

Abstract
Originally described over a decade ago as a T cell transcription factor regulating T helper 1 cell lineage commitment, T-bet is now recognized as having an important role in many cells of the adaptive and innate immune system. T-bet has a fundamental role in coordinating type 1 immune responses by controlling a network of genetic programmes that regulate the development of certain immune cells and the effector functions of others. Many of these transcriptional networks are conserved across innate and adaptive immune cells and these shared mechanisms highlight the biological functions that are regulated by T-bet.
T-bet (encoded by Tbx21) is an immune cell-specific member of the T-box family of transcription factors (FIG. 1). The adaptive immune system arose approximately 500 million years ago in jawed fish probably as a result of the emergence of the recombination-activating gene (RAG) transposon1. Prior to this event, primitive B cell-like and T cell-like cells could be identified in jawless fish, such as lampreys2. Intriguingly, a comparative evolutionary analysis indicates that the eomesodermin (EOMES), T-box brain protein 1 (TBR1) and T-bet subfamily of T-box genes arose very early in evolution before the appearance of classical adaptive immunity and can be identified in both lampreys (see ensembl database) and amphioxus3. Many of our current ideas about the key functions of T-bet derive from studies in T cells. The initial description of T-bet as a master regulator of commitment to the T helper 1 (TH1) cell lineage as well as the elucidation of intersecting T cell transcriptional pathways has enabled rapid progress to be made in defining the genetic modules that control T cell polarity. However, it is possible that these regulatory pathways may have been coopted from more ancient cell types, especially as it is now appreciated that innate immune cells also express T-bet.
Figure 1 |. Expression and functions of T-bet in immune cells.

T-bet is expressed in multiple cells of the innate and adaptive immune system. Its expression is required for the survival, development and proper functions of immune cells. In the innate immune system, T-bet is expressed in dendritic cells (DCs), natural killer (NK) cells, natural killer T (NKT) cells and innate lymphoid cells (ILCs). In the adaptive immune system, T-bet is expressed in CD4+ and CD8+ T effector cells, B cells, γδ T cells and a subset of regulatory T (TReg) cells. IFNγ, interferon-γ; IL-7Rα, interleukin-7 receptor-α; NKp46, NK cell p46-related protein; RORγt, retinoic acid receptor-related orphan receptor-γt; TFH, T follicular helper; TH, T helper; TNF, tumour necrosis factor.
The effects of T-bet on the regulation of adaptive immune functions are well recognized and the molecular mechanisms have mostly been elucidated by analysing the genomic transcriptional targets in different cell types. Transcription factors activate or repress their target genes by binding to accessible promoter and enhancer elements. The development of methodologies to identify the direct genomic targets of T-bet has transformed our ability to dissect the intracellular pathways that regulate T cell differentiation. Chromatin immunoprecipitation (ChIP) coupled with array-based proximal promoter analysis (ChIP-chip) provided insight into the genes that are directly regulated by T-bet. The advent of massively parallel DNA sequencing (also known as ChIP-sequencing (ChIP-seq)) has more recently facilitated both greater resolution of promoter targets and insight into the distal enhancer elements that T-bet regulates4,5. ChIP-seq data sets from other transcription factors, such as GATA-binding protein 3 (GATA3), retinoic acid receptor-related orphan receptor-γt (RORγt; encoded by Rorc) and signal transducer and activator of transcription 4 (STAT4), combined with knowledge of the chromatin architecture at specific loci, has increased the understanding of how these molecules interact to coordinate T cell function4–9. Interestingly, it has recently been shown that T-bet binds to large domains that have clusters of enhancers associated with important T cell genes — known as ‘super enhancers’ — which are characteristics of master regulatory genes that define cell identity10. However, in most cases, it remains to be formally proven that these specific interactions take place at a single-cell level and that they do not reflect the heterogeneity within the cell populations studied. By contrast, much less is known about the range of T-bet target genes in primary innate immune cells because of the relatively large cell numbers that are required for these types of studies. However, many of the transcriptional pathways that have been defined in T cells are also relevant in innate immune cells, which indicates that considerable insights can be gained by investigating the parallel roles of T-bet in innate and adaptive immunity.
In this Review, we discuss the shared transcriptional mechanisms that regulate the development and/or the effector functions of innate and adaptive immune cells. We review the role of T-bet in maintaining mucosal homeostasis through its action in dendritic cells (DCs) and innate lymphoid cells. Special emphasis is given to the role of T-bet in the biology of CD4+ TH cells. We discuss the intricate molecular mechanisms that are regulated by T-bet to promote TH1 cell differentiation and to suppress the development of opposing TH cell lineages. In addition, we describe how T-bet expression ‘tips the balance’ in favour of terminal differentiation of CD4+ and CD8+ effector T cells at the expense of memory cell formation.
T-bet in the innate immune system
T-bet in DCs.
Much less is known about the function of T-bet in the innate immune system than is known about its function in T cells. T-bet was initially shown to be expressed in human monocytes and myeloid DCs after stimulation with interferon-γ (IFNγ)11. Loss-of-function studies indicated that, in the absence of T-bet, DCs were unable to efficiently prime TH1 cell responses and they did not respond to the protective adjuvant activity of CpG DNA in vivo during Listeria spp. infection12,13 (FIG. 2). T-bet was also shown to directly regulate inflammatory arthritis via its expression in DCs and by regulating T cell activation14.
Figure 2 |. T-bet in innate immune cells.

a | T-bet expression in dendritic cells (DCs) is required to properly prime T helper 1 (TH1) cells. T-bet suppresses tumour necrosis factor (TNF) production in colonic DCs and this is required for the maintenance of mucosal homeostasis. b | T-bet is expressed in the innate lymphoid cell 1 (ILC1) subset, which is characterized by the sole production of interferon-γ (IFNγ). ILC3s are dependent on the expression of retinoic acid receptor-related orphan receptor-γt (RORγt) and can be subdivided into CC-chemokine receptor 6 (CCR6)+ and CCR6– subsets. CCR6− ILC3s express T-bet. High T-bet expression in these cells is associated with low RORγt and interleukin-7 receptor (IL-7R) expression, and high expression of NK cell p46-related protein (NKp46), CXC-chemokine receptor 3 (CXCR3) and IFNγ. Low T-bet expression in the CCR6− ILC3 subset is associated with IL-22 expression and low or no expression of NKp46 and IFNγ. CCR6+ ILC3s do not express T-bet but express IL-17 and IL-22. The lineage inter-relationships of these different subpopulations are incompletely defined. c | T-bet expression in invariant natural killer T (iNKT) cells promotes their survival through the regulation of CD122 (also known as IL-15Rβ) expression. d | T-bet and eomesodermin (EOMES) regulate the maturation process of NK cells in a coordinated fashion. T-bet is expressed at an immature stage of differentiation that is characterized by TNF-related apoptosis-inducing ligand (TRAIL) expression. EOMES expression is required to silence TRAIL expression and to complete the maturation process of NK cells. e | T-bet is not expressed in naive γδ T cells. Its expression is rapidly induced following T cell receptor (TCR) engagement. Together with EOMES, T-bet regulates IFNγ production in mature γδ T cells. Dashed line indicates that the developmental relationship between these cells is unclear. γc, common cytokine receptor γ-chain; IFNγR, IFNγ receptor.
T-bet has more recently been recognized as being an important regulator of intestinal homeostasis15. T-bet expression in DCs was shown to regulate the homing of mast cell progenitors to mucosal tissue through the control of expression of mucosal addressin cell adhesion molecule 1 (MADCAM1) and vascular cell adhesion molecule 1 (VCAM1)16. However, despite a marked reduction in the number of mucosal mast cells, Tbx21−/− mice were still able to vigorously respond to intestinal infection with Trichinella spiralis.
In order to focus solely on the role of T-bet in innate immunity, T-bet-deficient mice were crossed onto a Rag2−/− background. Remarkably, mice deficient in both T-bet and RAG2 (Tbx21−/−Rag2−/− mice) developed a spontaneous intestinal inflammation that resembled ulcerative colitis in humans17. Inflammation in this model was transmissible to T-bet-sufficient mice and was driven by a dysbiotic microbiota17,18. The model became known as the TRUC (Tbx21−/−Rag2−/− ulcerative colitis) mouse model and has proved useful in uncovering mechanisms of inflammation-associated colorectal carcinoma and in providing a novel conceptual framework for innate immune dysregulation in ulcerative colitis19,20. Dysregulated tumour necrosis factor (TNF) production from colonic DCs underlies disease in TRUC mice as it causes epithelial cell apoptosis (FIG. 2). In CD4+ T cells, T-bet transactivates Tnf, whereas in DCs, the Tnf locus is repressed by T-bet21. The generation of an isogenic colony of mice that did not develop colitis (TRnUC mice (Tbx21−/−Rag2−/− no ulcerative colitis)) enabled the identification of a crucial pathobiont, Helicobacter typhlonius. Metagenomic sequencing of the colonic microbiome from TRUC and TRnUC mice, combined with complementation and transmissibility studies, confirmed that H. typhlonius was responsible for the colitic phenotype of TRUC mice22.
T-bet in ILCs.
Innate lymphoid cells (ILCs) are a newly described type of cell that share many functional attributes with effector T cell subsets23. ILCs are important at mucosal sites, where they regulate epithelial homeostasis in relation to the intestinal microbiome, and they are dysregulated in inflammatory disease in both mice and humans24,25. It is increasingly recognized that shared transcriptional mechanisms are conserved between ILCs and CD4+ T cells and that ILC subsets mirror their TH cell counterparts in terms of their cytokine-producing capabilities. Thus, group 1 ILCs express T-bet, group 2 ILCs express GATA3 and group 3 ILCs express RORγt26. Natural killer (NK) cells have also been classified as group 1 ILCs, but given that they have substantial differences to this cell type, they will be considered separately. ILCs express the IL-7 receptor (IL-7R) and CD90 (also known as THY1) and are negative for all lineage markers and antigen (B cell and T cell) receptors. ILCs are dependent on IL-7R signalling through the common cytokine receptor γ-chain (γc). ILC1s, which are another subset of group 1 ILCs, are positive for NK cell p46-related protein (NKp46; also known as NCR1), as are a subset of group 3 ILCs (NKp46+ ILC3s), which also express IL-23R26.
The first description of a functional role for T-bet in ILC biology came from studies in the TRUC mouse model (FIG. 3). Colitis in TRUC mice was abrogated by genetic or antibody-mediated depletion of ILCs. Furthermore, T-bet seemed to control the plasticity of RORγt+ ILCs, by inducing IFNγ expression and by repressing IL-17A production (FIG. 2). These effects were partly mediated through direct repression of IL-7R expression by T-bet22. As IL-7 has been shown to stabilize RORγt expression in ILCs27, this provides a potential mechanism for the reciprocal expression pattern of T-bet and RORγt (FIG. 2). DC-derived TNF functioned with IL-23 to drive IL-17 production by ILCs; this indicates that there is a newly identified level of innate cellular crosstalk at the inflamed mucosal surface (FIG. 3).
Figure 3 |. T-bet expression in ILCs and DCs in the intestines.

Innate lymphoid cells (ILCs) and T-bet-expressing dendritic cells (DCs) interact in the colonic lamina propria to maintain epithelial integrity. T-bet (encoded by Tbx21) represses tumour necrosis factor (TNF) expression in colonic DCs. In the absence of T-bet, TNF from DCs causes colonic epithelial cell apoptosis in the context of the pathobiont, Helicobacter typhlonius. TNF from DCs cooperates with interleukin-23 (IL-23) to induce interferon-γ (IFNγ) and IL-17 expression from ILCs. T-bet expression in ILCs regulates the plasticity of cytokine responses by repressing IL-17 and by inducing IFNγ production. This pathology creates an inflammatory milieu that generates a dysbiotic microbiota that is capable of transmitting colonic inflammation to wild-type mice. Dashed line indicates that the developmental relationship between these ILC subsets is unclear. CCR6, CC-chemokine receptor 6; NKp46, NK cell p46-related protein; RORγt, retinoic acid receptor-related orphan receptor-γt.
Other studies have shown that T-bet has a crucial role in the development and function of group 1 and 3 ILCs. The absence of T-bet was shown to result in a loss of NKp46+ ILC3s in the small intestine28. T-bet was found to be expressed in CC-chemokine receptor 6 (CCR6)− ILC3s but not in CCR6+ ILC3s. Within the T-bet+CCR6− ILC3 subset, both NKp46+ and NKp46− ILC3s were identified, and T-bet expression was found to be increased in proportion to NKp46 expression, which also correlated with increasing IFNγ expression29. Other canonical T-bet target genes, such as CXC-chemokine receptor 3 (Cxcr3) and CD95 ligand (Cd95l; also known as Fasl) are also expressed in T-bet+CCR6− ILC3s. The signals that regulate T-bet expression in ILC3s are incompletely defined; IL-12 was shown not to be required but IL-23 induced T-bet expression29. IFNγ-producing T-bet+ ILC1 subsets have recently been reported in mice and humans and these cells were shown to accumulate in inflamed mucosal tissues of patients with Crohn’s disease24,25. Whether ILC1s represent a separate lineage or whether they are related to NK cells or ILC3s remains to be determined.
T-bet in NK cells.
In combination with the T-box transcription factor EOMES, T-bet regulates the development and the terminal maturation of NK cells30,31 (FIG. 2). In the absence of T-bet, susceptibility to metastatic cancer, including melanoma, was shown to be increased because of impaired NK cell function and survival in vivo32. T-bet deficiency alone only induces a partial defect in NK cell numbers as a result of a compensatory increase in EOMES, whereas deletion of both T-bet and EOMES results in a complete absence of NK cells30. The development of immature NK cells that express TNF-related apoptosis-inducing ligand (TRAIL; also known as TNFSF10) was shown to be dependent on T-bet, whereas maturation and loss of TRAIL expression was shown to be more strongly influenced by EOMES30.
T-bet in non-conventional T cells.
T-bet is also expressed in invariant NKT (iNKT) and γδ T cells. These thymus-derived cells share features of both innate and adaptive immune cells and require T-bet either for their development (in the case of iNKT cells) or for their production of effector cytokines (in the case of γδ T cells). In the absence of T-bet, the number of Vα14i NKT cells is significantly reduced, possibly because of impaired survival that results from reduced IL-15R signalling secondary to low expression of the T-bet target gene Cd122 (also known as Il15rb)31,33. Despite the marked reduction in numbers of iNKT cells in the absence of T-bet, the remaining iNKT cells were able to induce airway hyper-reactivity through their augmented IL-4 and IL-13 production34.
T-bet is not expressed in naive γδ T cells but its expression is induced in γδ T cells following T cell receptor (TCR) signalling35 (FIG. 2). A high percentage of γδ T cells will produce IFNγ after TCR stimulation, and this is regulated by inducible T-bet and constitutive EOMES expression36. T-bet deficiency results in a 50% reduction in the frequency of IFNγ-producing γδ T cells, which suggests that constitutive EOMES expression and the hypomethylation of intron 1 at the Ifng locus contribute to the remaining IFNγ production by T-bet-deficient γδ T cells36.
T-bet in TH cells
T-bet: the master regulator of TH1 cell differentiation.
Selective expression of T-bet accounts for TH1 cell development and for the TH1 cell-specific expression of IFNγ37 (BOX 1; FIG. 4). Ectopic expression of T-bet in differentiated TH2 or TH17 cells results in their conversion into IFNγ-producing TH1-like cells37–40. Conversely, T-bet-deficient CD4+ T cells only produce small amounts of IFNγ under TH1 cell-polarizing conditions and fail to mount effective TH1 cell responses during infections with Leishmania major, Mycobacterium tuberculosis and Salmonella enterica subsp. enterica serovar Typhimurium41–43.
Box 1 |. CD4+ TH subsets.
When a naive CD4+ T cell gets activated by an antigen-presenting cell (APC) in the periphery, it differentiates into one of several T helper (TH) cell subsets to become a TH1, TH2, TH17, T follicular helper (TFH) or peripherally derived regulatory T (pTReg) cell. The decision-making process is instructed by the cytokines that are present in the environment during the activation process. Each TH cell subset is distinguished by a specialized gene expression programme, which is under the control of a lineage-defining transcription factor. The lineage-defining transcription factors are T-bet for the TH1 cell lineage, GATA-binding protein 3 (GATA3) for the TH2 cell lineage, retinoic acid receptor-related orphan receptor-γt (RORγt) for the TH17 cell lineage, B cell lymphoma-6 (BCL-6) for TFH cells and forkhead box P3 (FOXP3) for pTReg cells (see the figure). CD4+ TH cell subsets are defined by the signature cytokines that they express, their distinct homing properties and their specialized effector functions, which make them best equipped to target a particular class of pathogen. Thus, TH1 cells produce interferon-γ (IFNγ) and are particularly effective at activating macrophage microbicidal mechanisms against intracellular bacteria, protozoa and viruses. By contrast, TH2 cells secrete interleukin-4 (IL-4), IL-5 and IL-13, which are required for the expulsion of extracellular parasites. TH17 cells have evolved to protect mucosal surfaces against extracellular bacteria and fungi through the production of IL-17A, IL-17F, IL-21 and IL-22, whereas TFH cells support antiviral humoral immunity by promoting antibody class-switching and affinity maturation in germinal centre B cells. In contrast to these protective functions of TH cells, inappropriate or overwhelming activation of TH cells can lead to the development of allergies and autoimmune diseases. The most important function of pTReg cells is to prevent inflammation-mediated tissue injury through the local secretion of suppressive cytokines such as IL-10, IL-35 and transforming growth factor-β (TGFβ), or through the cell–cell contact-mediated inhibition of CD4+ effector cell proliferation.
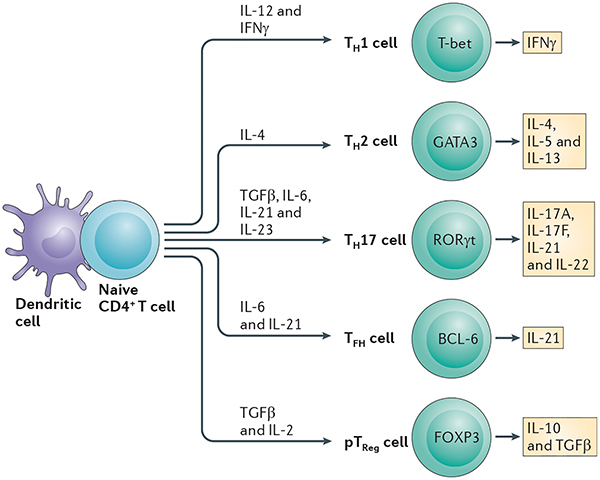
Figure 4 |. Role of T-bet in TH1 cell differentiation.

T cell receptor (TCR) and interferon-γ (IFNγ)-mediated signalling induces the first wave of T-bet (encoded by Tbx21) expression in an interleukin-12 (IL-12)independent manner, as signalling through TCR inhibits the expression of IL-12 receptor-β2 (Il12rb2) subunit. Cessation of TCR stimulation and IL-2 signalling induces expression of the IL-12Rβ2 subunit, which thus enables IL-12 signalling. IL-12-mediated activation of signal transducer and activator of transcription 4 (STAT4) prompts the second wave of T-bet expression. IFNγ produced by T helper 1 (TH1) cells functions in a feedforward loop, which further increases T-bet expression in TH1 cells. T-bet recruits enzymes (jumonji domain-containing protein 3 (JMJD3) and SET domain-containing protein 7 (SET7; also known as SET9)), which generate chromatin modifications that are associated with gene activation. T-bet-mediated transactivation of the Ifng gene is enhanced by H2.0-like homeobox protein 1 (HLX) and runt-related transcription factor 3 (RUNX3) transcription factors, both of which are encoded by T-bet target genes. T-bet facilitates CCCTC-binding factor (CTCF) binding and chromatin looping at the Ifng locus, which is required for optimal IFNγ expression in TH1 cells. T-bet induces chemokines, chemokine receptors and other effector molecules that are required for proper migration and function of TH1 cells. Sequestration of GATA-binding protein 3 (GATA3) by T-bet prevents the GATA3-mediated activation of the Il4–Il5–Il13 locus, and binding of a T-bet–RUNX3 complex to the Il4 silencer (DNase I hypersensitivity site 4 (HS4)) prevents the expression of Il4 in TH precursor cells. T-bet also binds to the Gata3 locus, and its binding is associated with the presence of repressive epigenetic marks. RUNX1 enhances the expression of retinoic acid receptor-related orphan receptor-γt (RORγt; encoded by Rorc) in TH17 cells and it functions as a RORγt-specific co-activator of the Il17a gene. T-bet binding to RUNX1 prevents Rorc and Il17a expression in TH precursor cells. Furthermore, T-bet together with STAT4, silences the Rorc locus. T-bet interacts with B cell lymphoma 6 (BCL-6) and recruits it to the promoters of genes that are repressed by T-bet in TH1 cells (for example, T cell factor (Tcf7) and suppressor of cytokine signalling (Socs)). By sequestering BCL-6 away from BCL-6 target genes (for example B lymphocyte-induced maturation protein (Blimp), CXC-chemokine receptor 5 (Cxcr5) and programmed cell death 1 (Pdcd1), T-bet effectively blocks T follicular helper (TFH) cell lineage commitment. Ccl, CC-chemokine ligand; CNS, conserved non-coding sequence; DC, dendritic cell; IFNγR, IFNγ receptor; NK, natural killer.
T-bet is not expressed in naive CD4+ T cells, but is readily induced in response to TCR, IFNγ–STAT1 and IL-12R–STAT4 signalling pathways37,44–47. TCR and IFNγ receptor (IFNγR) signalling induce the first wave of T-bet expression, which is independent of IL-12R signalling that is caused by the TCR-mediated inhibition of expression of IL-12Rβ2 subunit44–46 (FIG. 4). Cessation of both TCR stimulation and IL-2R–STAT5 signalling induces IL-12Rβ2 subunit expression, which thus enables IL-12R signalling46,48 (FIG. 4). IL-12 functions via STAT4 to induce a second wave of T-bet expression, which is required for the stabilization of the TH1 cell phenotype8,45–47. It is unclear whether imprinting of the TH1 cell developmental programme involves autoactivation of the Tbx21 gene by T-bet. Ectopic expression of T-bet induces endogenous expression of Tbx21, which suggests that autoactivation occurs4,44,45. However, in one reported study, endogenous Tbx21 induction was dependent on autocrine IFNγR signalling44. The question of whether T-bet autoregulation occurs in developing TH1 cells has been recently re-examined using T-bet–ZsGreen-reporter mice. In this study, it was shown that T-bet was not required for its own induction when IL-12 or IFNγ were present during differentiation; however, T-bet may be involved in directly promoting its own expression when induced by IL-12and IFNγ-independent pathways8. Another level of complexity with regard to the regulation of T-bet expression in TH1 cells has been added by the observations that T-bet expression in TH1 cells is fine-tuned post-transcriptionally by the microRNA-29 cluster and post-translationally by the ubiquitin-proteasomal degradation pathway49–51.
Together with STAT4, T-bet has a central role in the generation of transcriptionally competent TH1 cell-specific genes in CD4+ T cells. The molecular mechanisms by which T-bet modifies chromatin state have recently been reviewed52 and will be summarized here in general terms. T-bet-mediated chromatin changes are primarily dependent on the ability of T-bet to recruit enzymes that generate chromatin modifications associated with either gene activation (histone H3 or H4 acetylation, and H3 lysine 4 (H3K4) dimethylation) or gene repression (H3K27 trimethylation)53,54. CD4+ T cells that have been polarized under TH1 cell-associated conditions have increased expression of permissive marks at gene loci that are known to be positively regulated by T-bet. Conversely, genes that are normally repressed by T-bet in TH1 cells are characterized by the presence of repressive H3K27 trimethylation marks8,47,55–58 (FIG. 4). High levels of Ifng transcription are subject to change in TH1 cells depending on epigenetic modifications that remodel the Ifng locus; T-bet and STAT4 cooperate in this process8,47,56. T-bet directly activates the Ifng gene by binding to the Ifng promoter and to multiple distal regulatory elements located −54, −34, −22 and −6 kilobases (kb) upstream and +18−20 kb downstream of the Ifng gene, almost all of which serve as T-bet-dependent enhancers56–58. T-bet-mediated transactivation of the Ifng gene is further enhanced by H2.0-like homeobox protein (HLX) and runt-related transcription factor 3 (RUNX3) transcription factors, the expression of both of which is induced by T-bet in differentiating TH1 cells59,60.
Another key regulatory role of T-bet is to organize the three-dimensional architecture of the Ifng locus by enhancing occupancy of the transcriptional repressor CCCTC-binding factor (CTCF) between the boundaries of the Ifng locus (−70 kb and +66 kb) and a +1 kb site. T-bet binding at these locations promotes CTCF-dependent chromatin looping, which brings T-bet-binding enhancers and CTCF-binding sites in close proximity at the Ifng promoter. This configuration promotes robust Ifng expression in TH1 cells61. T-bet directly activates approximately 50% of TH1 cell-specific genes including cytokines (for example, Ifng, lymphotoxin-α (Lta) and Tnf), chemokines (for example, XC-chemokine ligand 1 (Xcl1), CC-chemokine ligand 3 (Ccl3) and Ccl4) and chemokine receptors (Cxcr3 and Ccr5), which are required for the proper effector function and the migration of TH1 cells, and for the recruitment of other immune cells to sites of inflammation8,21,47,62 (FIG. 4). Although most of the TH1 cell-specific gene expression is T-bet dependent, T-bet has limited effects in establishing global enhancer competence in TH1 cells. This function has been attributed to STAT4, which has a major effect on the activation of TH1 cell lineage-specific enhancers5.
Reinforcement of the TH1 cell differentiation programme relies not only on the T-bet-regulated activation of TH1 cell-specific genes but also on the concomitant inhibition of alternative TH cell differentiation pathways. T-bet regulates this either by suppressing the induction of other lineage-specifying transcription factors in TH pre-cursor cells or by interfering with their transcriptional activity (FIG. 4). Thus, T-bet deficiency is often accompanied by increased production of TH2 cell-specific or TH17 cell-specific cytokines38,43.
Inhibition of TH2 cell lineage commitment.
In contrast to T-bet, GATA3, which is the master regulator of the TH2 cell differentiation programme, is already expressed in naive CD4+ T cells63. Furthermore, activation of naive CD4+ T cells results in a transient, non-selective histone acetylation at both TH1 cell-specific and TH2 cell-specific cytokine genes, which persists for 17 to 20 hours after TCR stimulation55. It is at these early stages of TH cell lineage commitment that T-bet must compete with GATA3 to establish a TH1 cell-specific gene expression programme. Early TCR signalling induces T-bet expression, and IL-2-inducible T cell kinase (ITK)-mediated phosphorylation of T-bet at Tyr525 promotes the interaction of T-bet with GATA3 (REF. 64). This interaction redistributes GATA3 away from TH2 cell-specific genes to T-bet-binding sites at TH1 cell-specific loci4,64. Sequestration of GATA3 away from TH2 cell-specific genes (primarily the Il4–Il5–Il13 locus) and the binding of a T-bet–RUNX3 complex to the Il4 silencer (DNase I hypersensitivity site 4 (HS4)) prevents the expression of TH2 cell-specifc cytokine genes in developing TH1 cells4,60,64 (FIG. 4). Furthermore, T-bet binds to and promotes repressive chromatin modifications at the Gata3 locus, which thus inhibits the de novo expression of Gata3 in TH1 cells8. In addition to GATA3, nuclear factor of activated T cells 1 (NFAT1; also known as NFATC2) promotes TH2 cell differentiation through the activation of Il2, Il4, Il5 and Il13. T-bet phosphorylation at Thr302 is crucial for the inter action of T-bet with NFAT1 and loss of this interaction through mutation of the Thr302 residue abrogates the ability of T-bet to suppress NFAT1-dependent IL-2 and TH2-type cytokine expression51. T-bet can also limit IL-2 production by suppressing RELA activity through serine phosphorylation. T-bet phosphorylation at Ser508 by casein kinase I and glycogen synthase kinase 3 (GSK3) kinases promotes the interaction of T-bet with RELA, which impairs RELA binding to the Il2 promoter and the subsequent transcriptional activation of the Il2 gene by RELA65. Since IL-2 is required for upregulation of the IL-4Rα subunit, the negative regulatory effects of T-bet on Il2 gene expression efficiently limit IL-4 signalling and TH2 cell differentiation.
T-bet can also override a previously programmed TH2 cell differentiation state. Lymphocytic choriomeningitis virus (LCMV) infection, as well as a combination of type I or type II interferons and IL-12 in vitro, has been shown to reprogramme committed TH2 cells into TH1-like TH2 cells. These cells co-express GATA3 and T-bet and they produce TH1- and TH2-type cytokines following reactivation66. The conversion of TH2 cells into TH1-like cells was shown to be entirely dependent on T-bet both in vitro and in vivo. Sequential acquisition of T-bet and its downstream differentiation programme without extinguishing the previously acquired TH2 cell differentiation programme produced a population of optimally adapted TH cells, which were highly effective against LCMV but which protected tissues against immune-mediated pathology66.
Inhibition of TH17 cell lineage commitment.
T-bet expression in TH precursor cells inhibits commitment to the TH17 cell lineage by blocking RUNX1-mediated induction of the TH17 cell-specific transcription factor RORγt38. Thus, differentiation of Tbx21−/− CD4+ T cells under TH17 cell-skewing conditions produces a higher frequency of TH17 cells with a high expression of IL-17A, IL-17F and IL-21 (REFS 38,39). T-bet expression in mature TH17 cells results in their conversion into IFNγ-producing TH17 cells, which seems to be an important step in the pathophysiology of experimental autoimmune encephalomyelitis (EAE), as Tbx21−/− TH17 cells are not pathogenic in the EAE model67–70. Although, little is known about the downstream targets of T-bet that are responsible for the pathogenicity of TH17 cells, it has recently been shown that Tbx21−/− TH17 cells express lower levels of transforming growth factor-β3 (TGFβ3) than wild-type TH17 cells, and exogenous addition of TGFβ3 could restore the pathogenic potential of Tbx21−/− TH17 cells in the passive EAE model68.
A switch to IFNγ production is induced by the exposure of TH17 cells to IL-12 or IL-23 (REFS 67,71,72). The Tbx21 promoter has bivalent histone methylation marks in TH17 cells, which suggests that the Tbx21 locus is maintained in a poised state and is responsive to changes in environmental cues67,71–73. In addition to T-bet upregulation, the conversion of TH17 cells into TH1-like cells is accompanied by the downregulation of RORγt and TH17 cell-specific cytokines38,72 (FIG. 4). Silencing of Rorc in committed TH17 cells following IL-12R signalling has been linked with STAT4-induced and T-bet-induced epigenetic repression of the Rorc gene72. This is in contrast to what has been observed in TH2 cells, in which T-bet induction did not result in silencing of GATA3 expression. Although it is possible for opposing transcription factors to be present in the same cell during transitions, termination of Rorc expression in TH17 cells during conversion into TH1-like cells could reflect a greater susceptibility of the Rorc locus to epigenetic remodelling by T-bet and STAT4 transcription factors72.
Inhibition of TFH cell lineage commitment.
The early steps in TH1 cell commitment have many features in common with the T follicular helper (TFH) cell differentiation pathway74. IL-12 signals through STAT4 to induce a transitional stage of TFH cell-like TH1 cells, which express IL-21 and B cell lymphoma 6 (BCL-6). However, these TFH cell-like features are not sustained, as STAT4 also promotes T-bet expression, which inhibits the induction of TFH cell-specific genes by limiting the expression of BCL-6. Interestingly, the Bcl6 locus is not fully repressed in differentiated TH1 cells74. In fact, the low concentrations of BCL-6 that are present in TH1 cells are necessary for T-bet to effectively repress alternative TH cell gene programmes. T-bet interacts with BCL-6 and recruits it to the promoters of genes that are repressed by T-bet in TH1 cells75. When the concentrations of BCL-6 are low, T-bet sequesters BCL-6 away from its target genes, which thereby effectively blocks TFH cell lineage commitment (FIG. 4). The primary determinant of the relative ratio of T-bet to BCL-6 in TH1 cells is the concentration of IL-2 (REF. 76). In the presence of high levels of IL-2, the levels of BCL-6 are kept low. However, in low IL-2 concentrations, forkhead box O (FOXO) transcription factors induce BCL-6 and tip the balance in favour of expression of BCL-6 and the TFH cell gene expression profile. Thus, low doses of IL-2R signalling can change polarized TH1 cells into TFH cells, which suggests that TH1 cells retain a great degree of flexibility and have a TFH cell-like gene profile76. Similarly, TFH cells can acquire the properties of TH1 cells after exposure to TH1 cell-polarizing cytokines77. TFH cells have positive chromatin modifications at the Tbx21 locus, which maintains this gene in a state that is primed for re-expression. T-bet expression endows TFH cells with the ability to produce IFNγ without affecting their ability to produce IL-21 and to express TFH cell-specific markers77.
Peripherally derived TReg cells.
Similarly to what was observed in other TH cell subsets, in peripherally derived regulatory T (pTReg) cells (also known as induced or adaptive TReg cells) the Tbx21 locus is maintained in an open conformation and T-bet is upregulated in response to STAT1-mediated IFNγR and IL-27R signalling73,78–80. T-bet-deficient pTReg cells do not survive well after adoptive transfer into scurfy mice and fail to protect the mice from TH1 cell-mediated inflammatory responses. This suggests that T-bet is required for the functional fitness of pTReg cells80. It is incompletely understood how T-bet controls pTReg cell function and it is equally intriguing how pTReg cells maintain a FOXP3-regulated gene expression profile in the presence of T-bet. As adoptive transfers of pTReg cells have been considered as a potential therapy for inflammatory and autoimmune diseases, the plasticity of the pTReg cell transcriptional programme has gained increasing attention. There is the possibility that a pTReg cell could gain the effector profile of a TH1 cell in a dominating TH1 cell-associated environment. A recent study showed that T-bet+ pTReg cells express 10–20-fold less T-bet than conventional TH1 cells even in TH1-type inflammatory conditions80. The low level of T-bet expression in pTReg cells is maintained by the silencing of the Il12rb2 locus, which prevents T-bet+ pTReg cells from completing IL-12R–STAT4-dependent TH1 cell differentiation. This maintains the suppressive function of these cells without the production of pro-inflammatory cytokines79. However, T-bet expression in pTReg is absolutely essential for their proper migration. T-bet induces the expression of CXCR3 — a TH1 cell-associated chemokine receptor — which enables T-bet+ pTReg cells to migrate to sites of TH1 cell-mediated inflammation and to suppress local TH1 cell responses.
Although Tbx21 encodes the TH1 cell lineage-specifying transcription factor, the promoter of the Tbx21 gene is marked by permissive and repressive genetic marks in TH2, TH17, TFH and pTReg cells, and it is poised for subsequent activation or silencing71,73,77. Maintenance of the master transcription factor loci in a poised, bivalent epigenetic state in the opposing TH cell subsets is the basis for functional plasticity of CD4+ TH cells, which respond to changes in the environment by adopting the gene expression profiles of other TH cell lineages73. T-bet has a dominant role, often tipping the balance towards a TH1 cell phenotype. As described in the aforementioned examples, sometimes the acquisition of TH1 cell properties is accompanied by the repression of the existing gene expression profile (in the case of TH17 cells). In other cases, the responding TH cell subset gains features of the TH1 cell lineage, such as IFNγ and CXCR3 expression, but keeps its genetic profile (in the case of TH2, TFH and pTReg cells). Functional plasticity of CD4+ TH cells could be one mechanism to provide maximum host protection by generating large numbers of TH1 cells from pre-existing TH cell subsets following infection with intracellular pathogens. However, in the case of pTReg cells, increased T-bet expression is required for proper migration of pTReg cells to sites of TH1-cell dominated responses to minimize the amount of immune-mediated pathology.
Generation of B cell and T cell memory
T-bet in CD8+ T cell memory.
The acquisition of CD8+ T cell effector functions and the development of CD8+ T cell memory (BOX 2) are crucially dependent on transcriptional events that are regulated by T-bet and EOMES. CD8+ T cells that lack either T-bet or EOMES show a minor defect in their effector functions, which suggests that these two transcription factors have overlapping and partially redundant roles in establishing the differentiation programme of CD8+ T cells81,82. However, CD8+ T cells that are deficient in both T-bet and EOMES lose their cytotoxicity and aberrantly produce large amounts of IL-17A in response to infection with LCMV83. T-bet expression is rapidly induced by TCR and IL-12R signalling, and is required for the early production of IFNγ and granzyme B (encoded by Gzmb) by antigen-specific CD8+ T cells84,85 (FIG. 5). EOMES, which is induced later in a RUNX3-dependent manner, can substitute for T-bet to promote IFNγ and granzyme B expression and, together with STAT5, regulates perforin gene expression in CD8+ T cells84,86 (FIG. 5a). The concerted action of T-bet and EOMES results in the development of fully differentiated effector CD8+ T cells that migrate to inflamed tissues and that, following antigen recognition, secrete cytokines (for example, IFNγ and TNF) or lyse infected cells by releasing cytotoxic granules that contain granzymes and perforin.
Box 2 |. CD8+ T cell memory.
When a naive CD8+ T cell encounters an antigen, it undergoes a differentiation programme that can be divided into three main developmental stages: clonal expansion and differentiation, contraction, and memory formation. The differentiation process that occurs at the peak of the response generates quite a heterogeneous population of CD8+ T cells, which have different fates and memory cell developmental potential. Short-lived effector cells (SLECs) are terminally differentiated effector cells that express low levels of interleukin-7 receptor-α (IL-7Rα) and high levels of killer cell lectin-like receptor subfamily G member 1 (KLRG1) and IL-2Rα. By contrast, memory precursor effector cells (MPECs), which give rise to long-lived, self-renewing memory CD8+ T cells, express high levels of IL-7Rα and low levels of KLRG1 and IL-2Rα. After an infection is cleared, the vast majority of antigen-specific CD8+ T cells that are generated during clonal expansion die. The cells that survive the contraction phase, the MPECs, go on to generate a reservoir of memory CD8+ T cells. Memory T cells can be either effector memory (TEM) cells or central memory (TCM) cells. TEM cells (KLRG1hiIL-7RαhiCD62LlowCCR7low) monitor and guard peripheral tissues from reinfection by providing an immediate effector response, whereas TCM cells (KLRG1low IL-7RαhiCD62LhiCCR7hi) reside in lymphoid tissues and give rise to a large number of secondary effector cells during recall responses.
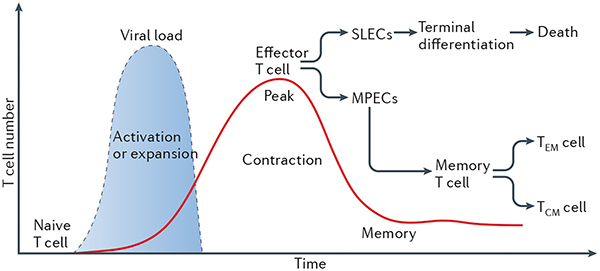
Figure 5 |. T-bet in the generation of B cell and T cell memory.

a | In CD8+ T cells, T-bet (encoded by Tbx21) expression is induced by T cell receptor (TCR) and interleukin-12 receptor (IL-12R) signalling, whereas eomesodermin (EOMES) is upregulated later in a runt-related transcription factor 3 (RUNX3)-dependent manner. RUNX3, T-bet and EOMES regulate the expression of interferon-γ (IFNγ), granzyme B (encoded by Gzmb), perforin (encoded by Prf) and CD122 in CD8+ effector cells. b | Strong TCR and IL-12R signalling promotes the terminal differentiation of short-lived effector cells (SLECs) by enhancing and maintaining the activity of the kinase mammalian target of rapamycin (mTOR) downstream of signal transducer and activator of transcription 4 (STAT4). mTOR simultaneously induces T-bet and represses EOMES expression through inhibition of forkhead box O1 (FOXO1). The IL-2R–STAT5 pathway induces the expression of B lymphocyte-induced maturation protein 1 (BLIMP1), inhibiting the expression of B cell lymphoma 6 (BCL-6), which is a transcriptional repressor of the Tbx21 gene. Inhibition of mTOR activity by rapamycin or by activators of AMP-activated protein kinase (AMPK), such as metformin, supports the development of memory precursor effector cells (MPECs). Similarly, enhanced MPEC formation is observed in low IL-2 and IL-12 conditions. c | EOMES expression increases and T-bet levels decline in memory CD8+ T cells. Minimal levels of T-bet are maintained for proper expression of CD122, which promotes the homeostatic proliferation of memory cells. T-bet expression is associated with a more differentiated phenotype of effector memory T (TEM) cells and secondary and tertiary memory cells. d | P-selectin glycoprotein ligand 1 (PSGL1)hi LY6ChiT-bethi cells are terminally differentiated T helper 1 (TH1) effector cells. PSGL1hiLY6ClowT-betint cells are less differentiated, are longer lived and give rise to TH1 effector cells after re-challenge. T-bet is expressed at very low levels in T follicular helper (TFH) and TFH memory cells, as a result of BCL-6 expression. e | T-bet is a selective inducer of IFNγ-mediated IgG2a class-switching in B cells. T-bet is required for the survival of IgG2a+CD38hi memory B cells and it promotes the migration of IgG2a+ B cells to inflammatory foci through the transcriptional regulation of CXC-chemokine receptor 3 (CXCR3) expression. CCR7; CC-chemokine receptor 7; IFNγR, IFNγ receptor; KLRG1; killer cell lectin-like receptor subfamily G member 1; PDK1, 3-phosphoinositide-dependent protein kinase 1; PI3K, phosphoinositide 3-kinase; TCM, central memory.
T-bet and EOMES have essential but reciprocal roles in regulating effector and memory T cell differentiation pathways87–89 (BOX 2; FIG. 5b). Inflammatory signals that are present during CD8+ T cell priming define a gradient of T-bet and EOMES expression and, consequently, influence the fate of effector CD8+ T cells87. Strong IL-12R signalling provides the instructive signal for the terminal differentiation of short-lived effector cells (SLECs) by enhancing and maintaining STAT4-mediated mammalian target of rapamycin (mTOR) kinase activity. This functions as a molecular switch to concomitantly induce T-bet expression and to repress EOMES expression through the inhibition of FOXO1 transcriptional activity85,88,89. Thus, T-bet deficiency, IL-12 deficiency or inhibition of mTOR activity results in a profound absence of SLECs and in the accumulation of long-lived memory precursor effector cells (MPECs)87,89 (FIG. 5b). In addition to cytokine signalling, asymmetric partitioning of key transcriptional regulators, such as T-bet, between the daughter cells during cell division may regulate the SLEC versus MPEC fate decision90. The cell that acquires more T-bet during cell division becomes a terminally differentiated effector cell, whereas the cell that has low T-bet levels has a higher memory cell developmental potential90.
Once the pathogen is cleared, the vast majority of antigen-specific CD8+ T cells that are generated during clonal expansion die. A small fraction of CD8+ T cells that survive the contraction phase, the MPECs, generate a reservoir of memory CD8+ T cells, which depend on the cytokines IL-7 and IL-15 for their long-term persistence (BOX 2). As CD8+ T cells acquire a memory phenotype, EOMES expression increases whereas T-bet levels decrease91–93 (FIG. 5c). Nevertheless, low levels of T-bet are maintained in memory CD8+ T cells, and T-bet and EOMES cooperate to sustain proper expression of CD122 (the β-subunit of IL-2R and IL-15R) to promote the longevity and the homeostatic proliferation of memory CD8+ T cells94. Effector memory T (TEM) cells express higher levels of T-bet and seem to be more differentiated than central memory T (TCM) cells92. By contrast, the expression of EOMES is higher in TCM cells than in TEM cells91,92 (FIG. 5c). EOMES deficient memory CD8+ T cells are less efficient at competing to populate the bone marrow niche and are defective in long-term persistence and secondary expansion after antigen re-challenge92. These findings suggest that EOMES expression is needed for the competitive fitness and the persistence of TCM cells, whereas T-bet expression is associated with a more differentiated phenotype of TEM cells.
Subsequent antigen encounters or the exposure of memory CD8+ T cells to inflammatory cytokines leads to a rapid upregulation of T-bet and to the terminal differentiation of memory CD8+ T cells during secondary and tertiary infections91 (FIG. 5c). Limited T-bet expression in Tbx21+/− CD8+ T cells enables memory CD8+ T cells to retain a less differentiated state during multiple rounds of antigen re-challenge, which thereby prevents the senescence of memory CD8+ T cells91. Furthermore, memory CD8+ T cells that develop in the absence of CD4+ T cell help seem to be more TEM cell-like, as they are characterized by elevated T-bet levels. In these cases, T-bet deletion has been found to prevent the aberrant differentiation of ‘unhelped’ memory CD8+ T cells93. Therefore, it may be possible to minimize terminal differentiation of secondary and tertiary memory CD8+ T cells and to correct the dysfunctional programming of CD8+ T cells that develop in the absence of CD4+ T cell help simply by limiting T-bet expression91,93.
During chronic infections, CD8+ T cell responses are quite different from those observed during acute infections. When they are exposed to persistent antigenic stimulation, CD8+ T cells become dysfunctional, exhausted and fail to differentiate into effective memory CD8+ T cells. The exhaustion of CD8+ T cells is accompanied by the upregulation of inhibitory molecules (such as programmed cell death protein 1 (PD1; encoded by Pdcd1), lymphocyte activation gene 3 protein (LAG3) and Band T-lymphocyte attenuator (BTLA)), as well as defective proliferation, cytokine production and cytotoxicity in these cells95,96. In this context, elevated T-bet levels improve the functionality and the long-term durability of antigen-specific CD8+ T cells, whereas EOMES expression enhances CD8+ T cell exhaustion97,98. T-bet represses the expression of inhibitory receptors, including PD1, and improves the fitness of exhausted CD8+ T cells97. Thus, therapeutic strategies that aim to increase T-bet expression in CD8+ cells could minimize CD8+ T cell exhaustion and improve CD8+ T cell responses during chronic infections97.
Collectively, these studies show the complex regulation of T-bet and EOMES expression that takes place during acutely resolved versus chronic infections. Our understanding of the differentiation pathways that are regulated by T-bet and EOMES will provide novel ideas for therapeutic interventions and will improve the efficacy of memory CD8+ T cell responses in the context of acute and chronic infections.
T-bet in CD4+ T cell memory.
Unlike the formation of CD8+ T cell memory, which has been intensely studied over many years, very little is known about the requirements for the formation, the proper function and the maintenance of CD4+ T cell memory. Similarly to CD8+ T cell responses, there is marked phenotypic heterogeneity in the antigen-specific CD4+ T cells at the peak of the response. However, the particular features that distinguish a population of committed memory precursor cells from the larger pool of diverse effector cells remain a matter of debate. Infection with intracellular bacteria or viruses induces the formation of both TH1 cells and TFH cells, and these effector CD4+ T cell subsets can be readily distinguished by their differential expression of CXCR5, P-selectin glycoprotein ligand 1 (PSGL1) and LY6C (FIG. 5d). TH1 cells do not express CXCR5 and are positive for PSGL1 and LY6C.
Among TH1 cells, two major subsets can be identified on the basis of LY6C expression: LY6Chi and LY6Clow cells. PSGL1hiLY6Chi cells have features of terminally differentiated effector cells, such as increased expression of T-bet, CXCR3, IFNγ and granzyme B. By contrast, PSGL1hiLY6Clow cells express intermediate levels of T-bet and are less differentiated than their PSGL1hiLY6Chi counterparts99. High levels of expression of CD62L (also known as L-selectin) and CCR7 are associated with CD4+ TCM cells, and these cells are preferentially enriched in the PSGL1hiLY6Clow memory compartment, which persists longer and proliferates better than the LY6Chi subset following antigen rechallenge99. Similarly to antigen-specific CD8+ T cells, elevated T-bet expression is required for maximal clonal expansion and for the formation of terminally differentiated PSGL1hiLY6ChiCD4+ T cells. Loss of one or two copies of T-bet results in a proportionally increased frequency of PSGL1hiLY6Clow TH1 memory precursor cells99. Thus, T-bet balances terminal differentiation and memory cell potential in both CD4+ and CD8+ T cells99. Following antigen re-challenge, TH1 memory cells efficiently produce secondary TH1 effector cells. These cells have high T-bet, IFNγ and granzyme B expression, which suggests that memory TH1 cells maintain a default TH1 cell lineage differentiation programme in the absence of antigens100 (FIG. 5d).
In contrast to TH1 effector and TH1 memory cells, T-bet is expressed at very low levels in TFH cells (CXCR5+PSGL1lowLY6Clow or CXCR5+PSGL1lowLY6Cint) and TFH memory cells99–101. Furthermore, TFH cells maintain repressive methylation of the granzyme B locus throughout effector and memory cell differentiation. They also produce limited amounts of IFNγ following reinfection with LCMV99–101. Silencing of the Gzmb locus and other TH1 cell-specific loci in TFH memory cells reinforces the TFH cell-lineage programme and prevents the expression of TH1 cell-specific genes following acute LCMV infection100. In contrast to these findings, CXCR5+CCR7+ memory CD4+ T cells described by Pepper et al.101 resemble TCM cells rather than TFH cells, as they have down-regulated TFH cell markers (such as high BCL-6 and PD1) and they have a potential to equally reconstitute TH1 and TFH effector cells following reinfection with Listeria monocytogenes101. It remains to be determined whether primary TH1 and TFH effector cells give rise to TH1 and TFH memory cells in a linear fashion and lineage-specific effector functions are maintained in these cells even in the absence of antigens by their master regulators T-bet and BCL-6, respectively; or, alternatively, whether PSGL1hiLY6ClowT-betint cells represent more terminally differentiated TEM cells than CXCR5+PSGL1lowLY6Clow T-betlow or CXCR5+PSGL1lowLY6CintT-betlow cells, which give rise to TCM cells. In the second example, the balance between T-bet and BCL-6 would be a principal mechanism regulating the generation of distinct memory cell populations100,101.
T-bet in B cell memory.
In response to antigen stimulation, activated B cells proliferate and undergo affinity maturation and class-switch recombination (CSR) in order to produce high-affinity antibodies that have different immune effector functions. IgG2a is the most prevalent isotype that is produced in response to intracellular bacterial and viral infections. Cytokines such as IFNγ and IL-27 increase T-bet expression in B cells in a STAT1-dependent and ETS1-dependent manner102–105 (FIG. 5e). T-bet subsequently induces the expression of Iγ2a transcripts and promotes IFNγ-mediated class-switching to the IgG2a isotype103. T-bet-deficient B cells show impaired IgG2a, IgG2b and IgG3 secretion. In addition, ectopic expression of T-bet is sufficient to induce the expression of germline Iγ2a transcripts in Tbx21−/− B cells even in the absence of IFNγR signalling103. T-bet expression persists in antigen-specific IgG2a+CD38hi memory B cells and it promotes the survival of this cell subset by regulating the transcription of the mature B cell receptor106. Furthermore, T-bet drives the migration of memory IgG2a+ B cells to sites of inflammation by controlling the expression of the chemokine receptor CXCR3 (REF. 107). These findings identify T-bet as a selective inducer of IFNγ-mediated IgG2a class-switching in B cells and a key regulator of B cell migration to inflammatory foci.
Conclusions and perspectives
T-bet has important roles in multiple aspects of a coordinated immune response. It regulates the development of components of the innate immune system, it affects the trafficking of both innate and adaptive immune cells and it controls the polarity of cytokine responses. In this way, T-bet functions as the key molecule in coordinating effective and anatomically appropriate type 1 immunity. Despite the intensive studies that have been ongoing for over a decade, much still remains to be learned about the cell-specific roles of T-bet. Indeed, the recent descriptions of the role of T-bet in the newly discovered ILCs have highlighted further functions of this protean transcription factor. There is still much to be learned about the upstream signals that regulate T-bet expression in specific cell types and how T-bet interacts with other lineage-determining transcription factors in vivo. It is tempting to speculate that modulation of T-bet expression may be a very powerful therapeutic target for the treatment of autoimmunity, transplant rejection, infectious diseases and cancer in the not too distant future.
Experimental autoimmune encephalomyelitis.
(EAE). A mouse model of multiple sclerosis. It can be induced by the active immunization of mice with central nervous system (CNS)-derived antigens emulsified in complete Freund’s adjuvant (in the case of active EAE) or by adoptive transfer of activated, T cell receptor (TCR)-transgenic CD4+ T cells, in which the TCRs recognize a CNS-derived antigen (in the case of passive EAE).
Scurfy mice.
Mice with a spontaneous mutation in forkhead box P3 (Foxp3), which leads to a rapidly fatal lymphoproliferative disease, causing death by ~4 weeks of age.
Mamammalian target of rapamycin.
(mTOR). A serine/threonine protein kinase that regulates cell metabolism, proliferation, survival, protein synthesis and transcription. It is activated by T cell receptor signalling and is sustained by inflammatory cytokines such as interleukin-12.
AMP-activated protein kinase.
(AMPK). A negative regulator of mammalian target of rapamycin (mTOR) kinase activity. It is activated in response to cellular stress and ATP deprivation. It can be also activated by the pharmacological agent metformin.
Affinity maturation.
The process by which B cells produce antibodies that have an increased affinity for antigens during an immune response.
Class-switch recombination.
(CSR). The process by which B cells produce antibodies of different isotypes without changing the antigen specificity of the variable region.
DATABASES
Ensembl database: www.ensembl.org
FURTHER INFORMATION
National Institute for Health Research: www.biomedicalresearchcentre.org
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
Acknowledgements
We would like to acknowledge support from grants awarded by the US National Institutes of Health (grants CA112663 and PO1NSO38037) to L.H.G.; the Irvington Institute to V.L.; and to G.M.L. the Medical Research Council, UK (grant G0802068), the Wellcome Trust, UK, (grant WT088747MA) and the British Heart Foundation, UK, (grant PG/12/36/29444). G.M.L. is also supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy’s and St Thomas’ National Health Service (NHS) Foundation Trust and King’s College London, UK. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Footnotes
Competing interests statement
The authors declare competing financial interests: see Web version for details.
References
- 1.Flajnik MF & Kasahara M Origin and evolution of the adaptive immune system: genetic events and selective pressures. Nature Rev. Genet 11, 47–59 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guo P et al. Dual nature of the adaptive immune system in lampreys. Nature 459, 796–801 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horton AC & Gibson-Brown JJ Evolution of developmental functions by the Eomesodermin, T-brain-1, Tbx21 subfamily of T-box genes: insights from amphioxus. J. Exp. Zool 294, 112–121 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Kanhere A et al. T-bet and GATA3 orchestrate Th1 and Th2 differentiation through lineage-specific targeting of distal regulatory elements. Nature Commun 3, 1268 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vahedi G et al. STATs shape the active enhancer landscape of T cell populations. Cell 151, 981–993 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ciofani M et al. A validated regulatory network for Th17 cell specification. Cell 151, 289–303 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yagi R et al. The transcription factor GATA3 actively represses RUNX3 protein-regulated production of interferon-γ. Immunity 32, 507–517 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu J et al. The transcription factor T-bet is induced by multiple pathways and prevents an endogenous Th2 cell program during Th1 cell responses. Immunity 37, 660–673 (2012).This is the first description of T-bet-specific reporter mice that can be used to address important questions about the regulation of T-bet expression during TH1 cell differentiation.
- 9.Wei G et al. Genome-wide analyses of transcription factor GATA3-mediated gene regulation in distinct T cell types. Immunity 35, 299–311 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Whyte WA et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lighvani AA et al. T-bet is rapidly induced by interferon-γ in lymphoid and myeloid cells. Proc. Natl Acad. Sci. USA 98, 15137–15142 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lugo-Villarino G, Maldonado-Lopez R, Possemato R, Penaranda C & Glimcher LH T-bet is required for optimal production of IFN-γ and antigen-specific T cell activation by dendritic cells. Proc. Natl Acad. Sci. USA 100, 7749–7754 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lugo-Villarino G, Ito S, Klinman DM & Glimcher LH The adjuvant activity of CpG DNA requires T-bet expression in dendritic cells. Proc. Natl Acad. Sci. USA 102, 13248–13253 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J et al. Transcription factor T-bet regulates inflammatory arthritis through its function in dendritic cells. J. Clin. Invest 116, 414–421 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Powell N, Canavan JB, MacDonald TT & Lord GM Transcriptional regulation of the mucosal immune system mediated by T-bet. Mucosal Immunol 3, 567–577 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Alcaide P et al. Dendritic cell expression of the transcription factor T-bet regulates mast cell progenitor homing to mucosal tissue. J. Exp. Med 204, 431–439 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garrett WS et al. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell 131, 33–45 (2007).This paper describes the microbiome-dependent spontaneous colitis that occurs in the absence of T-bet and RAG2 as a result of the derepression of TNF in mucosal DCs.
- 18.Garrett WS et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe 8, 292–300 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garrett WS et al. Colitis-associated colorectal cancer driven by T-bet deficiency in dendritic cells. Cancer Cell 16, 208–219 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hecht GA Inflammatory bowel disease — live transmission. N. Engl. J. Med 358, 528–530 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Jenner RG et al. The transcription factors T-bet and GATA-3 control alternative pathways of T-cell differentiation through a shared set of target genes. Proc. Natl Acad. Sci. USA 106, 17876–17881 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Powell N et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity 37, 674–684 (2012).This is the first description of a functional role of T-bet that is expressed in ILCs in the regulation of mucosal immunity.
- 23.Spits H & Cupedo T Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu. Rev. Immunol 30, 647–675 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Bernink JH et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nature Immunol 14, 221–229 (2013).This is the first description of human ILCs expressing T-bet that correlate with mucosal inflammation in Crohn’s disease.
- 25.Fuchs A et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12and IL-15-responsive IFN-γ-producing cells. Immunity 38, 769–781 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spits H et al. Innate lymphoid cells — a proposal for uniform nomenclature. Nature Rev. Immunol 13, 145–149 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Vonarbourg C et al. Regulated expression of nuclear receptor RORγt confers distinct functional fates to NK cell receptor-expressing RORγt+ innate lymphocytes. Immunity 33, 736–751 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sciume G et al. Distinct requirements for T-bet in gut innate lymphoid cells. J. Exp. Med 209, 2331–2338 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klose CS et al. A T-bet gradient controls the fate and function of CCR6-RORγt+ innate lymphoid cells. Nature 494, 261–265 (2013).This paper describes the mechanistic relationship of T-bet-expressing ILCs with RORγt and CCR6 expression and how this affects mucosal immunity.
- 30.Gordon SM et al. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity 36, 55–67 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Townsend MJ et al. T-bet regulates the terminal maturation and homeostasis of NK and Vα14i NKT cells. Immunity 20, 477–494 (2004).This paper is the first to describe the function of T-bet in NK and iNKT cells.
- 32.Werneck MB, Lugo-Villarino G, Hwang ES, Cantor H & Glimcher LH T-bet plays a key role in NK-mediated control of melanoma metastatic disease. J. Immunol 180, 8004–8010 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsuda JL et al. T-bet concomitantly controls migration, survival, and effector functions during the development of Vα14i NKT cells. Blood 107, 2797–2805 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim HY et al. The development of airway hyperreactivity in T-bet-deficient mice requires CD1d-restricted NKT cells. J. Immunol 182, 3252–3261 (2009). [DOI] [PubMed] [Google Scholar]
- 35.Yin Z et al. T-bet expression and failure of GATA-3 cross-regulation lead to default production of IFN-γ by γδ T cells. J. Immunol 168, 1566–1571 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Chen L et al. Epigenetic and transcriptional programs lead to default IFN-γ production by γδ T cells. J. Immunol 178, 2730–2736 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Szabo SJ et al. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669 (2000).This is the first report to show the role of T-bet as the master regulator of the TH1 cell differentiation programme.
- 38.Lazarevic V et al. T-bet represses TH17 differentiation by preventing Runx1-mediated activation of the gene encoding RORγt. Nature Immunol 12, 96–104 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mathur AN et al. T-bet is a critical determinant in the instability of the IL-17-secreting T-helper phenotype. Blood 108, 1595–1601 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Villarino AV, Gallo E & Abbas AK STAT1-activating cytokines limit Th17 responses through both T-bet-dependent and -independent mechanisms. J. Immunol 185, 6461–6471 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ravindran R, Foley J, Stoklasek T, Glimcher LH & McSorley SJ Expression of T-bet by CD4 T cells is essential for resistance to Salmonella infection. J. Immunol 175, 4603–4610 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Sullivan BM et al. Increased susceptibility of mice lacking T-bet to infection with Mycobacterium tuberculosis correlates with increased IL-10 and decreased IFN-γ production. J. Immunol 175, 4593–4602 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Szabo SJ et al. Distinct effects of T-bet in TH1 lineage commitment and IFN-γ production in CD4 and CD8 T cells. Science 295, 338–342 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Afkarian M et al. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nature Immunol 3, 549–557 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Mullen AC et al. Role of T-bet in commitment of TH1 cells before IL-12-dependent selection. Science 292, 1907–1910 (2001). [DOI] [PubMed] [Google Scholar]
- 46.Schulz EG, Mariani L, Radbruch A & Hofer T Sequential polarization and imprinting of type 1 T helper lymphocytes by interferon-γ and interleukin-12. Immunity 30, 673–683 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Thieu VT et al. Signal transducer and activator of transcription 4 is required for the transcription factor T-bet to promote T helper 1 cell-fate determination. Immunity 29, 679–690 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liao W, Lin JX, Wang L, Li P & Leonard WJ Modulation of cytokine receptors by IL-2 broadly regulates differentiation into helper T cell lineages. Nature Immunol 12, 551–559 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith KM et al. miR-29ab1 deficiency identifies a negative feedback loop controlling Th1 bias that is dysregulated in multiple sclerosis. J. Immunol 189, 1567–1576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steiner DF et al. MicroRNA-29 regulates T-box transcription factors and interferon-γ production in helper T cells. Immunity 35, 169–181 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jang EJ, Park HR, Hong JH & Hwang ES Lysine 313 of T-box is crucial for modulation of protein stability, DNA binding, and threonine phosphorylation of T-bet. J. Immunol 190, 5764–5770 (2013). [DOI] [PubMed] [Google Scholar]
- 52.Miller SA & Weinmann AS Molecular mechanisms by which T-bet regulates T-helper cell commitment. Immunol. Rev 238, 233–246 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miller SA, Mohn SE & Weinmann AS Jmjd3 and UTX play a demethylase-independent role in chromatin remodeling to regulate T-box family member-dependent gene expression. Mol. Cell 40, 594–605 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miller SA, Huang AC, Miazgowicz MM, Brassil MM & Weinmann AS Coordinated but physically separable interaction with H3K27-demethylase and H3K4-methyltransferase activities are required for T-box protein-mediated activation of developmental gene expression. Genes Dev 22, 2980–2993 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Avni O et al. TH cell differentiation is accompanied by dynamic changes in histone acetylation of cytokine genes. Nature Immunol 3, 643–651 (2002). [DOI] [PubMed] [Google Scholar]
- 56.Balasubramani A et al. Modular utilization of distal cis-regulatory elements controls Ifng gene expression in T cells activated by distinct stimuli. Immunity 33, 35–47 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shnyreva M et al. Evolutionarily conserved sequence elements that positively regulate IFN-γ expression in T cells. Proc. Natl Acad. Sci. USA 101, 12622–12627 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hatton RD et al. A distal conserved sequence element controls Ifng gene expression by T cells and NK cells. Immunity 25, 717–729 (2006).This study provides detailed analysis of the regulatory elements within the Ifng locus and maps T-bet-dependent enhancers.
- 59.Mullen AC et al. Hlx is induced by and genetically interacts with T-bet to promote heritable TH1 gene induction. Nature Immunol 3, 652–658 (2002). [DOI] [PubMed] [Google Scholar]
- 60.Djuretic IM et al. Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nature Immunol 8, 145–153 (2007). [DOI] [PubMed] [Google Scholar]
- 61.Sekimata M et al. CCCTC-binding factor and the transcription factor T-bet orchestrate T helper 1 cell-specific structure and function at the interferon-γ locus. Immunity 31, 551–564 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lord GM et al. T-bet is required for optimal proinflammatory CD4+ T-cell trafficking. Blood 106, 3432–3439 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zheng W & Flavell RA The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 89, 587–596 (1997). [DOI] [PubMed] [Google Scholar]
- 64.Hwang ES, Szabo SJ, Schwartzberg PL & Glimcher LH T helper cell fate specified by kinasemediated interaction of T-bet with GATA-3. Science 307, 430–433 (2005).This paper describes T-bet-mediated inhibition of GATA3 function during TH2 cell differentiation.
- 65.Hwang ES, Hong JH & Glimcher LH IL-2 production in developing Th1 cells is regulated by heterodimerization of RelA and T-bet and requires T-bet serine residue 508. J. Exp. Med 202, 1289–1300 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hegazy AN et al. Interferons direct Th2 cell reprogramming to generate a stable GATA-3+T-bet+ cell subset with combined Th2 and Th1 cell functions. Immunity 32, 116–128 (2010). [DOI] [PubMed] [Google Scholar]
- 67.Hirota K et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nature Immunol 12, 255–263 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee Y et al. Induction and molecular signature of pathogenic TH17 cells. Nature Immunol 13, 991–999 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ghoreschi K et al. Generation of pathogenic TH17 cells in the absence of TGF-β signalling. Nature 467, 967–971 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang Y et al. T-bet is essential for encephalitogenicity of both Th1 and Th17 cells. J. Exp. Med 206, 1549–1564 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee YK et al. Late developmental plasticity in the T helper 17 lineage. Immunity 30, 92–107 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mukasa R et al. Epigenetic instability of cytokine and transcription factor gene loci underlies plasticity of the T helper 17 cell lineage. Immunity 32, 616–627 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wei G et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30, 155–167 (2009).This study shows that the maintenance of master transcription factor loci in a bivalent epigenetic state, which is characterized by the presence of repressive and permissive marks, is the underlying mechanism for the functional plasticity of TH cells.
- 74.Nakayamada S et al. Early Th1 cell differentiation is marked by a Tfh cell-like transition. Immunity 35, 919–931 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Oestreich KJ, Huang AC & Weinmann AS The lineage-defining factors T-bet and Bcl-6 collaborate to regulate Th1 gene expression patterns. J. Exp. Med 208, 1001–1013 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Oestreich KJ, Mohn SE & Weinmann AS Molecular mechanisms that control the expression and activity of Bcl-6 in TH1 cells to regulate flexibility with a TFH-like gene profile. Nature Immunol 13, 405–411 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lu KT et al. Functional and epigenetic studies reveal multistep differentiation and plasticity of in vitro generated and in vivo-derived follicular T helper cells. Immunity 35, 622–632 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hall AO et al. The cytokines interleukin 27 and interferon-gamma promote distinct Treg cell populations required to limit infection-induced pathology. Immunity 37, 511–523 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Koch MA et al. T-bet+ Treg cells undergo abortive Th1 cell differentiation due to impaired expression of IL-12 receptor β2. Immunity 37, 501–510 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Koch MA et al. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nature Immunol 10, 595–602 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sullivan BM, Juedes A, Szabo SJ, von Herrath M & Glimcher LH Antigen-driven effector CD8 T cell function regulated by T-bet. Proc. Natl Acad. Sci. USA 100, 15818–15823 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pearce EL et al. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science 302, 1041–1043 (2003). [DOI] [PubMed] [Google Scholar]
- 83.Intlekofer AM et al. Anomalous type 17 response to viral infection by CD8+ T cells lacking T-bet and eomesodermin. Science 321, 408–411 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cruz-Guilloty F et al. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J. Exp. Med 206, 51–59 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Takemoto N, Intlekofer AM, Northrup JT, Wherry EJ & Reiner SL Cutting edge: IL-12 inversely regulates T-bet and eomesodermin expression during pathogen-induced CD8+ T cell differentiation. J. Immunol 177, 7515–7519 (2006). [DOI] [PubMed] [Google Scholar]
- 86.Pipkin ME et al. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity 32, 79–90 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Joshi NS et al. Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity 27, 281–295 (2007).This paper shows the importance of the T-bet expression levels in determining the terminal differentiation of CD8+ effector cells or in determining CD8+ memory cell formation.
- 88.Rao RR, Li Q, Gubbels Bupp MR & Shrikant PA Transcription factor Foxo1 represses T-bet-mediated effector functions and promotes memory CD8+ T cell differentiation. Immunity 36, 374–387 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rao RR, Li Q, Odunsi K & Shrikant PA The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity 32, 67–78 (2010).This paper describes the importance of mTOR kinase in determining effector versus memory CD8+ T cell fate through its effects on the regulation of T-bet expression.
- 90.Chang JT et al. Asymmetric proteasome segregation as a mechanism for unequal partitioning of the transcription factor T-bet during T lymphocyte division. Immunity 34, 492–504 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Joshi NS et al. Increased numbers of preexisting memory CD8 T cells and decreased T-bet expression can restrain terminal differentiation of secondary effector and memory CD8 T cells. J. Immunol 187, 4068–4076 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Banerjee A et al. Cutting edge: The transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J. Immunol 185, 4988–4992 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Intlekofer AM et al. Requirement for T-bet in the aberrant differentiation of unhelped memory CD8+ T cells. J. Exp. Med 204, 2015–2021 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Intlekofer AM et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nature Immunol 6, 1236–1244 (2005). [DOI] [PubMed] [Google Scholar]
- 95.Virgin HW, Wherry EJ & Ahmed R Redefining chronic viral infection. Cell 138, 30–50 (2009). [DOI] [PubMed] [Google Scholar]
- 96.Blackburn SD et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nature Immunol 10, 29–37 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kao C et al. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nature Immunol 12, 663–671 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Paley MA et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 338, 1220–1225 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Marshall HD et al. Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4+ cell properties during viral infection. Immunity 35, 633–646 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hale JS et al. Distinct memory CD4+ T cells with commitment to T follicular helper and T helper 1-cell lineages are generated after acute viral infection. Immunity 38, 805–817 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pepper M, Pagan AJ, Igyarto BZ, Taylor JJ & Jenkins MK Opposing signals from the Bcl6 transcription factor and the interleukin-2 receptor generate T helper 1 central and effector memory cells. Immunity 35, 583–595 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nguyen HV et al. The Ets-1 transcription factor is required for Stat1-mediated T-bet expression and IgG2a class switching in mouse B cells. Blood 119, 4174–4181 (2012). [DOI] [PubMed] [Google Scholar]
- 103.Peng SL, Szabo SJ & Glimcher LH T-bet regulates IgG class switching and pathogenic autoantibody production. Proc. Natl Acad. Sci. USA 99, 5545–5550 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Xu W & Zhang JJ Stat1-dependent synergistic activation of T-bet for IgG2a production during early stage of B cell activation. J. Immunol 175, 7419–7424 (2005). [DOI] [PubMed] [Google Scholar]
- 105.Yoshimoto T et al. Induction of IgG2a class switching in B cells by IL-27. J. Immunol 173, 2479–2485 (2004). [DOI] [PubMed] [Google Scholar]
- 106.Wang NS et al. Divergent transcriptional programming of class-specific B cell memory by T-bet and RORα. Nature Immunol 13, 604–611 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Serre K et al. CD8 T cells induce T-bet-dependent migration toward CXCR3 ligands by differentiated B cells produced during responses to alum-protein vaccines. Blood 120, 4552–4559 (2012). [DOI] [PubMed] [Google Scholar]