DOI] [PubMed] [Google Scholar]
- 41.Holub J.M., Kirshenbaum K. Tricks with clicks: modification of peptidomimetic oligomers via copper-catalyzed azide-alkyne [3 + 2] cycloaddition. Chem. Soc. Rev. 2010;39:1325–1337. doi: 10.1039/b901977b. [DOI] [PubMed] [Google Scholar]
- 42.Aragao-Leoneti V., Campo V.L., Gomes A.S., Field R.A., Carvalho I. Application of copper(I)-catalyzed azide/alkyne cycloaddition (CuAAC) click chemistry' in carbohydrate drug and neoglycopolymer synthesis. Tetrahedron. 2010;66:9475–9492. doi: 10.1016/j.tet.2010.10.001. [DOI] [Google Scholar]
- 43.Amblard F., Cho J.H., Schinazi R.F. Cu(I)-catalyzed huisgen azide-alkyne 1,3-dipolar cycloaddition reaction in nucleoside, nucleotide, and oligonucleotide chemistry. Chem. Rev. 2009;109:4207–4220. doi: 10.1021/cr9001462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li N., Binder W.H. Click-chemistry for nanoparticle-modification. J. Mater. Chem. 2011;21:16717–16734. doi: 10.1039/c1jm11558h. [DOI] [Google Scholar]
- 45.Fahrenbach A.C., Stoddart J.F. Reactions under the click chemistry philosophy employed in supramolecular and mechanostereochemical systems. Chem. Asian J. 2011;6:2660–2669. doi: 10.1002/asia.201100457. [DOI] [PubMed] [Google Scholar]
- 46.Qin A., Lam J.W.Y., Tang B.Z. Click polymerization. Chem. Soc. Rev. 2010;39:2522–2544. doi: 10.1039/b909064a. [DOI] [PubMed] [Google Scholar]
- 47.McDonald K.P., Hua Y., Flood A.H. 1,2,3-Triazoles and the expanding utility of charge neutral CH···anion interactions. Top. Heterocycl. Chem. 2010;24:341–366. doi: 10.1007/7081_2010_38. [DOI] [Google Scholar]
- 48.Crowley J.D., Goldup S.M., Lee A.-L., Leigh D.A., McBurney R.T. Active metal template synthesis of rotaxanes, catenanes and molecular shuttles. Chem. Soc. Rev. 2009;38:1530–1541. doi: 10.1039/b804243h. [DOI] [PubMed] [Google Scholar]
- 49.Crowley J.D., McMorran D.A. “Click-triazole” coordination chemistry: Exploiting 1,4-disubstituted-1,2,3-triazoles as ligands. Top. Heterocycl. Chem. 2012;28:31–83. doi: 10.1007/7081_2011_67. [DOI] [Google Scholar]
- 50.Crowley J.D., Lee A.-L., Kilpin K.J. 1,3,4-Trisubstituted-1,2,3-triazol-5-ylidene click carbene ligands. Synthesis, catalysis, and self-assembly. Aust. J. Chem. 2011;64:1118–1132. doi: 10.1071/CH11185. [DOI] [Google Scholar]
- 51.Schweinfurth D., Deibel N., Weisser F., Sarkar B. Getting new ligands with a click. Nachr. Chem. 2011;59:937–941. doi: 10.1002/nadc.201190062. [DOI] [Google Scholar]
- 52.Struthers H., Mindt T.L., Schibli R. Metal chelating systems synthesized using the copper(I) catalyzed azide-alkyne cycloaddition. Dalton Trans. 2010;39:675–696. doi: 10.1039/b912608b. [DOI] [PubMed] [Google Scholar]
- 53.Scott S.O., Gavey E.L., Lind S.J., Gordon K.C., Crowley J.D. Self-assembled palladium(II) “click” cages: Synthesis, structural modification and stability. Dalton Trans. 2011;40:12117–12124. doi: 10.1039/c1dt10551e. [DOI] [PubMed] [Google Scholar]
- 54.Kilpin K.J., Paul U.S.D., Lee A.-L., Crowley J.D. Gold(I) “click” 1,2,3-triazolylidenes: Synthesis, self-assembly and catalysis. Chem. Commun. 2011;47:328–330. doi: 10.1039/c0cc02185g. [DOI] [PubMed] [Google Scholar]
- 55.Gower M.L., Crowley J.D. Self-assembly of silver(I) metallomacrocycles using unsupported 1,4-substituted-1,2,3-triazole “click” ligands. Dalton Trans. 2010;39:2371–2378. doi: 10.1039/b923211g. [DOI] [PubMed] [Google Scholar]
- 56.Crowley J.D., Gavey E.L. Use of di-1,4-substituted-1,2,3-triazole “click” ligands to self-assemble dipalladium(II) coordinatively saturated, quadruply stranded helicate cages. Dalton Trans. 2010;39:4035–4037. doi: 10.1039/c003683h. [DOI] [PubMed] [Google Scholar]
- 57.Crowley J.D., Bandeen P.H. A multicomponent CuAAC “click” approach to a library of hybrid polydentate 2-pyridyl-1,2,3-triazole ligands: new building blocks for the generation of metallosupramolecular architectures. Dalton Trans. 2010;39:612–623. doi: 10.1039/b911276f. [DOI] [PubMed] [Google Scholar]
- 58.Garcia L., Maisonneuve S., Xie J., Guillot R., Dorlet P., Riviere E., Desmadril M., Lambert F., Policar C. Sugars to Control Ligand Shape in Metal Complexes: Conformationally Constrained Glycoligands with a Predetermination of Stereochemistry and a Structural Control. Inorg. Chem. 2010;49:7282–7288. doi: 10.1021/ic1002379. [DOI] [PubMed] [Google Scholar]
- 59.Najar A.M., Tidmarsh I.S., Ward M.D. Lead(II) complexes of bis- and tris-bidentate compartmental ligands based on pyridyl-pyrazole and pyridyl-triazole fragments: coordination networks and a discrete dimeric box. CrystEngComm. 2010;12:3642–3650. [Google Scholar]
- 60.The biological properties of “click” complexes have received considerable recent attention for selected examples see ref 61–69.
- 61.Yano S., Ohi H., Ashizaki M., Obata M., Mikata Y., Tanaka R., Nishioka T., Kinoshita I., Sugai Y., Okura I., et al. Syntheses, Characterization, and Antitumor Activities of Platinum(II) and Palladium(II) Complexes with Sugar-Conjugated Triazole Ligands. Chem. Biodivers. 2012;9:1903–1915. doi: 10.1002/cbdv.201100426. [DOI] [PubMed] [Google Scholar]
- 62.Clede S., Lambert F., Sandt C., Gueroui Z., Refregiers M., Plamont M.-A., Dumas P., Vessieres A., Policar C. A rhenium tris-carbonyl derivative as a single core multimodal probe for imaging (SCoMPI) combining infrared and luminescent properties. Chem. Commun. 2012;48:7729–7731. doi: 10.1039/c2cc32163g. [DOI] [PubMed] [Google Scholar]
- 63.Seridi A., Wolff M., Boulay A., Saffon N., Coulais Y., Picard C., Machura B., Benoist E. Rhenium(I) and technetium(I) complexes of a novel pyridyltriazole-based ligand containing an arylpiperazine pharmacophore: Synthesis, crystal structures, computational studies and radiochemistry. Inorg. Chem. Commun. 2011;14:238–242. doi: 10.1016/j.inoche.2010.11.002. [DOI] [Google Scholar]
- 64.Benoist E., Coulais Y., Almant M., Kovensky J., Moreau V., Lesur D., Artigau M., Picard C., Galaup C., Gouin S.G. A Click procedure with heterogeneous copper to tether technetium-99m chelating agents and rhenium complexes. Evaluation of the chelating properties and biodistribution of the new radiolabelled glucose conjugates. Carbohydr. Res. 2011;346:26–34. doi: 10.1016/j.carres.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 65.Urankar D., Pevec A., Kosmrlj J. Synthesis and characterization of platinum(II) complexes with a diazenecarboxamide-appended picolyl-triazole ligand. Eur. J. Inorg. Chem. 2011:1921–1929. doi: 10.1002/ejic.201001051. [DOI] [Google Scholar]
- 66.Bratsos I., Urankar D., Zangrando E., Genova-Kalou P., Kosmrlj J., Alessio E., Turel I. 1-(2-Picolyl)-substituted 1,2,3-triazole as novel chelating ligand for the preparation of ruthenium complexes with potential anticancer activity. Dalton Trans. 2011;40:5188–5199. doi: 10.1039/c0dt01807d. [DOI] [PubMed] [Google Scholar]
- 67.Chevry A., Teyssot M.-L., Maisonial A., Lemoine P., Viossat B., Traikia M., Aitken D.J., Alves G., Morel L., Nauton L., et al. Click chelators - The behavior of platinum and palladium complexes in the presence of guanosine and DNA. Eur. J. Inorg. Chem. 2010:3513–3519. [Google Scholar]
- 68.Obata M., Kitamura A., Mori A., Kameyama C., Czaplewska J.A., Tanaka R., Kinoshita I., Kusumoto T., Hashimoto H., Harada M., et al. Syntheses, structural characterization and photophysical properties of 4-(2-pyridyl)-1,2,3-triazole rhenium(I) complexes. Dalton Trans. 2008:3292–3300. doi: 10.1039/b718538c. [DOI] [PubMed] [Google Scholar]
- 69.Maisonial A., Serafin P., Traikia M., Debiton E., Thery V., Aitken D.J., Lemoine P., Viossat B., Gautier A. Click chelators for platinum-based anticancer drugs. Eur. J. Inorg. Chem. 2008:298–305. [Google Scholar]
- 70.Kilpin K.J., Gavey E.L., McAdam C.J., Anderson C.B., Lind S.J., Keep C.C., Gordon K.C., Crowley J.D. Palladium(II) complexes of readily functionalized bidentate 2-pyridyl-1,2,3-triazole “Click” ligands: A synthetic, structural, spectroscopic, and computational study. Inorg. Chem. 2011;50:6334–6346. doi: 10.1021/ic200789b. [DOI] [PubMed] [Google Scholar]
- 71.Crowley J.D., Bandeen P.H., Hanton L.R. A one pot multi-component CuAAC “click” approach to bidentate and tridentate pyridyl-1,2,3-triazole ligands: synthesis, x-ray structures and copper(II) and silver(I) complexes. Polyhedron. 2010;29:70–83. doi: 10.1016/j.poly.2009.06.010. [DOI] [Google Scholar]
- 72.The related bis(2,6-bis(1,2,3-triazol-4-yl)pyridine)) Fe(II) complexes had been reported by the Flood group in 2007 see ref. [73].
- 73.Li Y., Huffman J.C., Flood A.H. Can terdentate 2,6-bis(1,2,3-triazol-4-yl)pyridines form stable coordination compounds? Chem. Commun. 2007:2692–2694. doi: 10.1039/b703301j. [DOI] [PubMed] [Google Scholar]
- 74.Fletcher J.T., Bumgarner B.J., Engels N.D., Skoglund D.A. Multidentate 1,2,3-triazole-containing chelators from tandem deprotection/click reactions of (trimethylsilyl)alkynes and comparison of their ruthenium(II) complexes. Organometallics. 2008;27:5430–5433. [Google Scholar]
- 75.Happ B., Friebe C., Winter A., Hager M.D., Hoogenboom R., Schubert U.S. 2-(1H-1,2,3-Triazol-4-yl)-pyridine ligands as alternatives to 2,2′-bipyridines in ruthenium(II) complexes. Chem. Asian J. 2009;4:154–163. doi: 10.1002/asia.200800297. [DOI] [PubMed] [Google Scholar]
- 76.Howson S.E., Allan L.E.N., Chmel N.P., Clarkson G.J., van Gorkum R., Scott P. Self-assembling optically pure Fe(A-B)3 chelates. Chem. Commun. 2009:1727–1729. doi: 10.1039/b821573a. [DOI] [PubMed] [Google Scholar]
- 77.Spek A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003;36:7–13. doi: 10.1107/S0021889802022112. [DOI] [Google Scholar]
- 78.Stevenson K.A., Melan C.F.C., Fleischel O., Wang R., Petitjean A. Solid-state self-assembly of triazolylpyridine-based helicates and mesocate: control of the metal-metal distances. Cryst. Growth Des. 2012;12:5169–5173. doi: 10.1021/cg301197w. [DOI] [Google Scholar]
- 79.Albrecht M., Riether C. Self assembly of a triple-stranded meso-helicate from two iron(II) ions and three [CH2]3-bridged bis(2,2'-bipyridine) ligands. Chem. Ber. 1996;129:829–832. doi: 10.1002/cber.19961290714. [DOI] [Google Scholar]
- 80.Albrecht M., Kotila S. Formation of a “meso-helicate” by self-assembly of three bis(catecholate) ligands and two titanium(IV) ions. Angew. Chem. Int. Ed. 1995;34:2134–2137. doi: 10.1002/anie.199521341. [DOI] [Google Scholar]
- 81.Kerckhoffs J.M.C.A., Peberdy J.C., Meistermann I., Childs L.J., Isaac C.J., Pearmund C.R., Reudegger V., Khalid S., Alcock N.W., Hannon M.J., et al. Enantiomeric resolution of supramolecular helicates with different surface topographies. Dalton Trans. 2007:734–742. doi: 10.1039/b614093a. [DOI] [PubMed] [Google Scholar]
- 82.Anderson C.B., Elliott A.B.S., Lewis J.E.M., McAdam C.J., Gordon K.C., Crowley J.D. fac-Re(CO)3 complexes of 2,6-bis(4-substituted-1,2,3-triazol-1-ylmethyl)pyridine “click” ligands: Synthesis, characterisation and photophysical properties. Dalton Trans. 2012;41:14625–14632. doi: 10.1039/c2dt31569f. [DOI] [PubMed] [Google Scholar]
- 83.Brotherton W.S., Guha P.M., Phan H., Clark R.J., Shatruk M., Zhu L. Tridentate complexes of 2,6-bis(4-substituted-1,2,3-triazol-1-ylmethyl)pyridine and its organic azide precursors: an application of the copper(II) acetate-accelerated azide-alkyne cycloaddition. Dalton Trans. 2011;40:3655–3665. doi: 10.1039/c0dt01702g. [DOI] [PubMed] [Google Scholar]
- 84.Kilpin K.J., Crowley J.D. Palladium(II) and platinum(II) complexes of bidentate 2-pyridyl-1,2,3-triazole “click” ligands: Synthesis, properties and X-ray structures. Polyhedron. 2010;29:3111–3117. doi: 10.1016/j.poly.2010.08.021. [DOI] [Google Scholar]
- 85.Guha P.M., Phan H., Kinyon J.S., Brotherton W.S., Sreenath K., Simmons J.T., Wang Z., Clark R.J., Dalal N.S., Shatruk M., et al. Structurally diverse copper(II) complexes of polyaza ligands containing 1,2,3-triazoles: Site selectivity and magnetic properties. Inorg. Chem. 2012;51:3465–3477. doi: 10.1021/ic2021319. [DOI] [PubMed] [Google Scholar]
- 86.Young M.C., Johnson A.M., Gamboa A.S., Hooley R.J. Achiral endohedral functionality provides stereochemical control in Fe(II)-based self-assemblies. Chem. Commun. 2013;49:1627–1629. doi: 10.1039/c3cc37912d. [DOI] [PubMed] [Google Scholar]
- 87.Sakamoto S., Fujita M., Kim K., Yamaguchi K. Characterization of self-assembling nano-sized structures by means of coldspray ionization mass spectrometry. Tetrahedron. 2000;56:955–964. doi: 10.1016/S0040-4020(99)01092-3. [DOI] [Google Scholar]
- 88.Miras H.N., Wilson E.F., Cronin L. Unravelling the complexities of inorganic and supramolecular self-assembly in solution with electrospray and cryospray mass spectrometry. Chem. Commun. 2009:1297–1311. doi: 10.1039/b819534j. [DOI] [PubMed] [Google Scholar]
- 89.Hawes C.S., Fitchett C.M., Kruger P.E. Synthesis of an Fe(II) dinuclear triple helicate from a novel -bis-(N-pyrazolyl)pyridine ligand, [Fe2L3]4+: Solution and solid-state studies. Supramol. Chem. 2012;24:553–562. doi: 10.1080/10610278.2012.688129. [DOI] [Google Scholar]
- 90.Welby C.E., Grkinic S., Zahid A., Uppal B.S., Gibson E.A., Rice C.R., Elliott P.I.P. Synthesis, characterisation and theoretical study of ruthenium 4,4[prime or minute]-bi-1,2,3-triazolyl complexes: fundamental switching of the nature of S1 and T1 states from MLCT to MC. Dalton Trans. 2012;41:7637–7646. doi: 10.1039/c2dt30510k. [DOI] [PubMed] [Google Scholar]
- 91.Lamping E., Monk B.C., Niimi K., Holmes A.R., Tsao S., Tanabe K., Niimi M., Uehara Y., Cannon R.D. Characterization of Three Classes of Membrane Proteins Involved in Fungal Azole Resistance by Functional Hyperexpression in Saccharomyces cerevisiae. Eukaryotic Cell. 2007;6:1150–1165. doi: 10.1128/EC.00091-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Paul L.E.H., Therrien B., Furrer J. Investigation of the reactivity between a ruthenium hexacationic prism and biological ligands. Inorg. Chem. 2012;51:1057–1067. doi: 10.1021/ic2021935. [DOI] [PubMed] [Google Scholar]
- 93.Paul L.E.H., Therrien B., Furrer J. Interaction of a ruthenium hexacationic prism with amino acids and biological ligands: ESI mass spectrometry and NMR characterization of the reaction products. J. Biol. Inorg. Chem. 2012;17:1053–1062. doi: 10.1007/s00775-012-0918-8. [DOI] [PubMed] [Google Scholar]
- 94.Lavanant H., Hecquet E., Hoppilliard Y. Complexes of l-histidine with Fe2+, Co2+, Ni2+, Cu2+, Zn2+ studied by electrospray ionization mass spectrometry. Int. J. Mass Spectrom. 1999;185–187:11–23. doi: 10.1016/S1387-3806(98)14044-7. [DOI] [Google Scholar]
- 95.Fleischel O., Wu N., Petitjean A. Click-triazole: Coordination of 2-(1,2,3-triazol-4-yl)-pyridine to cations of traditional tetrahedral geometry (Cu(I), Ag(I)) Chem. Commun. 2010;46:8454–8456. doi: 10.1039/c0cc02899a. [DOI] [PubMed] [Google Scholar]
- 96.Jones G., Willett P., Glen R.C., Leach A.R., Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 97.Narayana N., Weiss M.A. crystallographic analysis of a sex-specific enhancer element: Sequence-dependent DNA structure, hydration, and dynamics. J. Mol. Biol. 2009;385:469–490. doi: 10.1016/j.jmb.2008.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McPhillips T.M., McPhillips S.E., Chiu H.J., Cohen A.E., Deacon A.M., Ellis P.J., Garman E., Gonzalez A., Sauter N.K., Phizackerley R.P., et al. Blu-Ice and the distributed control system: Software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat. 2002;9:401–406. doi: 10.1107/S0909049502015170. [DOI] [PubMed] [Google Scholar]
- 99.Blessing R.H. An empirical correction for absorption anisotropy? Acta Cryst. Sect. A. 1995;51:33–38. doi: 10.1107/S0108767394005726. [DOI] [PubMed] [Google Scholar]
- 100.Altomare A., Burla M.C., Camalli M., Cascarano G.L., Giacovazzo C., Guagliardi A., Moliterni A.G.G., Polidori G., Spagna R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Crystallogr. 1999;32:115–119. doi: 10.1107/S0021889898007717. [DOI] [Google Scholar]
- 101.Sheldrick G.M. A short history of SHELX. Acta Crystallogr. Sect. A: Found. Crystallogr. 2008;A64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 102.Farrugia L.J. ORTEP-3 for windows - a version of ORTEP-III with a graphical user interface (GUI) J. Appl. Crystallogr. 1997;30:565–565. doi: 10.1107/S0021889897003117. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.