

Abstract
Background & Aims:
Glycogen synthase kinase 3β (Gsk3β [Gsk3b]) is a ubiquitously expressed kinase with distinctive functions in different types of cells. Although its roles in regulating innate immune activation and ischaemia and reperfusion injuries (IRIs) have been well documented, the underlying mechanisms remain ambiguous, in part because of the lack of cell-specific tools in vivo.
Methods:
We created a myeloid-specific Gsk3b knockout (KO) strain to study the function of Gsk3β in macrophages in a murine liver partial warm ischaemia model.
Results:
Compared with controls, myeloid Gsk3b KO mice were protected from IRI, with diminished proinflammatory but enhanced anti-inflammatory immune responses in livers. In bone marrow-derived macrophages, Gsk3β deficiency resulted in an early reduction of Tnf gene transcription but sustained increase of Il10 gene transcription on Toll-like receptor 4 stimulation in vitro. These effects were associated with enhanced AMP-activated protein kinase (AMPK) activation, which led to an accelerated and higher level of induction of the novel innate immune negative regulator small heterodimer partner (SHP [Nr0b2]). The regulatory function of Gsk3β on AMPK activation and SHP induction was confirmed in wild-type bone marrowderived macrophages with a Gsk3 inhibitor. Furthermore, we found that this immune regulatory mechanism was independent of Gsk3β Ser9 phosphorylation and the phosphoinositide 3-kinase–Akt signalling pathway. In vivo, myeloid Gsk3β deficiency facilitated SHP upregulation by ischaemia–reperfusion in liver macrophages. Treatment of Gsk3b KO mice with either AMPK inhibitor or SHP small interfering RNA before the onset of liver ischaemia restored liver proinflammatory immune activation and IRI in these otherwise protected hosts. Additionally, pharmacological activation of AMPK protected wild-type mice from liver IRI, with reduced proinflammatory immune activation. Inhibition of the AMPK–SHP pathway by liver ischaemia was demonstrated in tumour resection patients.
Conclusions:
Gsk3β promotes innate proinflammatory immune activation by restraining AMPK activation.
Keywords: Glycogen synthase kinase 3 beta, AMP-activated protein kinase, Macrophage, Small heterodimer partner, Inflammation, Ischaemia–reperfusion injury, Toll-like receptor, IL-10
Lay summary:
Glycogen synthase kinase 3β promotes macrophage inflammatory activation by inhibiting the immune regulatory signalling of AMP-activated protein kinase and the induction of small heterodimer partner. Therefore, therapeutic targeting of glycogen synthase kinase 3β enhances innate immune regulation and protects liver from ischaemia and reperfusion injury.
Graphical abstract
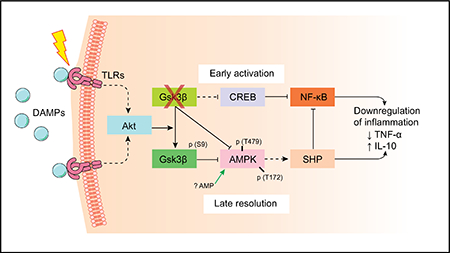
Introduction
Liver inflammation triggered by ischaemia–reperfusion (IR) is an innate immune-dominated response, mediated by the sentinel pattern recognition receptor (PRR) system.1,2 Damageassociated molecular patterns (DAMPs),3 such as high-mobility group box 1, released from necrotic/stressed cells,4–6 activate PRRs, including Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain-like receptors (NLRs),7–10 to initiate the proinflammatory immune response. At the cellular level, macrophages, the predominant innate immune cells in the liver, play a major role in reacting to DAMPs to trigger tissue inflammatory immune response.
Highlights.
Myeloid Gsk3β promotes liver pro-inflammatory immune responses to ischemia reperfusion.
Active Gsk3β inhibits AMPK activation and SHP induction in activated macrophages.
Gsk3β regulates AMPK/SHP independent of its S9 phosphorylation status.
Glycogen synthase kinase 3 (Gsk3) is a ubiquitously expressed serine/threonine kinase with a large number of substrates.11–14 There are two isoforms, α and β, which share extensive homology in the kinase domain, but are functionally distinctive possibly because of their unique N- and C-terminals. In mice, global Gsk3β (Gsk3b) knockout (KO) is embryonically lethal because of liver degeneration,15 whereas Gsk3α (Gsk3a) KO mice are viable and relatively normal.16 Most research on Gsk3 has focused on the β isoform using chemical inhibitors, which do not discriminate between the α and β isoforms.17 Gsk3β is a unique signalling kinase: constitutively active in resting cells and inhibited by Ser9 phosphorylation on stimulation. Gsk3β regulates diverse cellular activities in different cell types, including metabolism, proliferation, differentiation, apoptosis, and immune activation.11–14,18–20 Gsk3β inhibition suppresses proinflammatory gene programmes but promotes anti-inflammatory gene programmes in macrophages.12 TLR activation triggers Gsk3β inhibitory phosphorylation by the phosphoinositide 3-kinase (PI3K)–protein kinase B/Akt pathway, resulting in an increase of cyclic AMP response element-binding protein (CREB) activity but a decrease of NF-κB activity.12,21 In patients with cirrhosis, defects in Akt-mediated Gsk3 inhibitory phosphorylation in monocytes resulted in an excessive proinflammatory response in vitro.22 In vivo, Gsk3 inhibitors protected mice from endotoxin shock.21 We have shown that the Gsk3 inhibitor SB216763 protects mice from liver IR injury (IRI) via an IL-10 (Il10)-mediated immune regulatory mechanism.23
Gsk3β can play either pro- or anti-apoptotic roles, depending on cell death mechanisms. Active Gsk3β inhibits the extrinsic cell death pathway initiated by TNF-α (Tnf) or other death receptor ligands.11,15 Meanwhile, Gsk3β inactivation prevents the opening of mitochondrial permeability transition pore, a key step in the intrinsic cell death pathway initiated by oxidative or endoplasmic reticulum stress.11,24,25 Gsk3 inhibition has been shown to protect against tissue parenchymal cell death caused by IR,26–29 although the opposite effect has also been observed,30 possibly due to the difference in its targeted cell death processes.
Because of its diverse regulatory roles in many cell types and different cellular activities, Gsk3β may regulate liver IRI in both parenchymal and non-parenchymal cells (NPCs). The immune regulatory effects of global Gsk3 inhibition in vivo may result either from its direct regulation of macrophage activation by PRRs or via regulation of parenchymal cell death, which may impact innate responses via DAMPs, or a combination of both. Because of limitations in dosing/toxicities and tissue accessibility, pharmacological inhibition of Gsk3β may differ in different tissues/cells in vivo. Its effects on disease pathogenesis, therefore, may differ, depending on downstream pathways and/or substrates of Gsk3. Non-differential inhibition of both Gsk3 isoforms and off-target effects of chemical inhibitors are of concern as well in their use to define the functions of Gsk3β. Thus, genetic approaches are better suited in dissecting the functional mechanisms of Gsk3β in vivo. In this study, we created myeloidspecific Gsk3β gene KO mice to determine how Gsk3β regulates macrophage responses in liver against IR.
Materials and methods
Animals
Gsk3βb-LoxP mice (provided by Jim Woodgett of the University of Toronto, Samuel Lunenfeld Research Institute, Toronto, Canada) were bred with myeloid-specific Cre mice (Lyz2-Cre, The Jackson Laboratory, Bar Harbor, ME, USA) to create myeloid-specific Gsk3b KO mice. Briefly, homozygous Gsk3bloxP/loxP mice were first bred with homozygous Lyz2-Cre mice, and the heterozygous offspring (for both Gsk3β and Cre) were back-crossed with homozygous Gsk3bloxP/loxP mice. The primers used for genotyping Gsk3b-LoxP included 5ˊ-GGGGCAACCTTAATTTCATT-3ˊ as the forward primer and 5ˊ-GTGTCTGTATAACTGACTTCCTGTGGC-3ˊ as the reverse primer, which yielded 685 and 585 bp bands, respectively, for the floxed and wild-type alleles. Cre genotyping was performed by use of a standard protocol with primers described in the JAX Genotyping Protocol Database. Myeloid phosphatase and tensin homologue (Pten)-deficient mice were derived as described previously,31 and Gsk3β S9A mutant (alanine replaces serine at position 9, resistant to inhibitory phosphorylation) knock-in mice were provided by Dario Alessi of the University of Dundee (Medical Research Council, Dundee, UK). Mice were group-housed (from two to five mice per cage) in standard mouse cages and maintained under a 12 h light/dark cycle with free access to water and standard mouse chow in the University of California, Los Angeles, CA, USA animal facility under specific pathogen-free conditions, and received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institutes of Health.
Mouse liver IRI model
Male Gsk3b wild-type (WT) (including WT B6, Lyz-Cre, and FloxP-Gsk3b mice) and Gsk3b-deficient mice (6–8 weeks old) were used in the experiments. A model of partial hepatic warm IRI was used as described previously.32 Briefly, after successful anaesthesia with 2.5% isoflurane, heparin (100 mg/kg) was injected into the mice. An atraumatic clip was used to interrupt the arterial and portal venous blood supply to the cephalad lobes of the liver. After 90 min of partial hepatic warm ischaemia, the clip was removed, initiating hepatic reperfusion. Mice were maintained anesthetized with isoflurane and placed in a designed warm container (HTP-1500 heat therapy pump, Adroit Medical Systems, Loudon, TN, USA) to keep the temperature at 29°C. Mice were killed after various times of reperfusion (0–24 h). Sham controls underwent the same procedure, but without vascular occlusion. Anti-IL-10 antibody (0.25 mg per mouse, BioXcell, West Lebanon, NH, USA), compound C (dorsomorphin, 20 mg/kg, Tocris, Minneapolis, MN) or 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR; 100 mg/kg, i.p., Tocris), or phosphate-buffered saline (vehicle control) was administered intraperitoneally 1 h before the onset of liver ischaemia.
Serum alanine aminotransferase (sALT) levels were measured with an autoanalyser from ANTECH Diagnostics (Los Angeles, CA, USA). Some liver specimens were fixed in 10% buffered formalin and embedded in paraffin. Liver sections (4 μm) were stained with haematoxylin and eosin. The severity of liver IRI was graded blindly with use of Suzuki’s criteria on a scale from 0 to 4. No necrosis and no congestion/centrilobular ballooning was given a score of 0, whereas severe congestion and >60% lobular necrosis was given a score of 4.33
Myeloperoxidase assay
Liver myeloperoxidase (MPO) activities were measured. In brief, the frozen tissue sample was thawed and placed in iced 0.5% hexadecyltrimethylammonium bromide and 50 mM potassium phosphate buffer solution (pH 5). Each sample was homogenized and centrifuged at 15,000 rpm for 15 min at 4 °C. Supernatants were mixed with H2O2–sodium acetate and tetramethylbenzidine solutions. The change in absorbance was measured by spectrophotometry at 655 nm. One unit of MPO activity was defined as the quantity of enzyme degrading 1 μmol H2O2 per minute at 25 °C per gram of tissue.
Bone marrow-derived macrophage cultures
Bone marrow cells were isolated from mouse femurs and tibias. Cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 20% L929-conditioned medium for 7 days. Bone marrow-derived macrophages (BMMs) were then replated and cultured overnight in new culture dishes for further experiments.
BMMs were stimulated with lipopolysaccharide (LPS; 100 ng/mL; InvivoGen, San Diego, CA, USA) in the absence or presence of the AMPK inhibitor compound C (2 μM). Culture supernatants were collected at 0, 6, and 24 h after stimulation for measurement of cytokines. Cells were collected at various time points for western blot analysis.
Isolation of liver cells
Livers were perfused in situ via the portal vein with calcium- and magnesium-free Hanks balanced salt solution (HBSS) supplemented with 2% heat-inactivated FBS, followed by 0.27% collagenase IV (Sigma-Aldrich, St Louis, MO, USA). Perfused livers were dissected, and teased through 70 lm nylon mesh cell strainers (BD Biosciences, San Diego, CA, USA). Liver cells were suspended in 20 mL DMEM with 10% FBS and divided into NPCs, including Kupffer cells (KCs), and hepatocytes as follows. Liver cells were centrifuged at 50 g for 2 min, and the supernatant was collected. This step was repeated three times, and the NPCs in the supernatant were collected by centrifugation at 800 g for 5 min. NPCs were then suspended and allowed to attach to cell culture plates in DMEM with 10% FBS, 10 mM N-(2-hydroxyethyl)piperazine-Nˊ-ethanesulfonic acid, 2 mM GlutaMax, penicillin (100 U/mL), and streptomycin (100 μg/mL) for 15 min at 37 C. Non-adherent cells were removed by replacement of the culture medium. The attached cells were KCs of 80% purity as assessed by immunofluorescence staining of F4/80 antibody. Primary hepatocytes were pelleted after centrifugation at 50 g for 2 min. Cells were resuspended in 20 mL of 40% cold Percoll solution (P1644, Sigma) and centrifuged at 150 g for 7 min. The pelleted hepatocytes were washed once with DMEM plus 10% FBS, suspended in plating medium (Williams E medium with hepatocyte thawing and plating supplement pack), and plated in collagen type I-coated plates for 3 h. Maintenance medium (Williams E medium with hepatocyte maintenance supplement pack) was used for cultures overnight or longer.
Small heterodimer partner gene expression knockdown
Small heterodimer partner (SHP [Nr0b2]) siRNA with a pool of three target-specific siRNAs (sc-44870, Santa Cruz Biotechnology, Dallas, Texas) was used to knock down Nr0b2 gene expression. In vitro, BMMs were transiently transfected with siRNA with use of a LipoJet™ in vitro transfection kit (SignaGen Laboratories, Rockville, MD) according to the manufacturer’s protocol. In vivo, siRNA was mixed with mannose-conjugated polymers (Polyplus-transfection, New York, NY) in a ratio specified by the manufacturer, and administered intraperitoneally (siRNA 2 mg/kg) at 3 h before the onset of liver ischaemia.
Quantitative reverse transcription PCR
Total RNA (2.0 mg) was reverse transcribed into complementary DNA with use of an oligo(dT)12–18 primer and M-MLV reverse transcriptase kit (Invitrogen, Carlsbad, CA, USA). QuantitativePCR was performed with a QuantStudio 3 system (Thermo Fisher Scientific, Waltham, MA, USA) in a final reaction volume of 20 μL containing 1 ×PCR mix (Power SYBR Green PCR master mix, Life Technologies, Woolston, UK), complementary DNA, and each primer at 0.125 μM. The amplification conditions were as follows: 50 °C for 2 min, 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s, and 60 °C for 1 min. The Nr0b2 primers were CTCTGCAGGTCGTCCGACTATTCTG (5ˊ) and CCTCGAAGGTCACAGCATCCTG (3ˊ). The other primers used to amplify specific mouse gene fragments were the same as those described previously.34
Western blots
Tissue or cellular proteins were extracted with ice-cold lysis buffer (1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 10% glycerol, 137 mM sodium chloride, 20 mM Tris, pH 7.4). Proteins (20 μg) were subjected to 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane. Antibodies against total and phosphorylated Gsk3β (Ser9), phosphorylated signal transducer and activator of transcription 1 (Stat1), Stat3, Stat6, phosphorylated NF-κBp65 (Ser536), phosphorylated CREB (Ser133), phosphorylated Akt (Ser473 and Thr308), phosphorylated acetyl-CoA carboxylase (ACC) (Ser79), phosphorylated AMPK (Thr172), and β-actin (Cell Signaling Technology, San Diego, CA, USA), phosphorylated AMPK (Thr479) (ARP American Research Products, Waltham MA, USA), and SHP (NROB2; Abcam, Cambridge, MA, USA) were used for western blot analysis.
ELISA
Secretion of cytokines (TNF-α, IL-6, and IL-10) in cell culture supernatants was measured by ELISA, according to the manufacturer’s protocols (eBioscience, San Diego, CA, USA).
Patients and specimens
A total of five patients with hepatocellular carcinoma undergoing liver partial resection in the Department of Liver Surgery, the First Affiliated Hospital of Nanjing Medical University, were included in the current study. Human liver graft biopsy specimens were collected serially from tumour peripherals at 0, 10, 30, and 50 min after partial hepatic portal occlusion by ligation of branches of the hepatic artery and portal vein into the tumour lobes. The study protocol was approved by the Institutional Review Board of the First Affiliated Hospital of Nanjing Medical University (Institutional Review Board approval number 2017SRFA-138). Informed consent was obtained from each patient.
Statistical analysis
Results are shown as the mean ± SD. Multiple group comparisons were performed by one-way analysis of variance followed by Bonferroni’s post hoc test. All analyses were performed with Stata (Version 11.0; StataCorp LLC, College Station, Texas, USA). P values <0.05 (two-tailed) were considered statistically significant.
For further details regarding the materials used, please refer to the CTAT table.
Results
Myeloid Gsk3β deficiency protects mice from liver IRI
To better define the functions of Gsk3β in regulating liver inflammatory immune response against IRI, we used the Cre-LoxP system to create myeloid-specific Gsk3b KO mice. In these KO mice, the overall level of Gsk3β in livers was not much different from that in WT mice. Hepatocytes from KO and WT mice expressed the same levels of Gsk3β. Only liver NPCs from KO mice were deficient in Gsk3β expression (Fig. 1A). In liver IR experiments, myeloid Gsk3b KO mice were protected, with much reduced hepatocellular injuries, as evidenced by lower sALT levels at both 6 and 24 h after reperfusion, and better preserved liver histological architectures (lower Suzuki scores in haematoxylin and eosin stained tissue sections) as compared with WT controls (Fig. 1B,C). Liver proinflammatory immune responses against IR were diminished in myeloid Gsk3b KO mice, as evidenced by reductions in both neutrophil infiltration/activation (lower MPO activities; Fig. 1B) and proinflammatory cytokine/chemokine gene expression (Tnf, Il6, and Cxcl10; Fig. 1D). Liver anti-inflammatory Il10 gene induction, however, was enhanced in the KO mice (Fig. 1D). Consistent with our previous results with the Gsk3 inhibitor in WT mice,31 administration of anti-IL-10 antibody in myeloid Gsk3b KO mice abrogated their protective phenotype against liver IRI (Fig. S1). These results indicate that myeloid Gsk3β promotes liver proinflam-matory immune activation and development of hepatocellular damage by IR by inhibiting the IL-10-mediated regulatory mechanism.
Fig. 1. Myeloid glycogen synthase kinase 3β (Gsk3β) deficiency protects livers from ischaemia–reperfusion (IR) injury.

(A) Gsk3β expression in livers of myeloid Gsk3β wild-type (WT) and knockout (KO) mice. Protein lysates were prepared from liver tissues or hepatocytes or liver non-parenchymal cells (NPCs) and subjected to western blot analysis of Gsk3β and β-actin. (B) Myeloid Gsk3β WT or KO mice were subjected to liver partial warm ischaemia followed by 6 or 24 h of reperfusion, as described in “Materials and methods”. Average serum alanine aminotransferase (ALT) levels in different groups of mice are plotted, as well as average Suzuki scores and liver myeloperoxidase (MPO) activities. (C) Representative haematoxylin and eosin staining of liver tissue sections, sham livers, and ischaemic livers after 6 or 24 h of reperfusion. (D) Liver inflammatory gene expression was measured by quantitative reverse transcription PCR. Average target gene/Hprt1 gene ratios of different experimental groups are plotted. All results are representative of at least two independent experiments. There was from four to six mice per group. Data were tested for normal distribution and analysed by one-way analysis of variance with post-test. *p <0.05.
Gsk3β restrains AMPK activation in macrophages
To determine the mechanisms of Gsk3β regulation of liver inflammatory immune response, we compared WT and Gsk3β-deficient macrophages in vitro. BMMs or KCs were stimulated with LPS, and culture supernatants were harvested at 0, 6, and 24 h after stimulation. TNF-α, IL-6, and IL-10 levels were measured by ELISA. Gsk3b-deficient macrophages, including both BMMs and KCs, produced significantly lower levels of proinflammatory cytokines and much higher levels of IL-10 as compared with their controls (Fig. 2A). To determine whether Gsk3β differentially regulated pro- and anti-inflammatory gene transcription in macrophages, we measured Tnf and Il10 gene expression in BMMs after stimulation for 3 and 24 h. Gsk3b-deficient cells expressed lower levels of Tnf at 3 h but similar levels at 24 h; however, they expressed higher levels of Il10 at both early and late time points as compared with their controls (Fig. 2B).
Fig. 2. Glycogen synthase kinase 3β (Gsk3β) regulates macrophage activation.

(A) Bone marrow-derived macrophages (BMMs) and Kupffer cells (KCs) derived from myeloid Gsk3β wild-type (WT) and knockout (KO) mice were prepared and cultured as described in “Materials and methods”. These cells were stimulated with lipopolysaccharide (LPS; 100 ng/mL), and culture supernatants were harvested at 0, 6, and 24 h. TNF-α, IL-6, and IL-10 levels were measured by ELISA. Average concentrations of cytokines at each time point are plotted. (B) Gsk3β WT and KO BMMs were stimulated with LPS, as in (A), for 3 and 24 h. Total cellular RNA was prepared and reverse transcribed. Tnf and Il10 gene expression levels were measured by quantitative reverse transcription PCR. Average target gene/Hprt1 gene ratios of different experimental groups are plotted. All results are representative of at least two independent experiments. There were three mice per group. Data were tested for normal distribution and analysed by one-way analysis of variance with post-test. *p <0.05. Ctl., control.
To study how Gsk3β regulates macrophage activation, we measured activation of intracellular signalling pathways downstream of TLR4 in BMMs (Fig. 3A). Similar levels of Stat1 phosphorylation were induced in Gsk3b WT and KO cells by LPS. Stat3 phosphorylation was enhanced in KO cells, consistent with their higher IL-10 phenotype. The Stat6 phosphorylation profile in KO cells was altered as compared with that in WT cells: the initial downregulation on stimulation (at 30 min and 1 h) in WT cells was diminished in KO cells, indicating the enhancement of the M2 phenotype/function in KO cells. Nuclear NF-κBp65 and CREB levels measured by Ser536 or Ser133 phosphorylation respectively35,36 were similar in stimulated cells regardless of Gsk3b genotypes.
Fig. 3. Glycogen synthase kinase 3β (Gsk3β) regulates macrophage AMP-activated protein kinase (AMPK) activation and small heterodimer partner (SHP) induction.

(A) Gsk3β wild-type (WT) and knockout (KO) bone marrow-derived macrophages (BMMs) were stimulated with lipopolysaccharide (LPS) for 0 min, 30 min, 1 h, and 6 h. Total cellular proteins were extracted and analysed by western blotting with anti-phosphorylated signal transducer and activator of transcription 1 (p-Stat1), signal transducer and activator of transcription 3 (p-Stat3), signal transducer and activator of transcription 6 (p-Stat6), NF-κBp65 (Ser536) (p-NF-kBp65), cyclic AMP response element-binding protein (Ser133) (p-CREB), AMPK (Thr172) (p-AMPK(T172)), AMPK (Thr479) (p-AMPK(T479)), and acetyl-CoA carboxylase (p-ACC) (Ser79), anti-SHP, anti-Gsk3β, and anti-β-actin. (B) Gsk3β WT (top) and KO BMMs (bottom) were treated with either vehicle or compound C before LPS stimulation. Cellular proteins were harvested after 0, 3, 6, 12, and 24 h of stimulation and analysed by western blotting with anti-SHP or anti-β-actin. (C) Gsk3β WT BMMs were treated with vehicle control or the Gsk3 inhibitor SB216763 before the addition of LPS. Cellular proteins were harvested after 0, 3, 6, and 12 h of stimulation and analysed by western blotting with anti-p AMPK(T172) and anti-SHP or anti-β-actin. (D). Gsk3β WT and KO BMMs were stimulated with LPS (top panel), or Gsk3β WT BMMs were treated with vehicle control or SB216763 before the addition of LPS (middle panel), or Gsk3β KO BMMs were treated with either vehicle or compound C before LPS stimulation (bottom panel). Total cellular RNA was prepared and reverse transcribed. Nr0b2 and Hprt1 gene expression levels were measured by quantitative reverse transcription PCR. Average Nr0b2/Hprt1 gene ratios of different experimental groups are plotted. All results are representative of at least two independent experiments. There was three mice per group. Data were tested for normal distribution and analysed by one-way analysis of variance with post-test. *p <0.05. Comp. C, compound C; Ctl., control; SB, SB216763.
As Gsk3β is a key regulator of glycogenesis, its genetic deficiency may alter cellular metabolism, which is critical for macrophage activation/differentiation. We next explored the possibility that Gsk3β regulates AMPK activation, the master metabolic regulator, to impact macrophage activation and liver IRI. AMPKα has been shown to promote anti-inflammatory functions in macrophages,37–39 and it could be phosphorylated directly by Gsk3β at Thr479,40 which prohibits AMPK activation. Indeed, AMPK activation was increased in Gsk3b-deficient BMMs, as shown by a lower level of inhibitory phosphorylation at Thr479 and a higher level of activating phosphorylation at Thr172 (Fig. 3A). Downstream of AMPK, the phosphorylation of its direct substrate ACC at Ser79 was significantly increased in Gsk3b-deficient BMMs in the first 6 h of LPS stimulation, whereas in WT cells, ACC phosphorylation was transient and at much lower levels (Fig. 3A). Furthermore, SHP, which was recently shown to be a key negative regulator of innate immune activation downstream of AMPK,41,42 was upregulated significantly in Gsk3b KO cells, whereas the level in control cells was downregulated, in the first 6 h after LPS stimulation (Fig. 3A). We subsequently found in WT macrophages that SHP expression did not increase until 24 h on activation (Fig. 3B). To further establish the role of Gsk3β in regulating AMPK activation and SHP induction downstream of TLR, we treated WT BMMs with the Gsk3 inhibitor SB216763. Inhibition of Gsk3β enhanced AMPK activation (Thr172) and facilitated SHP induction in WT cells after LPS stimulation (Fig. 3C). Furthermore, pharmacological inhibition of AMPK by compound C The induction of SHP expression in macrophages was reguprevented the rapid SHP induction in Gsk3b KO BMMs in lated at the transcriptional level, as quantitative reverse tranresponse to TLR stimulation (Fig. 3B).
The induction of SHP expression in macrophages was regulated at the transcriptional level, as quantitative reverse transcription PCR (RT-qPCR) measurement of Nr0b2 transcripts showed that it was induced earlier (6 vs. 24 h) and at significantly higher levels in Gsk3b-deficient vs. Gsk3b-proficient BMMs in response to LPS stimulation (Fig. 3D, top panel). Inhibition of Gsk3β in WT cells promoted and inhibition of AMPK by compound C in Gsk3b KO BMMs inhibited the Nr0b2 gene transcription induced by TLR stimulation (Fig. 3D).
The Gsk3β–AMPK–SHP pathway regulates innate inflammatory immune activation in liver IRI
To determine whether the Gsk3β–AMPK–SHP signalling pathway regulates innate inflammatory immune activation in vivo, we first determined whether SHP was induced in liver in myeloid Gsk3b-deficient vs. WT mice in response to IR. As SHP is expressed in many types of liver cells, we isolated liver NPCs from sham and ischaemic livers after 6 h of reperfusion and measured Nr0b2 transcript levels by quantitative RT-PCR. SHP levels were upregulated significantly in NPCs from ischaemic livers of KO mice only (>10-fold vs. sham controls), but not in those of WT mice (Fig. 4A). This is consistent with the SHP induction profiles in BMMs in vitro (Fig. 3D). Thus, myeloid Gsk3β deficiency facilitates SHP induction in liver macrophages by IR.
Fig. 4. AMP-activated protein kinase (AMPK) inhibition increased liver ischaemia–reperfusion (IR) injury in myeloid glycogen synthase kinase 3β (Gsk3β) knockout (KO) mice.

(A) Liver non-parenchymal cells (NPCs) were isolated from sham or ischaemic livers of myeloid Gsk3β wild-type (WT) or KO mice after 6 h of reperfusion. Total cellular RNA was isolated, and Nr0b2 expression levels were analysed by quantitative reverse transcription PCR (qRT-PCR). (B) Myeloid Gsk3β WT or KO mice were treated with either vehicle control or compound C (AMPK inhibitor) before the onset of liver ischaemia, as described in “Materials and methods”. Average serum alanine aminotransferase (ALT) levels in different groups of mice are plotted. Representative haematoxylin and eosin (H&E) staining of tissue sections of ischaemic livers after 6 h of reperfusion is shown, as well as average Suzuki scores based on liver H&E-stained sections of different groups of mice. (C) Liver Nr0b2 and inflammatory gene expression levels were measured by qRT-PCR. Average target gene/Hprt1 gene ratios of different experimental groups are plotted. All results are representative of at least two independent experiments. Four to six mice per group. Data were tested for normal distribution and analysed by one-way analysis of variance with post-test. *p <0.05. Comp. C, compound C; PBS, phosphate-buffered saline.
To test the functional significance of macrophage AMPK activation in myeloid Gsk3b KO mice in response to IR, we treated mice with the AMPK inhibitor compound C before the onset of liver ischaemia. As AMPK was activated only in KO cells but not WT cells early in their response, AMPK inhibition did not affect liver IRI at 6 h after reperfusion in WT mice. However, it did abrogate the liver-protective phenotype in myeloid Gsk3b KO mice, as evidenced by both elevated sALT levels and worse damaged liver histological architectures with higher Suzuki scores (Fig. 4B). The inhibition of liver AMPK by treatment with compound C was confirmed by the western blot analysis (Fig. S2), which revealed the abolishment of SHP induction in KO mice by IR (Fig. 4C). Liver inflammatory immune response became more proinflammatory in the KO mice than in the WT mice in response to IR such that Tnf, Il6, and Cxcl10 gene expression increased, with a simultaneous decrease of Il10 gene expression (Fig. 4C).
Downstream of AMPK activation, we also determined the functional roles of SHP induction in liver inflammation and IRI by the siRNA-mediated knockdown approach. We used mannose-conjugated polymers to deliver siRNA specifically to phagocytes in vivo.43 SHP induction in Gsk3b KO mice by IR was inhibited by Nr0b2-specific siRNA but not control siRNA (Fig. 5B), which led to increased liver IRI in these mice, as evidenced by higher levels of sALT and worse damaged liver histological architecture with higher Suzuki scores (Fig. 5A). No such effects were seen in WT mice. This differential effect of SHP knockdown in Gsk3b-deficient mice vs. WT mice is consistent with our in vitro result that SHP was induced early only in Gsk3b-deficient macrophages (Fig. 3). Liver inflammatory immune response against IR became more proinflammatory in those Nr0b2 siRNA-treated Gsk3b KO mice such that liver Tnf, Il6, and Cxcl10 gene expression increased, with simultaneous decrease of Il10 gene expression, as compared with mice treated with control siRNA (Fig. 5B).
Fig. 5. Small heterodimer partner (SHP) knockdown increased liver ischaemia–reperfusion injury in myeloid glycogen synthase kinase 3β (Gsk3β) knockout (KO) mice.

Myeloid Gsk3β wild-type (WT) or KO mice were treated with either control or SHP small interfering RNA before the onset of liver ischaemia, as described in “Materials and methods”. (A) Average serum alanine aminotransferase (sALT) levels in different groups of mice are plotted. Representative haematoxylin and eosin (H&E) staining of tissue sections of ischaemic livers after 6 h of reperfusion is shown, as well as average Suzuki scores based on liver H&E-stained sections of different groups of mice. (B) Liver Nr0b2 and inflammatory gene expression levels were measured by quantitative reverse transcription PCR. Average target gene/hypoxanthine guanine phosphoribosyl transferase gene ratios of different experimental groups are plotted. All results are representative of at least two independent experiments. There was from four to six mice per group. Data were tested for normal distribution and analysed by one-way analysis of variance with post-test. *p <0.05. Ctl, control.
Lastly, we tested whether pharmacological activation of AMPK could protect livers from IRI in WT mice. Intravenous administration of AICAR before the onset of liver ischaemia increased liver AMPK activation (Fig. S2), which led to reduced liver IRI in WT mice, as shown by significantly lower sALT levels and better preserved liver histological architecture with lower Suzuki scores as compared with controls (Fig. 6A). In myeloid Gsk3b KO mice, the protective effect was not as significant as in WT mice, possibility because AMPK was already activated. Although AMPK activation was not sufficient to upregulate SHP expression in IR livers of WT mice, it prevented its downregulation after IR (Fig. 6B). Most strikingly, liver proinflammatory gene inductions were inhibited with a simultaneous increase of Il10 gene induction after IR by the treatment in WT mice (Fig. 6B). These results establish a novel regulatory axis downstream of TLRs whereby the Gsk3β–AMPK–SHP path-waammatory tissy controls innate macrophage activation and liver influe injury from IR.
Fig. 6. AMP-activated protein kinase (AMPK) activation protected livers from ischaemia–reperfusion injury in wild-type (WT) mice.

Myeloid glycogen synthase kinase 3β WT or knockout (KO) mice were treated with either vehicle control or 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) before the onset of liver ischaemia, as described in “Materials and methods”. (A) Average serum alanine aminotransferase (ALT) levels in different groups of mice are plotted. Representative haematoxylin and eosin (H&E) staining of tissue sections of ischaemic livers after 6 h of reperfusion is shown, as well as average Suzuki scores based on liver H&E-stained sections of different groups of mice. (B) Liver Nr0b2 and inflammatory gene expression levels were measured by quantitative reverse transcription PCR. Average target gene/hypoxanthine guanine phosphoribosyl transferase gene ratios of different experimental groups are plotted. All results are representative of at least two independent experiments. There was from four to six mice per group. Data were tested for normal distribution and analysed by one-way analysis of variance with post-test. *p <0.05. Ctl., control.
Gsk3β inhibition of AMPK/SHP is independent of the PI3K–Akt pathway
The kinase activity of Gsk3β is negatively regulated by the PI3K–Akt signalling pathway via Ser9 phosphorylation. In BMMs, TLR4 stimulation triggered rapid phosphorylation in both Akt at Thr308 and Ser473 and Gsk3β at Ser9, which all peaked at 6 h after stimulation (Fig. 7A). At the same time, AMPK inhibitory phosphorylation (Thr479) was at the highest level and AMPK activating phosphorylation (Thr172) was at the lowest level. The phosphorylation at Ser79 of ACC, the direct substrate of AMPK, was also at the lowest level at 6 h. This kinetic discrepancy between Gsk3β inhibitory phosphorylation and AMPK activities indicates that Gsk3β inhibits AMPK (at Thr479) in macrophages regardless of its Ser9 phosphorylation status. This is consistent with the initial finding by Suzuki et al. in epithelial cells.40 To further establish that Gsk3β regulates AMPK/SHP independently of the PI3K–Akt pathway, we studied Pten-deficient BMMs, in which PI3K was constitutively active, leading to increased activation of Akt and inhibitory phosphorylation of Gsk3β at Ser9.44 SHP induction in response to LPS stimulation in the presence of constitutively active PI3K–Akt was the same as that in the WT counterparts such that Nr0b2 gene expression was downregulated at 6 h and recovered at 24 h, despite Tnf expression being lower and Il10 expression being higher compared with the levels in WT cells (Fig. 7B, left panel). Furthermore, in Gsk3β S9A mutant knock-in BMMs, in which Gsk3β was resistant to PI3K–Akt-induced inhibitory phosphorylation, the Nr0b2 gene expression profile in response to LPS was the same as that in WT cells, although Tnf expression was higher and Il10 expression was lower than in WT cells (Fig. 7B, right panel). These data demonstrate that Gsk3β inhibits AMPK activation independently of its Ser9 phosphorylation and the PI3K–Akt signalling pathway.
Fig. 7. Glycogen synthase kinase 3β (Gsk3β) regulated AMP-activated protein kinase (AMPK)/small heterodimer partner (SHP) independently of the phosphoinositide 3-kinase–Akt pathway.

(A) Wild-type (WT) bone marrow-derived macrophages (BMMs) were stimulated with lipopolysaccharide (LPS) for 0, 3, 6, 12, or 24 h. Total cellular proteins were extracted and analysed by western blotting with anti-phosphorylated Akt (Thr308) (p-AKT(T308)), Akt (Ser473) (pAKT(S473)), Gsk3β (Ser9) (p-Gsk3β(S9)), AMPK (Thr479) (p-AMPK(T479)), AMPK (Thr172) (p-AMPK(T172), and acetyl-CoA carboxylase (p-ACC) (Ser79), and β-actin. (B) Phosphatase and tensin homologue (Pten) WT or knockout (KO) BMMs (left panel) or Gsk3β S9A mutant knock-in (KI) BMMs (right panel) were stimulated with LPS for 6 or 24 h. Total cellular RNA was prepared and reverse transcribed. Tnf, Il10, Nr0b2, and Hprt1 gene expression levels were measured by quantitative reverse transcription PCR. Average target gene/Hprt1 gene ratios of different experimental groups are plotted. All results are representative of at least two independent experiments. There were three mice per group. Data were tested for normal distribution and analysed by one-way analysis of variance with post-test. *p <0.05. Ctl, control.
Prolonged ischaemia inhibits AMPK in human livers
To test the clinical relevance of the Gsk3β–AMPK–SHP signalling pathway in liver ischaemia, we collected series of peritumoral tissues during hepatic tumour resection after various lengths of ischaemia (0–50 min) from five patients with hepatocellular carcinoma. Western blot analysis showed that AMPK phosphorylation at Thr172 was significantly downregulated by prolonged ischaemia (Fig. 8; 50 vs. 0 min). Downstream, both ACC phosphorylation at Ser79 and SHP levels were decreased in livers after 50 min of ischaemia. Gsk3β Ser9 phosphorylation, by contrast, was increased after prolonged ischaemia. These results indicate that prolonged liver ischaemia inhibits AMPK activities in a Gsk3β Ser9 phosphorylation-independent manner.
Fig. 8. Prolonged ischaemia inhibits AMP-activated protein kinase (AMPK) activities in human livers.

Human liver tissues peripheral of tumours were collected during hepatic tumour resection after various lengths of ischaemia (0, 10, 30, and 50 min) from five patients with hepatocellular carcinoma, as described in “Materials and methods”. Western blot analysis was performed to determine phosphorylated (Thr172) and total levels of AMPK, as well as phosphorylated acetyl-CoA carboxylase (Ser79) (p-ACC (Ser79)), phosphorylated glycogen synthase kinase 3β (Ser9) [p-Gsk3β (S9)], and total small heterodimer protein (SHP). β-Actin was used as a control. Two representative results are shown.
Discussion
This study focused on the immune regulatory role of Gsk3β in macrophages and in liver inflammatory responses against IR. By use of the Cre-LoxP system, we analysed Gsk3β in a kinase isoform-specific and cell type-specific manner in vitro and in vivo. The results demonstrate that Gsk3β controls liver innate immune response via the AMPK–SHP signalling pathway. Gsk3β was shown previously in vitro in fibroblast cell lines to directly phosphorylate the AMPK α subunit at Thr479 to reduce its activating phosphorylation at Thr172.40 AMPK activation is required for the induction of SHP in macrophages downstream of TLRs.42 On the basis of these findings, we have established in the current study both in vitro in macrophages and in vivo in livers that active Gsk3β promotes proinflammatory immune activation and liver IRI by restraining AMPK activation to prevent SHP induction. Thus, Gsk3b-deficient macrophages respond to inflammatory stimulation with enhanced AMPK activation and SHP induction, leading to inhibition of proinflammatory innate immune activation and protection of liver from IRI. Targeting the Gsk3β–AMPK–SHP pathway may provide novel therapeutic strategies to reduce liver inflammatory tissue injury from IR.
Gsk3β has been shown to regulate innate immune responses in both macrophages and dendritic cells.12,45 Gsk3 chemical inhibitors or siRNA selectively inhibited monocyte production of proinflammatory cytokines, including IL-12p40, TNF-β, and IL-6, while increasing IL-10 production on TLR stimulation.21 The major effector mechanism downstream of Gsk3β is mediated by the two prototypical transcription factors for pro- and anti-inflammatory cytokines (i.e. NF-κB and CREB).21 Gsk3β is able to directly phosphorylate CREB at Ser129, which is believed to decrease CREB binding with CREB-binding protein (CBP) and its transcriptional activities. With a limited amount of nuclear CBP, this results in the enhancement of NF-κB binding with CBP and its target gene transcription.46 Importantly, Gsk3β Ser9 phosphorylation inhibits its kinase activity towards CREB, which will shift the CBP binding balance towards CREB. In BMMs, TLR4 activation triggers PI3K signalling,47 leading to the Akt-mediated phosphorylation of Gsk3β at Ser9 within 1 h. Thus, the PI3K–Akt–Gsk3β Ser9 phosphorylation signalling pathway is a rapid autoregulatory mechanism of macrophage inflammatory activation. In our study, NF-κB and CREB activating phosphorylations in BMMs seemed to be normal in the absence of Gsk3β. It was the AMPK activation and SHP induction that were turned on earlier and at higher levels in Gsk3b-deficient vs. Gsk3b-proficient cells on stimulation. SHP has been documented recently in macrophages as an endogenous negative regulator in inflammatory signalling downstream of TLRs and NLRs.48 It suppresses NF-κB activities by physically binding to both the NF-κB p65 subunit in the resting state and TNF receptor-associated factor 6 on stimulation to inhibit its polyubiquitination and activation, leading to the downregulation of Tnf and Il6 transcription.42 SHP also interferes with the interaction between NLRP3 and ASC to intercept the assembly of the NLRP3 inflammasome complex, resulting in the downregulation of IL-1β and IL-18 production in NLRP3-activated macrophages.41 SHP could be translocated to the mitochondria with the NLRP3–ASC complex after inflammasome activation, and acts to regulate mitochondrial homeostasis through the recovery of inflammasome-induced mitochondrial reactive oxygen species generation and damage.42 The expression of SHP is regulated at the transcriptional level by multiple known transcriptional factors. Upstream stimulatory factor 1 is critical for SHP induction in macrophages downstream of AMPK activation on innate stimulation.42 Additionally, large numbers of nuclear receptors, including steroidogenic factor 1, liver receptor homologue 1, farnesoid X receptor, hepatocyte nuclear factor 4α, liver X receptor α, oestrogen receptor α, and peroxisome proliferatoractivated receptor, have been shown in hepatocytes to bind to the SHP promoter and regulate its expression.49
What we have found in the current study is that both the activation of AMPK and induction of SHP in macrophages are restrained by Gsk3β. TLR stimulation resulted in initial reductions of the SHP level at both the transcript level and the protein level. SHP upregulation occurs rather late (12–24 h) as compared with that of inflammatory cytokine/chemokines (from minutes to hour). In both Gsk3β-deficient cells and WT cells treated with a Gsk3 inhibitor, however, SHP is induced much earlier (30 min), and the level is significantly higher than in Gsk3β-proficient cells. This indicates that although the activation of the AMPK–SHP signalling pathway is able to regulate innate immune activation, its normal function in WT macrophages/mice is probably in inflammation resolution. This is supported by our in vivo results in WT mice indicting that pharmacological activation of AMPK inhibited liver inflammatory immune activation and protected livers from IRI at 6 h after reperfusion, but that neither AMPK inhibition nor SHP knockdown enhanced inflammatory immune activation or increased acute hepatocellular injuries at the same time point. Thus, the Gsk3β–AMPK–SHP signalling pathway functions distinctively from the Gsk3β–CREB pathway in regulation of innate immune activation and liver IRI, with the latter activated immediately after TLR stimulation to balance the expression of pro- and anti-inflammatory genes, and the former is activated late in the response to resolve inflammation/or repair damage.
Both Akt and Gsk3β have been shown to negatively regulate AMPK activation.40 As Gsk3β is constitutively active in resting cells and Akt is activated immediately on TLR stimulation, they prevent early AMPK activation and SHP induction in macrophages, which allows the proinflammatory immune activation. Although Akt activation results in inhibitory phosphorylation of Gsk3β at Ser9, it does not inhibit Gsk3β kinase activity towards AMPK.40 The independence of the Gsk3β regulation of AMPK/SHP from its inhibitory phosphorylation by PI3K/Akt is further supported by the finding that SHP induction was neither elevated by the constitutively active PI3K–Akt in Pten-deficient BMMs nor prevented by the constitutively active Gsk3β (in Gsk3β S9A mutant knock-in BMMs). Akt activation (measured by Thr308 and Ser473 phosphorylation) started to decline after 12 h of stimulation in BMMs. This fits with the delayed kinetics of AMPK activation and SHP induction. However, the mechanisms of Gsk3β inactivation (Ser9 phosphorylation independent) which will free AMPK from its Thr479 inhibitory phosphorylation remain to be determined. Potential candidates include inhibitory phosphorylation of Gsk3β at Ser389/Thr390 by p38 mitogen-activated protein kinase,50 and intracellular protein complex formation or distinctive subcellular organelle location which precludes Gsk3β from interacting with AMPK.51,52 Alternatively, the appearance of an AMPK activator, such as AMP, might simply overcome the Gsk3β-mediated AMPK inhibition, as shown in our in vivo experiment with AICAR.
Different from previous studies on molecular targets of Gsk3β and SHP regulation,21,42 we found that expression of IL10, rather than proinflammatory cytokines (such as TNF-α and IL-6), was altered most significantly in Gsk3b-deficient macro-phages. Its transcription was markedly increased and sensitive to AMPK inhibition and SHP knockdown. This is in sharp contrast to the result in SHP-deficient cells,42 where IL-10 induction was not changed at both the transcriptional and translational level. A possible reason is that Gsk3β is involved in regulating not only SHP induction but also CREB and AMPK activation, which will impact much more downstream effector pathway/molecules. They may synergistically regulate IL-10 production in macrophages in their response to innate immune stimulation. Indeed, AMPK has been documented recently to exert potent anti-inflammatory effects by altering cellular metabolic preferences from glycolysis to mitochondrial oxidation, which favours the development of M2 macrophages. Additionally, Gsk3β is a key enzyme in glycose metabolism. Its inactivation enhances glycogen synthesis, which may alter glucose flux and glycolysis. Thus, Gsk3β may regulate inflammatory immune response via not only direct transcriptional control of cytokine/chemokine genes but also regulation of intracellular metabolism to indirectly affect macrophage differentiation/activation.
In summary, our results demonstrate a novel innate immune regulatory axis mediated by Gsk3β in liver inflammatory response against IR. Targeting key molecules in this axis may provide a unique therapeutic strategy to reduce liver IRI in patients by actively modulating the innate immune response from proinflammatory to immune regulatory/proresolving. As Gsk3β, AMPK, and SHP also play important roles in cellular metabolism, our study of their functions in disease pathogenesis may shed light on the potential interplay of metabolism and inflammation, which will further our understanding of liver immunobiology and disease mechanisms.
Supplementary Material
Acknowledgments
Financial support
The study was supported by NIH grants R21AI126516 (Y.Z.) and R01DK102110 (J.W.K.-W.), the Dumont Research Foundations, and grants from the National Nature Science Foundation of China: 81600450 (H.Z.) and 81522020, 91442117, and 81571564 (L.L.).
Footnotes
Conflict of interest
The authors declare no conflicts of interest that pertain to this work.
Please refer to the accompanying ICMJE disclosure forms for further details.
Supplementary data
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.jhep.2018.01.036.
References
- [1].Zhai Y, Busuttil RW, Kupiec-Weglinski JW. Liver ischemia and reperfusion injury: new insights into mechanisms of innate-adaptive immunemediated tissue inflammation. Am J Transplant 2011;11:1563–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kaczorowski DJ, Tsung A, Billiar TR. Innate immune mechanisms in ischemia/reperfusion. Front Biosci 2009;1:91–98. [DOI] [PubMed] [Google Scholar]
- [3].Zhai Y, Qiao B, Shen XD, Gao F, Busuttil RW, Cheng G, et al. Evidence for the pivotal role of endogenous toll-like receptor 4 ligands in liver ischemia and reperfusion injury. Transplantation 2008;85:1016–1022. [DOI] [PubMed] [Google Scholar]
- [4].Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, et al. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med 2007;204:2913–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med 2005;201:1135–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Huang H, Evankovich J, Yan W, Nace G, Zhang L, Ross M, et al. Endogenous histones function as alarmins in sterile inflammatory liver injury through Toll-like receptor 9 in mice. Hepatology 2011;54:999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tsung A, Hoffman RA, Izuishi K, Critchlow ND, Nakao A, Chan MH, et al. Hepatic ischemia/reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J Immunol 2005;175:7661–7668. [DOI] [PubMed] [Google Scholar]
- [8].Zhai Y, Shen XD, O’Connell R, Gao F, Lassman C, Busuttil RW, et al. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J Immunol 2004;173:7115–7119. [DOI] [PubMed] [Google Scholar]
- [9].Bamboat ZM, Balachandran VP, Ocuin LM, Obaid H, Plitas G, DeMatteo RP. Toll-like receptor 9 inhibition confers protection from liver ischemiareperfusion injury. Hepatology 2010;51:621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Inoue Y, Shirasuna K, Kimura H, Usui F, Kawashima A, Karasawa T, et al. NLRP3 regulates neutrophil functions and contributes to hepatic ischemia-reperfusion injury independently of inflammasomes. J Immunol 2014;192:4342–4351. [DOI] [PubMed] [Google Scholar]
- [11].Beurel E, Jope RS. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog Neurobiol 2006;79:173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Beurel E, Michalek SM, Jope RS. Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3). Trends Immunol 2010;31:24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cohen P, Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat Rev Drug Discov 2004;3:479–487. [DOI] [PubMed] [Google Scholar]
- [14].Wu D, Pan W. GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem Sci 2010;35:161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3b in cell survival and NF-jB activation. Nature 2000;406:86–90. [DOI] [PubMed] [Google Scholar]
- [16].MacAulay K, Doble BW, Patel S, Hansotia T, Sinclair EM, Drucker DJ, et al. Glycogen synthase kinase 3a-specific regulation of murine hepatic glycogen metabolism. Cell Metab 2007;6:329–337. [DOI] [PubMed] [Google Scholar]
- [17].Avrahami L, Licht-Murava A, Eisenstein M, Eldar-Finkelman H. GSK-3 inhibition: achieving moderate efficacy with high selectivity. Biochim Biophys Acta 2013;1834:1410–1414. [DOI] [PubMed] [Google Scholar]
- [18].McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, et al. Role that phosphorylation of GSK3 plays in insulin. EMBO J 2005;24:1571–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Rayasam GV, Tulasi VK, Sodhi R, Davis JA, Ray A. Glycogen synthase kinase 3: more than a namesake. Br J Pharmacol 2009;156:885–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang H, Brown J, Martin M. Glycogen synthase kinase 3: a point of convergence for the host inflammatory response. Cytokine 2011;53:130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol 2005;6:777–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Coant N, Simon-Rudler M, Gustot T, Fasseu M, Gandoura S, Ragot K, et al. Glycogen synthase kinase 3 involvement in the excessive proinflammatory response to LPS in patients with decompensated cirrhosis. J Hepatol 2011;55:784–793. [DOI] [PubMed] [Google Scholar]
- [23].Ren F, Duan Z, Cheng Q, Shen X, Gao F, Bai L, et al. Inhibition of glycogen synthase kinase 3 beta ameliorates liver ischemia reperfusion injury by way of an interleukin-10-mediated immune regulatory mechanism. Hepatology 2011;54:687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chiara F, Gambalunga A, Sciacovelli M, Nicolli A, Ronconi L, Fregona D, et al. Chemotherapeutic induction of mitochondrial oxidative stress activates GSK-3α/β and Bax, leading to permeability transition pore opening and tumor cell death. Cell Death Dis 2012;3:e444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Miura T, Tanno M. The mPTP and its regulatory proteins: final common targets of signalling pathways for protection against necrosis. Cardiovasc Res 2012;94:181–189. [DOI] [PubMed] [Google Scholar]
- [26].Vigneron F, Dos Santos P, Lemoine S, Bonnet M, Tariosse L, Couffinhal T, et al. GSK-3β at the crossroads in the signalling of heart preconditioning:implication of mTOR and Wnt pathways. Cardiovasc Res 2011;90:49–56. [DOI] [PubMed] [Google Scholar]
- [27].Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, et al. Glycogen synthase kinase-3βeta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest 2004;113:1535–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Juhaszova M, Zorov DB, Yaniv Y, Nuss HB, Wang S, Sollott SJ. Role of glycogen synthase kinase-3β in cardioprotection. Circ Res 2009;104:1240–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fu H, Xu H, Chen H, Li Y, Li W, Zhu Q, et al. Inhibition of glycogen synthase kinase 3 ameliorates liver ischemia/reperfusion injury via an energy-dependent mitochondrial mechanism. J Hepatol 2014;61:816–824. [DOI] [PubMed] [Google Scholar]
- [30].Xia Y, Rao J, Yao A, Zhang F, Li G, Wang X, et al. Lithium exacerbates hepatic ischemia/reperfusion injury by inhibiting GSK-3β/NF-κB-mediated protective signaling in mice. Eur J Pharmacol 2012;697:117–125. [DOI] [PubMed] [Google Scholar]
- [31].Abdellatif M Sirtuinsandpyridinenucleotides. CircRes 2012;111:642–656. [Google Scholar]
- [32].Shen XD, Ke B, Zhai Y, Amersi F, Gao F, Anselmo DM, et al. CD154-CD40 T-cell costimulation pathway is required in the mechanism of hepatic ischemia/reperfusion injury, and its blockade facilitates and depends on heme oxygenase-1 mediated cytoprotection. Transplantation 2002;74:315–319. [DOI] [PubMed] [Google Scholar]
- [33].Suzuki S, Toledo-Pereyra LH, Rodriguez FJ, Cejalvo D. Neutrophil infiltration as an important factor in liver ischemia and reperfusion injury. Modulating effects of FK506 and cyclosporine. Transplantation 1993;55:1265–1272. [DOI] [PubMed] [Google Scholar]
- [34].Zhai Y, Shen XD, Gao F, Zhao A, Freitas MC, Lassman C, et al. CXCL10 regulates liver innate immune response against ischemia and reperfusion injury. Hepatology 2008;47:207–214. [DOI] [PubMed] [Google Scholar]
- [35].Johannessen M, Delghandi MP, Moens U. What turns CREB on? Cell Signal 2004;16:1211–1227. [DOI] [PubMed] [Google Scholar]
- [36].Oeckinghaus A, Ghosh S. The NF-jB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol 2009;1:a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Steinberg GR, Schertzer JD. AMPK promotes macrophage fatty acid oxidative metabolism to mitigate inflammation: implications for diabetes and cardiovascular disease. Immunol Cell Biol 2014;92:340–345. [DOI] [PubMed] [Google Scholar]
- [38].Mounier R, Theret M, Arnold L, Cuvellier S, Bultot L, Goransson O, et al. AMPKa1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab 2013;18:251–264. [DOI] [PubMed] [Google Scholar]
- [39].Sag D, Carling D, Stout RD, Suttles J. Adenosine 5’-monophosphateactivated protein kinase promotes macrophage polarization to an antiinflammatory functional phenotype. J Immunol 2008;181:8633–8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Suzuki T, Bridges D, Nakada D, Skiniotis G, Morrison SJ, Lin JD, et al. Inhibition of AMPK catabolic action by GSK3. Mol Cell 2013;50:407–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yang C-S, Kim J-J, Kim TS, Lee PY, Kim SY, Lee H-M, et al. Small heterodimer partner interacts with NLRP3 and negatively regulates activation of the NLRP3 inflammasome. Nat Commun 2015;6:6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yuk JM, Shin DM, Lee HM, Kim JJ, Kim SW, Jin HS, et al. The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nat Immunol 2011;12:742–751. [DOI] [PubMed] [Google Scholar]
- [43].Rao J, Yue S, Fu Y, Zhu J, Wang X, Busuttil RW, et al. ATF6 mediates a proinflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ischemia-reperfusion injury. Am J Transplant 2014;14:1552–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yue S, Rao J, Zhu J, Busuttil RW, Kupiec-Weglinski JW, Lu L, et al. Myeloid PTEN deficiency protects livers from ischemia reperfusion injury by facilitating M2 macrophage differentiation. J Immunol 2014;192:5343–5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Wang H, Kumar A, Lamont RJ, Scott DA. GSK3b and the control of infectious bacterial diseases. Trends Microbiol 2014;22:208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tullai JW, Chen J, Schaffer ME, Kamenetsky E, Kasif S, Cooper GM. Glycogen synthase kinase-3 represses cyclic AMP response elementbinding protein (CREB)-targeted immediate early genes in quiescent cells. J Biol Chem 2007;282:9482–9491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Li X, Jiang S, Tapping RI. Toll-like receptor signaling in cell proliferation and survival. Cytokine 2010;49:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yuk JM, Jin HS, Jo EK. Small heterodimer partner and innate immune regulation. Endocrinol Metab 2016;31:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhang Y, Wang L. Nuclear receptor small heterodimer partner in apoptosis signaling and liver cancer. Cancers 2011;3:198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Thornton TM, Pedraza-Alva G, Deng B, Wood CD, Aronshtam A, Clements JL, et al. Phosphorylation by p38 MAPK as an alternative pathway for GSK3βeta inactivation. Science 2008;320:667–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kaidanovich-Beilin O, Woodgett JR. GSK-3: functional insights from cell biology and animal models. Front Mol Neurosci 2011;4:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther 2015;148:114–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.