

Abstract
Linker histones bind to the nucleosome and regulate the structure and function of chromatin. We have previously shown that the globular domains of chicken H5 and Drosophila H1 linker histones bind to the nucleosome with on- or off-dyad modes, respectively. To explore the determinant for the distinct binding modes, we investigated the binding of a mutant globular domain of H5 to the nucleosome. This mutant, termed GH5_pMut, includes substitutions of five globular domain residues of H5 with the corresponding residues in the globular domain of Drosophila H1. The residues at these five positions play important roles in nucleosome binding by either H5 or Drosophila H1. NMR and spin-labeling experiments showed that GH5_pMut bound to the nucleosome off the dyad. We further found that the nucleosome array condensed by either the GH5_pMut or the globular domain of Drosophila H1 displayed a similar sedimentation coefficient, whereas the same nucleosome array condensed by the wild-type globular domain of H5 showed a much larger sedimentation coefficient. Moreover, NMR and spin-labeling results from the study of the nucleosome in complex with the full-length human linker histone H1.0, whose globular domain shares high sequence conservation with the corresponding globular domain of H5, are consistent with an on-dyad binding mode. Taken together, our results suggest that a small number of residues in the globular domain of a linker histone can control its binding location on the nucleosome and higher-order chromatin structure.
Keywords: chromatosome, linker histone, nucleosome, chromatin higher-order structure, methyl-TROSY
Introduction
Genomic DNA in eukaryotic cells is packaged into chromatin through association with core histones to form the nucleosome [1–4]. The canonical nucleosome core particle comprises an octamer of histones with two copies of H2A, H2B, H3, and H4, around which ~146-bp DNA winds in ~1.65 left-handed superhelical turns [5,6]. Further packaging involves the formation of the chromatosome, the repeating structural unit of chromatin in metazoans [1,7,8], which contains the nucleosome bound to a linker histone [9,10].
In contrast to the core histones, linker histones exchange rapidly between chromosomal locations [11,12]. As a chromatin factor, linker histones play important roles in regulating cellular functions [13], including gene expression [14,15], mitotic chromosome architecture and segregation [16], muscle differentiation [17], embryonic stem cell differentiation [18], genetic activity of heterochromatin [19], cell pluripotency [20], and ubiquitin signaling after DNA damage [21]. In addition, linker histones may also exist outside the nucleus and are involved in apoptosis [22].
Linker histones have a conserved tripartite structure, which typically consists of a short, flexible N-terminal tail (~25 residue); a central globular domain (~80 residue); and a long (~100 residue) intrinsically disordered, highly basic C-terminal tail. The short N-terminal tail of linker histones contributes little to nucleosome binding [23–25]. The central globular domain preferentially binds to the nucleosome core and linker DNA [23,26,27]. The long C-terminal tail interacts with linker DNA [28–30] and is important for the higher-affinity binding of linker histones to the nucleosome [27,31], the folding of 30-nm chromatin fibers [32], the association of linker histones with chromatin in vivo [24,33], and the stem structure formation of longer linker DNA in vitro [25,34,35].
Earlier studies of nucleosome recognition by linker histones have focused mainly on how the globular domain of chicken H5 (GH5, H524–98) binds to the nucleosome [23,36–38]. Either the full-length linker histone H5 or GH5 alone can protect the same native chromatin linker DNA from micrococcal nuclease digestion [23,39]. Recently, we combined X-ray crystallography and NMR to investigate the structural mechanism of nucleosome recognition by GH5 and found that it binds to the nucleosome on the dyad [40]. Using NMR, we have also shown that the globular domain of Drosophila linker histone H1 (GH1) binds to the nucleosome off the dyad [27]. Here, we explored the determinant for the distinct binding modes of linker histones and found that a small number of residues in the globular domain of linker histones play critical roles in determining the nucleosome-binding modes.
Results and Discussion
A penta mutant of GH5
To explore the determinant for the distinct binding modes of the globular domains of chicken H5 and Drosophila H1 linker histones, we compared the amino acid sequences of the globular domains and the structure/models of the globular domains in complex with the nucleosome. We observed differences in 33 aa, including five positively charged residues that interact with DNA (Fig. 1a). Three positively charged residues (Arg47, Arg74, and Lys97) in the H5 globular domain bind to the dyad and two DNA linkers (Fig. 1b). They correspond to neutral residues (Leu68, Ser96, and Ala119) in the globular domain of Drosophila H1 that do not have close contacts with DNA (Fig. 1c). Conversely, two positively charged residues (Lys102 and Lys109) in the globular domain of Drosophila H1 that interact with one DNA linker and nucleosomal DNA, respectively, correspond to neutral residues (Val80 and Val87) in the H5 globular domain. Residue Val80 of GH5 is fully exposed, whereas residue Val87 inserts into the minor groove of the dyad. We speculated that the difference in the residues at these five locations plays an important role in determining the binding modes of the two globular domains. To test this hypothesis, we replaced the five residues in the globular domain of H5 with the corresponding residues in the globular domain of H1 to create a penta mutant, termed GH5_pMut [GH5(R47L/R74S/K97A/V80K/V87K)]. We anticipated that GH5_pMut would bind to the nucleosome off the dyad.
Fig. 1.
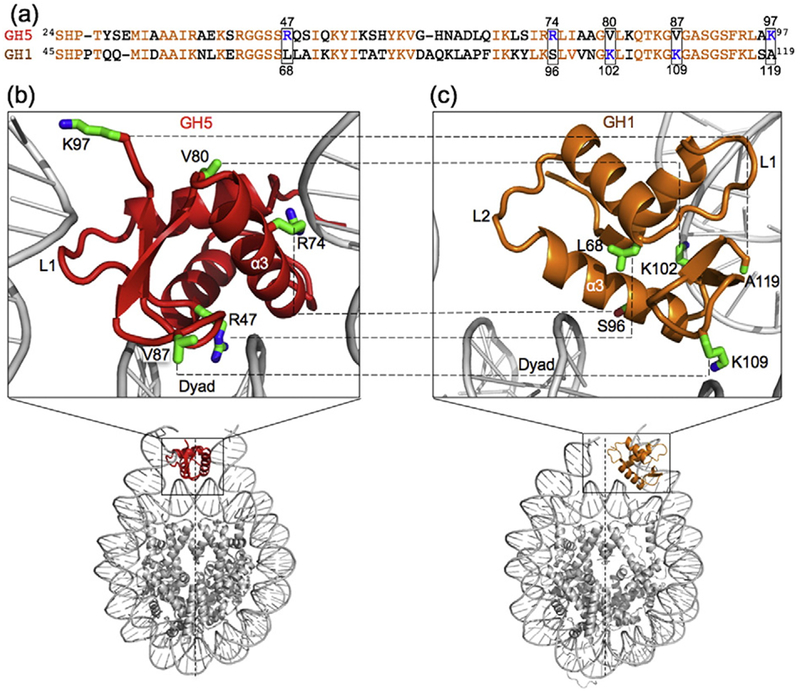
Comparison of the globular domains of chicken H5 and Drosophila H1 in complex with the nucleosome. (a)Sequence comparison of GH5 and GH1. The conserved residues are shown in orange. The boxes highlight the five positively charged resides that are important for the binding of the globular domain of either Drosophila H1 or chicken H5 but correspond to neutral residues in the corresponding globular domain. (b and c) Comparison of the locations of the five key residues in the chromatosome structure containing GH5 [40] and in the chromatosome structural model containing GH1 [27].
GH5_pMut binds to the nucleosome off the dyad
We used methyl-based NMR and spin-labeling methods to examine the binding mode of GH5_pMut on the nucleosome. We first assigned the chemical shifts of the 13C-labeled methyl groups in residues Ile, Leu, and Val [41] of GH5_pMut in complex with the nucleosome by comparing the spectra of the wild-type GH5 and the GH5_pMut in their free forms (Fig. 2a), by site-specific mutations (Fig. 2b–d), and by comparison of the spectra of GH5_pMut in complex with the nucleosome and the free GH5_pMut (Fig. 2e). The nucleosome contains 167-bp DNA centered with the 147-bp “601” nucleosome-positioning sequence [42]. We then mutated H2A Thr119 or H3 Lys37 in the nucleosome to Cys and linked it to the paramagnetic spin label {[S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl) methyl methanethiosulfonate] MTSL} through a disulfide bond. Residues H2A Thr119 and H3 Lys37 in the nucleosome are largely disordered but are close to the folded region. Spin labels at these positions should cause little perturbation to the structure of the nucleosome but have a relatively defined location [27,40]. The effects of the spin labels on the NMR peak intensities of the methyl groups are inversely dependent on their distances to the corresponding methyl groups of the globular domain [43].
Fig. 2.
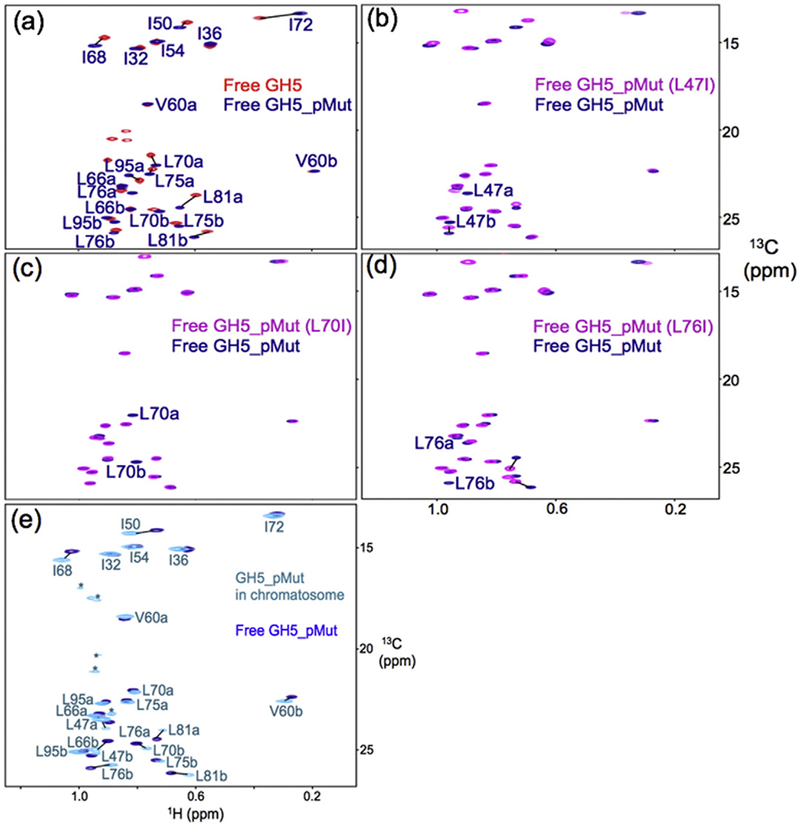
Illustration of chemical shift assignment of the methyl groups of GH5_pMut. (a) 1H–13C HMQC spectra of free wild-type GH5 and free GH5_pMut overlay, allowing the assignment of most of the methyl groups. (b–d) Specific mutations allow unambiguous assignment of the methyl groups of residues L47, L70, and L76. (e) 1H–13C HMQC spectra of GH5_pMut with (cyan) and without (blue) the nucleosome overlay for assigning the methyl groups of GH5_pMut in the chromatosome.
We previously found that the measured intensity changes of the methyl NMR peaks in GH5 are in agreement with the calculated distances between the methyl groups and the spin-label (MTSL) sites, in which the conformer of MTSL is modeled to be close to the GH5 in complex with the nucleosome [40]. Here, we measured the effects of the MTSL spin labels at H2A T119C or H3 Lys37C on the methyl groups of GH5_pMut bound to the nucleosome (Fig. 3). The spin labels at H2A T119 have strong effects on the methyl groups of residue Leu66, leading to a more than 80% decrease in its methyl peak intensities. Spin labels at H3 K37 have strong effects on the methyl groups of Leu47, Ile72, and Leu76 residues, causing a ~60% or greater decrease in their methyl peak intensities. The results from H3 K37 spin labels clearly indicate that GH5_pMut binds off the dyad. By contrast, any methyl group in the globular domain of H5 bound to the nucleosome on-dyad is more than 24 Å away from the atom with a paramagnetic electron and shows a no more than 20% decrease in peak intensity in the presence of MTSL at H3 K37 [40].
Fig. 3.
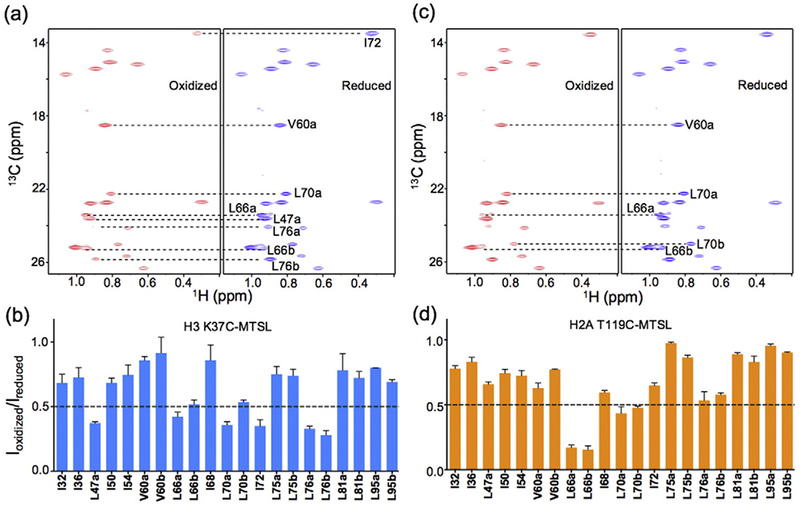
Spin-label results for the GH5_pMut chromatosome. (a) 1H–13C transverse relaxation-optimized spectroscopy spectra of the methyl groups of GH5_pMut with H3 K37C-MTSL in the reduced (red) and oxidized (blue) forms. (a) Spin-label effects on the methyl groups from H3 K37C-MTSL. The values represented by the bars are the means of the two independent experiments. The error bars represent the range of the measured values from the two experiments. (b) 1H–13C HMQC spectra of the methyl groups of GH5_pMut with H2A T119C-MTSL in the reduced (blue) and oxidized (red) forms. (d) Spin-label effects on the methyl groups from H2A T119C-MTSL. The values represented by the bars are the means of the two independent experiments. The error bars represent the deviation of the measured values from the mean or indicate the range of the measured values (with the same absolute values below or above the mean).
We also investigated the binding of GH5_pMut to the nucleosome using isothermal titration calorimetry (ITC) (Fig. 4). We found that GH5_pMut binds to the nucleosome with a dissociation constant (KD) of ~1.2 μM (Table 1), which is essentially the same KD value (~1.3 μM) for GH1 [27]. In contrast, GH5 binds to the nucleosome with a KD value of 0.3 μM [40]. In addition, GH5_pMut and GH1 bind to the nucleosome with a positive change in enthalpy, whereas GH5 binds to the nucleosome with a negative change in enthalpy (Fig. 4). These results further support that GH5_pMut binds to the nucleosome off the dyad.
Fig. 4.
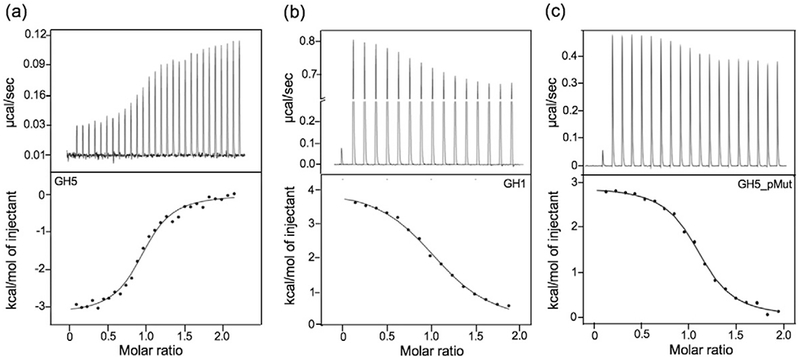
ITC measurements of nucleosome binding of GH5, GH1, or GH5_pMut to the nucleosome, respectively. (a) GH5. (b) GH1. (c) GH5_pMut. The nucleosome includes 167-bp DNA centered with “601” sequence. Data in (a) and (b) are from our earlier publications [27,40].
Table 1.
Parameters of nucleosome binding by GH5_pMut at 25 °C
| Expt_1 | Expt_2 | Average | |
|---|---|---|---|
| N | 1.1 | 1.2 | 1.15 |
| KD (μM) | 1.0 | 1.3 | 1.2 |
| ΔH (kcal/mol) | 2.9 | 3.1 | 3.0 |
| −TΔS (kcal/mol) | −11.1 | −11.1 | −11.1 |
A structural model for the GH5_pMut–nucleosome complex
To build a more detailed structural model for the GH5_pMut–nucleosome complex, we converted the above spin-label effects into distances using the empirical relationship based on the crystal structure of the GH5–nucleosome complex and the results from the spin labels at H2A Thr119 or H3 Lys37 positions [40]. With these distances as a guide, we used PyMol to mutate GH5 to GH5_pMut and to manually place GH5_pMut near the nucleosome with an orientation wherein residues Leu66 and Leu70 are close to the H2A T119 spin-label site and residue Leu76 is close to the H3 K37 spin-label site on one side of the nucleosome structure (Fig. 5a). We then used the HADDOCK program to dock the GH5_pMut to the DNA of the nucleosome with ambiguous restraints between the GH5_pMut and the DNA region near the dyad (Fig. 5a) [44–46]. We found that the distances derived from a docking model were in a good agreement with those calculated from the spin-label data (Fig. 5b and c). This docking model was chosen as the structural model for the GH5_pMut–nucleosome complex.
Fig. 5.
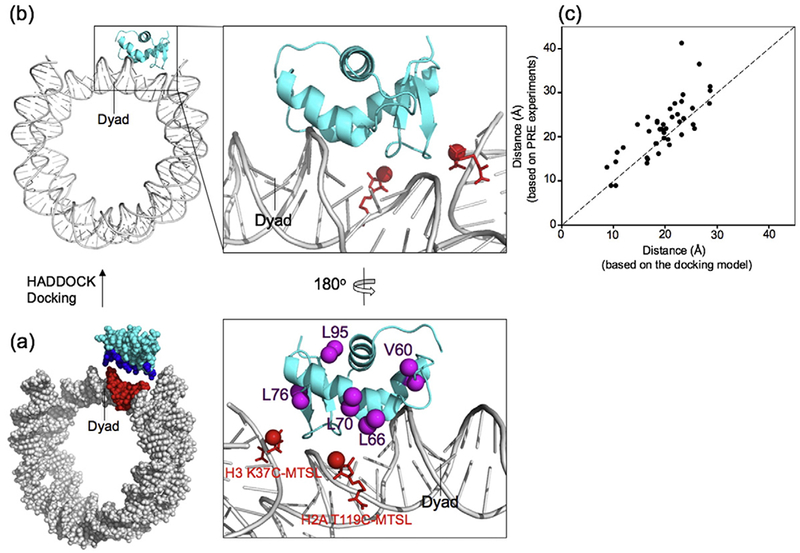
Structural modeling of GH5_pMut in complex with the nucleosome core particle. (a) The input structural model for HADDOCK docking calculations. The unambiguous residues are colored with blue for GH5_pMut and red for DNA. (b) The selected docking model and the enlarged region for GH5_pMut–DNA interactions. The red sticks represent the C-MTSL at H2A T119 and H3 K37 positions. The red sphere represents the paramagnetic oxygen atom in the MTSL, and the magenta spheres represent the methyl groups in the corresponding residues. (c) Comparison of the distances derived from the spin-label experiments and the corresponding distances measured from the structural model of GH5_pMut in complex with the nucleosome core particle. Note that the distances derived from the PRE experiments used the following empirical equation [40]: Ioxidized/Ireduced = exp(β(r + d)−6)/[1 + α(r + d)−6)], where the values of α, β, and d are 4.5 × 108, 3.4 × 107, and 9.0, respectively, in the range from 10 to 50 Å for r. r is the distance between the carbon atom of the methyl group and the oxygen atom in MTSL.
Our initial structural model of the globular domain of Drosophila H1 in complex with the nucleosome was built based on the pattern of the spin-label results and the effects of mutating positively charged surface residues of the globular domain on the binding affinity of the globular domain to the nucleosome [27]. Using the empirical equation describing the relationship between distances and NMR peak intensities [40], we derived the distances from earlier spin-label results and built a new docking model for the GH1–nucleosome complex using a method similar to the establishment of the structural model for the GH5_pMut–nucleosome complex. The new model is similar to the original model and shows a better correlation between the distances derived from spin-label results and those calculated from the structural model (Fig. 6a and b). In comparison with the GH5_pMut model (Fig. 5c), the distance correlation for the model of the GH1–nucleosome complex is weaker (Fig. 6a). It is possible that the structural model of the free globular domain of H1, which was built using the crystal structure of free GH5, may not be exactly the same as GH5 and could lead to the weaker correlation. A comparison of the structures of the GH5_pMut–nucleosome and GH1–nucleosome complexes showed that GH5_pMut and GH1 have similar locations on the nucleosome but have significantly different orientations (Fig. 6c).
Fig. 6.
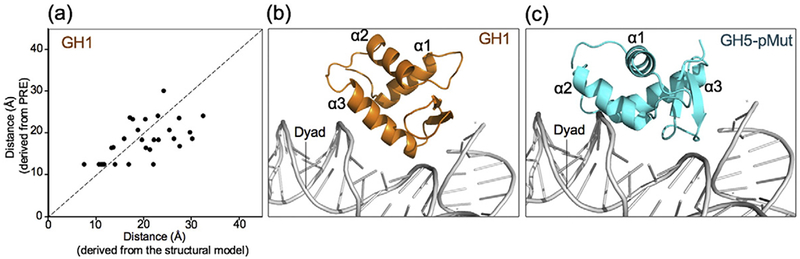
Structural model of the GH1–nucleosome complex and the comparison with the GH5_pMut–nucleosome complex. (a) Comparison of the distances derived from the spin-label experiments and the corresponding distances calculated from the new structural model GH1. (b and c) Comparison of the structural models of GH1 and GH5_pMut in complex with the nucleosome.
The nucleosome arrays condensed by GH5_pMut and GH1 show similar compaction
To examine the effect of GH5_pMut on the structure of chromatin, we performed analytical ultracentrifugation experiments on the nucleosome array condensed by GH1, GH5, or GH5_pMut in the presence of 0.3 mM Mg2+ (Fig. 7). The nucleosome array contains 12 nucleosomes with a nucleosome repeat length of 207-bp DNA centered with the “601” sequence. The sedimentation coefficient of the nucleosome array containing GH1 or GH5_pMut was similar but substantially smaller (by ~3 S) than that of the nucleosome array containing GH5 when the ratio of the globular domain to the nucleosome is at 1 (Fig. 7).
Fig. 7.
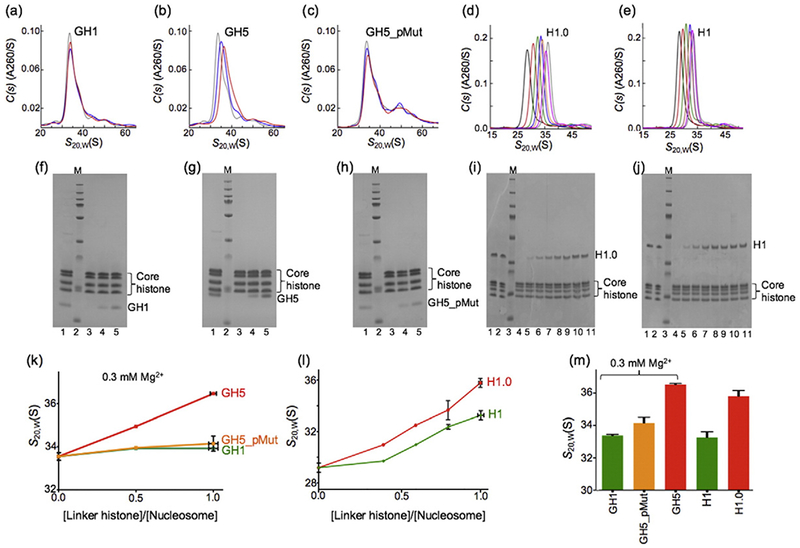
Sedimentation of nucleosome arrays condensed by the globular domain and full-length linker histones. (a–c) Sedimentation velocity c(s) profiles of the nucleosome arrays (12 × 207) condensed by the globular domains (GH1, GH5, and GH5_pMut) in the presence of 0.3 mM Mg2+ with the concentration ratio of the linker histones to the nucleosome at 0,0.5, and 1.0, respectively (see below). (d and e) Sedimentation velocity c(s) profiles of the nucleosome arrays (12 × 207) condensed by the full-length linker histones (Xenopus H1.0 and Drosophila H1) with increasing concentration ratios of the linker histones to the nucleosome from left to right. (f–j) SDS gels corresponding to samples in (a–e), respectively. The intensities of the bands were used to calculate the concentration ratios of the linker histones to the nucleosome. Lane 1 in (f–h) was the reference for which the linker histone to the core histone octamer ratio is 1. Lane 2 in (f–h) is the protein marker; lanes 3, 4, and 5 are nucleosome arrays in which the ratios of linker histone globular domain to the nucleosome are 0.0, 0.5, and 1.0, respectively [plotted in (k)]. (i and j) lanes 1 and 2 are the references for which the linker histone to the core histone octamer ratio is 1. Lane 3 is the protein marker. Lanes 4–11 are the nucleosome arrays with increasing ratios of full-length linker histone to the nucleosome [plotted in (l)]. (k) For the globular domains in the presence of 0.3 mM Mg2+. (l) For the full-length protein at five concentration ratios of linker histone to the nucleosome. (m) Values from (k) and (l) at a concentration ratio of the linker histones to the nucleosome of 1.
For comparison, we also performed analytical ultracentrifugation experiments on the same nucleosome array condensed by full-length Drosophila H1 or Xenopus H1.0. Here, we used Xenopus H1.0 to replace H5, since a full-length H5 cannot be expressed in Escherichia coli. The globular domains of Xenopus H1.0 and H5 share 84% sequence identity, and all of the residues important for nucleosome binding are conserved. Based on phylogeny analysis, it has been suggested that H5 be renamed H1.0 [47]. We found a similar difference in the sedimentation coefficients of the nucleosome arrays condensed by Drosophila H1 and Xenopus H1.0 when the two globular domains were used in the presence of 0.3 mM Mg2+ (Fig. 7m). These results are consistent with the suggestion that the tails of linker histones play a role in neutralizing the negative charges of the linker DNA and help the arrays to condense but do not dictate the specific structures of the folded nucleosome arrays [40].
Linker histone tails do not control the binding location of the globular domain in chromatosome
To specifically demonstrate that the globular domain in the full-length human H1.0 also binds to the nucleosome on the dyad, we investigated the effects of spin labels at H3 K37 on the 13C-labeled methyl groups of Ile residues of a mutant of human H1.0. There are seven Ile residues in human H1.0. Six of them are in the globular domain. The other one is in the C-terminal tail. The methyl group of the Ile residue in the C-terminal tail showed strong NMR signal, suggesting that this Ile residue has a disordered conformation in the chromatosome. To avoid the strong NMR signal, which reduces the sensitivity of other methyl groups, we mutated this Ile residue to Leu (I113L). We again used the 207-bp DNA centered with the “601” positioning sequence. We found that the spin labels at H3 K37 had little effect (< 15%) on the NMR peak intensity for any of the methyl groups of the six Ile residues (Fig. 8). In contrast, for the GH5_pMut, most of the Ile methyl groups showed a larger decrease in their peak intensities due to the spin labels at H3 K37 (Fig. 3b). In particular, the methyl group in one of the Ile residues (Ile72) showed a decrease of more than 60% in its peak intensity. Thus, these results provide direct evidence that the globular domain in the full-length human H1.0 binds to the dyad of the nucleosome.
Fig. 8.
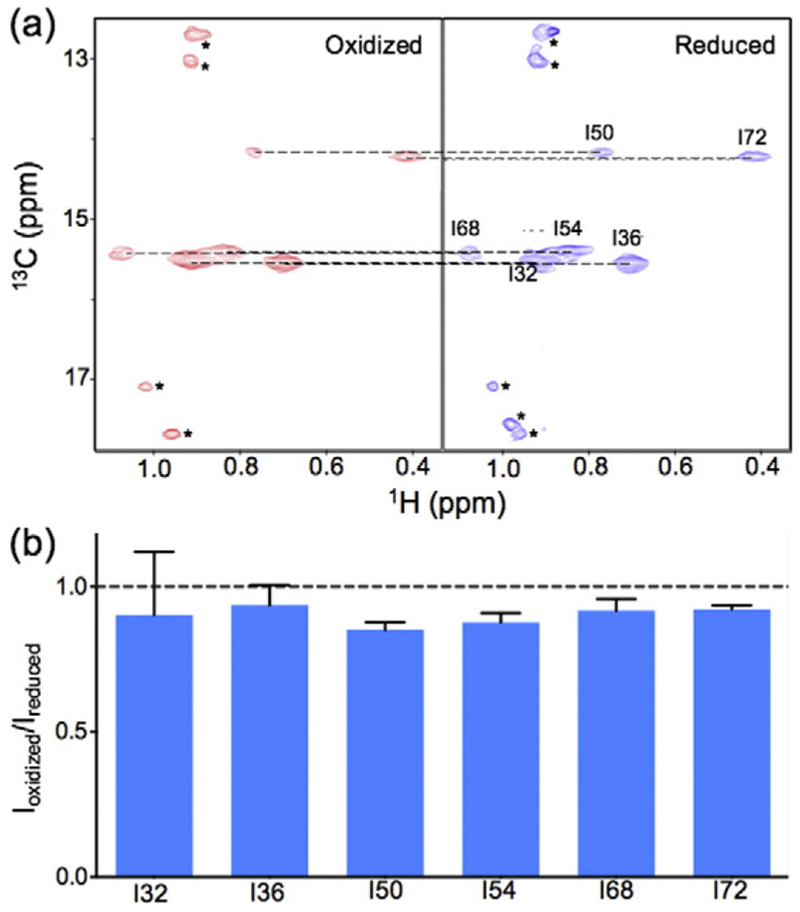
The globular domain in the full-length human H1.0 binds to the nucleosome on the dyad. (a) 1H–13C transverse relaxation-optimized spectroscopy NMR spectra of the Ile methyl groups in the globular domain of human H1.0 in the oxidized and reduced forms for spin-label H3 K37C-MTSL. (b) Bar graphs showing little spin-label effects on the methyl groups. The peaks with an asterisk reflect a natural abundance (13C) of the methyl groups in the core histone tails. The values represented by the bars are the means of the two independent experiments. The error bars represent the deviation of the measured values from the mean or indicate the range of the measured values (with the same absolute values below or above the mean).
Linker histones are known to bind to the nucleosome to form the chromatosome and can condense chromatin into 30-nm fibers in vitro [13]. However, the exact interactions between the linker histones and the nucleosome, and how the linker histones condense chromatin, only begin to be revealed by the structural details [27,40,48]. Our earlier NMR studies of Drosophila H1 showed that the globular domain, either alone or in the full-length protein, binds to the nucleosome in the same off-dyad manner [27], indicating that the determinant of the binding mode is within the globular domain. We have now shown that the five key residues in the globular domain of H5 largely determine the binding location of the globular domain on the nucleosome. We demonstrated that the globular domain in the full-length H1.0 also binds to the nucleosome on the dyad, suggesting that the tails of the linker histones do not play an important role in determining the location of the globular domain in the chromatosome. These results are further supported by our earlier observation that there is an excellent correlation between the effects of mutations on the Fluorescence recovery after photo bleaching (FRAP) residence time of the full-length mouse H1.0 in the chromatin [33] and the binding affinity of the globular domain of H5 to the nucleosome in vitro [40]. Our results are also consistent with the observation that the globular domain of linker histones preferentially binds to both the nucleosomal and the linker DNA, whereas the tails of linker histones do not have such preference [26]. Taken together, these results strongly suggest that the location of nucleosome binding of a linker histone is largely determined by the globular domain. Since the globular domains of linker histones with different binding modes can lead to distinct higher-order chromatin structures, they may also lead to different functions of chromatin.
In addition to condensing the chromatin into higher-order structures such as the 30-nm chromatin fibers, recent studies point to more diverse functions for linker histones that are likely associated with open chromatin [13]. For example, linker histones co-localize with RNA polymerase in uncondensed chromatin regions [49]. Linker histones H1.2 and H1× are recruited to sites of DNA damage, and their C-terminal tails are ubiquitinated [21]. Phosphorylation of the C-terminal tails of linker histones is also associated with chromatin decondensation in the S-phase of the cell cycle [50]. It is likely that ubiquitination and phosphorylation of linker histone tails compromise the role of the C-terminal tails in chromatin condensation. In such cases, the structural role of linker histones in the chromatin would be analogous to that of the core histones. The globular domain of linker histones behaves like the folded regions of the core histones, which are mainly responsible for the folding of DNA. The tails of the linker histones function like the tails of core histones, which can be post-translationally modified to interact with other proteins. In addition, linker histone C-terminal tails may modulate post-translational modifications of the core histone H3 [51].
Materials and Methods
Cloning and purification of linker histones
The DNA sequence of GH5_pMut was commercially synthesized (BioBasic, Ontario). Human H1.0 cDNA was obtained from Origen (Rockville, MD). These were cloned into the pET42b vector in frame with a C-terminal His6-tag using NdeI and BamHI sites. Linker histones were expressed in E. coli BL21(DE3) RIPL cells and purified using Ni-NTA beads (Qiagen, CA). The proteins were further purified using reverse phase HPLC (Protein-RP column, YMC, Japan; water/acetyl nitrile solvent; HPLC instrument 600, Waters) and were lyophilized. GH5_pMut and H1.0 were either 15N/13C or 15N/(Ile, Leu, Val) methyl-labeled by growing the cells in M9 medium containing the corresponding isotopes, as described in our earlier work [27]. Mutations were generated using the Quikchange kit (Stratagene, CA), and the DNA sequences were verified by DNA sequencing (Genewiz, NJ).
DNA construct, core histones, and nucleosome reconstitution
The 167- or 207-bp DNA with a central Widom “601” DNA sequence in the middle was released from the vector using ScaI or EcoRV digestion (New England Biolabs, MA) [52]. Further purification of the DNA followed the procedure described by Dyer et al. [53]. Purification of the Drosophila core histones followed the procedure described previously [54]. Nucleosome reconstitution and purification followed the procedures described previously [54]. Protein, DNA, and nucleosome concentrations were measured using a UV-vis spectrophotometer. The extinction coefficient at 276 nm for proteins was calculated using Protparam†. The extinction coefficient of ε(260 nm) = 1.3 × 104 N−1 cm−1 (N, number of DNA bp) was used for DNA and the nucleosome.
NMR and spin-label experiments
15N/(Ile, Leu, Val) methyl-labeled GH5_pMut or 15N/Ile methyl-labeled H1.0 was mixed with the nucleosome at 0.75:1 ratio for the 1H–13C Heteronuclear Multiple-Quantum Correlation (HMQC) NMR experiments. NMR spectra were collected on Bruker 700 and 900 MHz NMR instruments equipped with cryo-probes. The data were analyzed using NMRPipe [55]. The peak intensities of the well-separated peaks or the volumes of the merged peaks were measured using NMRViewJ (One Moon Scientific Inc. MD) or UCSF Sparky software‡.
To obtain spin-label-derived distance restraints from the nucleosome to the globular domain, we mutated H2A Thr119 or H3 Lys37 to Cys (the H3 Cys110 was mutated to Ala simultaneously) and linked it to MTSL as described previously [27]. Complete labeling was confirmed by mass spectrometry. Nucleosomes bearing the MTSL spin labels were reconstituted in the buffer without reducing agent. Leu, Val, and Ile methyl-labeled GH5_pMut was mixed with spin-labeled nucleosomes at a ratio of 0.75 in the NMR buffer [20 mM Tris–D11-DCl (pD 7.4), 0.1 mM EDTA, and 99.8% D2O]. 1H–13C HMQC spectra of the GH5_pMut–nucleosome complex were recorded before and after reducing the MTSL by adding 2 μl of 1 M DTT.
Assignment of methyl peaks of GH5_pMut and isoleucine methyl peaks of H1.0
To assign the chemical shifts of the methyl groups of GH5_pMut in the chromatosome, we first assigned the methyl groups of free GH5_pMut by overlaying the 1H–13C HMQC spectra of wild-type GH5 and GH5_pMut in their free forms. The new methyl group in residue Leu47 and the methyl groups with large chemical shift changes were assigned by introducing individual mutations (L47I or L70 I or L76I). We then assigned the chemical shifts of the GH5_pMut in complex with the nucleosome by overlaying the spectra of the GH5_pMut in the complex and in the free form. Because the globular domains of H1.0 and H5 are highly conserved, we assigned the H1.0 Ile methyl groups by overlaying the spectra of GH5 and H1.0 in complex with the nucleosome.
ITC experiments
ITC experiments were performed on a microcalorimeter (PEAQ-ITC, Malvern) at 25 °C. GH5_pMut and the nucleosome were extensively dialyzed against ITC buffer [20 mM Tris–HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA, and 1 mM DTT] and degassed before loading into the syringe and the cell. Of 30 μM of nucleosome, 200 μl was titrated with each injection of 1.6 μl of 395 μM of the globular domain in ITC buffer. The ITC data were analyzed using the software provided by the manufacturer. The heat change of the binding reaction was plotted against the ratio of the total concentration of the globular domain to the total concentration of the nucleosome to generate the titration thermographs, which were fitted with a model containing a single equilibrium association constant for binding.
Docking calculation
The globular domain of linker histones was first manually docked on the nucleosome using PyMol with the Paramagnetic relaxation enhancement (PRE) data as an approximate guide. The globular domain residues, which are close to the DNA, and the nucleotides, which are close to the globular domain, were chosen to generate ambiguous constraints for HADDOCK calculation. The manually docked globular domain and DNA were pulled away slightly using PyMol to generate two input pdb files for the globular domain and DNA. HADDOCK server was used for the final docking calculation§. Default parameters were used for the docking calculation. Structural clusters generated by the HADDOCK were inspected, and those that appear to be consistent with the PRE data were selected for further examination based on the correlation between the distances calculated from the PRE data and the distances calculated from the structure model by adopting the spin-label location from our earlier models [40]. The structural model that produced the best correlation was chosen as the final structural model.
Sedimentation experiments
The nucleosome arrays were reconstituted according to Dorigo et al. [52]. Drosophila histone octamer, 12 Hepes 207 tandem repeats of “601” sequence DNA (plasmid kindly provided by the Luger lab), and 147-bp competitor DNA (from pUC18 plasmid backbone, with lower histone octamer affinity) were mixed at a 12:0.5:6 ratio in Tris-EDTA (TE) 10/1 [10 mM Tris–HCl and 1 mM EDTA (pH 8.0)] buffer with 2 M NaCl and 10 mM DTT. The nucleosome array was assembled by dialyzing against Tris-EDTA (TE) buffer with the NaCl concentration gradually decreasing from 2 M to 0.6 M for 16 h, followed by a final 6-h dialysis step in HE 10/0.1 [10 mM Hepes, and 0.1 mM EDTA (pH 8.0)] buffer. The array was separated from mononucleosome and competitor DNA using Superose 6 chromatography in HE 10/0.1 buffer. The saturation of the reconstituted nucleosome array by histone octamers was confirmed by EcoRI digestion, which produced only mononucleosomes.
To reconstitute the nucleosome arrays containing linker histones, we mixed the globular domain or full-length linker histones with the purified array in HE 10/0.1 (pH 8.0) and 0.6 M NaCl, followed by a thorough dialysis in 10 mM Hepes buffer at pH 8.0, in the presence of 0.3 mM MgCl2 (for arrays containing globular domain) or in the absence of MgCl2 (for arrays containing full-length linker histones). The ratio of linker histone to nucleosome in the array was determined by SDS-PAGE gel imaging. The gel bands were imaged using a Fujifilm LAS-3000 imager (Fujifilm, Japan) and were analyzed using the ImageJ program. The real ratio of linker histone to nucleosome was measured using the optical density ratio of the linker histone band to the histone octamer bands divided by the corresponding value of the reference sample, which contains an equal molar amount of linker histone and histone octamer.
Sedimentation velocity data were collected at 15,000 rpm and at 20 °C using a Beckman XL-A analytical ultracentrifuge. The absorbance at 260 nm was monitored. Using SEDFIT [56], the data were analyzed in terms of a continuous c(s) distribution spanning a sedimentation coefficient range of 0 to 100 (or 150) S with a resolution of 200 (or 300) points and a confidence interval of 68%. The sedimentation coefficient was the value at the peak maximum. A partial specific volume of 0.65 cm3/g was used [57]. Solution densities and viscosities were calculated in SEDNTERP [58], and sedimentation coefficients were corrected to standard conditions s20,w.
Acknowledgments
The work is supported by the intramural research programs of the NIH, the National Cancer Institute (Y.B.), the National Institute of Diabetes and Digestive and Kidney Diseases (R.G.), and the Center for Information Technology (C.D.S.).
Abbreviations
- GH5
globular domain of chicken H5
- GH1
globular domain of Drosophila linker histone H1
- ITC
isothermal titration calorimetry
- MTSL
[S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl) methyl methanethiosulfonate]
- HMQC
Heteronuclear Multiple-Quantum Correlation
Footnotes
References
- [1].Kornberg RD, Chromatin structure: a repeating unit of histones and DNA, Science 184 (1974) 868–871. [DOI] [PubMed] [Google Scholar]
- [2].Olins AL, Olins DE, Spheroid chromatin units (v bodies), Science 183 (1974) 330–332. [DOI] [PubMed] [Google Scholar]
- [3].Kornberg RD, Lorch Y, Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome, Cell 98 (1999) 285–294. [DOI] [PubMed] [Google Scholar]
- [4].Khorasanizadeh S, The nucleosome: from genomic organization to genomic regulation, Cell 116 (2004) 259–272. [DOI] [PubMed] [Google Scholar]
- [5].Arents G, Burlingame RW, Wang BC, Love WE, Moudrianakis EN, The nucleosomal core histone octamer at 3.1 A resolution: a tripartite protein assembly and a left-handed superhelix, Proc. Natl. Acad. Sci. U. S. A 88 (1991) 10, 148–10, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ, Crystal structure of the nucleosome core particle at 2.8 A resolution, Nature 389 (1997) 251–260. [DOI] [PubMed] [Google Scholar]
- [7].Bates DL, Thomas JO, Histones H1 and H5: one or two molecules per nucleosome? Nucleic Acids Res 9 (1981) 5883–5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Woodcock CL, Skoultchi AI, Fan Y, Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length, Chromosom. Res 14 (2006) 7–25. [DOI] [PubMed] [Google Scholar]
- [9].Simpson RT, Structure of the chromatosome, a chromatin particle containing 160 base pairs of DNA and all the histones, Biochemistry 17 (1978) 5524–5531. [DOI] [PubMed] [Google Scholar]
- [10].Thoma F, Koller T, Influence of histone H1 on chromatin structure, Cell 12 (1977) 101–107. [DOI] [PubMed] [Google Scholar]
- [11].Misteli T, Gunjan A, Hock R, Bustin M, Brown DT, Dynamic binding of histone H1 to chromatin in living cells, Nature 408 (2000) 877–881. [DOI] [PubMed] [Google Scholar]
- [12].Lever MA, Th’ng JP, Sun X, Hendzel MJ, Rapid exchange of histone H1.1 on chromatin in living human cells, Nature 408 (2000) 873–876. [DOI] [PubMed] [Google Scholar]
- [13].Hergeth SP, Schneider R, The H1 linker histones: multifunctional proteins beyond the nucleosomal core particle, EMBO Rep 16 (2015) 1439–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fan Y, Nikitina T, Zhao J, Fleury TJ, Bhattacharyya R, Bouhassira EE, et al. , Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation, Cell 123 (2005) 1199–1212. [DOI] [PubMed] [Google Scholar]
- [15].Shen X, Gorovsky MA, Linker histone H1 regulates specific gene expression but not global transcription in vivo, Cell 86 (1996) 475–483. [DOI] [PubMed] [Google Scholar]
- [16].Maresca TJ, Freedman BS, Heald R, Histone H1 is essential for mitotic chromosome architecture and segregation in Xenopus laevis egg extracts, J. Cell Biol 169 (2005) 859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lee H, Habas R, Abate-Shen C, MSX1 cooperates with histone H1b for inhibition of transcription and myogenesis, Science 304 (2004) 1675–1678. [DOI] [PubMed] [Google Scholar]
- [18].Zhang Y, Cooke M, Panjwani S, Cao K, Krauth B, Ho PY, et al. , Histone h1 depletion impairs embryonic stem cell differentiation, PLoS Genet 8 (2012) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lu X, Wontakal SN, Kavi H, Kim BJ, Guzzardo PM, Emelyanov AV, et al. , Drosophila H1 regulates the genetic activity of heterochromatin by recruitment of Su(var)3-9, Science 340 (2013) 78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Christophorou MA, Castelo-Branco G, Halley-Stott RP, Oliveira CS, Loos R, Radzisheuskaya A, et al. , Citrullination regulates pluripotency and histone H1 binding to chromatin, Nature 507 (2014) 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Thorslund T, Ripplinger A, Hoffmann S, Wild T, Uckelmann M, Villumsen B, et al. , Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage, Nature 527 (2015) 389–393. [DOI] [PubMed] [Google Scholar]
- [22].Konishi A, Shimizu S, Hirota J, Takao T, Fan Y, Matsuoka Y, et al. , Involvement of histone H1.2 in apoptosis induced by DNA double-strand breaks, Cell 114 (2003) 673–688. [DOI] [PubMed] [Google Scholar]
- [23].Allan J, Hartman PG, Crane-Robinson C, Aviles FX, The structure of histone H1 and its location in chromatin, Nature 288 (1980) 675–679. [DOI] [PubMed] [Google Scholar]
- [24].Hendzel MJ, Lever MA, Crawford E, Th’ng JP, The C-terminal domain is the primary determinant of histone H1 binding to chromatin in vivo, J. Biol. Chem 279 (2004) 20,028–20,034. [DOI] [PubMed] [Google Scholar]
- [25].Syed SH, Goutte-Gattat D, Becker N, Meyer S, Shukla MS, Hayes JJ, et al. , Single-base resolution mapping of H1–nucleosome interactions and 3D organization of the nucleosome, Proc. Natl. Acad. Sci. U. S. A 107 (2010) 9620–9625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Singer DS, Singer MF, Studies on the interaction of H1 histone with superhelical DNA: characterization of the recognition and binding regions of H1 histones, Nucleic Acids Res 3 (1976) 2531–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhou BR, Feng H, Kato H, Dai L, Yang Y, Zhou Y, et al. , Structural insights into the histone H1–nucleosome complex, Proc. Natl. Acad. Sci. U. S. A 110 (2013) 19,390–19,395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Caterino TL, Hayes JJ, Structure of the H1 C-terminal domain and function in chromatin condensation, Biochem. Cell Biol 89 (2011) 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fang H, Clark DJ, Hayes JJ, DNA and nucleosomes direct distinct folding of a linker histone H1 C-terminal domain, Nucleic Acids Res 40 (2012) 1475–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lu X, Hansen JC, Identification of specific functional subdomains within the linker histone H10 C-terminal domain, J. Biol. Chem 279 (2004) 8701–8707. [DOI] [PubMed] [Google Scholar]
- [31].White AE, Hieb AR, Luger K, A quantitative investigation of linker histone interactions with nucleosomes and chromatin, Sci. Rep 6 (2016) 19, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Allan J, Mitchell T, Harborne N, Bohm L, Crane-Robinson C, Roles of H1 domains in determining higher order chromatin structure and H1 location, J. Mol. Biol 187 (1986) 591–601. [DOI] [PubMed] [Google Scholar]
- [33].Brown DT, Izard T, Misteli T, Mapping the interaction surface of linker histone H1(0) with the nucleosome of native chromatin in vivo, Nat. Struct. Mol. Biol 13 (2006) 250–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bednar J, Horowitz RA, Grigoryev SA, Carruthers LM, Hansen JC, Koster AJ, et al. , Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin, Proc. Natl. Acad. Sci. U. S. A 95 (1998) 14,173–14,178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hamiche A, Schultz P, Ramakrishnan V, Oudet P, Prunell A, Linker histone-dependent DNA structure in linear mononucleosomes, J. Mol. Biol 257 (1996) 30–42. [DOI] [PubMed] [Google Scholar]
- [36].Cui F, Zhurkin VB, Distinctive sequence patterns in metazoan and yeast nucleosomes: implications for linker histone binding to AT-rich and methylated DNA, Nucleic Acids Res 37 (2009) 2818–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fan L, Roberts VA, Complex of linker histone H5 with the nucleosome and its implications for chromatin packing, Proc. Natl. Acad. Sci. U. S. A 103 (2006) 8384–8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhou YB, Gerchman SE, Ramakrishnan V, Travers A, Muyldermans S, Position and orientation of the globular domain of linker histone H5 on the nucleosome, Nature 395 (1998) 402–405. [DOI] [PubMed] [Google Scholar]
- [39].Puigdomenech P, Jose M, Ruiz-Carrillo A, Crane-Robinson C, Isolation of a 167 basepair chromatosome containing a partially digested histone H5, FEBS Lett 154 (1983) 151–155. [DOI] [PubMed] [Google Scholar]
- [40].Zhou BR, Jiang J, Feng H, Ghirlando R, Xiao TS, Bai Y, Structural mechanisms of nucleosome recognition by linker histones, Mol. Cell 59 (2015) 628–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tugarinov V, Hwang PM, Kay LE, Nuclear magnetic resonance spectroscopy of high-molecular-weight proteins, Annu. Rev. Biochem 73 (2004) 107–146. [DOI] [PubMed] [Google Scholar]
- [42].Thastrom A, Bingham LM, Widom J, Nucleosomal locations of dominant DNA sequence motifs for histone–DNA interactions and nucleosome positioning, J. Mol. Biol 338 (2004) 695–709. [DOI] [PubMed] [Google Scholar]
- [43].Battiste JL, Wagner G, Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data, Biochemistry 39 (2000) 5355–5365. [DOI] [PubMed] [Google Scholar]
- [44].de Vries SJ, van Dijk M, Bonvin AM, The HADDOCK web server for data-driven biomolecular docking, Nat. Protoc 5 (2010) 883–897. [DOI] [PubMed] [Google Scholar]
- [45].de Vries SJ, van Dijk AD, Krzeminski M, van Dijk M, Thureau A, Hsu V, et al. , HADDOCK versus HADDOCK: new features and performance of HADDOCK2.0 on the CAPRI targets, Proteins 69 (2007) 726–733. [DOI] [PubMed] [Google Scholar]
- [46].Dominguez C, Boelens R, Bonvin AM, HADDOCK: a protein–protein docking approach based on biochemical or biophysical information, J. Am. Chem. Soc 125 (2003)1731–1737. [DOI] [PubMed] [Google Scholar]
- [47].Talbert PB, Ahmad K, Almouzni G, Ausio J, Berger F, Bhalla PL, et al. , A unified phylogeny-based nomenclature for histone variants, Epigenetics Chromatin 5 (2012) 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Song F, Chen P, Sun D, Wang M, Dong L, Liang D, et al. , Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units, Science 344 (2014) 376–380. [DOI] [PubMed] [Google Scholar]
- [49].Ricci MA, Manzo C, Garcia-Parajo MF, Lakadamyali M, Cosma MP, Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo, Cell 160 (2015) 1145–1158 [DOI] [PubMed] [Google Scholar]
- [50].Alexandrow MG, Hamlin JL, Chromatin decondensation in S-phase involves recruitment of Cdk2 by Cdc45 and histone H1 phosphorylation, J. Cell Biol 168 (2005) 875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Stutzer A, Liokatis S, Kiesel A, Schwarzer D, Sprangers R, Soding J, et al. , Modulations of DNA contacts by linker histones and post-translational modifications determine the mobility and modifiability of nucleosomal H3 tails, Mol. Cell 61 (2016) 247–259. [DOI] [PubMed] [Google Scholar]
- [52].Dorigo B, Schalch T, Kulangara A, Duda S, Schroeder RR, Richmond TJ, Nucleosome arrays reveal the two-start organization of the chromatin fiber, Science 306 (2004) 1571–1573. [DOI] [PubMed] [Google Scholar]
- [53].Dyer PN, Edayathumangalam RS, White CL, Bao Y, Chakravarthy S, Muthurajan UM, et al. , Reconstitution of nucleosome core particles from recombinant histones and DNA, Methods Enzymol 375 (2004) 23–44. [DOI] [PubMed] [Google Scholar]
- [54].Kato H, van Ingen H, Zhou BR, Feng H, Bustin M, Kay LE, et al. , Architecture of the high mobility group nucleosomal protein 2-nucleosome complex as revealed by methyl-based NMR, Proc. Natl. Acad. Sci. U. S. A 108 (2011) 12,283–12,288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A, NMRPipe: a multidimensional spectral processing system based on UNIX pipes, J. Biomol. NMR 6 (1995) 277–293. [DOI] [PubMed] [Google Scholar]
- [56].Schuck P, Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling, Biophys. J 78 (2000) 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ausio J, Borochov N, Seger D, Eisenberg H, Interaction of chromatin with NaCl and MgCl2. Solubility and binding studies, transition to and characterization of the higher-order structure, J. Mol. Biol 177 (1984) 373–398. [DOI] [PubMed] [Google Scholar]
- [58].Cole JL, Lary JW, Moody PT, Laue TM, Analytical ultracentrifugation: sedimentation velocity and sedimentation equilibrium, Methods Cell Biol 84 (2008) 143–179. [DOI] [PMC free article] [PubMed] [Google Scholar]