

Abstract
miR-150 functions as a tumor suppressor in malignancies of the lymphocyte lineage and its expression is significantly reduced in these cells. However, the mechanism of miR-150 repression is unknown and so are pharmacological interventions that can reverse it. Here, we report that reduced expression of miR-150 in human Jurkat T-cell acute lymphoblastic leukemia (T-ALL) cells is mediated by constitutive mTOR signaling, a common characteristic of T-ALL cell lines and clinical isolates. Activating mTOR signaling in non- malignant T cells also resulted in a significant miR-150 down-regulation. Conversely, treatment with a pharmacological mTOR inhibitor, rapamycin, increased miR-150 expression in a dose-dependent manner in Jurkat cells, as well as in other leukemia cells. Interestingly, ectopic over-expression of miR-150 acted in a feed-forward loop and further sensitized Jurkat cells to a rapamycin-induced cell cycle arrest by targeting a large network of cell cycle genes. These findings suggest that miR-150 is normally expressed in quiescent T lymphocytes to reinforce an anti-proliferative state, and that mTOR signaling promotes cell proliferation in part by inhibiting miR-150 expression. Restoration of the miR-150- dependent anti-proliferative loop constitutes a novel mechanism underlying the efficacy of rapamycin in a T-ALL cell line. Further investigation of this mechanism in clinical isolates of T-ALL and other hematopoietic malignancies could help better guide development of targeted therapies.
Keywords: microRNA, miR-150, mTOR, cell cycle, proliferation, rapamycin, acute lymphoblastic leukemia, Jurkat, gene network
1. Introduction
The mechanistic/mammalian target of rapamycin (mTOR) is a conserved protein kinase that induces cell growth, proliferation and changes in metabolism in response to mitogenic signals (1). Unsurprisingly, pathogenesis and progression of numerous cancers, including T-cell acute lymphoblastic leukemia (T-ALL), is associated with constitutive activation of mTOR, caused primarily by mutations in its upstream regulators, such as AKT and PTEN (2, 3). In the majority of T-ALL patients, constitutive mTOR activation negatively affects outcomes (4). Conversely, inhibition of mTOR signaling slows down T-ALL cell growth in vitro (5, 6) and improves survival in T-ALL mouse models (7). Clinical studies of the mTOR inhibitor everolimus, alone or in combination with chemotherapy, showed promising levels of toxicity and efficacy in non-Hodgkin lymphoma (8) and relapsed/refractory T-ALL (9) patients. Although several downstream targets of mTOR are known, the molecular basis for the efficacy of mTOR inhibition against T-ALL remains incompletely understood.
microRNAs (miRNAs) are small non-coding RNAs that post-transcriptionally regulate gene expression by inducing translational repression and/or degradation of mRNA molecules with partial sequence complementarity (10). Key pathways involved in oncogenesis are targeted by miRNAs (11, 12) and profound alterations in miRNA expression are well documented in malignantly transformed cells (13–15). It was reported recently that mTOR signaling can interfere with miRNA biogenesis (16).
miR-150 is a highly abundant miRNA in cells of the lymphoid lineage (17–19), but its expression is strongly reduced in hematopoietic malignancies, such as acute myeloid leukemias (20), NK/T cell lymphoma (21), advanced cutaneous T-cell lymphoma (22) and multiple T-ALL cell lines (23). Ectopic expression of miR-150 negatively affects survival of these cells (20, 21, 23) and reduces their metastatic potential (22), suggesting that it acts as a tumor suppressor. Indeed, restoring miR-150 expression in chronic myeloid leukemia cells significantly slows tumor growth in a xenograft model (20). However, the mechanism for miR-150 down-regulation in any type of hematopoietic malignancy is unknown. Given the high levels of constitutive mTOR activation in T-ALL cells, we set out to test a hypothesis that mTOR signaling induces oncogenic down-regulation of miR-150 in these cells. We show that two different classes of mTOR inhibitors, rapamycin and PP242, rescue miR-150 expression in Jurkat T-ALL cells. This result connects mTOR signaling to the regulation of miRNA expression in a T-ALL cell line, specifically to the down-regulation of a tumor suppressor miRNA. We also report a novel feed-forward regulatory loop, whereby miR-150 reinforces a rapamycin-induced cell cycle arrest by targeting a functional gene network linked to the cell cycle. We propose that restoration of this novel miRNA- dependent regulatory loop by rapamycin is a novel mechanism underlying the efficacy of mTOR inhibitors against T-ALL and suggest that it should be studied further in primary cells.
2. Methods
2.1. Ethics statement
All experiments were covered by Human Subjects Research Protocols approved by the Institutional Review Board of The Scripps Research Institute.
2. 2. Cells
Jurkat (ATCC, Cat # TIB-152) cells were grown in RPMI-1640 with 2mM L-glutamine, 10mM HEPES and 1mM sodium pyruvate; primary T cells were grown in RPMI-1640 with 2mM L-glutamine. All media contained 10% heat-inactivated FBS, penicillin and streptomycin. Primary naïve (CD4+CD45RA+CD45RO-) and memory (CD4+CD45RA- CD45RO+) T cells were isolated from peripheral blood collected from healthy donors ages 18–30 years. Briefly, peripheral blood mononuclear cells were enriched by Ficoll- histopaque (Sigma-Aldrich) density gradient centrifugation, and T cells were purified from these cells by negative selection using magnetic beads (Miltenyi Biotec). Cells were stimulated with anti-CD3/CD28 beads (Life Technologies) using manufacturer’s instructions for 48 h. All other cell lines were obtained from ATCC and grown following the recommended culture methods. All cells were grown at 37°C in a 5% CO2 humidified chamber. Rapamycin (Sigma-Aldrich) and PP242 (Tocris) were reconstituted in DMSO. Rapamycin was used at 100 nM unless otherwise noted. Anti-CD95 antibody (Millipore) was used at 0.1ug/ml.
2.3. qRT-PCR
Total RNA was extracted using Trizol (Life Technologies), cleaned up using the miRNeasy kit (Qiagen) and subjected to on-column DNase digestion (Qiagen). Mature miRNAs were detected with the qScript system and PerfeCTa miRNA assays (Quanta BioSciences). Quantification was done using the delta Cq method relative to RNU6. For pri-miRNA quantification, cDNA was synthesized using high capacity cDNA reverse transcriptase kit (Life Technologies) and used to template qPCR reactions with Taqman assays (Life Technologies) specific for pri-miR-150. Cq values were normalized to 18S RNA. For pre- miRNA quantification, primers were designed to the pre-miRNA region directly upstream of the mature sequence (pre-miR-150: TCCCCATGGCCCTGTCTC). Total RNA was fractionated (miRNeasy kit, Qiagen) to only contain molecules < 200 nt, converted to cDNA using qScript microRNA cDNA Synthesis Kit (Quanta BioSciences) and used to template SYBR green qPCR reactions with a specific 5’ primer and the universal Quanta 3’ primer. Cq values were normalized to RNU6. Because pri-miRNAs can be detected by pre-miRNA primers, we confirmed that pri-miRNAs were absent in the < 200 nt RNA fraction.
2.4. microRNA-seq
Small RNA sequencing libraries were prepared using the TruSeq kit (Illumina) and sequenced on an Illumina HiSeq instrument with 100-base single-end reads. QC and read filtering was done using the FASTX toolkit (v.0.0.13; http://hannonlab.cshl.edu/fastx_toolkit/) and sequencing adapters were trimmed off using a custom python script. Reads that did not contain any adapter sequence were discarded and trimmed reads were aligned to mature miRNA sequences (miRbase v.17 (24) ) using bowtie (v.0.12.9 (25)) with -v 0 -a options. On average, 1.6 million reads were mapped to mature miRNAs. miRNA read counts were normalized to the median tRNA expression (26). miRNAs with less than 10 reads were considered not expressed and removed from further analysis.
2.5. Microarrays
Jurkat cells were grown in 4 independent cultures for several passages, transiently transfected with pre-miR-150 or control (scrambled) pre-miRNA and then treated with rapamycin or DMSO for 72 h. Total RNA was analyzed on the Agilent Bioanalyzer to confirm integrity. cDNA was synthesized and labeled using the Ambion whole transcript (WT) expression kit (Life Technologies), hybridized to 24-array HuGene 1.1ST plates and run on the GeneTitan Multi-Channel Instrument (Affymetrix). Data analysis was done in Genomics Suite (Partek). Probe intensities were normalized using the RMA method. Expression changes were calculated using 1-way ANOVA with the p-value < 0.05 as a cutoff for significance. Network and pathway enrichment analyses were performed using Ingenuity Pathway Analysis software and GO term enrichment was performed using GOrilla: http://cbl-gorilla.cs.technion.ac.il (27).
2.6. Western blotting
All antibodies were obtained from Cell Signaling and were used with the ODYSSEY CLx infrared imaging system (LI-COR): phospho-Ser473-Akt (D9E), polyclonal Akt, phospho- Thr389-p70S6K (108D2), p70S6K (49D7), alpha-tubulin (DM1A), and LC3B (D11).
2.7. miR-150 over-expression and knockdown
Pre-miR-150 or control pre-miRNA vectors (PMIR150-PA-1 and PMIRH000-PA-1, System Biosciences) were transfected into Jurkat cells at 18 nM using Amaxa 4-D Nucleofector. MiR-150 LNA or control LNA (426828–00 and 199020–00, Exiqon) were transfected at 200 nM. Transfection efficiencies were monitored using copGFP co-expressed on the pre- miRNA vector or by transfection with FITC-labeled LNA probes. Transfection efficiencies ranged between 94–96%. Cells were recovered in complete media for 3–4 h prior to drug treatment.
2.8. Proliferation and cell cycle analysis
Cells were plated at 80,000 cells per well in 96-well plates and BrdU was added to the media for the final 18 h of culture at 0.01 mM. Cells were lysed and BrdU incorporation quantified using Ziva Cell Proliferation Assay kit (Jaden BioScience Inc). For cell cycle analysis, 0.01 mM BrdU was added to culture medium for 24 h, cells were stained with anti- BrdU antibodies and 7-AAD (BD Biosciences) and analyzed by flow cytometry.
2.9. Luciferase assays
3’UTRs were amplified from Jurkat genomic DNA using the following primers - ARRB1: CACATGGGCGATCGCGAAGTGAGGATGGGTGTC and GAGGCGGCCGCAAGCTTGGAAACATGACCTGC; FADS1: CAACAGCGATCGCGCCCAGTCTGGAAGAAGAGGAGG and CACAGCGGCCGCGGTGCTTTGAGGACTTGGTCTTGG; CDK2: TAGCGCGATCGCAGCCCCCAGCCCTAATCTCACCC and TCCGCGGCCGCAGGAGGTGGACGTCAGAGGAAAATGGG and cloned into the psiCheck2 Dual- luciferase reporter vector (Promega). To make the positive control reporter, oligonucleotides bearing 2 complementary miR-150 sequences separated by an EcoRI site were annealed and inserted into the psiCheck2 vector (CGCCACTGGTACAAGGGTTGGGAGAGAATTCCACTGGTACAAGGGTTGGGAGAGC and GGCCGCTCTCCCAACCCTTGTACCAGTGGAATTCTCTCCCAACCCTTGTACCAGTGGCGAT). MiR- 150 binding sites were sequentially mutated to miR-135 sites (miR-135 is not expressed in Jurkat) using QuikChange II XL Site-Directed Mutagenesis kit (Agilent). Luciferase assays were carried out using the Dual-Glo Luciferase Assay System (Promega).
3. Results
3.1. Expression of miR-150 is repressed by constitutive mTOR signaling and is rescued by rapamycin in Jurkat T-ALL cells
miR-150 levels are severely reduced in several T-ALL cell lines (MOLT-3, DND-41, CCRF- HSB-2, TALL-1 and Jurkat) relative to immature thymocytes (23). We found that mature T cells isolated from normal peripheral blood expressed high levels of miR-150 (almost 3 orders of magnitude higher than a T-ALL line; Figure 1A). This was true for both major T- cell subpopulations, naïve (CD45RA+CD45RO-) and memory (CD45RA-CD45RO+) CD4 T cells, that we analyzed. Crosslinking the T-cell receptor and the surface co-stimulatory molecule CD28 mimics antigen-induced activation signals and turns on the phosphatidylinositol-3-kinase (PI3K) signaling pathway, leading to activation of mTOR, which in turn induces rapid cell growth and proliferation (28). Activation of quiescent peripheral blood T cells by CD3/CD28 crosslinking led to a strong reduction of miR-150 levels (10-fold; Figure 1A). These results demonstrate that miR-150 is down-regulated in proliferating T lymphocytes. Because lymphocyte proliferation requires mTOR signaling and because mTOR signaling is generally elevated in T-ALL cells (4, 29), we hypothesized that miR-150 expression is repressed by mTOR.
Figure 1. Regulation of miR-150 expression in Jurkat T-cell acute lymphoblastic leukemia cells.
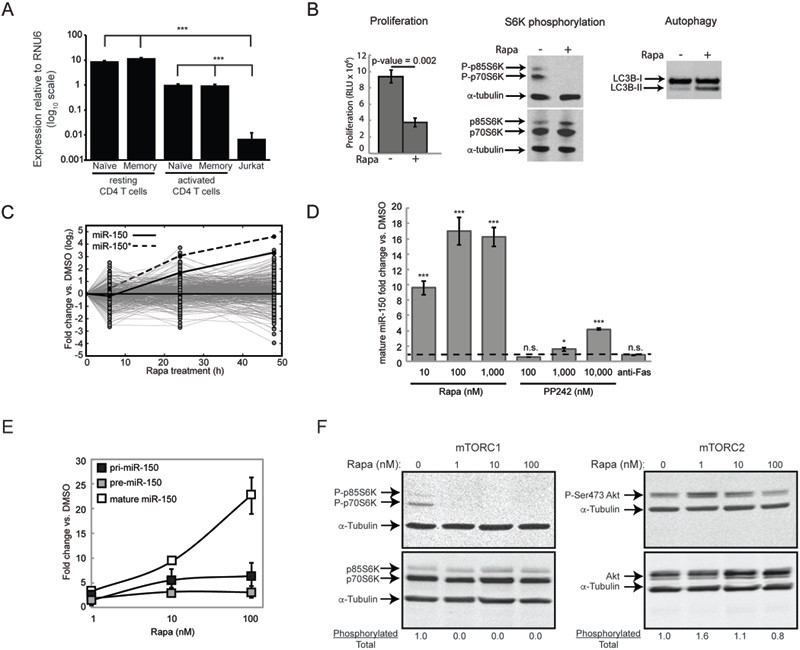
(A) Expression of miR-150 in resting and activated (CD3/CD28 co-stimulated for 48 h) naïve and memory CD4 T cells isolated from normal donor blood and Jurkat T-ALL cells determined by qRT-PCR. dCq values are calculated relative to RNU6 reference small RNA; note logio scale. (B) mTOR signaling is constitutively active in Jurkat cells and is inhibited by rapamycin treatment (100 nM; 48 h). Cell proliferation (determined by BrdU incorporation), phosphorylation of the canonical mTOR substrate S6K at Thr389 and low ratio of lipidated (II) to unlipidated (I) form of the autophagic protein LC3B are all makers of active mTOR signaling. (C) Expression of the 255 miRNAs detected in Jurkat cells by deep sequencing shown as log-fold-changes between cells treated with 100 nM rapamycin for 0, 6, 24 or 48 h and the corresponding DMSO-treated control cells. Data are shown as means of quadruplicates and are representative of 2 independent sequencing experiments. (D) Changes in miR-150 expression in Jurkat cells treated with rapamycin, PP242 or antiFas activating antibody (for 48 h) determined by qRT-PCR. Dashed line marks no change in expression. (E) Changes in pri-, pre- and mature miR-150 expression in response to rapamycin. Data are normalized to the corresponding form of miR-150 in DMSO-treated cells. (F) mTORC1 and mTORC2 activation status determined by phosphorylation of their respective canonical substrates, S6K-Thr389 and Akt-Ser473. Fraction of phosphorylated to total protein is indicated. All data are displayed as mean ± S.D, n = 4 or are representative of at least 3 independent experiments, unless otherwise noted. * p < 0.05, ** p < 0.005, *** p < 0.0005, n.s. - not significant (Student’s t-test).
In order to test this hypothesis we wanted to use a T-ALL cell line that has high constitutive levels of mTOR activity. Jurkat cells harbor a loss-of-function mutation in PTEN, a phosphatase that inhibits PI3K signaling (30), suggesting that mTOR signaling is constitutively active in these cells. To measure mTOR activity in Jurkat cells we assayed cell proliferation, phosphorylation of a canonical mTOR substrate, S6 kinase, and lipidation of LC3B, a protein involved in autophagy. As shown in Figure 1B, Jurkat cells exhibited constitutive phosphorylation of S6 kinase and this phosphorylation was completely removed by treatment with rapamycin, an mTOR inhibitor. In addition, rapamycin significantly reduced cell proliferation and induced post-translational changes in LC3B in a manner consistent with induction of autophagy. These results show that mTOR signaling is constitutively activated in Jurkat cells.
To test the hypothesis that miR-150 expression in T-ALL cells is repressed by mTOR signaling, we inhibited mTOR with rapamycin in Jurkat cells and examined miRNA expression over a 48-hour time period. It was reported recently that in some cells mTOR signaling can lead to global down-regulation of miRNA levels by interfering with miRNA biogenesis (16). Therefore, in order to differentiate between specific regulation of miR-150 expression and global regulation of miRNA levels by mTOR in Jurkat cells, we quantified genome-wide miRNA changes using deep sequencing. A total of 255 miRNAs were detected in Jurkat cells. Rapamycin treatment did not induce a concordant global change in miRNA expression, suggesting that mTOR signaling does not broadly remodel the miRNA repertoire of Jurkat cells. We did, however, identify 29 miRNAs that were either significantly up or down-regulated by rapamycin in a time-dependent manner (Table S1). Of these miRNAs, miR-150 (which consists of the miR-150 guide and the miR-150* passenger strands) exhibited the largest increase in response to rapamycin (Figure 1C, Table S1). This up-regulation showed dose-dependency and was not a rapamycin-specific phenomenon, because a structurally dissimilar mTOR inhibitor, PP242, also increased miR- 150 expression (Figure 1D). Induction of Fas-mediated apoptosis by CD95 crosslinking had no effect on miR-150 expression, suggesting that miR-150 is not induced by cell death. These results show that mTOR signaling inhibits miR-150 expression in Jurkat cells and regulates the expression of 27 other miRNAs.
Up-regulation of a mature miRNA can be accomplished in three ways: by increasing its transcription, processing or stability (31). To determine whether mTOR regulates miR- 150 transcription, the levels of pri-miR-150 (primary transcript), pre-miR-150 (a discrete processing intermediate) and mature miR-150 were measured in response to increasing doses of rapamycin. There was a modest but significant dose-dependent increase in pri- miR-150 levels indicating transcriptional induction (Figure 1E). Interestingly, at the concentration where the strongest up-regulation of mature miR-150 was observed (100 nM), the increase in mature miR-150 was not matched by a proportional increase in pri- or pre-miR-150. This result is consistent with the increased transcription of miR-150 being accompanied by enhanced pri- and pre- miRNA processing or enhanced stability of the mature miRNA at high rapamycin concentrations.
mTOR exists in two multi-protein complexes, mTORC1 and mTORC2, each with different downstream targets and functions (1). Although originally described as an mTORC1 inhibitor, higher doses and/or prolonged treatment with rapamycin can inhibit both complexes (32). Accordingly, treatment with 100 nM rapamycin for 48 hours (as in Figure 1E) inhibited both mTOR complexes in Jurkat cells, although mTORC2 inhibition was partial (Figure 1F). At lower concentrations (1 and 10 nM) mTORC1 was completely inhibited while mTORC2 was not inhibited at all. Coincidentally, it is at the highest concentration that we observed a disproportionate increase in the mature miR-150 suggestive of increased processing and/or stability (see Figure 1E). These data suggest a model wherein mTORC1 inhibition is sufficient to increase transcription of miR-150 but inhibition of both complexes also enhances post-transcriptional processing and/or increases stability of the mature miRNA.
3.2. miR-150 inhibits cell proliferation and reinforces a rapamycin-induced cell cycle arrest
Next, we wanted to determine the functional consequences of restoring miR-150 expression in Jurkat cells. Because our results placed regulation of miR-150 expression downstream of mTOR, and because one of the main processes controlled by mTOR signaling is cell proliferation (33, 34), we hypothesized that miR-150 is also involved in regulating cell proliferation. Consistent with this idea, over-expression of miR-150 has been shown to reduce T-ALL cell number in culture (23). We ectopically expressed the miR-150 precursor (pre-miR-150) using a lentiviral vector in Jurkat cells. Introduction of pre-miR-150 or treatment with rapamycin increased mature miR-150 levels by a similar proportion (about 13-fold; Figure 2A). When combined, the two treatments resulted in a nearly 100-fold expression increase, yielding miR-150 expression levels equivalent to those in activated non-malignant T cells (compare Figure 2A to 1A).
Figure 2. miR-150 inhibits proliferation and reinforces a rapamycin-induced cell cycle arrest.
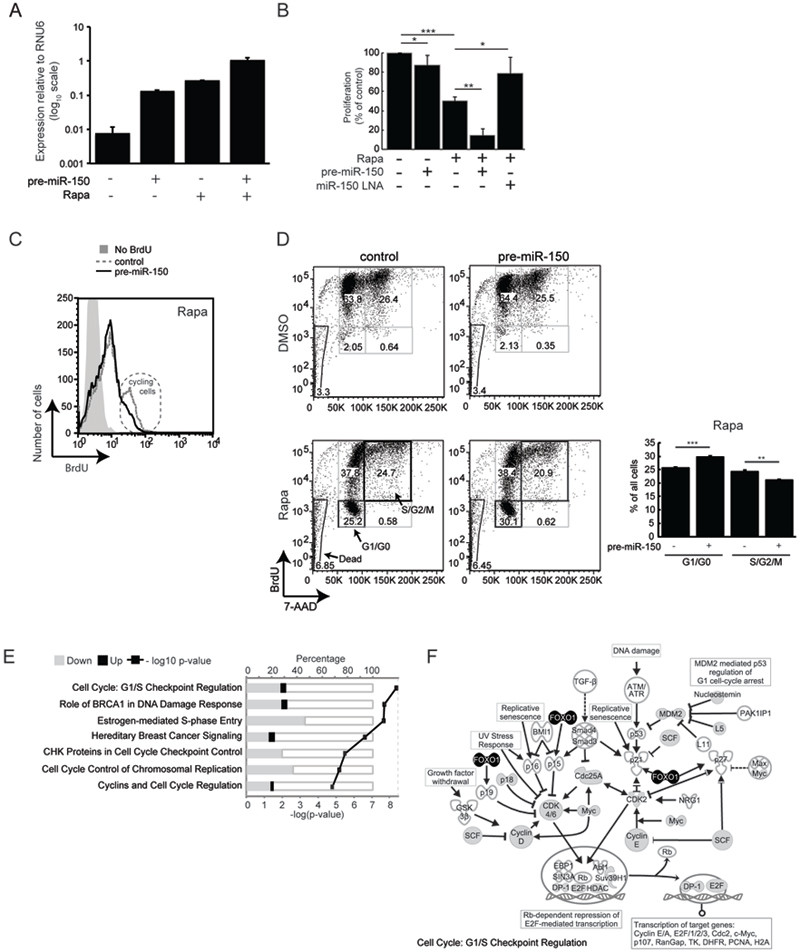
(A) Over-expression of miR-150 was achieved by transfecting Jurkat cells with a lentiviral vector containing the pre-miR-150 sequence flanked by ~400 bp of native genomic DNA and a GFP reporter. Cells were treated with DMSO or rapamycin 3–4 h after transfection and expression of mature miR-150 was quantified by qRT-PCR 48 h later. (B) Cell proliferation in response to rapamycin treatment, miR-150 over-expression, and/or miR- 150 knockdown. Proliferation was measured by BrdU ELISA and normalized to DMSO- treated cells transfected with a scrambled pre-miRNA hairpin or a scrambled LNA. (C) BrdU incorporation was quantified by flow cytometry. Plots are gated by forward and side scatter to exclude dead cells and mean fluorescence intensity (MFI) is displayed on the x-axis. Control MFI = 9.32; pre-miR-150 MFI = 7.53, p-value = 0.0082. The population of cycling cells (2x higher BrdU content) is circled. Data are representative of 3 experiments. (D) Cell cycle analysis of cells over-expressing miR-150 or a scrambled hairpin control and treated with DMSO or rapamycin for 48 h; BrdU was added in the last 24 h. All acquired cells are displayed and numbers within gates indicate percentages of the total cells. Bar graph summarizes percentages of rapamycin-treated cells in the G0/G1 and S/G2/M phases. (E) Genes that changed expression in response to miR-150 over-expression are enriched within these signaling pathways. Enrichment was calculated using Ingenuity Pathway Analysis software and the top 7 pathways are shown (Fisher’s exact t-test p-value < 1.6e-05). (F) Specific molecules in the “Cell Cycle: G1/S Checkpoint Regulation” pathway from (E). Gray fill - genes down-regulated by miR-150 over-expression, black fill - genes up-regulated by miR-150 over-expression. Data are shown as mean ± S.D, n = 4. * p < 0.05, ** p < 0.005, *** p < 0.0005 (Student’s t-test). All data are representative of at least 3 independent experiments.
Although ectopic expression of pre-miR-150 and treatment with rapamycin increased miR-150 levels by equivalent amounts (see Figure 2A), ectopic expression resulted in a modest decrease in cell proliferation (12% decrease) compared to rapamycin (50% decrease; Figure 2B). Thus, re-expression of miR-150 in the context of active mTOR does not recapitulate the full effect of rapamycin on cell proliferation. However, ectopic expression of miR-150 combined with rapamycin treatment reduced cell proliferation significantly further than would be expected from the combination of these two treatments (85% total decrease; Figure 2B), indicating a synergistic interaction. Flow cytometry also showed a marked contraction of the proliferating cell population, when rapamycin-treated cells were subjected to miR-150 over-expression (Figure 2C). To rule out off-target effects of miR-150 over-expression, we reduced miR-150 levels using antisense LNA probes. Concordantly, this treatment dampened the anti-proliferative effects of rapamycin (Figure 2B).
Rapamycin inhibits cell proliferation by blocking the G1 to S phase transition in the cell cycle (33). In untreated cells, ectopic expression of miR-150 did not yield a notable cell cycle phenotype (Figure 2D), consistent with the modest effect it had on proliferation (see Figure 2B). However, in rapamycin-treated cells, ectopic miR-150 expression resulted in a significant accumulation of G1/G0 cells accompanied by a proportional reduction in the percentage of actively cycling cells (Figure 2D). We did not observe any changes in the proportion of dead cells, suggesting that miR-150 does not induce apoptosis or necrosis. Together, these results strongly suggest that ectopic miR-150 expression sensitizes Jurkat cells to a rapamycin-induced cell cycle arrest. The effect of miR-150 on cell proliferation is modest in the context of active mTOR, but when mTOR is inhibited, additional miR-150 expression synergistically reduces cell proliferation, consistent with feed-forward regulation.
To better understand how miR-150 cooperates with rapamycin to reduce proliferation, we measured genome-wide mRNA expression changes that arise when miR- 150 is ectopically over-expressed in rapamycin-treated cells. Differentially expressed genes showed significant enrichment of cell cycle regulatory pathways, and “G1/S checkpoint regulation” was the most highly enriched pathway (Figure 2E). Genes than make up this pathway are shown in Figure 2F. Positive cell cycle regulators (e.g. Cyclin D, Cyclin E, Cdk 4/6, MDM2 and c-Myc) were down-regulated by miR-150, while the negative regulator, FOXO1, was up-regulated. Together, these results suggest that miR-150 sensitizes Jurkat T-ALL cells to rapamycin by interfering with expression of genes needed for cell cycle progression.
3.3. Genes targeted by miR-150 constitute a cell cycle regulatory network
We showed that ectopic expression of miR-150 in T-ALL cells modulates the expression of cell cycle genes (see Figure 2E-F). While some of these observed changes are likely due to direct interactions between miR-150 and its target mRNA molecules, others may represent indirect effects. To enrich for direct molecular targets of miR-150, we implemented the following filtering strategy.
Mammalian miRNAs achieve regulation primarily by reducing the levels of mRNA molecules they bind to (35). Because rapamycin increases miR-150 expression, the levels of mRNAs targeted by miR-150 should decrease when cells are treated with rapamycin. Microarray analysis of rapamycin-treated and untreated Jurkat cells revealed 3866 genes significantly down-regulated by rapamycin (Figure 3A). To further enrich for genes that were down-regulated due to the concomitant increase in miR-150 rather than other rapamycin-induced changes, we asked which of these genes would change if miR-150 were over-expressed further. A total of 727 genes changed significantly, the vast majority (634, or 90%) in the down direction (Figure 3A), indicating that ectopic miR-150 further reduces expression of genes down-regulated by rapamycin (Figure 3B). Finally, to increase the stringency of target enrichment even further, we removed any gene that did not contain a computationally predicted canonical miR-150 binding site, leaving 147 genes (Figure 3A, Table S2). In summary, these 147 genes are down-regulated by rapamycin, further down- regulated when miR-150 is over-expressed, and contain at least one canonical miR-150 binding site. Thus, these genes should be strongly enriched for direct biological targets of miR-150 in rapamycin-treated Jurkat cells.
Figure 3. Identification and analysis of genes directly targeted by miR-150.
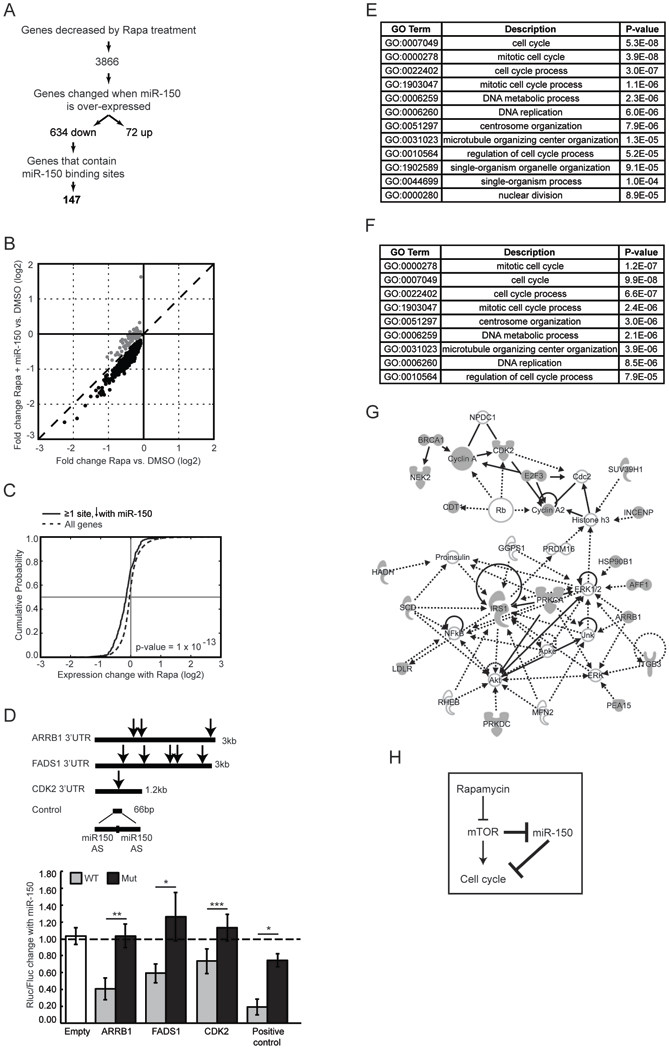
(A) Experimental flow chart of miR-150 target enrichment. Gene expression was determined in Jurkat cells at three conditions: control miRNA plus DMSO, control miRNA plus rapamycin, and miR-150 plus rapamycin. miRNA targets were predicted using Targetscan (51). Because miR-150 has relatively few predicted targets with a conservation score in the 50th percentile or above, we used low stringency criteria by imposing no conservation or context score cut offs. (B) Ectopic miR-150 expression prior to rapamycin treatment induces further down-regulation of genes that are normally decreased by rapamycin (black circles). Dashed line denotes an identity line. (C) Cumulative distribution function (CDF) plots of gene expression changes induced by rapamycin treatment. Genes that contain at least one miR-150 binding site and that also decreased in response to miR- 150 over-expression are plotted using a solid line; all genes expressed in Jurkat cells are plotted using a dashed line. Vertical line marks no change in expression; horizontal line marks mean expression change. A shift to the left indicates a higher probability of down- regulation in response to rapamycin. P-value was calculated using the Kolmogorov- Smirnov nonparametric test. (D) Luciferase reporter assay for selected miR-150 targets. Renilla luciferase luminescence was measured 24 h after transfection into Jurkat cells and normalized to the signal from firefly luciferase encoded on the same vector. Note, perfect complementarity between the positive control reporter and miR-150 triggers siRNA- mediated silencing. AS - antisense; Empty - reporter without a 3’UTR; Rluc - Renilla luciferase; Fluc - firefly luciferase; arrow - miR-150 site; dashed line - no change between cells transfected with miR-150 and control miRNA; Mut - mutated miR-150 sites. Data are shown as means ±S.D (n = 4) and represent at least 3 independent experiments. * p < 0.05, ** p < 0.005, *** p < 0.0005, n.s. - not significant (Student’s t-test). (E) GO terms enriched within the 147 miR-150 target genes relative to all genes expressed in Jurkat cells and (F) relative to genes differentially expressed in response to rapamycin. All terms with FDR < 0.1 are shown. (G) Network with the highest number of functional connections between the 147 target genes created using Ingenuity Pathway Analysis software. Target genes are shaded in gray; interpolated nodes are not shaded. (H) A model suggested by the present work; novel connections are bolded.
To computationally validate this target enrichment approach, we tested whether genes with a canonical miR-150 binding site that showed a decrease upon miR-150 over-expression are more likely to be down-regulated in response to rapamycin, as would be predicted if these genes are enriched for real biological targets of miR-150. We found a significant shift in the cumulative probability function, which indicates that these genes indeed have a higher likelihood of being down-regulated in response to rapamycin (Figure 3C).
We also experimentally tested whether miR-150 interacts with its novel predicted target genes. Three genes were tested using the standard dual-luciferase reporter assay: beta-arrestin 1 (ARRB1), fatty acid desaturase 1 (FADS1) and cyclin-dependent kinase 2 (CDK2). Wild-type 3’UTRs or 3’UTRs with mutated miR-150 binding sites were inserted downstream of a Renilla luciferase reporter and transfected into Jurkat cells. Due to the large size of the ARRB1 3’UTR (6 kb), only the first 3 kb containing 3 of the 5 predicted sites were tested (Figure 3D). Co-transfection with miR-150 resulted in reduced reporter activity. This effect was abolished when miR-150 sites were mutated, confirming that ARRB1, FADS1 and CDK2 3’UTRs are directly silenced by miR-150.
Individual miRNAs often regulate multiple genes that share a common biological function (36, 37). Likewise, the 147 candidate miR-150 targets showed significant enrichment for GO terms associated with cell cycle regulation (Figure 3E). Because one of the pre-requisites in the filtering process used to identify these genes in the first place was their down-regulation in response to rapamycin, we wanted to make sure that the cell cycle GO term enrichment was not simply a reflection of rapamycin-induced cell cycle arrest. To address this, we calculated GO term enrichment within the 147 candidate targets relative to genes that changed in response to rapamycin and found a similar significant enrichment for cell cycle GO terms (Figure 3F). To better visualize functional relationships between the candidate miR-150 targets, we constructed gene networks using known functional connections between them as edges. Genes within the largest such network include direct mediators of the G1/S transition (E2F, CDK2), molecules in the ERK/MAPK pathway (ARRB1, IRS1) that control the G1/S cell cycle transition in parallel with the mTOR pathway, and genes involved in the regulation of DNA replication and cell division (Figure 3G). Together, these results suggest that miR-150 inhibits cell cycle progression at multiple levels through down-regulation of direct and indirect cell cycle mediators.
3.4. Regulation of miR-150 by mTOR is specific to cells of T lymphocyte and leukemic origins
Most of the work connecting mTOR to miR-150 levels presented thus far has been conducted in the T-ALL cell line Jurkat, making use of its high levels of constitutive mTOR activity. Our results showing a decrease in miR-150 levels following antigenic stimulation of primary CD4 T cells (see Figure 1a) suggested that mTOR-mediated regulation of miR- 150 levels also occurs in normal T lymphocytes. To directly test this, we treated antigen- stimulated CD4 T cells with rapamycin. This treatment significantly inhibited mTOR signaling and, as predicted, significantly increased miR-150 levels (Figure 4), indicating that mTOR signaling does indeed repress miR-150 expression in non-malignant activated CD4 T cells.
Figure 4. Rapamycin increases miR-150 expression in cells of T lymphocyte and leukemic origin.
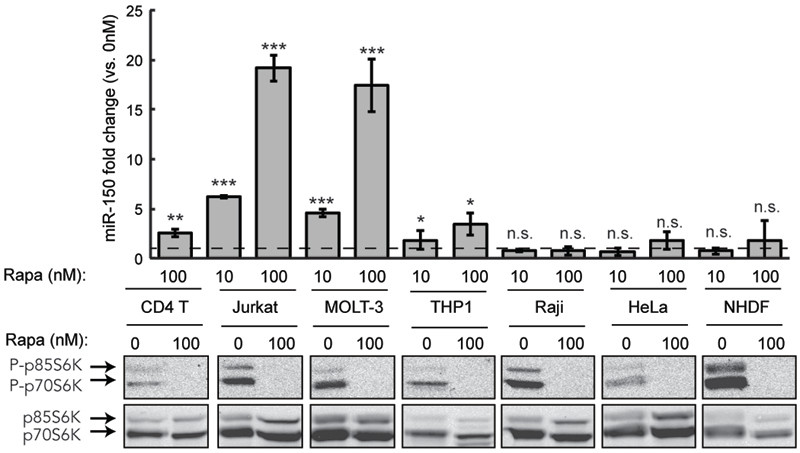
Expression of miR-150 in response to a 48 h rapamycin treatment was determined using qRT-PCR in the following cells: naïve CD4 T lymphocytes stimulated by CD3/CD28 crosslinking, T-ALL line Jurkat, T-ALL line MOLT-3, acute monocytic leukemia line THP-1, Burkitt’s lymphoma line Raji, epithelial carcinoma line HeLa and normal human dermal fibroblast line NHDF. Dashed line marks no change in expression relative to vehicle-treated cells. Phosphorylation of S6K was determined in each cell line to confirm constitutive mTOR activity and inhibition of this activity by rapamycin. Data are representative of 3 independent experiments and shown as mean ± S.D, n = 4. *p < 0.05, **p < 0.005, ***p < 0.0005, n.s. not significant (Student’s t-test).
We also tested whether mTOR-mediated control of miR-150 expression can be generalized to other forms and isolates of leukemia. Similar to the data obtained using TALL Jurkat cells, inhibition of mTOR by rapamycin resulted in a significant and dose- dependent increase in miR-150 levels in a different T-ALL line, MOLT-3 (Figure 4). Cells from a different type of leukemia, the acute monocytic leukemia line THP-1, showed a more modest yet still significant increase in miR-150 levels in response to increasing doses of rapamycin. Thus, at least two other leukemia lines seem to reduce miR-150 levels by activating mTOR signaling. Interestingly, while rapamycin was able to inhibit mTOR signaling in B cell lymphoma (Raji) and epithelial cancer cells (HeLa), it failed to induce any miR-150 expression. The same was true when rapamycin was applied to a fibroblast cell line NHDF. These results show that the mTOR/miR-150 regulatory loop seems to be specifically active in cells of the T lymphocyte origin and in leukemia cells, possibly due to a requirement for other lineage-specific factors, such as epigenetic modifiers, in order for miR-150 to be expressed.
4. Discussion
Both mTOR and miRNA pathways play important roles in oncogenesis (11, 34); so understanding how they are linked will provide important mechanistic insights into the steps that drive oncogenic transformation. In this study we found that mTOR regulates expression of a tumor suppressor miRNA in leukemia cells. There are three main points of interest regarding the novel connection between mTOR and miRNA expression uncovered here.
First, T-ALL is an aggressive T-cell malignancy, accounting for 15% of pediatric and 25% of adult ALL cases (4, 38). While significant progress has been made in treating ALL with combination chemotherapy, mortality rates remain at 20% in children and 40–50% in adults, mainly due to poor outcomes in relapsed patients (38–40). Therefore, targeted therapies hold the potential to improve treatment outcomes in refractory and poor prognosis patients, as well as to minimize toxicity associated with chemotherapy. A better understanding of signaling pathways involved in T-ALL pathogenesis and drug responses is critical for the development of such therapies. Here, we propose that miR-150 is normally expressed when mTOR signaling is off (e.g. in resting T cells) and it acts to reinforce the state of quiescence, likely by helping repress cell cycle genes beyond a critical threshold (Figure 3H). This feed-forward loop is turned off in T-ALL cells due to constitutively active mTOR signaling. Re-engagement of the miR-150-dependent feed-forward loop by rapamycin constitutes a novel mechanism by which this drug inhibits proliferation. The next step toward a better understanding of this mechanism would be to identify molecules downstream of mTORC1 and mTORC2 that directly regulate miR-150 transcription and processing. Further studies are also needed to test whether rapamycin rescues miR-150 expression in clinical isolates of T-ALL cells and whether these findings can be extended to other malignancies of the T-cell lineage.
Second, identification of a large number of miR-150 targets with a common role in cell cycle regulation supports the emerging paradigm that most mammalian miRNAs regulate gene networks rather than individual genes, thus providing buffering capacity and stability to biological processes (41, 42). It follows that misregulation of a single miRNA, such as miR-150, would lead to only a small change in expression of any one particular gene, but combined changes in all of its targets could have profound effects on the cell due to destabilization of a gene network. In addition to the cell cycle network identified here, miR-150 likely has other targets in T-ALL. For instance, Ghisi et al. (23) found that miR-150 regulates translation of the NOTCH3 mRNA in T-ALL cell lines. Our comprehensive miR- 150 target identification strategy based on detection of mRNA changes would have missed any targets, such as NOTCH3, whose expression is solely controlled at the level of protein synthesis. Furthermore, although miR-150 achieves anti-proliferative effects in multiple types of leukemias, its direct targets may differ in different cellular contexts. For example, targets we identified in T-ALL cells are largely different from those identified by Fang et al. in chronic myeloid leukemia cells (20). This is not surprising in light of a large body of evidence that a given miRNA can target different genes in different cells due to differences in target gene expression, 3’UTR splicing, 3’UTR truncation and/or miRNA binding site accessibility (43–48). It is also important to point out that other studies, including the Fang et al. study, aimed to identify miR-150 targets by looking for genes whose levels decreased in response ectopic expression of miR-150. In contrast, we focused our search specifically on genes that are normally involved in the miR-150/mTOR regulatory loop, or in other words, genes that are repressed by rapamycin due to its effect on miR-150 levels. Indeed, if we expand our definition of a miR-150 target to any gene with a predicted binding site that went down when miR-150 was ectopically expressed, 77% of targets identified by Fang et al. end up on that list. Thus, genes that are repressed when miR-150 is ectopically expressed in leukemia cells may not all normally be regulated by miR-150 in the context of its physiological function.
Finally, while mTOR inhibitors have had limited success in the clinic on their own (1), a large interest remains in combining mTOR inhibitors with other therapies (9). Given recent progress in the field of miRNA-based therapeutics (12, 49, 50) and our results showing that ectopic expression of miR-150 sensitizes Jurkat cells to rapamycin, we suggest that administration of miR-150 mimics in concert with rapamycin is a co-therapy worth investigating further using in vitro and possibly in vivo studies of T-cell leukemia.
Supplementary Material
Highlights.
Rapamycin restores miR-150 expression in leukemia Jurkat T cells
Acting in a feed-forward loop, miR-150 further sensitizes cells to rapamycin
miR-150 regulates a large network of cell cycle genes
A mechanism of miR-150 down-regulation in T-ALL cells is described
A novel microRNA-dependent mechanism of rapamycin efficacy is proposed
Acknowledgements
We thank Dr. Filip van Nieuwerburgh and Terri Gelbart for help with microarrays and deep sequencing. This work was supported by NIH grants: R01 AI081757, U19 A1063603, U01 GM094653 and U01 AI084146 to DRS and NIH TL1 RR025772 to KP.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zoncu R Efeyan A, and Sabatini DM 2011. mTOR: from growth signal integration to cancer, diabetes and ageing. Nature reviews. Molecular cell biology 12: 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kharas MG, Okabe R, Ganis JJ, Gozo M, Khandan T, Paktinat M, Gilliland DG, and Gritsman K 2010. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood 115: 1406–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peng C, Chen Y, Li D, and Li S 2010. Role of Pten in leukemia stem cells. Oncotarget 1: 156–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao WL 2010. Targeted therapy in T-cell malignancies: dysregulation of the cellular signaling pathways. Leukemia 24: 13–21. [DOI] [PubMed] [Google Scholar]
- 5.Baraz R, Cisterne A, Saunders PO, Hewson J, Thien M, Weiss J, Basnett J, Bradstock KF, and Bendall LJ 2014. mTOR inhibition by everolimus in childhood acute lymphoblastic leukemia induces caspase-independent cell death. PloS one 9: e102494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bressanin D, Evangelisti C, Ricci F, Tabellini G, Chiarini F, Tazzari PL, Melchionda F, Buontempo F, Pagliaro P, Pession A, McCubrey JA, and Martelli AM 2012. Harnessing the PI3K/Akt/mTOR pathway in T-cell acute lymphoblastic leukemia: eliminating activity by targeting at different levels. Oncotarget 3: 811–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crazzolara R, Cisterne A, Thien M, Hewson J, Baraz R, Bradstock KF, and Bendall LJ 2009. Potentiating effects of RAD001 (Everolimus) on vincristine therapy in childhood acute lymphoblastic leukemia. Blood 113: 3297–3306. [DOI] [PubMed] [Google Scholar]
- 8.Witzig TE, Reeder CB, LaPlant BR, Gupta M, Johnston PB, Micallef IN, Porrata LF, Ansell SM, Colgan JP, Jacobsen ED, Ghobrial IM, and Habermann TM 2011. A phase II trial of the oral mTOR inhibitor everolimus in relapsed aggressive lymphoma. Leukemia 25: 341–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daver N, Boumber Y, Kantarjian H, Ravandi F, Cortes J, Rytting ME, Kawedia JD, Basnett J, Culotta KS, Zeng Z, Lu H, Richie MA, Garris R, Xiao L, Liu W, Baggerly KA, Jabbour E, O’Brien S, Burger J, Bendall LJ, Thomas D, and Konopleva M 2015. A Phase I/II Study of the mTOR Inhibitor Everolimus in Combination with HyperCVAD Chemotherapy in Patients with Relapsed/Refractory Acute Lymphoblastic Leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research 21: 2704–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bartel DP 2009. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jansson MD, and Lund AH 2012. MicroRNA and cancer. Molecular oncology 6: 590–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayes J, Peruzzi PP, and Lawler S 2014. MicroRNAs in cancer: biomarkers, functions and therapy. Trends in molecular medicine 20: 460–469. [DOI] [PubMed] [Google Scholar]
- 13.Gaur A, Jewell DA, Liang Y, Ridzon D, Moore JH, Chen C, Ambros VR, and Israel MA 2007. Characterization of microRNA expression levels and their biological correlates in human cancer cell lines. Cancer research 67: 2456–2468. [DOI] [PubMed] [Google Scholar]
- 14.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, and Golub TR 2005. MicroRNA expression profiles classify human cancers. Nature 435: 834–838. [DOI] [PubMed] [Google Scholar]
- 15.Xie B, Ding Q, Han H, and Wu D 2013. miRCancer: a microRNA-cancer association database constructed by text mining on literature. Bioinformatics 29: 638–644. [DOI] [PubMed] [Google Scholar]
- 16.Ye P, Liu Y, Chen C, Tang F, Wu Q, Wang X, Liu C-G, Liu X, Liu R, Liu Y, and Zheng P 2015. An mTORC1-Mdm2-Drosha Axis for miRNA Biogenesis in Response to Glucose- and Amino Acid-Deprivation. Molecular cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bezman NA, Chakraborty T, Bender T, and Lanier LL 2011. miR-150 regulates the development of NK and iNKT cells. The Journal of experimental medicine 208: 2717–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiao C, Calado DP, Galler G, Thai TH, Patterson HC, Wang J, Rajewsky N, Bender TP, and Rajewsky K 2007. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell 131: 146–159. [DOI] [PubMed] [Google Scholar]
- 19.Zhou B, Wang S, Mayr C, Bartel DP, and Lodish HF 2007. miR-150, a microRNA expressed in mature B and T cells, blocks early B cell development when expressed prematurely. Proceedings of the National Academy of Sciences of the United States of America 104: 7080–7085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fang ZH, Wang SL, Zhao JT, Lin ZJ, Chen LY, Su R, Xie ST, Carter BZ, and Xu B 2016. miR-150 exerts antileukemia activity in vitro and in vivo through regulating genes in multiple pathways. Cell death & disease 7: e2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watanabe A, Tagawa H, Yamashita J, Teshima K, Nara M, Iwamoto K, Kume M, Kameoka Y, Takahashi N, Nakagawa T, Shimizu N, and Sawada K 2011. The role of microRNA-150 as a tumor suppressor in malignant lymphoma. Leukemia 25: 1324–1334. [DOI] [PubMed] [Google Scholar]
- 22.Ito M, Teshima K, Ikeda S, Kitadate A, Watanabe A, Nara M, Yamashita J, Ohshima K, Sawada K, and Tagawa H 2014. MicroRNA-150 inhibits tumor invasion and metastasis by targeting the chemokine receptor CCR6 in advanced cutaneous T- cell lymphoma. Blood. [DOI] [PubMed] [Google Scholar]
- 23.Ghisi M, Corradin A, Basso K, Frasson C, Serafin V, Mukherjee S, Mussolin L, Ruggero K, Bonanno L, Guffanti A, De Bellis G, Gerosa G, Stellin G, D’Agostino DM, Basso G, Bronte V, Indraccolo S, Amadori A, and Zanovello P 2011. Modulation of microRNA expression in human T-cell development: targeting of NOTCH3 by miR- 150. Blood 117: 7053–7062. [DOI] [PubMed] [Google Scholar]
- 24.Kozomara A, and Griffiths-Jones S 2011. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic acids research 39: D152–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langmead B, Trapnell C, Pop M, and Salzberg SL 2009. Ultrafast and memory- efficient alignment of short DNA sequences to the human genome. Genome biology 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bronevetsky Y, Villarino AV, Eisley CJ, Barbeau R, Barczak AJ, Heinz GA, Kremmer E, Heissmeyer V, McManus MT, Erle DJ, Rao A, and Ansel KM 2013. T cell activation induces proteasomal degradation of Argonaute and rapid remodeling of the microRNA repertoire. The Journal of experimental medicine 210: 417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eden E, Navon R, Steinfeld I, Lipson D, and Yakhini Z 2009. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC bioinformatics 10: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Powell JD, and Delgoffe GM 2010. The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity 33: 301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silva A, Yunes JA, Cardoso BA, Martins LR, Jotta PY, Abecasis M, Nowill AE, Leslie NR, Cardoso AA, and Barata JT 2008. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. The Journal of clinical investigation 118: 3762–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shan X, Czar MJ, Bunnell SC, Liu P, Liu Y, Schwartzberg PL, and Wange RL 2000. Deficiency of PTEN in Jurkat T cells causes constitutive localization of Itk to the plasma membrane and hyperresponsiveness to CD3 stimulation. Molecular and cellular biology 20: 6945–6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Finnegan EF, and Pasquinelli AE 2013. MicroRNA biogenesis: regulating the regulators. Critical reviews in biochemistry and molecular biology 48: 51–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, and Sabatini DM 2006. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Molecular cell 22: 159–168. [DOI] [PubMed] [Google Scholar]
- 33.Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, and Blenis J 2004. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E- BP1/eukaryotic translation initiation factor 4E. Molecular and cellular biology 24: 200–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cargnello M, Tcherkezian J, and Roux PP 2015. The expanding role of mTOR in cancer cell growth and proliferation. Mutagenesis 30: 169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo H, Ingolia NT, Weissman JS, and Bartel DP 2010. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466: 835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Linsley PS, Schelter J, Burchard J, Kibukawa M, Martin MM, Bartz SR, Johnson JM, Cummins JM, Raymond CK, Dai H, Chau N, Cleary M, Jackson AL, Carleton M, and Lim L 2007. Transcripts targeted by the microRNA-16 family cooperatively regulate cell cycle progression. Molecular and cellular biology 27: 2240–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsang JS, Ebert MS, and van Oudenaarden A 2010. Genome-wide dissection of microRNA functions and cotargeting networks using gene set signatures. Molecular cell 38: 140–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pui CH, Robison LL, and Look AT 2008. Acute lymphoblastic leukaemia. Lancet 371: 1030–1043. [DOI] [PubMed] [Google Scholar]
- 39.Einsiedel HG, von Stackelberg A, Hartmann R, Fengler R, Schrappe M, Janka- Schaub G, Mann G, Hahlen K, Gobel U, Klingebiel T, Ludwig WD, and Henze G 2005. Long-term outcome in children with relapsed ALL by risk-stratified salvage therapy: results of trial acute lymphoblastic leukemia-relapse study of the Berlin- Frankfurt-Munster Group 87. Journal of clinical oncology: official journal of the American Society of Clinical Oncology 23: 7942–7950. [DOI] [PubMed] [Google Scholar]
- 40.Fielding AK, Richards SM, Chopra R, Lazarus HM, Litzow MR, Buck G, Durrant IJ, Luger SM, Marks DI, Franklin IM, McMillan AK, Tallman MS, Rowe JM, Goldstone AH, Medical ALLWP Research Council of the United Kingdom Adult, and G. Eastern Cooperative Oncology. 2007. Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood 109: 944–950. [DOI] [PubMed] [Google Scholar]
- 41.Ebert MS, and Sharp PA 2012. Roles for microRNAs in conferring robustness to biological processes. Cell 149: 515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Podshivalova K, and Salomon DR 2013. MicroRNA regulation of T-lymphocyte immunity: modulation of molecular networks responsible for T-cell activation, differentiation, and development. Critical reviews in immunology 33: 435–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thiele A, Nagamine Y, Hauschildt S, and Clevers H 2006. AU-rich elements and alternative splicing in the beta-catenin 3’UTR can influence the human beta-catenin mRNA stability. Experimental cell research 312: 2367–2378. [DOI] [PubMed] [Google Scholar]
- 44.Sandberg R, Neilson JR, Sarma A, Sharp PA, and Burge CB 2008. Proliferating cells express mRNAs with shortened 3’ untranslated regions and fewer microRNA target sites. Science 320: 1643–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kedde M, Strasser MJ, Boldajipour B, Oude Vrielink JA, Slanchev K, le Sage C, Nagel R, Voorhoeve PM, van Duijse J, Orom UA, Lund AH, Perrakis A, Raz E, and Agami R 2007. RNA-binding protein Dnd1 inhibits microRNA access to target mRNA. Cell 131: 1273–1286. [DOI] [PubMed] [Google Scholar]
- 46.Weinmann L, Hock J, Ivacevic T, Ohrt T, Mutze J, Schwille P, Kremmer E, Benes V, Urlaub H, and Meister G 2009. Importin 8 is a gene silencing factor that targets argonaute proteins to distinct mRNAs. Cell 136: 496–507. [DOI] [PubMed] [Google Scholar]
- 47.Bhattacharyya SN, Habermacher R, Martine U, Closs EI, and Filipowicz W 2006. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 125: 1111–1124. [DOI] [PubMed] [Google Scholar]
- 48.Nam JW, Rissland OS, Koppstein D, Abreu-Goodger C, Jan CH, Agarwal V, Yildirim MA, Rodriguez A, and Bartel DP 2014. Global analyses of the effect of different cellular contexts on microRNA targeting. Molecular cell 53: 1031–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Broderick JA, and Zamore PD 2011. MicroRNA therapeutics. Gene therapy 18: 1104–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.van Rooij E, Purcell AL, and Levin AA 2012. Developing microRNA therapeutics. Circulation research 110: 496–507. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.