

Summary
Gamma-interferon-inducible lysosomal thiol reductase (GILT), expressed in antigen-presenting cells (APCs), facilitates the reduction of disulfide bonds of endocytosed proteins in the endocytic pathway and they are further processed for presentation of immunogenic peptides loaded on major histocompatibility complex (MHC) class II. Although the constitutive and IFNγ-inducible expression of GILT was observed in various APCs, such as dendritic cells, monocytes/macrophages, and B cells, GILT-expressing cell types remain unknown in the human central nervous system (CNS). Nasu-Hakola disease (NHD) is a rare autosomal recessive disorder characterized by sclerosing leukoencephalopathy and multifocal bone cysts, caused by a loss-of-function mutation of either TYROBP (DAP12) or TREM2, both of which are expressed on microglia. A rare heterozygous variant of the TREM2 gene encoding p.Arg47His causes a 3-fold increase in the risk for late-onset Alzheimer's disease (LOAD), suggesting that both NHD and AD are induced by dysfunction of the microglial TREM2 signaling pathway in the brains. We studied by immunohistochemistry GILT expression in NHD and AD brains. GILT was expressed on amoeboid microglia with the highest levels of expression in AD brains, compared with those in non-neurological control (NC) brains and in NHD brains. In AD brains, the clusters of amoeboid microglia surrounding amyloid-beta (Aꞵ) deposition strongly expressed GILT. Furthermore, a human microglial cell line expressed GILT in response to IFNγ. These results indicate that microglia, expressing constitutively high levels of GILT, act as a principal cell type of APCs in AD brains, in contrast to baseline levels of GILT expression in NHD brains.
Keywords: Alzheimer's disease, GILT, IFI30, microglia, Nasu-Hakola disease
1. Introduction
Gamma-interferon-inducible lysosomal thiol reductase (GILT), alternatively named IFI30, lysosomal thiol reductase, is the only known enzyme to catalyze disulfide bond reduction in the endocytic pathway (1-4). GILT enhances the major histocompatibility complex (MHC) class II-restricted presentation of a subset of epitopes derived from disulfide bond-containing antigens. GILT-dependent epitopes tend to be buried and they require reduction to be exposed for MHC class II binding. GILT also plays a role in the cross-presentation of exogenous viral antigens on MHC class I. In GILT-/- mice, antigen-presenting cells (APCs) displayed impaired MHC class II-restricted presentation and MHC class I-restricted cross-presentation (5,6).
Following delivery into the endosomal/lysosomal system by the mannose 6-phosphate receptor, N-terminal and C-terminal propeptides of the precursor 35-kDa GILT are cleaved in the early endosome, and the mature 28-kDa protein resides in late endosomes and lysosomes, where it is optimally active at acidic pH (1,7). Furthermore, an enzymatically active dimer of GILT precursors is secreted by activated macrophages/ monocytes in response to bacterial stimuli (1,8). GILT catalyzes the reduction of disulfide bonds through the active site consisting of a thioredoxin-like CXXC motif, corresponding to Cys-46 and Cys-49 in human GILT (1,3). GILT also regulates the cellular redox state. In GILT-/- cells, there is a shift from the reduced to the oxidized form of glutathione, resulting in reduced mitochondrial membrane potential, increased mitochondrial autophagy, decreased superoxide dismutase 2, and elevated superoxide levels (4,9). The absence of GILT in fibroblasts and T cells results in increased phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2) and increased cellular proliferation (4). GILT maintains the proteolytic activity of cathepsin S in phagosomes in alternatively activated macrophages, while GILT's reductase activity facilitates the degradation of cathepsin S in B cells (10,11).
GILT is constitutively expressed in most antigen presenting cells (APCs), such as dendritic cells, macrophages, and activated B cells, and induced by exposure to IFNγ in many cell types with maximal protein expression at 48 h (1,4). Signal transducer and activator of transcription 1 (STAT1) but not class II transactivator (CIITA) is responsible for IFNγ- inducible GILT expression, and it negatively regulates constitutive GILT expression (12).
Nasu-Hakola disease (NHD) is a rare autosomal recessive disorder characterized by sclerosing leukoencephalopathy and multifocal bone cysts, caused by a loss-of-function mutation of TREM2 or DAP12, both of which are expressed as a receptor-adaptor complex exclusively on the microglia in the central nervous system (CNS) (13). Pathologically, NHD brains exhibit extensive demyelination, astrogliosis, an accumulation of axonal spheroids, and activation of microglia predominantly in the white matter of frontal and temporal lobes (14). Alzheimer's disease (AD) is characterized by the hallmark pathology comprised of widespread amyloid-ꞵ (Aꞵ) deposition, neurofibrillary tangle (NFT) formation, extensive neurodegeneration, and profound activation of microglia in the brain (15). A single nucleotide polymorphism (SNP) p.Arg47His (R47H) of TREM2 elevates approximately three-fold the risk of AD, suggesting that the partial loss of function of TREM2 in microglia causes the AD pathology (16,17). Microglia is postulated to be the principal cell type of APCs (18,19), although it remains unknown whether microglia express GILT in NHD and AD brains. In the present study, we characterized GILT expression in NHD and AD brains by immunohistochemistry.
2. Materials and Methods
2.1. Human brain tissues
The brain autopsies were performed at the National Center Hospital, National Center of Neurology and Psychiatry (NCNP), Japan, Kohnodai Hospital, National Center for Global Health and Medicine (NCGM), Japan, and affiliated hospitals of Research Resource Network (RRN), Japan. The comprehensive examination by established neuropathologists (YS and TI) validated the pathological diagnosis. In all cases, written informed consent was obtained. The Ethics Committee of the NCNP for the Human Brain Research, the Ethics Committee of the NCGM on the Research Use of Human Samples, and the Human Research Ethics Committee (HREC) of the Meiji Pharmaceutical University (MPU) approved the present study.
For immunohistochemical studies, serial sections of the frontal cortex and the hippocampus were prepared from four subjects who died of non-neurological causes (NC), composed of a 63-year-old man who died of prostate cancer and acute myocardial infarction (NC1), a 67-year-old man who died of dissecting aortic aneurysm (NC2), a 57-year-old man who died of alcoholic liver cirrhosis (NC3), and a 61-year-old man who died of rheumatoid arthritis with interstitial pneumonia (NC4), ten AD patients, composed of a 68-year-old woman (AD1), a 70-year-old woman (AD2), a 68-year-old woman (AD3), a 56-year-old man (AD4), a 59-year-old man (AD5), an 81-year-old man (AD6), a 68-year-old woman (AD7), an 80-year-old man (AD8), a 72 year-old man (AD9), and a 77-year-old woman (AD11), and five NHD patients, composed of a 42-year-old man (NHD1), a 48-year-old woman (NHD2), a 44-year-old man (NHD3), a 32-year-old woman (NHD4), and a 38-year-old man (NHD5). The homozygous mutation of a single base deletion of 141G (c.141delG) in exon 3 of DAP12 was identified in NHD1, NHD2, and NHD5, while the genetic analysis was not performed in NHD3 or NHD4. All AD cases were satisfied with the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) criteria for diagnosis of definite AD (20). They were categorized into the stage C of amyloid deposition and the stage VI of neurofibrillary degeneration, following the Braak's staging (21).
2.2. Immunohistochemistry
After deparaffination, tissue sections were heated in 10 mM citrate sodium buffer, pH 6.0 by autoclave at 110℃ for 15 min in a temperature-controlled pressure chamber (Biocare Medical, Pacheco, CA, USA). They were treated at room temperature (RT) for 15 min with 3% hydrogen peroxide-containing methanol to block the endogenous peroxidase activity. They were then incubated with phosphate-buffered saline (PBS) containing 10% normal rabbit or goat serum at RT for 15 min to block non-specific staining, followed by incubation in a moist chamber at 4℃ overnight with goat polyclonal anti-GILT antibody (T-18, sc- 21827, Santa Cruz Biotechnology, Santa Cruz, CA, USA) or rabbit polyclonal anti-Iba1 antibody (Wako Pure Chemical, Tokyo, Japan) for a marker specific for microglia. The specificity of anti-GILT antibody was validated by western blot analysis of recombinant human GILT protein expressed in HEK293 cells, which were transfected with the pcDNA4/HisMax TOPO vector (Thermo Fisher Scientific, Carlsbad, CA, USA) containing the mature GILT sequence. After washing with PBS, tissue sections were incubated at RT for 30 min with horseradish peroxidase (HRP)- conjugated anti-goat or anti-rabbit secondary antibody (Nichirei, Tokyo, Japan), followed by incubation with diaminobenzidine tetrahydrochloride (DAB) substrate (Vector, Burlingame, CA, USA). They were processed for a counterstain with hematoxylin. Negative controls underwent all the steps except for exposure to primary antibody. In limited experiments, double immunolabeling was performed using T18 followed by incubation with HRP-conjugated anti-goat secondary antibody, and rabbit anti-Iba1 antibody (Wako), or mouse monoclonal antibody against MHC class II (TAL.1B5; HLA-DR, Dako, Tokyo, Japan) or amyloid-ꞵ peptide (12B2; Immunobiological Laboratories, Gunma, Japan), followed by incubation with alkaline phosphatase-conjugated anti-rabbit or anti-mouse secondary antibody (Nichirei) and exposure to DAB substrate and Warp Red chromogen (Biocare Medical).
2.3. Quantification of GILT immunoreactivity
To quantify immunolabeled areas, the images derived from three fields of the frontal cortex per each section were captured at a 200 X magnification on the Olympus BX51 universal microscope. They were then processed for quantification by using ImageJ software (National Institute of Health, Bethesda, MD, USA). The GILT-immunolabeled area was calibrated by the Iba1- immunolabeled area. The differences in the GILT/Iba1 ratio among NC, AD, and NHD subjects were evaluated statistically by one-way analysis of variance (ANOVA) followed by post-hoc Tukey's test.
2.4. Quantitative RT-PCR analysis and western blot analysis
To investigate the effects of inflammatory mediators on GILT expression, v-myc-immortalized human microglial cells named HMO6 (22), incubated in 10% fetal bovine serum (FBS)-containing Dulbecco's Modified Eagle's Medium (DMEM), were exposed for 24 hours to 1 μg/mL lipopolysaccharide (LPS; Sigma, St. Louis, MO, USA), recombinant human IFNγ, IL-4, IL-13 or TGFꞵ1 (50 ng/mL each; Peprotech, London, UK), followed by extraction of total cellular RNA for RT-PCR analysis or total cellular protein for western blot analysis. For quantitative RT-PCR (qPCR) analysis, cDNA was amplified by PCR on LightCycler 96 (Roche Diagnostics, Tokyo, Japan) with SYBR Green I and a primer set composed of 5'agtgtggagaccatcaaggaagac3' and 5'cagttcagccatcacttggatgag3' for detection of an 156 bp product of the GILT gene (NM_006332.4). The expression levels of GILT were standardized against the levels of glyceraldehyde-3-phosphate dehydrogenase (G3PDH) detected in the corresponding cDNA samples. All the assays were performed in triplicate.
3. Results
First, we validated the specificity of anti-GILT antibody T-18 by western blot of an Xpress-tagged recombinant mature GILT protein expressed in HEK293 cells (Figure 1A). Next, by qPCR and western blot, we found that human immortalized microglial cells HMO6 express high levels of GILT after stimulation for 24 hours with IFNγ but not with LPS, IL-4, IL-13, or TGFꞵ (Figure 1B and 1C). Then, by immunohistochemistry, we found that GILT is intensely expressed predominantly in Iba1- immunoreactive amoeboid microglia and occasionally in perivascular macrophages in the frontal cortex and the hippocampus of AD brains (Figure 2, panels a, c, d), while the much smaller number of microglia was labelled with GILT in both NHD and NC brains (Figure 3, panels a, c). In NC brains, most of the GILT-positive cells were perivascular macrophages. Since GILT is a marker of APCs, Iba-1 immunoreactive microglia and perivascular macrophages expressing GILT might represent a subset of professional APCs. The levels of GILT immunoreactivity were significantly elevated in AD brains, compared with NC brains (p = 0.0396) and NHD brains (p = 0.0057) (Figure 4). The levels of GILT expression were not elevated in NHD brains and not significantly different between NC and NHD brains (p = 0.8183) (Figure 4). In AD brains, clusters of amoeboid microglia strongly expressing GILT were accumulated in and around of amyloid plaques and these cells coexpressed HLA-DR, a MHC class II molecule and Iba1 (Figure 5a-5d).
Figure 1.
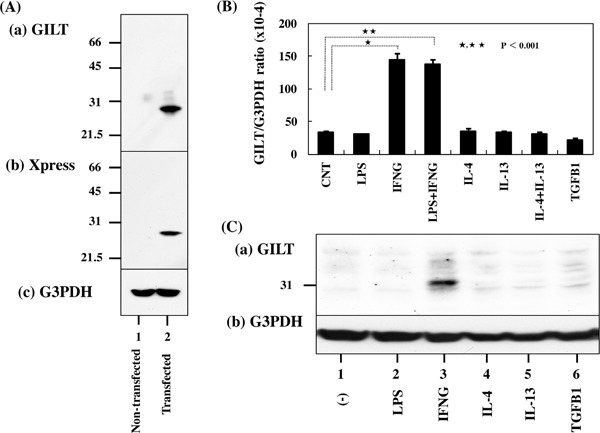
GILT expression in human microglial cells. Panel A. The specificity of GILT antibody. Western blot of non-transfected HEK293 cells (lane 1) and the cells transfected with the vector containing the mature GILT sequence (lane 2). (a) GILT, (b) Xpress tag, and (c) G3PDH as a loading control. Panel B. Quantitative RT-PCR analysis of GILT expression in HMO6 microglia in culture. HMO6 cells were exposed for 24 hours to 1 μg/mL lipopolysaccharide (LPS), recombinant human IFNγ(IFNG), IL-4, IL-13 or TGFꞵ1 (TGFB1) (50 ng/mL each), followed by extraction of total cellular RNA that is processed for qRT-PCR. The expression levels of GILT were standardized against the levels of G3PDH. Panel C. Western blot analysis. HMO6 is exposed for 24 hours to 1 μg/mL LPS, recombinant human IFNγ(IFNG), IL-4, IL-13 or TGFꞵ1 (TGFB1) (50 ng/mL each). Then, total cellular protein was processed for western blot analysis.
Figure 2.
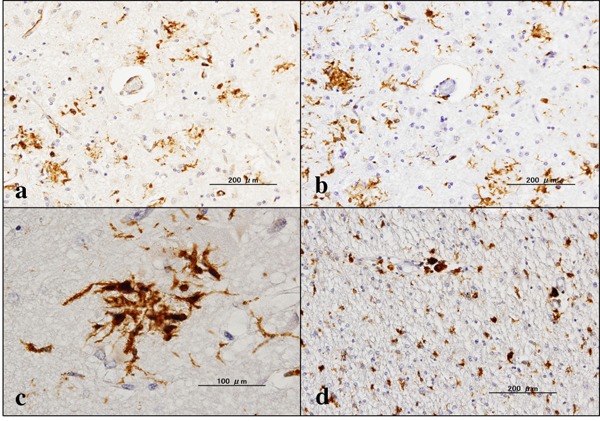
Expression of GILT in AD brains. (a) the frontal cortex, GILT, (b) the same area of (a), Iba1, (c) the hippocampus, GILT, and (d) the hippocampus white matter, GILT.
Figure 3.
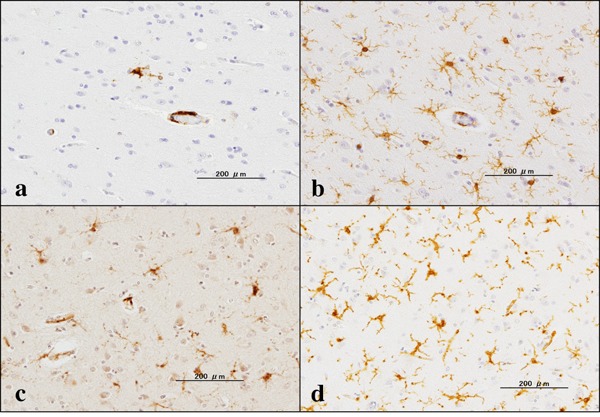
Expression of GILT in NC and NHD brains. (a) the frontal cortex of NC, GILT, (b) the same area of (a), Iba1, (c) the frontal cortex of NHD, GILT, and (d) the same area of (c), Iba1.
Figure 4.
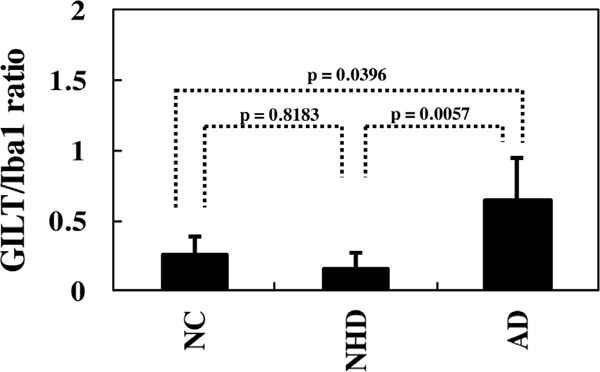
Quantitative analysis of GILT expression on microglia in NC, NHD, and AD brains. The GILT-immunolabeled area/Iba1-immunolabelled area ratio in the frontal cortex is shown.
Figure 5.
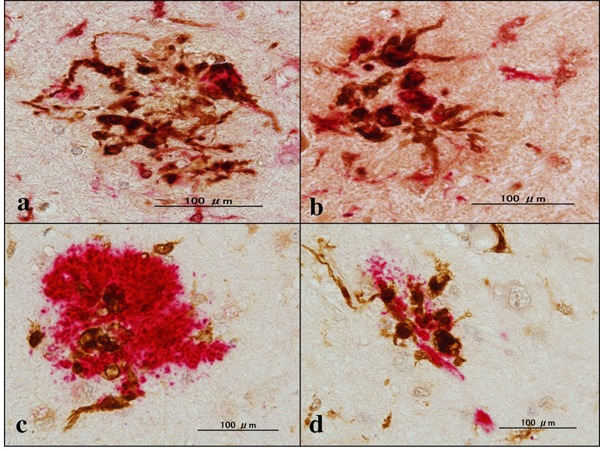
Expression of GILT, Iba1, HLA-DR and Aβ in AD brains. (a) the hippocampus, GILT (dark brown), Iba1 (red), (b) the hippocampus, GILT (dark brown), HLA-DR (red), (c) the hippocampus, GILT (dark brown), Aβ (red), and (d) the hippocampus, GILT (dark brown), Aβ (red).
4. Discussion
GILT is a lysosomal thiol reductase optimally active at acidic pH capable of catalyzing reduction of inter-and intrachain disulfide bonds (1-4). GILT is the only known enzyme to catalyze disulfide bond reduction in the endocytic pathway. GILT plays a key role in MHC class II-restricted antigen processing (5). GILT facilitates the presentation of a subset of epitopes from disulfide bond-containing antigens. Enhanced presentation of MHC class II-restricted epitopes alters immunological tolerance and modulates CD4+ T-cell mediated autoimmunity (2,3).
We found that microglia express GILT in AD and NHD brains. Both NHD and AD are induced by dysfunction of the microglial TREM2 signaling pathway in the brains. GILT immunoreactivity was upregulated on microglia in AD brains compared with NHD brains. This is attributable to a difference in demand for disulfide bond reduction by microglia between AD and NHD brains. Microglia, resident myeloid cells in the CNS, play a pivotal role in maintenance of brain homeostasis, along with progression of neurodegenerative disease (23). Microglia originate from erythromyeloid progenitor cells in the yolk sac and populate the CNS during early embryonic development (24). Microglia actively survey the surrounding microenvironment with dynamic processes. Microglial phagocytosis utilizes different types of receptors to initiate function, such as Toll-like receptors (TLRs) for foreign microbial pathogens and TREM2 for apoptotic cellular substrates (25). Microglia adopt two distinct activation phenotypes, composed of a proinflammatory and neurotoxic "classical" activation (M1) phenotype by exposure to IFNγ or LPS and an anti-inflammatory and neuroprotective "alternative" activation (M2) phenotype following treatment with IL4 or IL-13 (26). However, at present, a definite marker capable of clearly separating M1 and M2 phenotypes in human microglia remains largely unidentified (27). HMO6 human microglial cells expressed high levels of GILT after stimulation with IFNγ but not with LPS, IL-4, IL-13, or TGFꞵ, suggesting that GILT serves as a valid marker for detection of IFNγ-activated M1 microglia. A previous study showed that CD4+ and CD8+ T cells are accumulated in the hippocampal parenchyma of AD brains (28). We previously reported a discernible infiltration of CD3+ T cells in NHD brains (14). Therefore, the possibility exists that primed T cells in AD and NHD brains are locally reactivated by recognizing antigens presented by APCs in the CNS. Although the precise antigens presented by microglia in the brain for CD4+ and CD8+ T cells remain totally unknown, the adaptive immune response mediated by T cells plays a key role in exacerbation of the existing inflammation in AD, to a lesser extent, NHD brains (29). Both CD4+ T and CD8+ T cells produce large amount of IFNγ, a potent inducer of the M1 phenotype. IFNγ also increases the expression of MHC class II, CD86, CD40, and ICAM-1 on the cell surface of microglia that enhance their APC function. These results suggest that GILT-expressing M1 microglia observed in close proximity to amyloid plaques of AD brains are capable of efficiently presenting certain antigens enriched with disulfide bonds derived from the plaques in a MHC class II-restricted manner. Furthermore, GILT might regulate the cellular redox state in these cells under Aꞵ- induced oxidative stress conditions in AD brains (30).
Disulfide bonds are the most common covalent link between amino acids in proteins. Disulfide bonds are present in 15% of the human proteome (31). Disulfide bonds stabilize proteins to confine conformational changes. They exist in 55% of the proteins involved in pathological amyloid formation. They play an important role in the kinetics of aggregation and the structure and toxicity of the formed aggregates. Several proteins related to AD pathogenesis have disulfide bonds. Beta-secretase 1 (BACE1), a type I transmembrane aspartic protease responsible for the ꞵ-secretase cleavage of amyloid beta precursor protein (APP) has three intramolecular disulfide bonds in the catalytic domain that regulate protein maturation (32). APP has three intramolecular disulfide bonds in the copper-binding domain (CuBD) (33). Tau, whose phosphorylation induces neurofibrillary tangle (NFT) formation, exists as six different isoforms and the longest isoform of Tau has two cysteine residues that form both intramolecular and intermolecular disulfide bonds (34). Intermolecular disulfide bonds promote Tau aggregation, while intramolecular disulfide bonds prevent Tau aggregation. A detergent-insoluble disulfide-linked form of G3PDH is found in brain tissues of AD patients (35). Aꞵ42 and oxidative stress promote the nuclear and cytoplasmic accumulation of insoluble aggregates of disulfide-bonded G3PDH that cause neuronal cell death. It remains unknown whether disulfide bonds of these proteins are reduced by GILT in the endocytic pathway for antigen presentation on the MHC class II (36).
In conclusion, GILT, serving as a M1 marker for IFNγ-activated microglia, is intensely expressed by a subset of microglia and perivascular macrophages, although GILT expression levels are significantly increased in AD brains, compared with NC and NHD brains. We found a coexpression of GILT and MHC class II antigen in microglia. The class II-MHC-expressing microglia are increased in AD brains (37). GILT facilitates the MHC class II-restricted presentation of epitopes derived from disulfide bond-rich antigens in the endocytic pathway (1-4). These results suggest a pivotal role of GILT in antigen presentation by M1 microglia in AD brains, and to a lessor extent, NHD brains. The precise antigens presented by GILT-expressing microglia in AD brains and NHD brains remain to be investigated.
Acknowledgements
The authors thank Drs. Kenji Jinnai, Nobutaka Arai, Kiyotaka Nakamagoe, Nobutaka Motohashi, and Saburo Yagishita for providing us brain samples. This work was supported by grants from the Research on Intractable Diseases, entitled "Clinicopathological and genetic studies of Nasu-Hakola disease" (H21-Nanchi- Ippan-201; H22-Nanchi-Ippan-136), the Ministry of Health, Labour and Welfare of Japan, and grants from the JSPS KAKENHI (C25430054 and 16K07043) and the Dementia Drug Development Research Center (DRC) project (S1511016), the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
References
- 1. Hastings KT, Cresswell P. Disulfide reduction in the endocytic pathway: Immunological functions of gamma-interferon-inducible lysosomal thiol reductase. Antioxid Redox Signal. 2011; 15:657-668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hastings KT. GILT: Shaping the MHC class II-restricted peptidome and CD4+ T cell-mediated immunity. Front Immunol. 2013; 4:429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. West LC, Cresswell P. Expanding roles for GILT in immunity. Curr Opin Immunol. 2013; 25:103-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rausch MP, Hastings KT. Diverse cellular and organismal functions of the lysosomal thiol reductase GILT. Mol Immunol. 2015; 68:124-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maric M, Arunachalam B, Phan UT, Dong C, Garrett WS, Cannon KS, Alfonso C, Karlsson L, Flavell RA, Cresswell P. Defective antigen processing in GILT-free mice. Science. 2001; 294; 1361-1365. [DOI] [PubMed] [Google Scholar]
- 6. Sigh R, Cresswell P. Defective cross-presentation of viral antigens in GILT-free mice. Science. 2010; 328:1394-1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Phan UT, Arunachalam B, Cresswell P. Gamma-interferon-inducible lysosomal thiol reductase (GILT). Maturation, activity, and mechanism of action. J Biol Chem. 2000; 275:25907-25914. [DOI] [PubMed] [Google Scholar]
- 8. Lackman RL, Jamieson AM, Griffith JM, Geuze H, Cresswell P. Innate immune recognition triggers secretion of lysosomal enzymes by macrophages. Traffic. 2007; 8:1179-1189. [DOI] [PubMed] [Google Scholar]
- 9. Chiang HS, Maric M. Lysosomal thiol reductase negatively regulates autophagy by altering glutathione synthesis and oxidation. Free Radic Biol Med. 2011; 51:688-699. [DOI] [PubMed] [Google Scholar]
- 10. Phipps‐Yonas H, Semik V, Hastings KT. GILT expression in B cells diminishes cathepsin S steady‐state protein expression and activity. Eur J Immunol. 2013; 43:65-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Balce DR, Allan ER, McKenna N, Yates RM. γ-Interferon-inducible lysosomal thiol reductase (GILT) maintains phagosomal proteolysis in alternatively activated macrophages. J Biol Chem. 2014; 289:31891-31904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Srinivasan P, Maric M. Signal transducer and activator of transcription 1 negatively regulates constitutive gamma interferon-inducible lysosomal thiol reductase expression. Immunology. 2011; 2011; 132; 209-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Satoh J. Possible role of microgliopathy in the pathogenesis of Nasu-Hakola disease. Clin Exp Neuroimmunol. 2013; 4(Suppl. 17-26. [Google Scholar]
- 14. Satoh J, Tabunoki H, Ishida T, Yagishita S, Jinnai K, Futamura N, Kobayashi M, Toyoshima I, Yoshioka T, Enomoto K, Arai N, Arima K. Immunohistochemical characterization of microglia in Nasu-Hakola disease brains. Neuropathology. 2011; 31:363-375. [DOI] [PubMed] [Google Scholar]
- 15. Sarlus H, Heneka MT. Microglia in Alzheimer's disease. J Clin Invest. 2017; 127:3240-3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. 2013; 368:107-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ulrich JD, Ulland TK, Colonna M, Holtzman DM. Elucidating the role of TREM2 in Alzheimer's disease. Neuron. 2017; 94:237-248. [DOI] [PubMed] [Google Scholar]
- 18. Chastain EM, Duncan DS, Rodgers JM, Miller SD. The role of antigen presenting cells in multiple sclerosis. Biochim Biophys Acta. 2011; 1812:265-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schetters STT, Gomez-Nicola D, Garcia-Vallejo JJ, Van Kooyk Y. Neuroinflammation: Microglia and T cells get ready to tango. Front Immunol. 2018; 8:1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991; 41:479-486. [DOI] [PubMed] [Google Scholar]
- 21. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006; 112:389-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nagai A, Nakagawa E, Hatori K, Choi HB, McLarnon JG, Lee MA, Kim SU. Generation and characterization of immortalized human microglial cell lines: Expression of cytokines and chemokines. Neurobiol Dis. 2001; 8:1057-1068. [DOI] [PubMed] [Google Scholar]
- 23. Hickman S, Izzy S, Sen P, Morsett L, El Khoury J. Microglia in neurodegeneration. Nat Neurosci. 2018; 21:1359-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ginhoux F, Prinz M. Origin of microglia: Current concepts and past controversies. Cold Spring Harb Perspect Biol. 2015; 7:a020537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fu R, Shen Q, Xu P, Luo JJ, Tang Y. Phagocytosis of microglia in the central nervous system diseases. Mol Neurobiol. 2014; 49:1422-1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. 2016. 1181-1194. [DOI] [PubMed] [Google Scholar]
- 27. Walker DG, Lue LF. Immune phenotypes of microglia in human neurodegenerative disease: Challenges to detecting microglial polarization in human brains. Alzheimers Res Ther. 2015; 7:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Togo T, Akiyama H, Iseki E, Kondo H, Ikeda K, Kato M, Oda T, Tsuchiya K, Kosaka K. Occurrence of T cells in the brain of Alzheimer's disease and other neurological diseases. J Neuroimmunol. 2002; 124:83-92. [DOI] [PubMed] [Google Scholar]
- 29. McManus RM, Mills KH, Lynch MA. T cells-protective or pathogenic in Alzheimer's disease? J Neuroimmune Pharmacol. 2015; 10:547-560. [DOI] [PubMed] [Google Scholar]
- 30. Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, Collin F. Oxidative stress and the amyloid beta peptide in Alzheimer's disease. Redox Biol. 2018; 14:450-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mossuto MF, Bolognesi B, Guixer B, Dhulesia A, Agostini F, Kumita JR, Tartaglia GG, Dumoulin M, Dobson CM, Salvatella X. Disulfide bonds reduce the toxicity of the amyloid fibrils formed by an extracellular protein. Angew Chem Int Ed Engl. 2011; 50:7048-7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fisher F, Molinari M, Bodendorf U, Paganetti P. The disulphide bonds in the catalytic domain of BACE are critical but not essential for amyloid precursor protein processing activity. J Neurochem. 2002; 80:1079-1088. [DOI] [PubMed] [Google Scholar]
- 33. Reinhard C, Hébert SS, De Strooper B. The amyloid-ꞵ precursor protein: Integrating structure with biological function. EMBO J. 2005; 24:3996-4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Barghorn S, Mandelkow E. Toward a unified scheme for the aggregation of tau into Alzheimer paired helical filaments. Biochemistry. 2002; 41:14885-14896. [DOI] [PubMed] [Google Scholar]
- 35. Cumming RC, Schubert D. Amyloid-ꞵ induces disulfide bonding and aggregation of GAPDH in Alzheimer's disease. FASEB J. 2005; 19:2060-2062. [DOI] [PubMed] [Google Scholar]
- 36. Solé-Domènech S, Cruz DL, Capetillo-Zarate E, Maxfield FR. The endocytic pathway in microglia during health, aging and Alzheimer's disease. Ageing Res Rev. 2016; 32:89-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Perlmutter LS, Scott SA, Barrón E, Chui HC. MHC class II-positive microglia in human brain: Association with Alzheimer lesions. J Neurosci Res. 1992; 33:549-558. [DOI] [PubMed] [Google Scholar]