

Abstract
The Immunomodulatory Drugs (IMiDs) Thalidomide, Pomalidomide and Lenalidomide have been approved for the treatment of multiple myeloma for many years. Recently, their usage as E3 ligase recruiting elements for small molecule induced protein degradation has led to a resurgence in interest in ImiD synthesis and functionalization. Traditional ImiD synthesis follows a step-wise route employing multiple purification steps. Herein, we describe a novel one-pot synthesis without purification giving rapid access to a multitude of IMiD analogues. Binding studies with the IMiD target protein Cereblon (CRBN) reveals a narrow SAR with only a few compounds showing sub-micromolar binding affinity in the range of Pomalidomide and Lenalidomide. However, anti-proliferative activity as well as Aiolos degradation could be identified for two ImiD analogues. This study provides useful insight into the structure degradation relationships for molecules of this type as well as a rapid and robust method for IMiD synthesis.
Keywords: Protein Degradation, Immunomodulatory Drugs, Imides, Condensation Reactions, Cereblon
Graphical Abstract
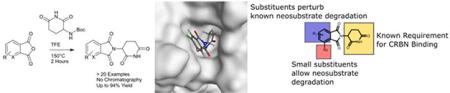
Thalidomide, Pomalidomide and Lenalidomide, collectively known as Immunomodulatory Drugs (IMiDs), have been employed to treat multiple myeloma for many years.1–4 For the majority of this time, the molecular mechanism of action was unknown rendering attempts to explore the structure activity relationships challenging.5, 6 In recent years, the elucidation of the unique mechanism of action of these compounds has caused a resurgence of interest in IMiDs.7–11 These compounds function by inducing ectopic protein-protein interactions between Cereblon, a substrate recognition component of the CRL4 E3 ligase complex, and un-natural substrates (Ikaros, Aiolos and CK1α)9–11, resulting in ubiquitination and subsequent degradation of the neo-substrate (See Figure 1). The fundamental understanding of the mechanism has enabled the discovery of analogues which induce degradation of additional proteins, namely the translation termination factor GSPT1.12, 13 Recently, a family of aryl sulfonamide molecules which function via a distinct but similar mechanism have also been reported.14, 15 Molecules which bind to an E3 ubiquitin ligase are also of great interest to the field of targeted protein degradation, particularly as recruiting elements for proteolysis targeting chimera (PROTACs).16, 17 Indeed ImiD drugs have been widely employed as E3 recruiting elements to enable the degradation of a wide variety of targets.18–25
Figure 1–

Crystal structure of the complex between CRBN (blue) and CK1α (green) with Lenalidomide mediating the interface (PDB ID: 5FQD) and the structures of the ImiDs.
Intrigued by the “molecular glue” mechanism of these compounds we sought to explore the structure degradation relationships of ImiD analogues with an aim to elucidate the requirements for neo-substrate recruitment and the development of high affinity recruiting elements for use in PROTACs. Previous SAR studies have proven challenging due to both time-consuming synthesis and insufficient knowledge surrounding the mechanism of action prior to 2014.
Confronted by the challenge of developing a rapid and robust synthesis of IMiD analogues we surveyed the literature. The majority of routes employ the cyclisation of glutamine and condensation with an anhydride to yield the desired scaffold. Preparation of Boc-2-aminoglutarimide can be achieved by the treatment of N-α-(tert-butoxycarbonyl)-L-glutamine (Boc-Gln) with 1,1-Carbonyldiimidazole (CDI) and catalytic 4-(Dimethylamino)pyridine (DMAP) in THF at reflux temperature as previously reported.†26 Previous routes have employed step-wise deprotection and condensation reactions to yield the desired scaffold in moderate yields following purification.26, 27 Alternatively, the condensation reaction can be performed before cyclisation of the glutarimide.28, 29
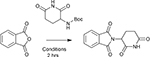
To expedite the process, we sought to combine a thermal BOC deprotection with the condensation step. Since 2,2,2-trifluoroethanol (TFE) can afford t-butoxycarbamate (BOC) deprotection at high temperature, we attempted the one pot procedure shown in Scheme 1, entry 1.30 Gratifyingly, the reaction proceeded to completion under these conditions and, upon cooling to room temperature, the product precipitated to provide analytically pure compound in high yield. Exploring the role of the reagent/solvent in this reaction exemplifies the advantage of employing TFE as a solvent in this reaction. Switching to ethanol results in a significantly reduced yield as the deprotection step is considerably less efficient in the less acidic solvent. Notably addition of trifluoroacetic acid results in a complex mixture of unidentified products. Heating of the reaction to reflux in TFE for 2 hours results in no deprotection of the BOC group and hence no subsequent condensation. Submersion of the reaction vessel in an oil bath at 150 °C for 2 hours rather than microwave irradiation results in a slightly reduced yield which can likely be attributed to less efficient heating. Interestingly, the use of phthalic acid in place of the anhydride is tolerated due to in-situ formation of the anhydride. ‡
Scheme 1–
Synthesis of Thalidomide and screening of reaction conditions.
 | |||
|---|---|---|---|
| Entry | Solvent | Temperature / °C | Isolated Yield |
| 1 | Trifluoroethanol | 150 | 91%(a) |
| 2 | Ethanol | 150 | 24%(a) |
| 3 | 10% TFA in Ethanol | 150 | Complex Mixture(a) |
| 4 | Trifluoroethanol | Reflux | No Reaction(b) |
| 5 | Trifluoroethanol | 150 | 72%(c) |
| 6 | Trifluoroethanol | 150 | 62%(a)(d) |
heated under microwave conditions
heated in an oil bath
heated in an oil bath at 150° in a seal tube
phthalic acid used in place of phthalic anhydride
Having explored conditions for the synthesis of Thalidomide, we employed these optimized conditions to prepare a library of analogues in a rapid and chromatography-free manner (see Figure 2) from commercially available anhydrides. These conditions proved to be relatively general and tolerate a variety of functional groups. Hetero-aromatic anhydrides were well tolerated as were substitutions in both the 3- and 4- positions. Bicyclic and fused ring systems as well as various degrees of saturation are also tolerated. Notably, we were able to prepare Pomalidomide (compound 13) in one step from commercially available starting materials in an 80% yield with no purification step required. To the best of our knowledge, this is the first synthesis of Pomalidomide avoiding the use of nitro-group reduction as the aniline is tolerated under these conditions. Furthermore, this approach can also be employed to prepare SEM protected 21 or N-methylated 22 analogues.
Figure 2–

Structures of compounds prepared in this study with corresponding reaction yields. [a] Prepared by hydrogenation of compound 16.
Having unprecedented access to novel analogues of ImiDs we sought to explore the structure-activity relationships with respect to CRBN binding and the degradation of Aiolos (IKZF3) and CK1α. To assess binding of ImiD analogues to CRBN directly, we employed an SPR assay with His-tagged CRBN immobilized. Dose-response curves were measured for each analogue and Kd values calculated. The results are summarized in Table 1. This direct binding analysis revealed that many of the newly synthesized ImiDs retain the ability to bind CRBN, indeed several of them (Compounds 8 and 12) have a higher affinity than the FDA approved compounds employed as positive controls. For example, the 1,8-Naphthalic anhydride derived compound 12 shows a higher affinity for CRBN presumably due to increased hydrophobic interactions with the protein surface. Molecular modelling revealed that compound 12 is able to adopt the same binding conformation as pomalidomide (see S.I.)
Table 1.
Binding constants for compounds to CRBN measured by surface plasmon resonance and their ability to induce degradation of neo-substrates Aiolos and CK1α at 10 μM.
| Degradation | |||
|---|---|---|---|
| Compound | CRBN Kd / nM[a] | Aiolos | CK1α |
| Lenalidomide | 445 ± 19 | + | + |
| Pomalidomide | 264 ± 18 | + | - |
| 1 | >11000 | - | - |
| 2 | N.B. | - | - |
| 3 | N.D. | - | - |
| 4 | N.D. | - | - |
| 5 | N.D. | - | - |
| 6 | 1450 ± 49 | - | - |
| 7 | 1400 ± 375 | - | - |
| 8 | 55 ± 18 | - | - |
| 9 | N.B. | - | - |
| 10 | N.D. | - | - |
| 11 | 221 ± 52 | - | - |
| 12 | 111 ± 6 | - | - |
| 13 | 271 ± 110 | + | - |
| 14 | N.D. | - | - |
| 15 | N.D. | - | - |
| 16 | >11000 | - | - |
| 17 | 558 ± 51 | + | - |
| 18 | 2100 ± 400 | - | - |
| 19 | 325 ± 24 | + | - |
| 20 | 549 ± 66 | - | - |
N.B – no binding observed, N.D. – Not determined due to reference cell binding
Average of at least 2 experiments. Errors represent the standard error of the mean.
In addition to measuring the binding to CRBN, we sought to explore the ability of these new analogues to induce degradation of the neo-substrates Aiolos and CK1α.31 To this end MM-1S cells were treated with 10 μM of each compound for 24 hours before immunoblotting for the target proteins. Pomalidomide, Lenalidomide and Thalidomide were included as positive controls while DMSO served as a negative control.
Compounds 17 and 19 which were capable of inducing degradation of Aiolos at 10 μM were further evaluated in dose-response experiments via immunoblotting. Quantification of the bands and normalization to tubulin loading controls allows the calculation of DC50 values (the concentration at which half maximal degradation is observed) and DMax (the maximal degradation achieved) (Fig 3A). Replacement of the aniline moiety in Pomalidomide (DC50 – 8.7 nM, Dmax > 95%) results in a loss of activity in the case of the methyl derivative compound 19 (DC50 – 120 nM, Dmax - 85%) and a more pronounced loss of activity in the fluoride compound 17 (DC50 – 1400 nM, Dmax - 83% ). Compounds 17 and 19, which displayed interesting degradation characteristics, were progressed to cell proliferation assays in MM1S cells and compared to Lenalidomide and Pomalidomide (Fig. 3B). Both the new compounds were able to inhibit cell proliferation at similar doses to their corresponding DC50 values (Compound 19 IC50 – 128 nM, Compound 17 IC50 – 3568 nM).
Figure 3–

Cellular evaluation of selected compounds. A – Normalized Aiolos levels in MM1S cells after 24 hours treatment with the indicated dose as assessed by Western Blotting and normalized to Tubulin. The dashed line represents non-linear regression for the calculation of DC50. B – Inhibition of cell proliferation of selected compounds after 72 hours as measured by MTS assay.
This small analogue library reveals the exquisite requirements of compounds which induce interactions between CRBN and the neo-substrates, Aiolos and CK1α. Small substituents are tolerated at the 4 position with the curious exception of a hydroxy group (Compound 14). The surprisingly lack of activity with Compound 14 could conceivably be due to perturbation of a local hydrogen bonding network. Substitution elsewhere on the aromatic ring or deviation from an isoindoline core perturbs recruitment of the known neo-substrates. Perhaps unsurprisingly given the mechanism of action, affinity for CRBN appears not to be the sole determinant for neo-substrate degradation, as some molecules binds to CRBN with an increased affinity yet fail to induce degradation. Indeed, even amongst the compounds which do induce degradation, CRBN affinity appears not to be predictive of cellular activity – Compound 17 binds with roughly 2-fold less affinity than Pomalidomide and Compound 19 however both Compounds 17 and 19 have greatly reduced effect on protein degradation and proliferation when compared to Pomalidomide. More important are structural changes which, even when subtle, can completely abrogate the degradation of the neo-substrate. This is most likely due to perturbation of the required protein-protein interaction as even subtle changes, such as fluorination in the 5 position (Compound 8), drastically effecting hydrophobicity of the combined protein/ligand surface (see S.I.) and presumably therefore prevent trimer formation. A similar change in surface characteristics may also be responsible for the reduced activity of compound 17 compared to thalidomide, although to a lesser extent since the 4 position is less buried at the interface. These subtle differences highlight the inherent difficulty in designing molecular glues.
In summary, we report a rapid, chromatography free synthesis of 2 FDA approved drugs as well as more than 20 additional analogues, along with the structure degradation relationships (SDR) for these new compounds which highlight the inherent challenge in developing molecules which function via this mechanism. We identified two previously uncharacterized thalidomide analogues with anti-proliferative activity in a cellular multiple myeloma model. Furthermore, we have identified several compounds with improved pharmacological properties and/or increased CRBN binding affinity which may be useful as recruiting elements for PROTACs. It is also conceivable that these new molecules are able to induce the degradation of additional neo-substrates or indeed inhibit the binding of natural substrates to CRBN, perhaps providing a useful tool to discover currently unknown substrates of CRBN. Finally, we hope this efficient synthetic method will prove useful to those engaged in the exciting field of targeted protein degradation.
Supplementary Material
Acknowledgements
We thank members of the Crews lab for useful discussions. G.M.B. is Fellow of The Leukemia & Lymphoma Society. P.M.C. is thankful to the Alexander von Humboldt Foundation for a Feodor Lynen research fellowship. C.M.C. gratefully acknowledges the US National Institutes of Health for their support (R35CA197589).
Footnotes
Conflicts of interest
C.M.C. is founder, consultant and shareholder in Arvinas, LLC, which partially supports research in his lab. A. M. is an employee of Arvinas, LLC. Other authors declare no conflict of interests.
Alternatively, tert-Butyl 2,6-dioxopiperidin-3-ylcarbamate is commercially available.
Phthalic acid heated to 150°C in TFE for 2 hours under microwave conditions is converted into the anhydride.
References
- 1.Kyle RA and Rajkumar SV, Clinical Lymphoma and Myeloma, 2009, 9, 278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lacy MQ and Rajkumar SV, American Journal of Hematology, 2010, 85, 95–96. [DOI] [PubMed] [Google Scholar]
- 3.Bartlett JB, Dredge K and Dalgleish AG, Nature Reviews Cancer, 2004, 4, 314–322. [DOI] [PubMed] [Google Scholar]
- 4.Dredge K, Marriott JB, Macdonald CD, Man HW, Chen R, Muller GW, Stirling D and Dalgleish AG, British Journal Of Cancer, 2002, 87, 1166–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muller GW, Shire MG, Wong LM, Corral LG, Patterson RT, Chen Y and Stirling DI, Bioorganic & Medicinal Chemistry Letters, 1998, 8, 2669–2674. [DOI] [PubMed] [Google Scholar]
- 6.Pandit B, Hu Z, Chettiar SN, Zink J, Xiao Z, Etter JP, Bhasin D and Li P-K, Bioorganic & Medicinal Chemistry Letters, 2013, 23, 6902–6904. [DOI] [PubMed] [Google Scholar]
- 7.Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, Yamaguchi Y and Handa H, Science, 2010, 327, 1345–1350. [DOI] [PubMed] [Google Scholar]
- 8.Fischer ES, Bohm K, Lydeard JR, Yang H, Stadler MB, Cavadini S, Nagel J, Serluca F, Acker V, Lingaraju GM, Tichkule RB, Schebesta M, Forrester WC, Schirle M, Hassiepen U, Ottl J, Hild M, Beckwith RE, Harper JW, Jenkins JL and Thoma NH, Nature, 2014, 512, 49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krönke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, Ciarlo C, Hartman E, Munshi N, Schenone M, Schreiber SL, Carr SA and Ebert BL, Science, 2014, 343, 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kronke J, Fink EC, Hollenbach PW, MacBeth KJ, Hurst SN, Udeshi ND, Chamberlain PP, Mani DR, Man HW, Gandhi AK, Svinkina T, Schneider RK, McConkey M, Jaras M, Griffiths E, Wetzler M, Bullinger L, Cathers BE, Carr SA, Chopra R and Ebert BL, Nature, 2015, 523, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu G, Middleton RE, Sun H, Naniong M, Ott CJ, Mitsiades CS, Wong K-K, Bradner JE and Kaelin WG, Science, 2014, 343, 305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hansen JD, Condroski K, Correa M, Muller G, Man H-W, Ruchelman A, Zhang W, Vocanson F, Crea T, Liu W, Lu G, Baculi F, LeBrun L, Mahmoudi A, Carmel G, Hickman M and Lu C-C, Journal of Medicinal Chemistry, 2018, 61, 492–503. [DOI] [PubMed] [Google Scholar]
- 13.Matyskiela ME, Lu G, Ito T, Pagarigan B, Lu C-C, Miller K, Fang W, Wang N-Y, Nguyen D, Houston J, Carmel G, Tran T, Riley M, Nosaka LA, Lander GC, Gaidarova S, Xu S, Ruchelman AL, Handa H, Carmichael J, Daniel TO, Cathers BE, Lopez-Girona A and Chamberlain PP, Nature, 2016, 535, 252–257. [DOI] [PubMed] [Google Scholar]
- 14.Han T, Goralski M, Gaskill N, Capota E, Kim J, Ting TC, Xie Y, Williams NS and Nijhawan D, Science, 2017, 356. [DOI] [PubMed] [Google Scholar]
- 15.Uehara T, Minoshima Y, Sagane K, Sugi NH, Mitsuhashi KO, Yamamoto N, Kamiyama H, Takahashi K, Kotake Y, Uesugi M, Yokoi A, Inoue A, Yoshida T, Mabuchi M, Tanaka A and Owa T, Nature Chemical Biology, 2017, 13, 675. [DOI] [PubMed] [Google Scholar]
- 16.Burslem GM and Crews CM, Chem. Rev, 2017, 117, 11269–11301. [DOI] [PubMed] [Google Scholar]
- 17.Cromm PM and Crews CM, Cell Chemical Biology, 24, 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lai AC, Toure M, Hellerschmied D, Salami J, Jaime-Figueroa S, Ko E, Hines J and Crews CM, Angew Chem Int Ed Engl, 2016, 55, 807–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S and Bradner JE, Science, 2015, 348, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler James D., Crew Andrew P, Coleman K and Crews Craig M., Chemistry & Biology, 2015, 22, 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bondeson DP, Smith BE, Burslem GM, Buhimschi AD, Hines J, Jaime-Figueroa S, Wang J, Hamman BD, Ishchenko A and Crews CM, Cell Chemical Biology, 2018, 25, 78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou B, Hu J, Xu F, Chen Z, Bai L, Fernandez-Salas E, Lin M, Liu L, Yang C-Y, Zhao Y, McEachern D, Przybranowski S, Wen B, Sun D and Wang S, Journal of Medicinal Chemistry, 2018, 61, 462–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chessum NEA, Sharp SY, Caldwell JJ, Pasqua AE, Wilding B, Colombano G, Collins I, Ozer B, Richards M, Rowlands M, Stubbs M, Burke R, McAndrew PC, Clarke PA, Workman P, Cheeseman MD and Jones K, Journal of Medicinal Chemistry, 2018, 61, 918–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ishoey M, Chorn S, Singh N, Jaeger MG, Brand M, Paulk J, Bauer S, Erb MA, Parapatics K, Müller AC, Bennett KL, Ecker GF, Bradner JE and Winter GE, ACS Chemical Biology, 2018, 13, 553–560. [DOI] [PubMed] [Google Scholar]
- 25.Schiedel M, Herp D, Hammelmann S, Swyter S, Lehotzky A, Robaa D, Oláh J, Ovádi J, Sippl W and Jung M, Journal of Medicinal Chemistry, 2018, 61, 482–491. [DOI] [PubMed] [Google Scholar]
- 26.Capitosti SM, Hansen TP and Brown ML, Organic Letters, 2003, 5, 2865–2867. [DOI] [PubMed] [Google Scholar]
- 27.Lohbeck J and Miller AK, Bioorganic & Medicinal Chemistry Letters, 2016, 26, 5260–5262. [DOI] [PubMed] [Google Scholar]
- 28.Muller GW, Konnecke WE, Smith AM and Khetani VD, Organic Process Research & Development, 1999, 3, 139–140. [Google Scholar]
- 29.Muller G, W., Saindane M, T., Ge Cand Chen R, (Celgene Corporation, New Jersey, USA: ), PCT/US2006/026210, 2007. [Google Scholar]
- 30.Choy J, Jaime-Figueroa S, Jiang L and Wagner P, Synthetic Communications, 2008, 38, 3840–3853. [Google Scholar]
- 31.Matyskiela ME, Zhang W, Man H-W, Muller G, Khambatta G, Baculi F, Hickman M, LeBrun L, Pagarigan B, Carmel G, Lu C-C, Lu G, Riley M, Satoh Y, Schafer P, Daniel TO, Carmichael J, Cathers BE and Chamberlain PP, Journal of Medicinal Chemistry, 2018, 61, 535–542. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.