

Abstract
Hepatocellular carcinoma (HCC) is the second leading cause of cancer deaths worldwide largely due to lack of effective targeted drugs to simultaneously block multiple cancer-driving pathways. The identification of all-trans retinoic acid (ATRA) as a potent Pin1 inhibitor provides a promising candidate for HCC targeted therapy because Pin1 is overexpressed in most HCC and activates numerous cancer-driving pathways. However, the efficacy of ATRA against solid tumors is limited due to its short half-life of 45 min in humans. A slow-releasing ATRA formulation inhibits solid tumors such as HCC, but can be used only in animals. Here, we developed a one-step, cost-effective route to produce a novel biocompatible, biodegradable, and non-toxic controlled release formulation of ATRA for effective HCC therapy. We used supercritical carbon dioxide process to encapsulate ATRA in largely uniform poly L-lactic acid (PLLA) microparticles, with the efficiency of 91.4% and yield of 68.3%, and ~4-fold higher Cmax and AUC over the slow-releasing ATRA formulation. ATRA-PLLA microparticles had good biocompatibility, and significantly enhanced the inhibitory potency of ATRA on HCC cell growth, improving IC50 by over 3-fold. ATRA-PLLA microparticles exerted its efficacy likely through degrading Pin1 and inhibiting multiple Pin1-regulated cancer pathways and cell cycle progression. Indeed, Pin1 knock-down abolished ATRA inhibitory effects on HCC cells and ATRA-PLLA did not inhibit normal liver cells, as expected because ATRA selectively inhibits active Pin1 in cancer cells. Moreover ATRA-PLLA microparticles significantly enhanced the efficacy of ATRA against HCC tumor growth in mice through reducing Pin1, with a better potency than the slow-releasing ATRA formulation, consistent with its improved pharmacokinetic profiles. This study illustrates an effective platform to produce controlled release formulation of anti-cancer drugs, and ATRA-PLLA microparticles might be a promising targeted drug for HCC therapy as PLLA is biocompatible, biodegradable and nontoxic to humans.
Keywords: Liver cancer, Pin1, ATRA, targeted therapy, controlled release, supercritical carbon dioxide
1. Introduction
Although molecularly targeted drugs have changed cancer treatment, it has become evident that blocking a single pathway may not be as effective in solid tumors as in leukemias, especially aggressive or drug-resistant tumors, due to the feedback and simultaneous activation of a wide range of interactive and/or redundant pathways [1–4], A notable example is hepatocellular carcinoma (HCC), which has extraordinarily high inter- and intra-tumor heterogeneity and complexity of etiology, with multiple cancer-driving pathways being often activated at the same time [5], As a result, HCC is the second leading cause of cancer-related deaths in the world, although it is the sixth most common cancer [6], However, so far there is no effective targeted therapy to block multiple activated signaling pathways at the same time [7], Thus the development of novel molecularly targeted drugs to block multiple cancer-driving pathways at the same time is urgently needed for treating HCC and other aggressive cancers.
A common signalling mechanism in cell proliferation and transformation is Pro-directed Ser/Thr phosphorylation (pSer/Thr-Pro), which is regulated by a huge and diverse family of Pro-directed kinases and phosphatases and their upstream regulators [8, 9], The structure and function of these phosphorylated proteins are further controlled by a unique peptidyl-prolyl cis/trans isomerase (PPIase), Pin1 [10], Pin1 is widely overexpressed and/or overactivated in most of human cancers, with its high levels being correlated with poor clinical prognosis [11, 12], For example, Pin1 is overexpressed in about 70% HCC [13–15] and high Pin1 expression is an independent factor for poor prognosis [16], Pin1 overexpression promotes tumorigenesis by activating over 40 oncogenes or growth-promoting regulators, and inactivating over 20 tumor suppressors or growth-inhibitory regulators [10], In contrast, PIN1 single nucleotide polymorphisms (SNPs) that lower Pin1 expression are associated with reduced cancer risk in humans [17–21], Pin1-null mice, which develop normally [22, 23], are highly resistant to tumorigenesis even after overexpression of oncogenes such as HER2 [24], RAS [24], Myc [25], Notch3 [26], or mutation [27] or ablation [28] of tumor suppressors such as p53. Thus, targeting Pin1 represents a novel anticancer strategy to block multiple cancer pathways simultaneously without general toxic effects on normal tissues [10, 12, 29], However, because the available Pin1 inhibitors lacked the required specificity and/or potency, or cannot enter cells [30–32], it was challenging to evaluate Pin1 targeted therapy until our recent discovery of all-trans retinoic acid (ATRA) as a potent inhibitor of Pin1 via high throughput screening [33], ATRA inhibits and ultimately degrades active Pin1 selectively in cancer cells, thereby blocking multiple Pin1-regulated cancer-driving pathways at the same time, an attractive property for treating aggressive and drug-resistant solid tumors [33].
ATRA, one of the active derivatives of vitamin A, is becoming as a promising compound for cancer therapy and prevention [34–36], Nowadays ATRA has become the standard frontline drug for acute promyelocytic leukemia (APL) therapy with almost complete remission, however, its therapeutic efficacy on solid tumors remains poor [37], Conventional systemic delivery such as oral administration of ATRA to these tumors is inefficient which always lead to side effects like drug resistance, plasma drug concentration reduction, and cancer relapse after a brief remission [37–39], The short half-life of 45 min in humans [40] and poor aqueous solubility of 0.21 μM under physiological conditions [41] are two main obstacles for delivery ATRA to tumors. In addition, ATRA is chemically unstable and susceptible to light, heat and oxidants, which further limit its clinical application.
To overcome these problems, it is needed to develop new formulations to deliver ATRA at a sustained rate to tumors while maintaining its activity and stability. Micro/nano-particles provide powerful tools to deliver anti-cancer molecules into cancer tissues [42–44], Some formulations for ATRA delivery including liposomes, solid lipid nanoparticles, and polymeric material based particles have been developed by a number of techniques such as hot melting homogenization method and emulsification–solvent evaporation [45–50], Although most of them demonstrated improved anti-cancer activities, almost none of them had been performed in clinical application especially in solid tumor therapy. A possible exception is liposomal ATRA, which has been shown to have some promising antitumor activity against renal cancer in phase I/II clinical trials, but further evaluation was stopped due to halt of liposomal ATRA production [51–53], In our previous study, we showed that ATRA slow-releasing pellets exerted potent anticancer activity against both APL and aggressive triple negative breast cancer by inhibiting and ablating Pint and thereby turning off and on numerous oncogenes and tumor suppressors, respectively, at the same time [33], However, this formulation of slow-releasing ATRA pellets can be used only in animals but not humans. In addition, some issues such as low ATRA encapsulation efficiency and stability and fast release rate are still needed to be addressed. What’s more, these preparation processes are lengthy and additional procedures are needed for organic solvent removal and product drying which may result in damage to the physical structure of carriers. Thus, it is highly desirable to develop a convenient and cost-effective route to prepare a biocompatible and biodegradable formulation for efficient sustained release of ATRA that can be used in humans.
Supercritical fluid technology, in particular of supercritical carbon dioxide (sc-CO2) process is growing into an attractive method for production of drug delivery carriers [54–56], Comparing to conventional methods for particle preparation, the sc-CO2 process has many inherent advantages: operation at moderate temperature (above 31,2°C) and in an inert medium that avoid degradation and oxidation of the products, efficient phase separation, direct obtaining solvent-free dry products, non-toxicity and environmental acceptability. However, to date, there is no report on preparation of ATRA controlled release formulation by sc-CO2 process. Poly lactic acid is a biocompatible, biodegradable and non-toxic material, and has been used in drug carrier preparation [48, 57].
The present study encapsulated ATRA into PLLA microparticles by sc-CO2 process for the first time and further evaluated its efficacy in treating HCC cell growth in vitro and tumor growth in vivo. Our results show that our novel ATRA-loaded PLLA microparticles have significantly enhanced the efficacy of ATRA against HCC through reducing Pin1 in vitro and in vivo, with a better potency than the commercial available ATRA slow-releasing pellets, which can be used only in animals. Given that PLLA is biocompatible, biodegradable and nontoxic to humans, these results suggest that the ATRA-PLLA microparticles might be a promising targeted drug for HCC therapy.
2. Materials and methods
2.1. Materials
ATRA (CAS 302-79-4, purity ≥ 98%), acitretin (CAS 55079-83-9, purity ≥ 98%), fluorescein isothiocyanate isomer I (FITC) (CAS 3326-32-7, purity ≥ 90%), and corn oil (CAS 8001-30-7, d: 0.9 g/mL) were purchased from Sigma-Aldrich (MO, USA). Ester-terminated PLLA (Mw 50,000 g/mol) was obtained from Jinan Daigang Biomaterial Co., Ltd. (Jinan, China). CO2 (purity > 99.9%, v/v) was supplied by Xiamen Rihong Co., Ltd. (Xiamen, China). ATRA slow-releasing pellets were purchased from Innovative Research of America (Florida, USA). All other chemical reagents were of analytical purity.
2.2. Cell culture and animal
293T cells, human HCC cell lines HuH7 and PLC/PRF/5 (hereinafter referred to as PLC), and normal human liver cell L-02 were purchased from the cell bank of Chinese Academy of Sciences (Shanghai, China). Cell lines were checked for contamination by Mycoplasma detection, and were authenticated by Short Tandem Repeat profiling. 293T, HuH7 and L-02 cells were cultured with Dulbecco’s Modified Eagle Medium (DMEM) (HyClone), and PLC cells were cultured in Minimum Essential Medium (MEM) (HyClone). These mediums were supplemented with 10% (v/v) fetal bovine serum (FBS) (Gibco) and 1% (v/v) penicilline-streptomycin (HyClone). All cells were routinely maintained at 37°C under 5% CO2 in humidified incubator. The medium was refreshed every 3 days. When confluent, the cells were harvested with trypsin.
4-weeks-old male BALB/c nu/nu mice were purchased from Shanghai SLAC laboratory Animal Co., Ltd. (Shanghai, China), and were raised in specific pathogen-free conditions. Animal care and experimental protocols were performed according to the Experimental Animal Ethics Committee of Fujian Medical University.
2.3. Preparation of ATRA-loaded PLLA particles, blank PLLA particles and FITC-loaded PLLA microparticles by supercritical anti-solvent (SAS) process
2.3.1. Apparatus and procedure
Fig. 1a shows a schematic diagram of the SAS apparatus and the ATRA-PLLA particles preparation process. The apparatus consists of three major sections: a CO2 supply system, an organic solution delivery system, and a high-pressure vessel. The running of the experiment was performed according to the procedure described in reference with some modifications [58], Briefly, in this process, when desired experimental conditions were reached, ATRA and PLLA dissolved in dichloromethane (DCM) was placed into the front chamber of a stainless steel cylinder, and the rear chamber was filled with ethanol which was pressurized by a High Performance Liquid Chromatography (HPLC) pump. The DCM solution was then injected through a stainless steel nozzle into the high pressure vessel where the sc-CO2 was already present. On the basis of our pilot experiments, the pressure, temperature and flow rate of CO2 in this process were kept at 7.6 MPa, 45°C and 40 g/min, respectively. When the spraying of the DCM solution was finished, the SC-CO2 was continually pumped to wash precipitated particles for about 30 minutes to eliminate residual organic solvent. During the washing process, the system operating conditions were maintained as described above. After the washing process, the CO2 flow was stopped and the high-pressure vessel was slowly depressurized to atmospheric pressure. Finally, the ATRA-PLLA particles were collected for use. The blank PLLA particles and FITC-loaded PLLA particles were also prepared using the above process.
Fig. 1.
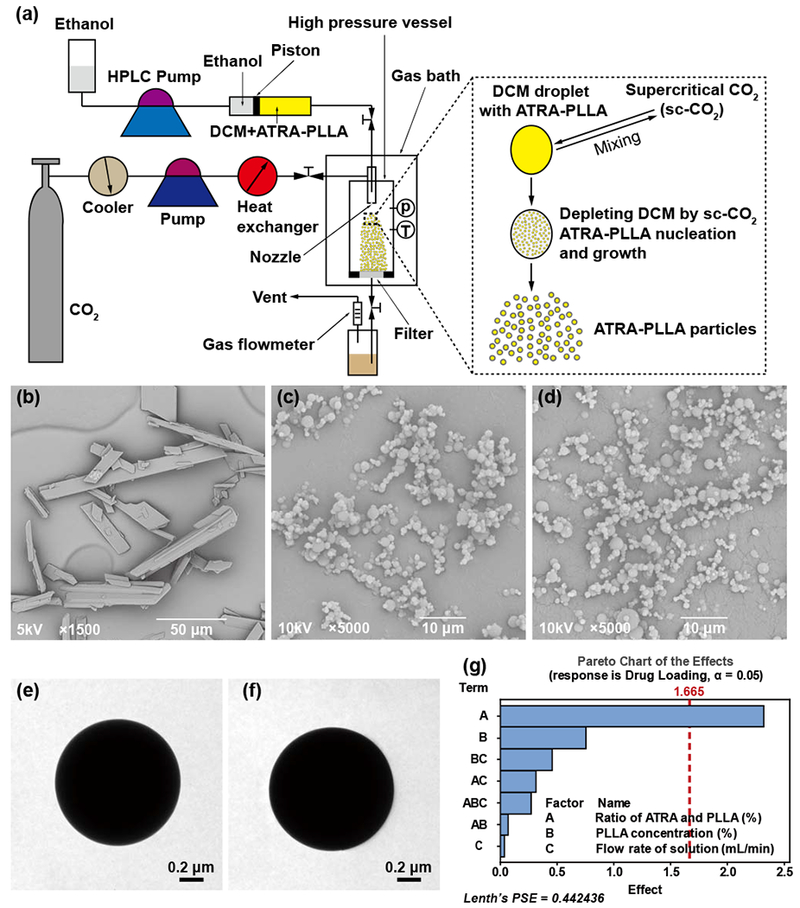
(a) Schematic diagram of the apparatus for the SAS process. SEM images for (b) original ATRA powders, (c) blank PLLA particles and (d) ATRA-PLLA particles. TEM images for (e) blank PLLA particles and (f) ATRA-PLLA particles, (g) Effect of the factors on drug loading. Factors A, B and C represent the ratio of ATRA and PLLA (%), PLLA concentration (%), and flow rate of solution (mL/min) in the SAS process, respectively.
2.3.2. Experimental design and data analysis
The ratio of ATRA and PLLA, PLLA concentration, and flow rate of solution are key parameters in SAS process, to investigate the influence of three parameters on the performance of ATRA-PLLA particles, a 23 factorial experiment was designed according to Table 1 by MINITAB software version 17. Since our pilot experiments suggested the suitable range for the ratio of ATRA and PLLA, PLLA concentration, and flow rate of solution in SAS process were 1% to 3%, 1% to 3%, and 0.5 to 1.5 mL/min, respectively, the low (coded level −1), middle (coded level 0), and high (coded level 1) factor settings for the ratio of ATRA and PLLA, and PLLA concentration were 1%, 2%, and 3%, while for the flow rate of solution were 0.5, 1.0, and 1.5 mL/min. To evaluate the variances in the SAS process, two experiments under middle factor setting were also performed in this study. The effect of the three parameters on the particle size, drug loading and encapsulation efficiency, and yield of the ATRA-PLLA particles prepared under different experimental conditions were statistically analyzed using MINITAB software. What’s more, the optimum experimental condition for producing ATRA-PLLA particles with excellent performances was predicted by further analysis of the above data using Minitab’s Response Optimizer.
Table 1.
Experimental factors and levels.
| Symbols | Factors | Coded level |
||
|---|---|---|---|---|
| −1 | 0 | +1 | ||
| A | Ratio of ATRA and PLLA (%) | 1 | 2 | 3 |
| B | PLLA concentration (%) | 1 | 2 | 3 |
| C | Flow rate of solution (mL/min) | 0.5 | 1 | 1.5 |
2.3.3. Confirmation experiments
Blank PLLA particles and ATRA-PLLA particles were further prepared according to the predicted optimum experimental condition. The particle size, drug loading and encapsulation efficiency, and yield of the prepared ATRA-PLLA particles were investigated and compared to the predicted values.
2.4. Characterization of surface morphology, structure and particle size
Surface morphology of the samples was examined using a scanning electron microscope (SEM) (Phenom Pro, Phenom-World, Netherlands). Before observation, the samples were adhered onto an aluminum sample holder with a thin self-adherent carbon film and then were coated with a thin layer of gold. The morphology and structure of the particles were further characterized by transmission electron microscope (TEM) (H-7650, HITACHI, Japan). A drop of the particle suspension was placed on a 400 mesh copper grid and then dried in air. Observation was done at 60 kV. Particle sizes of the samples were analyzed by SmileView software from the SEM photographs. For each sample, the sizes of 400 particles were measured.
2.5. Determination of drug loading and encapsulation efficiency and yield of productions
To determine the drug loading, 4 mg of ATRA-PLLA particles were dissolved in 4 mL dimethylsulfoxide (DMSO), and the amount of ATRA was analyzed using Multiskan Go microplate reader (Thermo Scientific, USA) at 360 nm. ATRA concentration was obtained by a calibration curve of standard ATRA solution. Blank PLLA particles were used as the background control and no significant absorbance at 360 nm was detected. In the measurement of encapsulation efficiency, another 4 mg of ATRA-PLLA particles was suspended in 4 mL 75% ethanol which was a solvent for ATRA but an anti-solvent for PLLA to wash off the unencapsulated or loosely surface-bound ATRA. The suspension was then filtrated through a 0.22 μM membrane and the amount of ATRA in the ethanol solution was analyzed as described above. The drug loading (DL%), encapsulation efficiency (EE%), and yield (Y%) of ATRA-PLLA particles were calculated by the following equations, respectively:
2.6. Assessment of drug release profiles
Release behaviors of ATRA from the ATRA-PLLA particles were studied in phosphate-buffered saline (PBS; pH 7.4), PBS supplemented 5% FBS, MEM, and MEM supplemented 5% FBS, respectively. In brief, 2 mg of ATRA-PLLA particles were placed in micro-centrifuge tube and dispersed in 1 mL of the medium. The micro-centrifuge tubes were then placed in a shaking incubator at 37°C and 100 rpm. At each experimental time point (2, 6, 12, 24 h; every other day thereafter), media in the tubes were centrifuged at 10,000 rpm for 5 minutes, and the supernatants were collected for analysis of ATRA concentration. After that, the ATRA-PLLA particles were re-dispersed in fresh media and placed back in the shaking incubator. The concentration of ATRA released into the media was determined as described in the above section. Drug release profiles were calculated in terms of cumulative release percentages of ATRA with release time. Each experiment was performed at least in triplicate.
2.7. Cell uptake study
HuH7 cells were seeded on coverslips and cultured for overnight. The cells were then incubated with FITC-loaded PLLA particles (0.13 mg/mL) for 4 h and 24 h. At each time point, the coverslips were removed, washed with phosphate-buffer saline (PBS), and fixed with paraformaldehyde. After washing with PBS three times, the cells were stained with 4’, 6-diamidino-2-phenylindole (DAPI) staining solution (Beyotime, Haimen, China) and examined using a laser scanning microscope (AXIO Observer Z1, Zeiss, Germany).
2.8. Generation of stable PIN1 knockdown cell lines
To knockdown PIN1, a validated PIN1 shRNA (5’-CCACCGTCACACAGTATTTAT-3’) was subcloned into the pLKO.1 lentiviral vector. All lentiviral plasmids were provided by Dr. Kun Ping Lu (Department of Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, USA). For producing lentiviruses, the pLKO.1 vector carrying PIN1 shRNA or scrambled shRNA was transfected into 293T cells with helper plasmid pVSVG pREV, and pMDL. The resulting supernatant containing virus was then collected and used for infection of HuH7 and PLC cells. Stably knockdown cells were selected with 1 μg/mL puromycin and validated by Western blotting analysis.
2.9. Cell viability assay
HuH7, PLC, and L-02 cells were seeded on 96-well plates at a density of 3000 cells/well, 6000 cells/well, and 5000 cells/well, respectively. After an overnight culture, HuH7 and PLC cells were treated with blank PLLA particles, free ATRA, and ATRA-PLLA particles for different times. L-02 cells were treated with free ATRA and ATRA-PLLA particles. Untreated cells were served as control. At each experimental time point, cell viability was measured using methylthiazoltetrazolium (MTT) assay. Briefly, 20 μL of MTT (5 mg/mL) was added to 96-well plates and cultured for additional 3 hours. The formazan crystals were then dissolved with DMSO and the absorbance value (OD) was determined using Multiskan Go microplate reader (Thermo Scientific, USA) at 490 nm. The percentage of cell viability was calculated by the following equation:
The half maximal inhibitory concentration (IC50) of free ATRA and ATRA-PLLA particles were further calculated from their inhibition rates on cell viability by using SPSS 20.0 software.
2.10. Western blotting
Total proteins from cells or xenograft tumors were extracted with RIPA lysis buffer and quantified using BCA protein assay kit (Beyotime, Haimen, China). Proteins were separated with sodium dodecyl sulfate polyacrylamide gel electrophoresis and then transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% non-fat-dry milk and incubated with primary antibodies. After washing, the membranes were incubated with horseradish peroxidase (HRP) conjugated secondary antibodies to probe the primary antibodies. Immunoreactive bands were visualized with a chemiluminescence HRP substrate (Millipore, Billerica, MA, USA). β-Actin was used as a loading control. The primary antibodies used were as follows: anti-Pin1 (1:3000) was provided by Dr. Kun Ping Lu (Department of Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, USA); anti-β-catenin (1:1000), anti-c-JUN (1:1000), anti-NF-κB (1:1000), anti-Cyclin D1 (1:1000), anti-CDK2 (1:1000), and anti-CDK6 (1:2000) were purchased from Cell Signaling technology (Danvers, MA, USA).
2.11. Focus formation assay
Focus formation assay was conducted to evaluate the long-term anti-proliferative effect of ATRA-PLLA particles. 500 or 2000 HuH7 cells were seeded on 12-well or 6-well plates. After an overnight culture, the cells were treated with blank PLLA particles, free ATRA, and ATRA-PLLA particles for 72 h. At 72 h, the drug-containing medium was replaced with fresh medium and the cells were cultured for another 10~14 days to form colonies. After that, cells were fixed with 0.5% solution of methylene blue in methanol and photographed. Viable cell colonies containing more than 50 cells were counted. The total area of foci was quantified by Image-Pro Plus 6.0 software. The percentage of foci area was calculated by the following equation:
2.12. Cell cycle and apoptosis analysis
HuH7 cells were seeded at a density of 0.9 × 104 cells/cm2 on dishes. After an overnight culture, the cells were treated with blank PLLA particles (0.25 mg/mL), free ATRA (10 μM and 20 μM), and ATRA-PLLA particles (containing ATRA 10 μM and 20 μM) for 72 h. Untreated cells were used as control. Then the cells were collected by trypsinization, and the cell cycle and apoptosis were determined by a cell cycle and apoptosis analysis kit (Beyotime, Haimen, China) according to the manufacturer’s protocol. In brief, the collected cells were fixed in 70% ethanol at 4°C for 2 h, washed in PBS and stained with propidium iodide solution for 30 min at 37°C in darkness, then analyzed by flow cytometer (BD FACSVerse, BD Biosciences, USA).
2.13. In vivo antitumor effect
For xenograft experiments, 4× 106 of HuH7 cells were injected subcutaneously in the hind limb of BALB/c nu/nu mice. After tumor grew to about 50 mm3, the mice were randomly divided into saline group, free ATRA group, blank PLLA particles group, ATRA slow-releasing pellet group, and ATRA-PLLA particles group. Free ATRA was dissolved in ethanol and diluted with corn oil. Blank PLLA particles and ATRA-PLLA particles were dispersed in saline. These drugs were then injected intraperitoneally into mice at a dose of 15 mg/kg (concentration of ATRA) twice a week for three weeks (i.e. each mouse was received a total of about 2 mg ATRA). 5 mg over 21 days of ATRA slow-releasing pellet was implanted subcutaneously in the lateral side of the neck between the ear and the shoulder (i.e. each mouse was received a total of 5 mg ATRA). Tumor sizes were recorded weekly by a caliper and tumor volumes were calculated using the formula: L × W2 × 0.52, where L and W represent length and width, respectively. The weight of mice was also recorded weekly. At the end of third week since the drug treatment, mice were sacrificed and tumors were isolated and weighed.
2.14. In vivo pharmacokinetic studies
2.14.1. Pharmacokinetic experiments
The pharmacokinetics of ATRA-PLLA particles were evaluated in mice by quantitative determination of ATRA in plasma. The commercial available ATRA slow-releasing pellets were used as a control. 8-weeks-old male BALB/c nu/nu mice were used for these experiments. The mice were randomly divided into ATRA-PLLA particles group and ATRA slow-releasing pellet group. Each group had 4 mice at each experimental time. For ATRA-PLLA particles group, each mouse was received an injection of ATRA-PLLA particles at a dose of 15 mg/kg (i.e. each mouse was received about 0.34 mg ATRA). ATRA-PLLA microparticles were dispersed in saline and injected intraperitoneally into mouse. For the ATRA slow-releasing pellet group, each mouse was received an implantation of a 5 mg over 21 days ATRA slow-releasing pellet (i.e. each mouse was received 5 mg ATRA). The pellet was implanted subcutaneously in the lateral side of the neck between the ear and the shoulder. Blood samples (about 200 μL) were collected from retro-orbital venous plexus puncture at 0.5, 1, 3, 4, 6, 24 and 48 h after drug administration with heparinized tubes. The collected blood was further centrifuged at 3000 rpm for 10 min at 4°C, and the separated plasma was collected and stored at −80°C until analysis.
2.14.2. LC-MS/MS analysis
The concentrations of ATRA in plasma samples were determined by liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis, and acitretin was used as the internal standards (IS). The LC-MS/MS system consisted of an Agilent 1260 series liquid chromatography system and an Agilent 6410 triple-quadruple mass spectrometer equipped with an Electrospray Ionization (ESI) source (Agilent Technologies, California, USA). Agilent MassHunter workstation (version B. 03. 01) was used for the control of LC-MS/MS system and data acquisition. The chromatographic analysis of ATRA and acitretin was performed on Welch ultimate XB-c18 column (2.1 × 50 mm, 3.5 μm) at 30°C. An isocratic elution was adopted with phase A (water containing 0.00625% ammonia solution) and phase B (methanol) in the ratio of 33:67. The total run time was 5 min and the injection volume was 3 μL. After chromatographic separation, the eluent was introduced to the mass spectrometer and determined in positive multiple reactions monitoring (MRM) mode with a dwell time of 500 ms. The precursor ion and product ion for ATRA and IS were m/z 299.2–255.2, and m/z 325.2–266.2, respectively. The optimized MS/MS conditions were as follows: fragmentor, 115 V; collision energy, 8 eV; capillary voltage, 3500 V; nebulizer gas pressure, 30 PSI; gas flow, 10 L/min; gas temperature, 325°C.
2.14.3. Pharmacokinetic analysis
Pharmacokinetic parameters, including the maximal plasma concentration (Cmax), the area under the plasma concentration–time curve (AUC), and the half life time (t1/2) were calculated using the non-compartmental method with DAS 3.2.3 pharmacokinetic software (Anhui Provincial Center for Drug Clinical Evaluation, Anhui, China)
2.15. Immunofluorescence staining
Frozen tumor tissue samples embedded in optimal cutting temperature compound were cut into sections with a thickness of 10 μm, fixed in acetone for 10 min at room temperature, washed with PBS containing 0.1% Triton X-100 and then permeabilized and blocked with 1% Triton X-100 and 10% donkey serum in PBS for 40 min at room temperature. Subsequently, the sections were incubated with primary antibodies for 24 h at 4°C in a humidified chamber. After three washes with PBS containing 0.1% Triton X-100, the sections were incubated with secondary antibodies for 1 h at room temperature. Nuclei were stained with DAPI staining solution. Then the sections were washed three times with PBS and mounted with mounting medium. Images were obtained using a laser scanning microscope (AXIO Observer Z1, Zeiss, Germany). The primary antibodies used were as follows: anti-Pin1 (1:250) was provided by Dr. Kun Ping Lu (Department of Medicine, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, USA); and anti-Ki67 (1:400) was purchased from Abcam (Cambridge, UK). The secondary antibodies were Alexa Fluor 488-conjugated anti-mouse IgG and Alexa Fluor 555-conjugated anti-rabbit IgG (all from Abcam).
2.16. Statistical analysis
Statistical analysis was performed using SPSS 20.0 software. Statistical significance was measured using t tests or one-way analysis of variance (ANOVA) followed by Student–Newman–Keuls or Dunnett C post-hoc tests for all paired combinations. The mean difference was considered to be significant while p value < 0.05, and very significant while p value < 0.01.
3. Results
3.1. Surface morphology, structure and particle size of ATRA-PLLA microparticles
The SEM imaging showed that the original ATRA powders were cuboid, and their length varied obviously from 10 μm to over 100 μm (Fig. 1b). The blank PLLA microparticles and ATRA-PLLA microparticles prepared by the SAS processing exhibited good spherical shape and smooth surface (Fig. 1c and d), meanwhile, distinctly differences in size for ATRA-PLLA microparticles prepared under different conditions were observed (Supplementary Fig. S1). Fig. 1e and f present representative TEM images of blank PLLA microparticles and ATRA-PLLA microparticles. Both the blank PLLA and ATRA-PLLA microparticles had very good spherical shape and compact structure, indicating they are solid sphere. Table 2 shows the particle sizes of ATRA-PLLA microparticles prepared under different conditions. The mean particle size of these ATRA-PLLA microparticles ranged from 1.12 ± 0.49 μm to 2.70 ± 1.45 μm. Statistical analysis shows that factor B (PLLA concentration) had a significant effect on the mean particle size (p < 0.05), while other factors had no significant effects (Supplementary Fig. S3a). Within the range of the parameters studied, the particle size increased with an increasing concentration of PLLA, flow rate of solution, or ratio of ATRA and PLLA (Supplementary Fig. S3b).
Table 2.
Experimental results from the factorial design (factor A: ratio of ATRA and PLLA (%), factor B: PLLA concentration (%), and factor C: flow rate of solution (mL/min); −1, 0 and 1 represent low, middle, and high factor settings in the experimental design, respectively).
| Run order | A | B | c | Particle size (μm) | DL (%) | EE (%) | Total Y (%) |
|---|---|---|---|---|---|---|---|
| 1 | −1 | 1 | −1 | 1.83±0.94 | 0.8±0.1 | 82.4±0.7 | 36.5 |
| 2 | −1 | 1 | 1 | 2.08±1.20 | 1.3±0.0 | 88.7±0.5 | 43.8 |
| 3 | 1 | 1 | −1 | 1.96±1.00 | 3.0±0.0 | 62.3±1.3 | 44.0 |
| 4 | −1 | −1 | −1 | 1.12±0.49 | 0.1±0.0 | 96.1±1.0 | 38.5 |
| 5 | 1 | −1 | 1 | 1.43±0.71 | 2.1±0.0 | 93.2±0.4 | 45.4 |
| 6 | 1 | −1 | −1 | 1.26±0.53 | 3.1±0.0 | 84.2±0.7 | 29.5 |
| 7 | −1 | −1 | 1 | 1.38±0.60 | 0.3±0.0 | 73.1±2.6 | 44.7 |
| 8 | 1 | 1 | 1 | 2.70±1.45 | 3.5±0.2 | 87.2±0.7 | 46.5 |
| 9 | 0 | 0 | 0 | 1.73±0.97 | 2.2±0.0 | 81.2±1.1 | 47.6 |
| 10 | 0 | 0 | 0 | 1.75±0.78 | 2.3±0.0 | 78.5±1.5 | 42.5 |
3.2. Drug loading, encapsulation efficiency and yield of ATRA-PLLA microparticles
Table 2 shows the drug loading, encapsulation efficiency and yield of ATRA-PLLA microparticles prepared under different conditions. The drug loading values varied significantly from 0.1% to 3.5%. The effects of the factors on drug loading and the main effects plot for drug loading were obtained. As shown in Fig. 1g, the ratio of ATRA and PLLA (Factor A) significantly affected the drug loading (p < 0.05), while the rest factors had no significant effect. The drug loading increased with increased ratio of ATRA and PLLA, or PLLA concentration, although the flow rate of solution almost had no effect on drug loading (Supplementary Fig. S2). The encapsulation efficiency ranged from 62.3% to 96.1% (Table 2). We also examined factors that had no significant effects on encapsulation efficiency (Supplementary Fig. S4a). The encapsulation efficiency increased with increased flow rate of solution, and decreased ratio of ATRA and PLLA or PLLA concentration (Supplementary Fig. S4b). Production yields obtained ranged from 29.5% to 47.6% (Table 2). Although the effects of factors on the yield values were not statistically significant, factor C (flow rate of solution) exhibited the maximal effect on the yield (Supplementary Fig. S5a). The yield increased with increased flow rate of solution or PLLA concentration (Supplementary Fig. S5b). The increase of ratio of ATRA and PLLA only slightly increased the yield.
3.3. Optimum experimental condition and results of confirmation experiments
Fig. 2a shows the optimization plot for the effects of various factors on the predicted responses. The goal of the optimization was to minimize particle size, and maximize drug loading, encapsulation efficiency and production yield. According to the goal, the optimum experimental condition obtained for preparing ATRA-PLLA microparticles with excellent performances was as follows: ratio of ATRA and PLLA = 3%; PLLA concentration = 1%; flow rate of solution =1.0 mL/min. The predicted values of particle size, drug loading, encapsulation efficiency and yield for ATRA-PLLA microparticles prepared under this condition were 1.40 μm, 2.2%, 91.7%, and 42.8%, respectively. Results of confirmation experiments show that both the prepared blank PLLA and ATRA-PLLA microparticles had quite good spherical shape and smooth surface (Fig. 1c and d). The particle size, drug loading and encapsulation efficiency of ATRA-PLLA microparticles were 1.24 ± 0.57 μm, 2.4 ± 0.2%, and 91.4 ± 4.0%, respectively, which were comparable to the predictive values (Fig. 2b, c and d). The yield of ATRA-PLLA microparticles was 68.3 ± 3.3%, which was higher than the predictive value (Fig. 2e). Overall, these experimental results confirm that the experimental conditions can produce desired ATRA-PLLA microparticles with excellent performance.
Fig. 2.
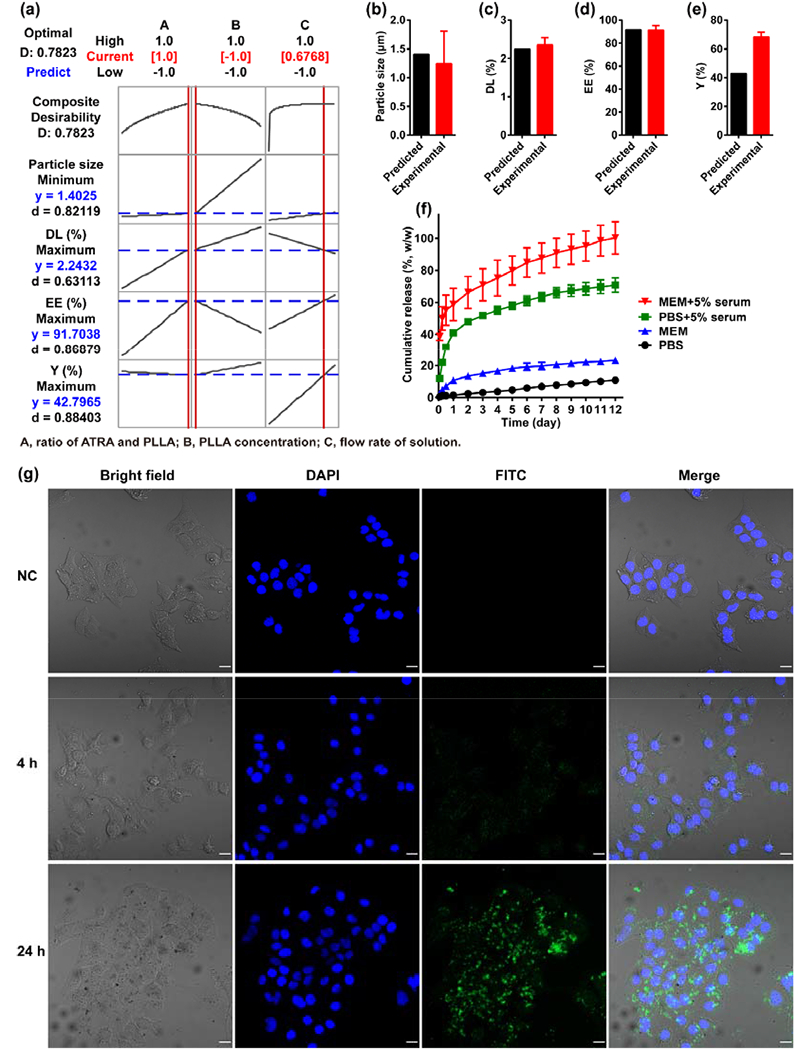
(a) The optimization plot for the effects of factors on the predicted responses. Factors A, B and C represent the ratio of ATRA and PLLA (%), PLLA concentration (%), and flow rate of solution (mL/min) in the SAS process, respectively. The numbers displayed at the top of a column show the current factor settings (in red) and the high and low factor settings in the experimental design; The Predict link in the top left of the graph calculates the prediction (in blue) for the current factor settings; The vertical red lines on the graph represent the current settings; The horizontal blue lines represent the current response values, (b) Particle size, (c) Drug loading, (d) Encapsulation efficiency, and (e) Yield of the ATRA-PLLA particles prepared according to the predicted optimum experimental condition were investigated and compared to the predicted values, (f) In vitro release profile of ATRA from ATRA-PLLA particles in different mediums, (g) Cellular uptake of FITC-loaded PLLA particles. HuH7 cells were incubated with 0.13 mg/mL of the FITC-loaded PLLA particles for the indicated time, then stained with 4’, 6-diamidino-2-phenylindole (DAPI) staining solution and examined using a laser scanning microscope. Untreated cells were used as negative control. Bright field image shows the morphology of HuH7 cells, blue fluorescence (DAPI staining) indicates cell nucleus, and green fluorescence indicates the localization of FITC or FITC-loaded PLLA particles. Scale bars, 20 μm. Values are the mean ± standard deviation of triplicate determinations.
3.4. In vitro drug release profiles
Fig. 2f shows the ATRA release profiles from ATRA-PLLA microparticles in PBS, serum-containing PBS, MEM, and serum-containing MEM up to 12 days. In general, the release of ATRA from ATRA-PLLA microparticles in the four media gradually increased with time. Meanwhile, it was observed that the release rates of ATRA were distinctly different in the four media. ATRA-PLLA microparticles only released 1.5% of the ATRA payload in the first day and 10.9% in 12 days in PBS. In comparison, in serum-containing PBS, 41.0% of ATRA payload was released in the first day and 70.9% in 12 days from ATRA-PLLA microparticles. Similar ATRA release behaviors were detected in MEM and serum-containing MEM. ATRA-PLLA microparticles released 10.8% of the ATRA payload in the first day and 23.4% in 12 days in MEM. However, in serum-containing MEM, 58.7% of ATRA payload was released in the first day from ATRA-PLLA microparticles. After that, it continued to gradually release the encapsulated ATRA up to 71.0% in the third day and 100% at the end of the twelfth day. These results indicate that the ATRA-PLLA microparticles can effectively slow release ATRA. In addition, serum and release medium itself have great effect on ATRA release from ATRA-PLLA microparticles, which might be due to the presence of retinol binding proteins in the serum.
3.5. Cell uptake
FITC-loaded PLLA microparticles were used to evaluate their uptake in cells. Fig. 2g shows the distribution of FITC-loaded PLLA microparticles in HuH7 cells at different times. After 4 h incubation with FITC-loaded PLLA microparticles, the bright filed images showed that the cells almost as clean as the negative control cells. However, the fluorescent images showed that some green dots clustered around the cell nucleus. This suggests that some FITC had been released from the microparticles, and entered into the cytoplasm. As the incubation time increased to 24 h, dark spots were observed in the cells in contrast to the negative control cells (Fig. 2g). The fluorescent images confirmed that many green spots clustered around cell nucleus. This indicates the ATRA-PLLA microparticles can be uptake by the cells, as shown previously [59].
3.6. ATRA-PLLA microparticles enhance the ability of ATRA to degrade Pin1 and inhibit its multiple oncogenic substrates in HCC cells
We have shown that ATRA induces degradation of Pin1 and many of its oncogenic substrates in breast cancer [33], To examine whether ATRA-PLLA microparticles would have similar effects, we examined expression of Pin1 and a set of Pin1 substrate oncogenic proteins in HuH7 and PLC cells treated with different concentrations of free ATRA or ATRA-PLLA microparticles for 72 h. Whereas the blank PLLA microparticles almost had no effect on Pin1 expression within the tested concentrations even as high as 0.5 mg/mL (Fig. 3a, b and c), ATRA-PLLA microparticles exhibited a dose-dependent down-regulation of Pin1 and its substrates including β-catenin, c-JUN, NF-κB, Cyclin D1, CDK2, and CDK6 in both HuH7 and PLC cells (Fig.3d-r). In comparison with free ATRA, the ATRA-PLLA microparticles induced much more significant down-regulation of Pin1 and its substrate proteins. These results indicate that although the PLLA particle itself has no effect on Pin1 expression, the ATRA-PLLA microparticles can increase the inhibitory effects of ATRA on Pin1 and its oncogenic substrates.
Fig. 3.
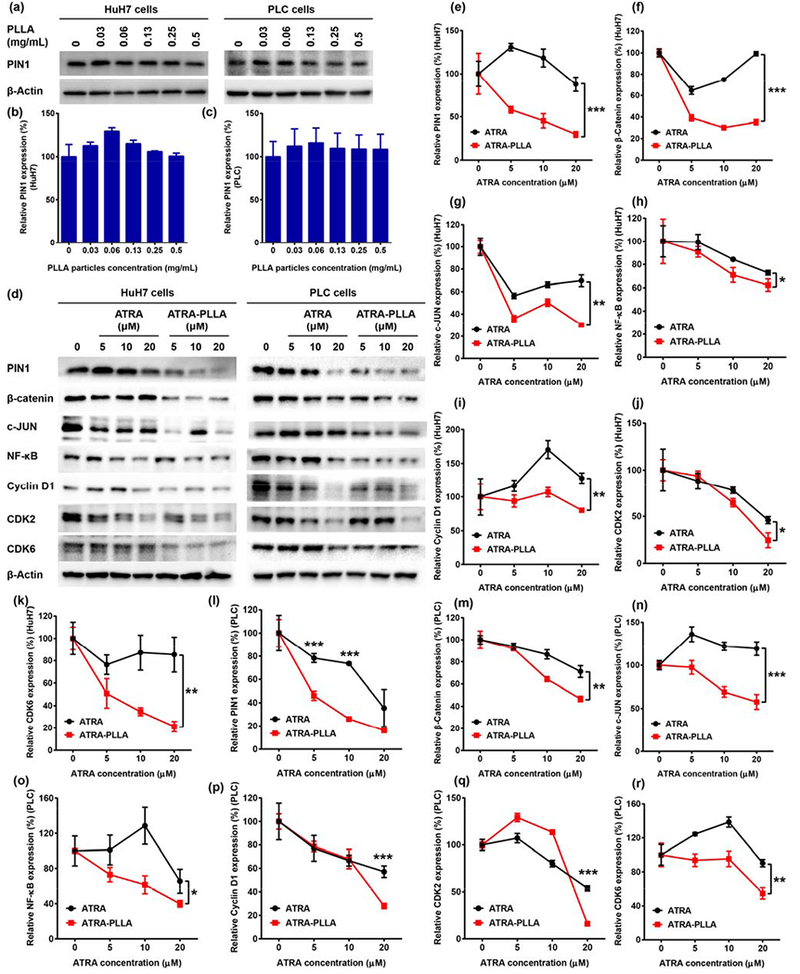
(a-c) Relative expression value of PIN1 in HuH7 and PLC cells after treating with different concentration of blank PLLA particles for 72 h were determined by western blot analysis, (d-r) Immunoblotting of proteins expressed in HuH7 and PLC cells treated with different concentration of free ATRA or ATRA-PLLA particles for 72 h. The related concentration of PLLA in ATRA-PLLA microparticle for 5, 10, 20 μM of ATRA was 0.06, 0.12, and 0.24 mg/mL, respectively. β-Actin was used as an internal control. Untreated cells were set as control. Each value is the mean ± standard deviation of triplicate determinations; *p < 0.05, **p < 0.01, ***p < 0.001.
3.7. ATRA inhibits the growth of HCC cells, but not normal human liver cells
Fig. 4 shows effects of blank PLLA particles, free ATRA, and ATRA-PLLA microparticles on viability of human HCC cells (HuH7 and PLC) and normal human liver L-02 cells. The blank PLLA particles almost had no inhibition on HuH7 cell viability within the tested concentrations even as high as 0.5 mg/mL (Fig. 4a). Dose-dependent inhibition on HuH7 cell viability was observed when the cells were treated with free ATRA or ATRA-PLLA microparticles for 24 h, 48 h, and 72 h, respectively (Fig. 4b, c and d). After 24 h and 48 h treatment, the ATRA-PLLA microparticles exhibited greater inhibition on HuH7 cell viability as compared to the free ATRA, but the difference had not reached a statistically significant level (Fig. 4b and c). However, after 72 h treatment, the inhibition exerted by ATRA-PLLA microparticles was significantly (p < 0.05) higher than free ATRA (Fig. 4d). The corresponding IC50 value of the ATRA-PLLA microparticles for 72 h treatment was highly significantly (p < 0.01) decreased by approximately 2.3 fold as compared to free ATRA (Fig. 4i). Similar inhibition behaviors of the blank PLLA particles, free ATRA, and ATRA-PLLA microparticles were observed in PLC cells. As shown in Fig. 4e, the blank PLLA particles did not exert obvious inhibition on PLC cell viability. When the PLC cells were treated with free ATRA or ATRA-PLLA microparticles, dose-dependent inhibition on PLC cell viability was detected at 24 h, 48 h, and 72 h (Fig. 4f, g and h). In comparison, the inhibition on PLC cell viability by the ATRA-PLLA microparticles was significantly (p < 0.05) higher than that by free ATRA for the 48 h and 72 h treatments (Fig. 4g and h). The IC50 value of the ATRA-PLLA microparticles for treating PLC cells 72 h was highly significantly (p < 0.01) decreased by approximately 3.3 fold when compared to free ATRA (Fig. 4i). These results indicate that the PLLA particle itself has good biocompatibility, and the ATRA-PLLA microparticles enhance the ability of ATRA to inhibit human HCC cells.
Fig. 4.
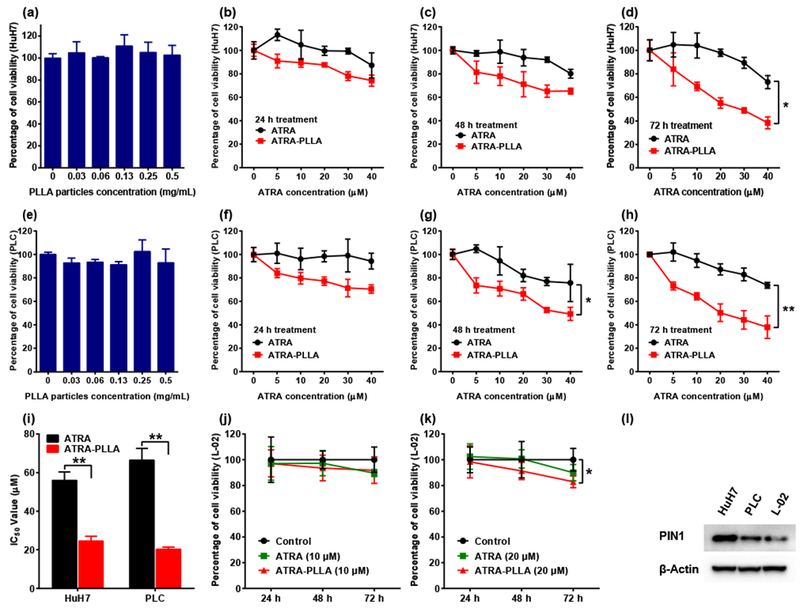
Effects of blank PLLA particles, free ATRA, and ATRA-PLLA microparticles on viability of HuH7, PLC and L-02 cells after treating for the indicated times were assayed by MTT method, (a) Viability of HuH7 cells after treating with blank PLLA particles (0–0.5 mg/mL) for 72 h. Viability of HuH7 cells after treating with free ATRA or ATRA-PLLA particles (ATRA concentration ranging from 0 to 40 μM) for (b) 24 h, (c) 48 h, and (d) 72 h. (e) Viability of PLC cells after treating with blank PLLA particles (0–0.5 mg/mL) for 72 h. Viability of PLC cells after treating with free ATRA or ATRA-PLLA particles (ATRA concentration ranging from 0 to 40 μM) for (f) 24 h, (g) 48 h, and (h) 72 h. (I) Corresponding IC50 values of free ATRA and ATRA-PLLA particles for treating HuH7 or PLC cells for 72 h. Viability of L-02 cells after treating with free ATRA or ATRA-PLLA particles at a dose of (j) 10 μM and (k) 20 μM for the indicated times. (1) Expression of PIN1 in normal cultured HuH7, PLC and L-02 cells were determined by western blot analysis. β-Actin was used as an internal control. Each value is the mean ± standard deviation of triplicate determinations; *p < 0.05, **p < 0.01.
In contrast to HCC cells, the free ATRA had no significant inhibition on viability of normal liver L-02 cells even at 20 μM (Fig. 4j and k). The 10 μM ATRA-PLLA microparticles had not generated significant inhibition on viability of L-02 after 72 h treatment (Fig. 4j). When the dose of ATRA-PLLA microparticles increased to 20 μM, the inhibition on viability of L-02 was still not significant at 24 h and 48 h, and the maximum inhibition exerted by ATRA-PLLA microparticles at 72 h was only 17% (Fig. 4k). Moreover, there was no significant difference in the inhibition between free ATRA and ATRA-PLLA microparticles even at 20 μM (Fig. 4j and k). These results indicate that ATRA almost has no inhibition on normal human liver cell, and ATRA-PLLA microparticles do not enhance the effect of ATRA on viability of normal human liver cell. Taken together, the above results are consistent with the previous findings that ATRA inhibits only the activated Pin1 selectively in cancer cells with little effects on normal cells [33].
3.8. ATRA-PLLA microparticles enhance the ability of ATRA to inhibit HCC cell focus formation
The blank PLLA particles did not affect the number and area of foci of HuH7 cells within the tested concentrations (Supplementary Fig. S6). However, when treated with different concentrations of free ATRA or ATRA-PLLA microparticles for 72 h, significant dose-dependent reductions in the number and area of foci were detected in treated HuH7 cells when compared to the control cells (Fig. 5b and c) (p < 0.01). In comparison, the number and area of foci of HuH7 cells treated by the ATRA-PLLA microparticles were significantly (p < 0.05) lower than that by the free ATRA. These results suggest that the PLLA particles themselves do not affect focus formation, and the ATRA-PLLA microparticles can enhance the inhibition of ATRA on focus formation of HCC cells.
Fig. 5.
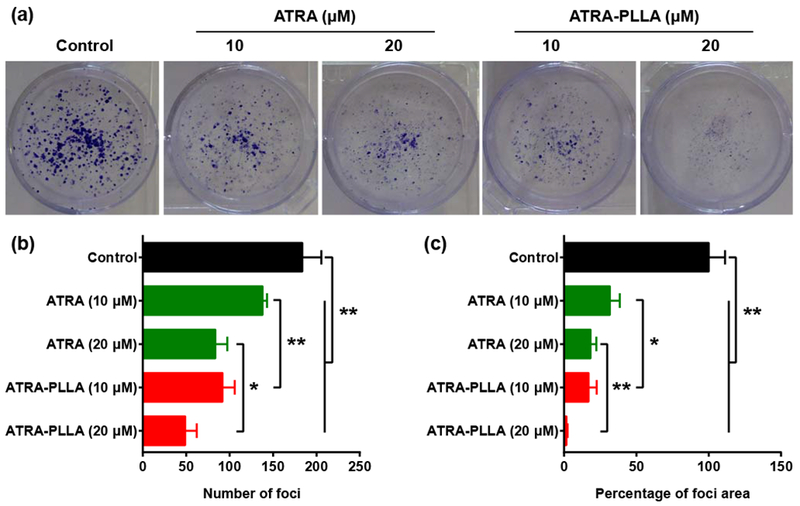
(a) Image, (b) number, and (c) area of foci formed by HuH7 cells treated with the indicated concentration of free ATRA or ATRA-PLLA particles for 72 h. Untreated cells were used as control. Each value is the mean ± standard deviation of triplicate determinations; *p < 0.05, **p < 0.01.
3.9. ATRA-PLLA microparticles enhance the ability of ATRA to inhibit cell cycle progression
The HuH7 cells treated with the blank PLLA particles within the tested concentrations did not show significant changes in cell cycle and apoptosis as compared to the control cells (Supplementary Fig. S7). However, the treatment with free ATRA or ATRA-PLLA microparticles significantly increased the proportion of cells in G0/G1 phase, and decreased the proportion of cells in G2/M phase (Supplementary Fig. S7b). In addition, the 20 μM ATRA-PLLA microparticles significantly decreased the proportion of cells in S phase (Supplementary Fig. S7b). In comparison, the increased proportion of cells in G0/G1 phase by the ATRA-PLLA microparticles was significantly higher than that by the free ATRA (Supplementary Fig. S7b). Moreover, both the free ATRA and ATRA-PLLA microparticles did not induce significant apoptosis (Supplementary Fig. S7c). These results indicate that the PLLA particles themselves do not affect cell cycle progression, but the ATRA-PLLA microparticles can increase the potency of ATRA to inhibit cell cycle progression.
3.10. Pin1 knockdown abolished the inhibitory effects of ATRA-PLLA microparticles on HCC cells
The above results show that ATRA-PLLA induced Pin1 degradation and inhibited the growth of HCC cells. If ATRA-PLLA would inhibit HCC cell growth through degrading Pin1 protein, Pin1 knockdown would remove the ATRA drug target Pin1 so that ATRA-PLLA should not inhibit the growth of Pin1 knockdown HCC cells. To examine this possibility, we stably knocked down Pin1 in HuH7 and PLC cells using lentiviruses expressing a validated Pin1 shRNA [60], followed by examining their sensitivity to ATRA inhibition. PIN1 knockdown significantly (p < 0.01) decreased cell growth when compared to the HuH7 and PLC cells expressing empty vector (shV) (Fig. 6a and d). Fig. 6b and c shows the viability of shV and shPin1–HuH7 cells treated with 20 μM free ATRA or ATRA-PLLA microparticles for 24 h, 48 h and 72 h. For shV–HuH7 cells, the treatment with free ATRA or ATRA-PLLA microparticles resulted in very significant (p < 0.001) time-dependent inhibition on cell viability. In comparison with the free ATRA, the ATRA-PLLA microparticles exerted much higher inhibition. For instance, free ATRA decreased cell viability by approximately 36% at 72 h, while ATRA-PLLA microparticles reduced cell viability by 61%. However, for shPin1–HuH7 cells, the treatment with ATRA and ATRA-PLLA microparticles did not decrease cell viability at 24 h and 48 h, with cell viability being reduced only about 15% at 72 h.
Fig. 6.
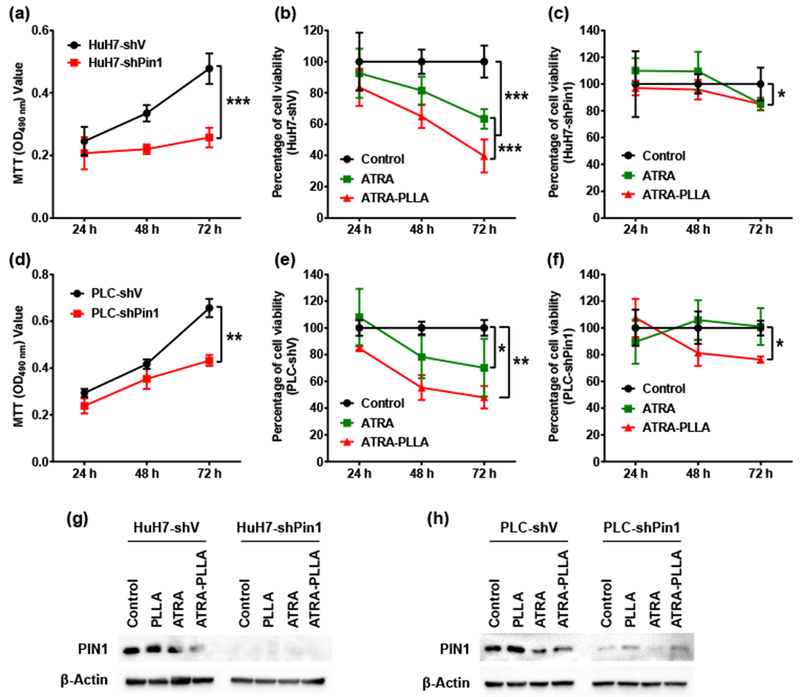
Effects of PIN1 knock-down on inhibition of ATRA against HuH7 and PLC cell growth. Growth curve of (a) HuH7 and (d) PLC cells expressing empty vector or PIN1 shRNA was established by MTT assay. HuH7 cells expressing empty vector (b) or PIN1 shRNA (c), and PLC cells expressing empty vector (e) or PIN1 shRNA (f) were treated with 20 μM free ATRA or ATRA-PLLA particles for 24 h, 48 h, and 72 h, and then the cell viability was assayed using MTT method, (g) HuH7 cells expressing empty vector or PIN1 shRNA, and (h) PLC cells expressing empty vector or PIN1 shRNA were treated with blank PLLA particles (0.25 mg/mL), free ATRA and ATRA-PLLA particles (ATRA concentration 20 μM) for 72 h, and then the PIN1 expression was determined by Western blotting analysis. β-Actin was used as an internal control. Untreated cells were set as control. Values are the mean ± standard deviation of triplicate determinations; *p < 0.05, **p < 0.01, ***p < 0.001.
Similar effects of ATRA and ATRA-PLLA microparticles on cell viability were observed in shV and shPin1–PLC cells. As shown in Fig. 6e, the treatment with ATRA or ATRA-PLLA microparticles resulted in significant time-dependent inhibition on cell viability, and the inhibition generated by ATRA-PLLA microparticles was higher than that by ATRA in shV–PLC cells. However, for shPin1–PLC cells, ATRA treatment did not decrease cell viability, and ATRA-PLLA microparticles treatment only decreased cell viability by about 20% at 72 h (Fig. 6f). The treatment with ATRA or ATRA-PLLA microparticles significantly down-regulated Pin1 expression in shV–HuH7 and shV–PLC cells, but did not affect Pin1 expression in shPin1–HuH7 and shPin1–PLC cells (Fig. 6g and h). These results further support that Pin1 plays a pivotal role in mediating inhibition of ATRA against HCC cell growth. These results are consistent with the previous findings that Pin1−/− cells are much more resistant to ATRA and such ATRA resistance in Pint1−/− cells is restored by re-expressing Pin1, but not Pin1 mutant that cannot bind ATRA [33].
3.11. ATRA-PLLA microparticles enhance the pharmacokinetic profiles and ability of ATRA to inhibit HCC tumor growth in mice
Fig. 7 shows the size, growth volume and weight of tumors, mice weight, and protein expression in xenograft tumors from nude mice treated with saline, blank PLLA particles, free ATRA, ATRA slow-releasing pellet (5 mg over 21 days), and ATRA-PLLA microparticles for 3 weeks. In comparison with the saline group, no notable differences in the size, growth volume and weight of tumors were detected in the blank PLLA particle or free ATRA group (Fig. 7a, b and c), suggesting that the anti-tumor activity of free ATRA was not obvious and the blank PLLA particles had no anti-tumor effect. In contrast, smaller tumor size, growth volume and weight were detected in the ATRA pellet group (5 mg over 21 days), and especially the ATRA-PLLA group (Fig. 7a, b and c). Remarkably, the tumor growth volume and weight of the ATRA-PLLA group were significantly (p < 0.05) smaller than that of both the saline and free ATRA groups (Fig. 7b and c). What’s more, tumors treated with ATRA-PLLA microparticles at about 2 mg ATRA were significantly (p < 0.05) smaller than that treated with commercial ATRA pellets at 5 mg ATRA (Fig. 7b and c). This indicates the ATRA-PLLA microparticles had much stronger anti-tumor effect than not only the free ATRA but also the commercially available slow-release ATRA pellets. Notably, the treatments with blank PLLA particles, free ATRA, ATRA slow-releasing pellets or ATRA-PLLA microparticles did not induce significant changes in weights of mice as compared to the saline group (Fig. 7d), indicating good biocompatibility.
Fig. 7.
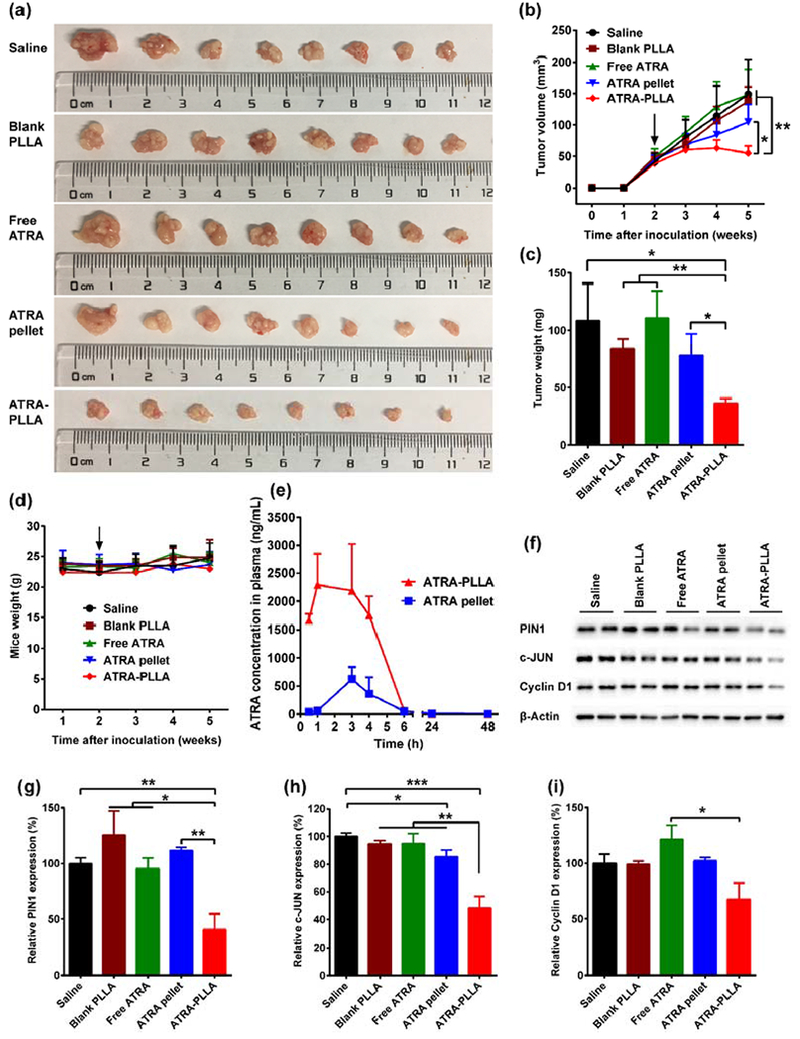
Anti-tumor effect of ATRA-encapsulated PLLA particles in vivo, (a) Tumor size, (b) Quantitative curves of tumor volume, (c) Tumor weight, and (d) Curves of mice weight for nude mice inoculated with 4 × 106 HuH7 cells and treated with saline, free ATRA, blank PLLA particles, ATRA slow-releasing pellet, and ATRA-PLLA particles 2 weeks later (arrow). For the treatment, free ATRA, blank PLLA particles and ATRA-PLLA particles were injected intraperitoneally into mice at a dose of 15 mg/kg (concentration of ATRA) twice a week for three weeks (i.e. each mouse was received a total of about 2 mg ATRA). 5 mg over 21 days of ATRA slow-releasing pellet was implanted subcutaneously in the lateral side of the neck between the ear and the shoulder (i.e. each mouse was received a total of 5 mg ATRA). Mice injected saline were used as control, n = 8. (e) Plasma concentrations of ATRA in nude mice after implanting an ATRA slow-releasing pellet (5 mg over 21 days) or injecting ATRA-PLLA particles (containing ATRA about 0.34 mg) once. n=4. (f–i) Relative expression value of PIN1, c-JUN, and Cyclin D1 in the xenograft tumors. *p < 0.05, **p < 0.01, ***p < 0.001.
To determine the basis of enhanced anti-tumor activity, we performed pharmacokinetic analysis of the ATRA-PLLA microparticles and commercial ATRA pellet. Under optimized LC-MS/MS conditions, ATRA and the internal control acitretin in plasma samples were well separated and eluted at about 2 and 3.5 min, respectively (Supplementary Fig. S8). This indicates the LC-MS/MS method used in the present study has good specificity and sensitivity. Fig. 7e shows the plasma concentration-time curves of ATRA after implantation of ATRA pellet or injection of ATRA-PLLA microparticles to BALB/c nu/nu mice. After implantation of an ATRA pellet (containing 5mg ATRA), ATRA concentrations in plasma gradually increased to the maximum concentration of 620 ng/mL at 3 h, and then decreased to 49 ng/mL at 6 h, and maintained at about 7 ng/mL at 48 h. This ATRA release behaviors are consistent with previous study [61], indicating good accuracy of our pharmacokinetic assay. For mouse injected of ATRA-PLLA microparticles (containing 0.34 mg ATRA), ATRA concentrations in plasma kept at a high level of about 2000 ng/mL during the first 4 h, and then decreased to about 60 ng/mL at 6 h, and still maintained at 2 ng/mL at 48 h. The major pharmacokinetic parameters of ATRA were further calculated from these data (Supplementary Table S1). The Cmax value for ATRA-PLLA microparticles was significantly higher than that for ATRA pellet (2704.21 ± 495.42 vs. 622.17 ± 216.56). Similarly, the AUC value for ATRA-PLLA microparticles was also significantly higher than that for ATRA pellet (10498.52 ± 3061.31 vs. 2633.29 ± 664.01). There was no statistical difference between ATRA-PLLA microparticles and ATRA pellet for t1/2 of ATRA from plasma.
Finally, we performed immunoblotting and immunostaining analyses to determine whether ATRA-PLLA microparticles inhibit Pin1 and its substrate oncogenes as well as cell proliferation of HCC tumor cells in mice. The treatments with blank PLLA particles or free ATRA did not result in significant down-regulation of Pin1 and its oncogenic substrates including c-JUN and Cyclin D1 in tumors as compared to the saline group, whereas the treatment of ATRA-PLLA microparticles significantly (p < 0.01) reduced the expression of Pin1 and its oncogenic substrates (Fig. 7f–i). In addition, the down-regulation of Pin1 and its oncogenic substrates by the ATRA-PLLA microparticles was significantly (p < 0.01) higher than that by ATRA slow-releasing pellets. Moreover, immunofluorescence staining results clearly showed that the treatment with blank PLLA particles or free ATRA did not induce obvious changes in expression of Pin1 and Ki67 in tumors when compared to the saline group (Fig. 8a). However, ATRA-PLLA microparticle treatment greatly decreased Pin1 and Ki67 expression (Fig. 8a), further supporting downregulation of Pin1 and inhibition of tumor growth.
Fig. 8.
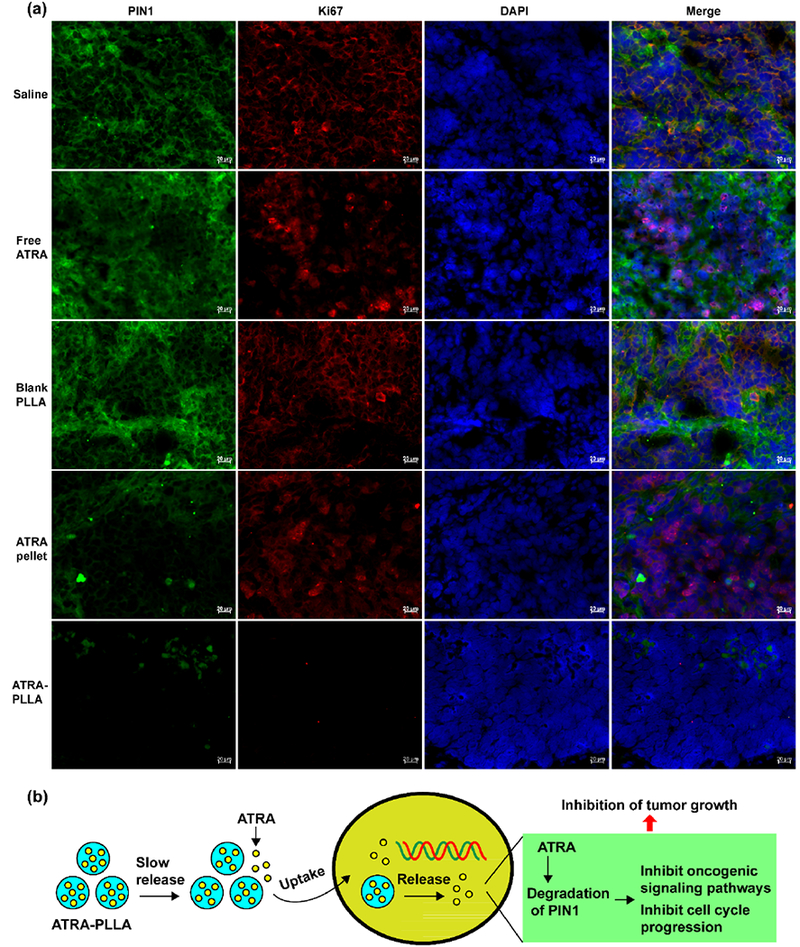
(a) Immunofluorescence images of PIN1 and Ki67 in xenograft tumors from nude mice inoculated with 4 × 106 HuH7 cells and treated with saline, free ATRA, blank PLLA particles, ATRA slow-releasing pellet, and ATRA-PLLA particles for three weeks, (b) The mechanism of anti-tumor effects of ATRA-PLLA microparticles.
4. Discussion
To develop a one-step, cost-effective procedure to produce a new biocompatible and biodegradable controlled release formulation of ATRA, this study produced ATRA-encapsulated PLLA microparticles by sc-CO2 process for the first time (Fig. 1a). Particle size, drug loading, encapsulation efficiency and yield are important performance indicators for drug delivery system. Especially, a higher encapsulation efficiency means more drugs encapsulated by the carrier, and a higher yield suggests better cost-effectiveness, which are crucial for industrial production.
Since parameters in sc-CO2 process had significant effects on product performance [62, 63], this study systematically evaluated the influence of the ratio of ATRA and PLLA, PLLA concentration, and flow rate of solution on the particle size, drug loading, encapsulation efficiency and yield of ATRA-PLLA microparticles. The results show that the three parameters affect the ATRA-PLLA microparticle’s performance in different manner. Specifically, the particle size increased significantly with increasing PLLA concentration (Supplementary Fig. S3), while the drug loading increased dramatically with increasing ratio of ATRA and PLLA (Fig. 1g and Supplementary Fig. S2). However, the encapsulation efficiency decreased with increase of the ratio of ATRA and PLLA (Supplementary Fig. S4). The yield increased with increasing flow rate of solution (Supplementary Fig. S5).
For the effects of liquid solution concentration on particle size, similar observations had been reported in literatures [64, 65], This phenomenon can be explained in terms of nucleation and growth in sc-CO2 process. The increase of solution concentration enhanced the viscosity and surface tension of the quaternary mixture (ATRA-PLLA-DCM-CO2), and then resulted in formation of larger primary droplets and consequent larger particles. For the opposite effects of drug dosage on drug loading and encapsulation efficiency, similar results were also observed in other study [66], The possible reasons for this phenomenon might be as follows. On one hand, the increase of drug dosage resulted in more ATRA precipitated in the ATRA-PLLA microparticles, thereby increasing the drug loading. On the other hand, with the increase of drug dosage, the percentage of ATRA absorbed or combined loosely on the surface of the particles was also increased, consequently decreasing the encapsulation efficiency. The increase of the flow rate of solution resulted in more solution content involved in the precipitation within the same time and decreased the anti-solvent effect of sc-CO2 [63], which reduced the dissolution and loss of ATRA and PLLA by SC-CO2. Hence, the yield of ATRA-PLLA microparticles was increased.
To minimize particle size and simultaneously maximize drug loading, encapsulation efficiency and yield of ATRA-PLLA microparticles, response optimization analysis was further employed to obtain optimum experimental conditions. As expected, the ATRA-PLLA microparticles prepared under the predicted optimum experimental condition (ratio of ATRA and PLLA = 3%; PLLA concentration = 1%; flow rate of solution =1.0 mL/min) had well-defined solid spherical shape and smooth surface with mean particle size 1.24 μm, and high encapsulation efficiency (91.4 ± 4.0%) and yield (68.3 ± 3.3%), and relatively high drug loading (actual drug loading 2.4 ± 0.2% versus theoretical drug loading 3%) (Fig. 1d and f, and Fig. 2b–e). Hence, this optimum experimental condition was selected to produce a new formulation of ATRA for in vitro and in vivo anti-cancer assays.
To understand the release behavior of ATRA-PLLA microparticles, this study investigated the in vitro release profile of ATRA-PLLA microparticles in PBS and MEM with or without serum. The results show that the ATRA-PLLA microparticles can slow release ATRA at least 12 days even in serum-containing MEM (Fig. 2f). No burst release of ATRA was detected in the mediums without serum, which further confirmed the effective encapsulation of ATRA by PLLA. This suggests that the ATRA-PLLA microparticles might be an effective controlled release formulation of ATRA. To confirm whether the ATRA-PLLA microparticles can slow release ATRA in vivo, the pharmacokinetics of ATRA-PLLA microparticles were evaluated in mice, and the commercial ATRA slow-releasing pellet (5 mg over 21 days) was used as control. Indeed, the ATRA-PLLA microparticles are able to slow release ATRA like the commercial ATRA slow-releasing pellet (Fig. 7e). More importantly, the pharmacokinetic properties of ATRA-PLLA microparticles are superior to the commercial ATRA slow-releasing pellet (Supplementary Table SI). Moreover, the ATRA release behaviors of the commercial ATRA pellet are consistent with previous study [61], indicating good accuracy of the pharmacokinetic assay. It is known that the release mechanisms for most nano/micro-particles include three types: (1) desorption of drugs on particle surface; (2) drug diffusion from particle matrix or through copolymer wall; and (3) degradation of particle matrix or combined process of degradation and diffusion [67], Based on these findings that ATRA-PLLA particles are solid sphere with high encapsulation efficiency of ATRA, and can slow release ATRA in vitro and in vivo without burst release, we believe that the release mechanism of ATRA-PLLA microparticles is mainly dependent on drug diffusion. In addition, the accelerated release of ATRA from ATRA-PLLA particles by serum might be attributed to the presence of ATRA binding proteins in serum, which facilitate the release of hydrophobic ATRA into aqueous solution [68, 69].
Good biocompatibility and more effective efficacy are two key requirements for drug delivery system in clinical applications. It is known that poly lactic acid has good biocompatibility [57] and ATRA has anti-proliferative activity against tumor cells [35], To determine whether the ATRA-PLLA particles have more effective efficacy against HCC cell growth, this study investigated the inhibition of ATRA-PLLA microparticles on two HCC cell lines. As expected, the ATRA-PLLA microparticles significantly enhanced the ATRA’s inhibition on cancer cell growth and dramatically decreased the dose of ATRA in both HuH7 and PLC cells. Moreover, the PLLA particle itself has good biocompatibility (Fig. 4a–i).
To confirm whether the ATRA and ATRA-PLLA microparticles selectively act on cancer cells, we further investigated the effects of ATRA and ATRA-PLLA microparticles on viability of normal human liver cells. As expected, the ATRA almost has no inhibition on viability of normal liver cells, and the ATRA-PLLA microparticles do not enhance the inhibition (Fig. 4j and k). What’s more, the inhibition of ATRA-PLLA microparticles on cell viability correlates with the Pin1 level in cells (Fig. 4l). To understand how the ATRA-PLLA microparticles act on cells, the cell uptake of FITC-loaded PLLA microparticles was investigated. The results show that both the released FITC and FITC-loaded PLLA microparticles can be taken into HCC cells in time-dependent manner (Fig. 2g). Our result is consistent with the previous findings that PLA-based particles can be taken into cytoplasm through endocytosis and endo-lysosomal escape [59], Hence, ATRA-PLLA microparticles not only can slow release ATRA in vitro and in vivo, but also can be taken into cytoplasm. This may explain why the ATRA-PLLA microparticles can significantly enhance the ability of ATRA against HCC cell growth. Therefore, application of ATRA-PLLA microparticles might reduce the ATRA’s dose and frequency of administration in cancer therapy.
Although it is known that ATRA has inhibition on liver cancer cell growth [70], the underlying molecular mechanism is still largely unknown. Considering the fact that Pin1 plays pivotal role in hepatocarcinogenesis [13, 14] and ATRA is a potent inhibitor of Pin1 [33], we hypothesize that ATRA exerts its efficacy against HCC mainly through acting on Pin1. This study investigated the effect of ATRA on expression of Pint and related proteins in both HuH7 and PLC cells. As expected, the treatment of free ATRA especially ATRA-PLLA microparticles induced obvious dose-dependent down-regulations of Pint, whereas the particle itself had no effect on Pin1 expression (Fig. 3). Since Pin1 can activate numerous cancer pathways by activating more than 40 oncogenic molecules [10], the down-regulation of Pin1 would lead to down-regulation of these oncogenic molecules in cancer cells. Indeed, the expressions of β-catenin, c-JUN, NF-κB, and Cyclin D1, oncogenic molecules activated by Pin1, were dose-dependently down-regulated in both HuH7 and PLC cells after the ATRA treatment. In addition, obvious down-regulation of CDK2 and CDK6 were also detected in these cells (Fig. 3). β-Catenin is a key component of the WNT signaling pathway, and the Wnt/β-catenin pathway is frequently upregulated in HCC and involved in the development of hepatocellular carcinoma [71, 72], c-JUN N-terminal kinase (JNK) pathway is required for the regulation of cell proliferation, and the dysregulated JNK pathway is contributed to many diseases such as cancer [73], The nuclear factor κB (NF-κB) pathway plays important role in many biological responses, and the aberrant activation of the pathway involved in the development and progression of cancer such as proliferation [16, 74], Cyclin D1, CDK2 and CDK6 are important regulatory factors in the progression of G1-S phase of cell cycle [75], and the excessive expression of Cyclin D1 is contributed to cancer development and maintenance of cancer phenotype [76, 77], The down-regulation of β-catenin, c-JUN, NF-κB, Cyclin D1, CDK2 and CDK6 could lead to inhibition of these above cancer pathways and inhibition of cell cycle progression, which resulted in inhibition of cancer cell growth. As expected, the ATRA treatment induced significant dose-dependent inhibition on focus formation of HCC cells and cell cycle progression where the ATRA-PLLA microparticles significantly enhanced this inhibition (Fig. 5 and Supplementary Fig. S7a and b). Our results are consistent with the previous findings that inhibiting Pint expression in cells lead to inhibition of multiple cancer pathways [33, 78, 79] and inhibition of cell cycle progression [29, 80], In addition, our results show that the treatments of ATRA or ATRA-PLLA microparticles do not induce significant apoptosis (Supplementary Fig. S7a and c), as shown previously [33], Although ATRA has ability to induce APL cell differentiation, our previous study has demonstrated that loss of Pin1 do not lead to APL cell differentiation, but rather lead to cytoreduction [33], These results consistently suggest that ATRA inhibits HCC cell growth mainly by degrading Pin1 and thereby inhibiting multiple oncogenic signaling pathways and cell cycle progression.
To confirm whether Pin1 is a key target of ATRA in HCC, this study investigated the effect of Pin1 knockdown on inhibition of ATRA against proliferation of two HCC cells. The results show that Pin1 knockdown significantly inhibited HCC cell proliferation, and dramatically abolished the inhibition of ATRA on HCC cell proliferation (Fig. 6). This not only confirmed the important role of Pin1 in HCC cell growth, but also confirmed that ATRA exerts its anti-cancer efficacy mainly through acting on Pin1.
To further determine the anti-cancer efficacy and underlying mechanism of ATRA-PLLA microparticles in vivo, this study investigated the inhibition of ATRA-PLLA particles and their counterparts on HCC cell growth in xenograft mice and their effects on expression of Pin1 and it oncogenic substrates in tumors. The results confirmed that the ATRA-PLLA microparticles significantly enhanced the efficacy of ATRA against tumor growth (Fig. 7a–c). Surprisingly, the anti-cancer efficacy of the ATRA-PLLA microparticles was significantly better than the commercial ATRA slow-releasing pellets, especially given the fact that the dose of the ATRA-PLLA microparticles was lower than the ATRA slow-releasing pellets (ATRA concentration is about 2 mg versus 5 mg). The better anti-cancer efficacy of ATRA-PLLA microparticles is attributed to the fact that ATRA-PLLA microparticles have superior pharmacokinetic properties to the commercial ATRA slow-releasing pellet (Fig. 7e and Supplementary Table SI). What’s more, the commercial ATRA slow-releasing pellets can only be used in animal. Moreover, neither the ATRA nor ATRA-PLLA microparticles generate effect on mice weight (Fig. 7d), indicating good biocompatibility. The PLLA particle itself did not inhibit Pin1 expression in tumor, whereas the ATRA-PLLA microparticles significantly enhanced the inhibition of ATRA on expression of Pin1 and its oncogenic substrates (Fig. 7f–i and Fig. 8a). Ki67 is a well-known cell proliferation marker, present during all active phases of the cell cycle (G1, S, G2, and mitosis), but absent in resting (quiescent) cells (GO) [81], Our results show that ATRA-PLLA microparticles dramatically enhance the inhibition of ATRA on Ki67 expression in tumors (Fig. 8a), confirming that the ATRA-PLLA microparticles increase the inhibitory effect of ATRA on tumor growth and cell cycle progression. These in vivo results were consistent with the results obtained from in vitro anti-cancer assays, which further confirmed the anti-cancer efficacy and underlying mechanism of the ATRA-PLLA microparticles. Taking together, the new ATRA-PLLA formulation not only greatly improves the therapeutic efficacy of ATRA on liver cancer, but also might hold great promise for clinical use in human cancer patients because of PLLA is biocompatible, biodegradable and nontoxic to humans.
5. Conclusions
This study reported the development of a novel controlled release formulation of ATRA against hepatocellular carcinoma using a one-step, cost-effective sc-CO2 process. The optimum experimental conditions to generate ATRA-PLLA microparticles with excellent performance were as follows: ratio of ATRA and PLLA = 3%; PLLA concentration = 1%; flow rate of solution =1.0 mL/min. The prepared ATRA-PLLA microparticles (mean size 1.24 μm) have well-defined solid spherical shape and smooth surface, high encapsulation efficiency (91.4%) and yield (68.3%). ATRA-PLLA microparticles can effectively slow release ATRA in vitro and in vivo, and can be taken into HCC cells. The ATRA-PLLA microparticles have good biocompatibility, and can significantly enhance the inhibition of ATRA on HCC cell growth and dramatically decrease the dose of ATRA in both in vitro and in vivo systems. ATRA-PLLA microparticles exerts the potent efficacy against HCC cell and tumor growth in vitro and in vivo through degrading Pin1 and thereby blocking multiple oncogenic signaling pathways and cell cycle progression at the same time (Fig. 8b). Importantly, pharmacokinetic profiles as well as anti-Pin1 and antitumor activities of ATRA-PLLA microparticles are much better than the commercially available slow-releasing ATRA formulation that can be used only in animals. Since PLLA is biocompatible, biodegradable and nontoxic to humans, this study suggests that the sc-CO2 process is a promising platform technique to produce controlled release formulation of anti-cancer drugs and that ATRA-PLLA microparticles might be a promising targeted drug for HCC therapy.
Supplementary Material
Acknowledgments
This paper was supported by the National Natural Science Foundation of China (Nos. 81502115, U1205024), the Natural Science Foundation of Fujian Province of China (No. 2017J01529), the Joint Funds for the Innovation of Science and Technology, Fujian Province (Nos. 2016Y9041, 2016Y9045), the Outstanding Young Scientific Research Personnel Training Plan in Colleges and Universities of Fujian Province (No. 2016B022), the Fujian Provincial Health and Family Planning Commission Fund (No. 2015-1-74), the Research Foundation of Educational Commission of Fujian Province (No. JA15221), the Collaborative Innovation Center for Stem Cells Translational Medicine (Fujian 2011 Program) and NIH grant R01CA167677 to K. P. L.
Footnotes
Conflict of Interest
Dr. Lu and Dr. Zhou are inventors of Pin1 technology, which was licensed by BIDMC to Pinteon Therapeutics. Both Dr. Lu and Dr. Zhou own equity in, and consult for, Pinteon. Their interests were reviewed and are managed by BIDMC in accordance with its conflict of interest policy.
References
- [1].Aggarwal S, Targeted cancer therapies, Nat. Rev. Drug Discov 9 (2010) 427–428. [DOI] [PubMed] [Google Scholar]
- [2].Hanahan D, Weinberg RA, Hallmarks of cancer: the next generation, Cell 144 (2011) 646–674. [DOI] [PubMed] [Google Scholar]
- [3].Luo J, Solimini NL, Elledge SJ, Principles of cancer therapy: oncogene and non-oncogene addiction, Cell 136 (2009) 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Vogelstein B, Papadopoulos N, Velculescu VE, Zhou SB, Diaz LA, Kinzler KW, Cancer genome landscapes, Science 339 (2013) 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lu LC, Hsu CH, Hsu C, Cheng AL, Tumor heterogeneity in hepatocellular carcinoma: facing the challenges, Liver cancer 5 (2016) 128–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F, Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012, Int. J. Cancer 136 (2015) E359–E386. [DOI] [PubMed] [Google Scholar]
- [7].Li L, Wang HY, Heterogeneity of liver cancer and personalized therapy, Cancer Lett. 379 (2016) 191–197. [DOI] [PubMed] [Google Scholar]
- [8].Blume-Jensen P, Hunter T, Oncogenic kinase signalling, Nature 411 (2001) 355–365. [DOI] [PubMed] [Google Scholar]
- [9].Pawson T, Scott JD, Protein phosphorylation in signaling - 50 years and counting, Trends Biochem. Sci 30 (2005) 286–290. [DOI] [PubMed] [Google Scholar]
- [10].Zhou XZ, Lu KP, The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target, Nature reviews. Cancer 16 (2016) 463–478. [DOI] [PubMed] [Google Scholar]
- [11].Lee TH, Chen CH, Suizu F, Huang PY, Schiene-Fischer C, Daum S, Zhang YJ, Goate A, Chen RH, Zhou XZ, Lu KP, Death-associated protein kinase 1 phosphorylates Pin1 and inhibits its prolyl isomerase activity and cellular function, Mol. Cell 42 (2011) 147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lu ZM, Hunter T, Prolyl isomerase Pin1 in cancer, Cell research 24 (2014) 1033–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pang R, Lee TK, Poon RT, Fan ST, Wong KB, Kwong YL, Tse E, Pin1 interacts with a specific serine-proline motif of hepatitis B virus X-protein to enhance hepatocarcinogenesis, Gastroenterology 132 (2007) 1088–1103. [DOI] [PubMed] [Google Scholar]
- [14].Pang RW, Lee TK, Man K, Poon RT, Fan ST, Kwong YL, Tse E, PIN1 expression contributes to hepatic carcinogenesis, The Journal of pathology 210 (2006) 19–25. [DOI] [PubMed] [Google Scholar]
- [15].Pang R, Yuen J, Yuen MF, Lai CL, Lee TK, Man K, Poon RT, Fan ST, Wong CM, Ng IO, Kwong YL, Tse E, PIN1 overexpression and beta-catenin gene mutations are distinct oncogenic events in human hepatocellular carcinoma, Oncogene 23 (2004) 4182–4186. [DOI] [PubMed] [Google Scholar]
- [16].Shinoda K, Kuboki S, Shimizu H, Ohtsuka M, Kato A, Yoshitomi H, Furukawa K, Miyazaki M, Pin1 facilitates NF-kappa B activation and promotes tumour progression in human hepatocellular carcinoma, Br. J. Cancer 113 (2015) 1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lu JC, Hu ZB, Wei S, Wang LE, Liu ZS, El-Naggar AK, Sturgis EM, Wei QY, A novel functional variant (−842G > C) in the PIN1 promoter contributes to decreased risk of squamous cell carcinoma of the head and neck by diminishing the promoter activity, Carcinogenesis 30 (2009) 1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Han CH, Lu JC, Wei QY, Bondy ML, Brewster AM, Yu TK, Buchholz TA, Arun BK, Wang LE, The functional promoter polymorphism (−842G > C) in the PIN1 gene is associated with decreased risk of breast cancer in non-Hispanic white women 55 years and younger, Breast Cancer Res. Treat. 122 (2010) 243–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lu JC, Yang L, Zhao HJ, Liu B, Li YY, Wu HX, Li QC, Zeng BH, Wang YN, Ji WD, Zhou YF, The polymorphism and haplotypes of PIN1 gene are associated with the risk of lung cancer in southern and eastern chinese populations, Hum. Mutat. 32 (2011) 1299–1308. [DOI] [PubMed] [Google Scholar]
- [20].Lu Y, Huang GL, Pu XX, He YX, Li BB, Liu XY, Dong ZG, He ZW, Association between PIN1 promoter polymorphisms and risk of nasopharyngeal carcinoma, Mol. Biol. Rep 40 (2013) 3777–3782. [DOI] [PubMed] [Google Scholar]
- [21].Li Q, Dong Z, Lin Y, Jia XY, Li Q, Jiang H, Wang LW, Gao Y, The rs2233678 polymorphism in PIN1 promoter region reduced cancer risk: a meta-analysis, PloS one 8 (2013) e68148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fujimori F, Takahashi K, Uchida C, Uchida T, Mice lacking Pin1 develop normally, but are defective in entering cell cycle from G(0) arrest, Biochemical and Biophysical Research Communications 265 (1999) 658–663. [DOI] [PubMed] [Google Scholar]
- [23].Lee TH, Pastorino L, Lu KP, Peptidyl-prolyl cis-trans isomerase Pin1 in ageing, cancer and Alzheimer disease, Expert Rev. Mol. Med. 13 (2011) e21. [DOI] [PubMed] [Google Scholar]
- [24].Wulf G, Garg P, Liou YC, Iglehart D, Lu KP, Modeling breast cancer in vivo and ex vivo reveals an essential role of Pin1 in tumorigenesis, Embo J. 23 (2004) 3397–3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].D’Artista L, Bisso A, Piontini A, Doni M, Verrecchia A, Kress TR, Morelli MJ, Del Sal G, Amati B, Campaner S, Pin1 is required for sustained B cell proliferation upon oncogenic activation of Myc, Oncotarget 7 (2016) 21786–21798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Franciosa G, Diluvio G, Del Gaudio F, Giuli MV, Palermo R, Grazioli P, Campese AF, Talora C, Bellavia D, D’Amati G, Besharat ZM, Nicoletti C, Siebel CW, Choy L, Rustighi A, Del Sal G, Screpanti I, Checquolo S, Prolyl-isomerase Pin1 controls notch3 protein expression and regulates T-ALL progression, Oncogene 35 (2016) 4741–4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Girardini JE, Napoli M, Piazza S, Rustighi A, Marotta C, Radaelli E, Capaci V, Jordan L, Quinlan P, Thompson A, Mano M, Rosato A, Crook T, Scanziani E, Means AR, Lozano G, Schneider C, Del Sal G, A Pin1/mutant p53 axis promotes aggressiveness in breast cancer, Cancer cell 20 (2011) 79–91. [DOI] [PubMed] [Google Scholar]
- [28].Takahashi K, Akiyama H, Shimazaki K, Uchida C, Akiyama-Okunuki H, Tomita M, Fukumoto M, Uchida T, Ablation of a peptidyl prolyl isomerase Pin1 from p53-null mice accelerated thymic hyperplasia by increasing the level of the intracellular form of Notch1, Oncogene 26 (2007) 3835–3845. [DOI] [PubMed] [Google Scholar]
- [29].Lu KP, Zhou XZ, The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease, Nat. Rev. Mol. Cell Biol. 8 (2007) 904–916. [DOI] [PubMed] [Google Scholar]
- [30].Moore JD, Potter A, Pin1 inhibitors: pitfalls, progress and cellular pharmacology, Bioorg. Med. Chem. Lett 23 (2013) 4283–4291. [DOI] [PubMed] [Google Scholar]
- [31].Guo CX, Hou XJ, Dong LM, Dagostino E, Greasley S, Ferre R, Marakovits J, Johnson MC, Matthews D, Mroczkowski B, Parge H, VanArsdale T, Popoff I, Piraino J, Margosiak S, Thomson J, Los G, Murray BW, Structure-based design of novel human Pin1 inhibitors (I), Bioorg. Med. Chem. Lett. 19 (2009) 5613–5616. [DOI] [PubMed] [Google Scholar]
- [32].Hennig L, Christner C, Kipping M, Schelbert B, Rucknagel KP, Grabley S, Kullertz G, Fischer G, Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone, Biochemistry 37 (1998) 5953–5960. [DOI] [PubMed] [Google Scholar]
- [33].Wei S, Kozono S, Kats L, Nechama M, Li WZ, Guarnerio J, Luo ML, You MH, Yao YD, Kondo A, Hu H, Bozkurt G, Moerke NJ, Cao SG, Reschke M, Chen CH, Rego EM, Lo-Coco F, Cantley LC, Lee TH, Wu H, Zhang Y, Pandolfi PP, Zhou XZ, Lu KP, Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer, Nature Medicine 21 (2015) 457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Schenk T, Stengel S, Zelent A, Unlocking the potential of retinoic acid in anticancer therapy, Br. J. Cancer 111 (2014) 2039–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Altucci L, Gronemeyer H, The promise of retinoids to fight against cancer, Nat. Rev. Cancer 1 (2001) 181–193. [DOI] [PubMed] [Google Scholar]
- [36].Arce F, Gatjens-Boniche O, Vargas E, Valverde B, Diaz C, Apoptotic events induced by naturally occurring retinoids ATRA and 13-cis retinoic acid on human hepatoma cell lines Hep3B and HepG2, Cancer Lett. 229 (2005) 271–281. [DOI] [PubMed] [Google Scholar]
- [37].Masetti R, Vendemini F, Zama D, Biagi C, Gasperini P, Pession A, All-trans retinoic acid in the treatment of pediatric acute promyelocytic leukemia, Expert review of anticancer therapy 12 (2012) 1191–1204. [DOI] [PubMed] [Google Scholar]
- [38].Degos L, Dombret H, Chomienne C, Daniel M, Miclea J, Chastang C, Castaigne S, Fenaux P, All-trans-retinoic acid as a differentiating agent in the treatment of acute promyelocytic leukemia, Blood 85 (1995) 2643–2653. [PubMed] [Google Scholar]
- [39].Adamson PC, Pitot HC, Balis FM, Rubin J, Murphy RF, Poplack DG, Variability in the oral bioavailability of all-trans-retinoic acid, Journal of the National Cancer Institute 85 (1993) 993–996. [DOI] [PubMed] [Google Scholar]
- [40].Muindi J, Frankel SR, Miller WH, Jakubowski A, Scheinberg DA, Young CW, Dmitrovsky E, Warrell RP, Continuous treatment with all-trans retinoic acid causes a progressive reduction in plasma drug concentrations: implications for relapse and retinoid “resistance” in patients with acute promyelocytic leukemia, Blood 79 (1992) 299–303. [PubMed] [Google Scholar]
- [41].Szuts EZ, Harosi FI, Solubility of retinoids in water, Arch. Biochem. Biophys. 287 (1991) 297–304. [DOI] [PubMed] [Google Scholar]
- [42].Peng Y, Zhu X, Qiu L, Electroneutral composite polymersomes self-assembled by amphiphilic polyphosphazenes for effective miR-200c in vivo delivery to inhibit drug resistant lung cancer, Biomaterials 106 (2016) 1–12. [DOI] [PubMed] [Google Scholar]
- [43].Li L, Song L, Yang X, Li X, Wu Y, He T, Wang N, Yang S, Zeng Y, Yang L, Wu Q, Wei Y, Gong C, Multifunctional “core-shell” nanoparticles-based gene delivery for treatment of aggressive melanoma, Biomaterials 111 (2016) 124–137. [DOI] [PubMed] [Google Scholar]
- [44].Hu Q, Wang K, Sun X, Li Y, Fu Q, Liang T, Tang G, A redox-sensitive, oligopeptide-guided, self-assembling, and efficiency-enhanced (ROSE) system for functional delivery of microRNA therapeutics for treatment of hepatocellular carcinoma, Biomaterials 104 (2016) 192–200. [DOI] [PubMed] [Google Scholar]
- [45].Silva EL, Carneiro G, Caetano PA, Goulart G, Costa DF, de Souza-Fagundes EM, Gomes DA, Ferreira LAM, Nanostructured lipid carriers loaded with tributyrin as an alternative to improve anticancer activity of all-trans retinoic acid, Expert review of anticancer therapy 15 (2015) 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Li RJ, Ying X, Zhang Y, Ju RJ, Wang XX, Yao HJ, Men Y, Tian W, Yu Y, Zhang LA, Huang RJ, Lu WL, All-trans retinoic acid stealth liposomes prevent the relapse of breast cancer arising from the cancer stem cells, J. Control. Release 149 (2011) 281–291. [DOI] [PubMed] [Google Scholar]
- [47].Narvekar M, Xue HY, Tran NT, Mikhael M, Wong HL, A new nanostructured carrier design including oil to enhance the pharmaceutical properties of retinoid therapy and its therapeutic effects on chemo-resistant ovarian cancer, Eur. J. Pharm. Biopharm 88 (2014) 226–237. [DOI] [PubMed] [Google Scholar]
- [48].Almouazen E, Bourgeois S, Boussaid A, Valot P, Malleval C, Fessi H, Nataf S, Briancon S, Development of a nanoparticle-based system for the delivery of retinoic acid into macrophages, Int. J. Pharm 430 (2012) 207–215. [DOI] [PubMed] [Google Scholar]
- [49].Drach J, Lopezberestein G, McQueen T, Andreeff M, Mehta K, Induction of differentiation in myeloid leukemia cell lines and acute promyelocytic leukemia cells by liposomal all-trans-retinoic acid, Cancer research 53 (1993) 2100–2104. [PubMed] [Google Scholar]
- [50].Mehta K, Sadeghi T, McQueen T, Lopezberestein G, Liposome encapsulation circumvents the hepatic clearance mechanisms of all-trans-retinoic acid, Leukemia research 18 (1994) 587–596. [DOI] [PubMed] [Google Scholar]
- [51].Boorjian SA, Milowsky MI, Kaplan J, Albert M, Cobham MV, Coll DM, Mongan NP, Shelton G, Petrylak D, Gudas LJ, Nanus DM, Phase 1/2 clinical trial of interferon alpha 2b and weekly liposome-encapsulated all-trans retinoic acid in patients with advanced renal cell carcinoma, J. Immunother 30 (2007) 655–662. [DOI] [PubMed] [Google Scholar]
- [52].Goldberg JS, Vargas M, Rosmarin AS, Milowsky MI, Papanicoloau N, Gudas LJ, Shelton G, Feit K, Petrylak D, Nanus DM, Phase I trial of interferon alpha 2b and liposome-encapsulated all-trans retinoic acid in the treatment of patients with advanced renal cell carcinoma, Cancer 95 (2002) 1220–1227. [DOI] [PubMed] [Google Scholar]
- [53].David KA, Mongan NP, Smith C, Gudas LJ, Nanus DM, Phase I trial of ATRA-IV and depakote in patients with advanced solid tumor malignancies, Cancer Biol. Ther. 9 (2010) 678–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Cocero MJ, Martin A, Mattea F, Varona S, Encapsulation and co-precipitation processes with supercritical fluids: Fundamentals and applications, J. Supercrit. Fluids 47 (2009) 546–555. [Google Scholar]
- [55].Yeo SD, Kiran E, Formation of polymer particles with supercritical fluids: A review, J. Supercrit. Fluids 34 (2005) 287–308. [Google Scholar]
- [56].Xie M, Fan D, Chen Y, Zhao Z, He X, Li G, Chen A, Wu X, Li J, Li Z, Hunt JA, Li Y, Lan P, An implantable and controlled drug-release silk fibroin nanofibrous matrix to advance the treatment of solid tumour cancers, Biomaterials 103 (2016) 33–43. [DOI] [PubMed] [Google Scholar]
- [57].Lasprilla AJR, Martinez GAR, Lunelli BH, Jardini AL, Maciel R, Poly-lactic acid synthesis for application in biomedical devices - A review, Biotechnol. Adv 30 (2012) 321–328. [DOI] [PubMed] [Google Scholar]
- [58].Chen AZ, Li Y, Chen D, Hu JY, Development of core-shell microcapsules by a novel supercritical CO2 process, J. Mater. Sci.-Mater. Med 20 (2009) 751–758. [DOI] [PubMed] [Google Scholar]
- [59].Panyam J, Zhou WZ, Prabha S, Sahoo SK, Labhasetwar V, Rapid endo-lysosomal escape of poly(DL-lactide-co-glycolide) nanoparticles: implications for drug and gene delivery, Faseb J. 16 (2002) 1217–1226. [DOI] [PubMed] [Google Scholar]
- [60].Luo ML, Gong C, Chen CH, Lee DY, Hu H, Huang PY, Yao YD, Guo WJ, Reinhardt F, Wulf G, Lieberman J, Zhou XZ, Song EW, Lu KP, Prolyl isomerase Pin1 acts downstream of miR200c to promote cancer stem-like cell traits in breast cancer, Cancer research 74 (2014) 3603–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Westervelt P, Pollock JL, Oldfather KM, Walter MJ, Ma MK, Williams A, DiPersio JF, Ley TJ, Adaptive immunity cooperates with liposomal all-trans-retinoic acid (ATRA) to facilitate long-term molecular remissions in mice with acute promyelocytic leukemia, Proc. Natl. Acad. Sci. U. S. A. 99 (2002) 9468–9473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chen AZ, Li Y, Chau FT, Lau TY, Hu JY, Zhao Z, Mok DKW, Application of organic nonsolvent in the process of solution-enhanced dispersion by supercritical CO2 to prepare puerarin fine particles, J. Supercrit. Fluids 49 (2009) 394–402. [Google Scholar]
- [63].Zhao Z, Xie MB, Li Y, Chen AZ, Li G, Zhang J, Hu HW, Wang XY, Li SP, Formation of curcumin nanoparticles via solution-enhanced dispersion by supercritical CO2, Int. J. Nanomed 10 (2015) 3171–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Wang Q, Guan YX, Yao SJ, Zhu ZQ, Microparticle formation of sodium cellulose sulfate using supercritical fluid assisted atomization introduced by hydrodynamic cavitation mixer, Chem. Eng. J 159 (2010) 220–229. [Google Scholar]
- [65].Reverchon E, De Marco I, Supercritical antisolvent precipitation of Cephalosporins, Powder Technol. 164 (2006) 139–146. [Google Scholar]
- [66].Chen AZ, Li L, Wang SB, Lin XF, Liu YG, Zhao C, Wang GY, Zhao Z, Study of Fe3O4-PLLA-PEG-PLLA magnetic microspheres based on supercritical CO2: Preparation, physicochemical characterization, and drug loading investigation, J. Supercrit. Fluids 67 (2012) 139–148. [Google Scholar]
- [67].Xiao RZ, Zeng ZW, Zhou GL, Wang JJ, Li FZ, Wang AM, Recent advances in PEG-PLA block copolymer nanoparticles, Int. J. Nanomed 5 (2010) 1057–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Di Muzio E, Polticelli F, di Masi A, Fanali G, Fasano M, Ascenzi P, All-trans-retinoic acid and retinol binding to the FA1 site of human serum albumin competitively inhibits heme-Fe(III) association, Arch. Biochem. Biophys 590 (2016) 56–63. [DOI] [PubMed] [Google Scholar]
- [69].Karnaukhova E, Interactions of human serum albumin with retinoic acid, retinal and retinyl acetate, Biochemical pharmacology 73 (2007) 901–910. [DOI] [PubMed] [Google Scholar]
- [70].Cui JJ, Gong MJ, He Y, Li QL, He TC, Bi Y, All-trans retinoic acid inhibits proliferation, migration, invasion and induces differentiation of hepa1–6 cells through reversing EMT in vitro, International journal of oncology 48 (2016) 349–357. [DOI] [PubMed] [Google Scholar]
- [71].Vilchez V, Turcios L, Marti F, Gedaly R, Targeting Wnt/beta-catenin pathway in hepatocellular carcinoma treatment, World J. Gastroenterol. 22 (2016) 823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Monga SP, Beta-catenin signaling and roles in liver homeostasis, injury, and tumorigenesis, Gastroenterology 148 (2015) 1294–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Johnson GL, Nakamura K, The c-jun kinase/stress-activated pathway: regulation, function and role in human disease, Biochim. Biophys. Acta-Mol. Cell Res 1773 (2007) 1341–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Dolcet X, Llobet D, Pallares J, Matias-Guiu X, NF-kB in development and progression of human cancer, Virchows Arch. 446 (2005) 475–482. [DOI] [PubMed] [Google Scholar]
- [75].Bertoli C, Skotheim JM, de Bruin RAM, Control of cell cycle transcription during G1 and S phases, Nat. Rev. Mol. Cell Biol 14 (2013) 518–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Tashiro E, Tsuchiya A, Imoto M, Functions of cyclin D1 as an oncogene and regulation of cyclin D1 expression, Cancer Sci. 98 (2007) 629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Knudsen KE, Diehl JA, Haiman CA, Knudsen ES, Cyclin D1: polymorphism, aberrant splicing and cancer risk, Oncogene 25 (2006) 1620–1628. [DOI] [PubMed] [Google Scholar]
- [78].Yan XX, Zhu ZD, Xu SM, Yang LN, Liao XH, Zheng M, Yang DY, Wang JC, Chen DM, Wang L, Liu XL, Liu JF, Chen RH, Zhou XZ, Lu KP, Liu HK, MicroRNA-140–5p inhibits hepatocellular carcinoma by directly targeting the unique isomerase Pin1 to block multiple cancer-driving pathways, Sci Rep 7 (2017) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Liao XH, Zhang AL, Zheng M, Li MQ, Chen CP, Xu HJ, Chu QS, Yang DY, Lu WX, Tsai TF, Liu HK, Zhou XZ, Lu KP, Chemical or genetic Pin1 inhibition exerts potent anticancer activity against hepatocellular carcinoma by blocking multiple cancer-driving pathways, Sci Rep 7 (2017) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Risal P, Shrestha N, Chand L, Sylvester KG, Jeong YJ, Involvement of prolyl isomerase PIN1 in the cell cycle progression and proliferation of hepatic oval cells, Pathol. Res. Pract 213 (2017) 373–380. [DOI] [PubMed] [Google Scholar]
- [81].Cuylen S, Blaukopf C, Politi AZ, Muller-Reichert T, Neumann B, Poser I, Ellenberg J, Hyman AA, Gerlich DW, Ki-67 acts as a biological surfactant to disperse mitotic chromosomes, Nature 535 (2016) 308–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.