

Abstract
Muraymycins belong to a family of nucleoside antibiotics that have a distinctive disaccharide core consisting of 5-amino-5-deoxyribofuranose (ADR) attached to 6′-N-alkyl-5′-C-glycyluridine (GlyU). Here we functionally assign and characterize six enzymes from the muraymycin biosynthetic pathway involved in the core assembly that starts from UMP. The biosynthesis is initiated by Mur16, a non-heme Fe(II)- and α-ketoglutarate-dependent dioxygenase, followed by four transferase enzymes—Mur17, a pyridoxal-5′-phosphate (PLP)-dependent transaldolase; Mur20, an aminotransferase; Mur26, a pyrimidine phosphorylase; and Mur18, a nucleotidylyltransferase. The pathway culminates in glycosidic bond formation in a reaction catalyzed by an additional transferase enzyme, Mur19, a ribosyltransferase. Analysis of the biochemical properties revealed several noteworthy discoveries including that (i) Mur16 and downstream enzymes can also process 2′-deoxy-UMP to generate a 2-deoxy-ADR, which is consistent with the structure of some muraymycin congeners; (ii) Mur20 prefers l-Tyr as the amino donor source; (iii) Mur18 activity absolutely depends on the amine functionality of the ADR precursor, consistent with the nucleotidyltransfer reaction occurring after the Mur20-catalyzed aminotransfer reaction; and (iv) the bona fide sugar acceptor for Mur19 is (5′S,6′S)-GlyU, suggesting that ribosyltransfer occurs prior to N-alkylation of GlyU. Finally, a one-pot, six-enzyme reaction was utilized to generate the ADR-GlyU disaccharide core starting from UMP.
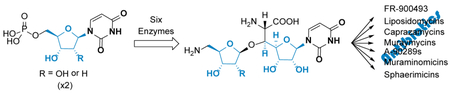
Graphical Abstract
INTRODUCTION
Several structural classes of nucleoside antibiotics have been discovered as a consequence of their ability to inhibit translocase I (MraY), an essential bacterial enzyme that initiates the lipid cycle of peptidoglycan.1–3 One class is characterized by a disaccharide core consisting of a 5-amino-5-deoxyribofuranose (ADR) attached to 6′-N-alkyl-5′-C-glycyluridine (GlyU) through a standard β-O-glycosidic bond, exemplified by the structurally simplest member, FR-900493 (1) from Bacillus cereus (Figure 1).4 Other members of this structural class, which notably differ in the alkyl substituent of the GlyU, are represented by the caprazamycins (represented by liposidomycin L-I (2) from Streptomyces sp. SN-1061M,5,6 caprazamycin B (3) from Streptomyces sp. MK730–62F2,7,8 A-90289 B (4) from Streptomyces sp. SANK 60405,9 and muraminomicin F (5) from Streptosporangium amethystogenes10), sphaerimicins [represented by sphaerimicin A (6) from Sphaerisporangium sp. SANK 60911],11 and muraymycins [represented by muraymycin C1 (7) from Streptomyces sp. NRRL 30471].12 The producing strains of 2-7 biosynthesize several congeners that vary in the length and functionality of the fatty acyl side chain or by the substitution pattern of the ADR-GlyU disaccharide core. As an example of the latter, the isolated muraymycins can contain a methylated ADR as in 7, a standard 2′′-OH variant [exemplified with muraymycin C2 (8)], or a 2′′-deoxy variant, exemplified by muraymycin C3 (9).12–14
Figure 1.

Structure of representative nucleoside antibiotics containing an ADR-GlyU disaccharide core.
The biosynthetic mechanism for GlyU and ADR has previously been defined using the enzymes involved in the biosynthesis of 4 (Figure 2).15–18 Both components start from UMP (10a) in a reaction catalyzed by LipL, a non-heme Fe(II), α-ketoglutarate (αKG)-dependent dioxygenase that catalyzes an unusual oxidative dephosphorylation via stereospecific 5′-hydroxylation of 10a to yield uridine-5′-aldehyde (11a).16,19 The two pathways diverge after the formation of 11a. In one pathway, a pyridoxal-5′-phosphate-dependent (PLP) l-Thr:11a transaldolase LipK catalyzes the stereospecific generation of the nonproteinogenic β-hydroxy amino acid, 5′S,6′S-GlyU (12).17 In the other pathway, a distinct PLP-dependent enzyme LipO initiates the synthesis of ADR by catalyzing the transamination of 11a to 5′-amino-5′-deoxyuridine (13a) using l-methionine as the amino group donor.18 The nucleoside phosphorylase LipP subsequently generates uracil (14) and 5-amino-5-deoxy-α-d-ribose-1-phosphate, which is activated to UDP-5-amino-2,5-dideoxyribose (15a) in a reaction catalyzed by the nucleotidyltransferase, LipM. Although this mechanism of sugar activation, i.e. generation of an NDP-sugar, is standard during the biosynthesis of glycosylated natural products, a unique feature of LipM is the strict selectivity toward the amine functionality of the ribofuranose substrate. Finally, using uridine as a surrogate sugar acceptor, the ribosyltransferase LipN was shown to catalyze the formation of the β-O-glycosidic bond of the unnatural disaccharide 16. By extension, LipN was proposed to catalyze the formation of the ADR-GlyU disaccharide 17a using the more hindered LipK-product 12 as an acceptor, although this remains to be verified.
Figure 2.

Biosynthesis of the ADR-GlyU disaccharide core 17a. Enzymes from the 4 biosynthetic pathway (annotated as Lip proteins in bold) were previously functionally assigned using the probable native substrates with the lone exception being LipN, which was characterized using a surrogate sugar acceptor, uridine. The Mur proteins involved in 7-9 biosynthesis have moderate sequence homology to the corresponding Lip protein and hence putatively catalyze the same reaction.
As expected, the biosynthetic gene clusters for 2, 3, and 5-7 encode for homologous proteins involved in the biosynthesis of the ADR-GlyU disaccharide of 4.10,11,20–22 For muraymycins this includes mur16 (encoding a putative αKG:10a dioxygenase related to LipL), mur17 (l-Thr:11a transaldolase, LipK), mur20 (l-Met:11a aminotransferase, LipO), mur26 (13a phosphorylase, LipP), mur18 (nucleotidylyltransferase, LipM) and mur19 (15a:12 ribosyltransferase, LipN), with sequence identities ranging from 34% to 46% based on pairwise alignments (Figure S1).22 Unlike in 4, the muraymycin disaccharide core is not modified with a 2′-O-sulfate and, as previously noted, the 2′′-OH of the ADR is differentially modified, suggesting the possibility that the homologous proteins have unique biochemical properties that direct these structural variations. In this paper, the function and substrate specificity for Mur16–20 and Mur26 are defined using recombinant enzymes in vitro, and the results compared with the homologs involved in 4 biosynthesis. Additionally, the ribosyltransferase Mur19 from the 7-9 biosynthetic pathway and LipN from the 4 biosynthetic pathway were tested, for the first time, with the hypothesized genuine ribosyl acceptor, 12. Finally, based on information garnered from substrate specificity studies, a one-pot enzymatic reaction was employed to produce 17a and its 2″-deoxy analogue.
RESULTS AND DISCUSSION
Mur16, a non-heme, Fe(II)-dependent αKG:10 dioxygenase.
The mur16 gene was cloned and expressed in Escherichia coli BL21(DE3) to yield soluble protein (Figure 3A). The activity of Mur16 was tested with 10a under optimized conditions previously reported for LipL.16 Using HPLC for analysis, a new peak appeared that co-eluted with the product of the LipL-catalyzed reaction and synthetic uridine-5′-aldehyde, 11a (Figure 3B).16 The identity of the product was further supported by LCMS analysis, yielding an (M + H)+ ion at m/z = 243.1 and an (M + H3O)+ ion at m/z = 261.1, which are consistent with the molecular formula for 11a [expected (M + H)+ and (M + H3O)+ ions at m/z = 243.1 and 261.1, respectively, for C9H10N2O6] (Figure S2A). Similar to LipL, conversion of 10a to 11a was only detected when αKG and FeCl2 were included, and the activity was stimulated by the inclusion of ascorbate (Figure S2B). Mur16 was subsequently determined to have optimal activity with 40 µM FeCl2 and 2 mM ascorbate. Based on LCMS analysis, succinate was identified as a co-product (Figure S2C), which was indirectly supported by monitoring the oxidation of NADH using an enzyme-coupled assay (Figure S2D). Similar to several other enzymes of this dioxygenase superfamily, succinate was formed in the absence of 10a via uncoupled oxidative decarboxylation of αKG, although the rate of formation was significantly enhanced when 10a was included (Figure S2D). Mur16 was previously proposed to initiate the biosynthesis of l-epi-capreomycidine of 7-9 by oxidation of l-Arg.22 However, incubation of Mur16 with l-Arg did not yield a product based on LCMS analysis, nor was the rate of succinate formation increased when compared to the uncoupled oxidation reaction control (Figure S2D). Thus, in contrast to prior predictions, l-Arg is not a substrate for Mur16 under the conditions tested.
Figure 3.

Characterization of Mur16. (A) Reaction catalyzed by Mur16. (B) HPLC analysis of 12-h incubations of 10a with (i) no enzyme and (ii) Mur16. (C) HPLC analysis of 12-h incubations of 10b with (i) no enzyme and (ii) Mur16. (D) HPLC analysis of 12-h incubations of 10c with (i) no enzyme and (ii) Mur16. (E) Single-substrate kinetic analysis for the indicated substrate with near saturating αKG and 40 nM Mur16. A260, absorbance at 260 nm; αKG, α-ketoglutarate; succ, succinate.
The substrate specificity of Mur16 was next examined. Using HPLC for detection, 2′-deoxy-UMP (10b) was revealed to be a substrate for Mur16 (Figure 3C), generating the respective aldehyde product 11b based on LCMS analysis (Figure S2E). The hypothetical substrate 2′-methoxy-UMP (10c) was synthesized,23 and the identity was confirmed by HRMS and NMR spectroscopy (Figures S3 and S4). HPLC analysis of the Mur16-catalyzed reaction with 10c revealed a new peak (Figure 3D), and LCMS analysis was consistent with the aldehyde product 11c (Figure S2F). The results for Mur16 with 10b and 10c are in contrast to LipL, which is highly specific for 10a,16 but are entirely consistent with the isolation of the different muraymycin congeners represented by 7-9. Single-substrate kinetic analysis of Mur16 with 10a and 10b revealed typical Michaelis-Menten kinetics, yielding kinetic constants of Km = 21 ± 4 µM and kcat = 4.4 ± 0.3 s−1 with respect to 10a and Km = 6.3 ± 2.2 µM and kcat = 2.3 ± 0.2 s−1 with respect to 10b (Figure 3E). Comparison of the second order rate constants (2.1 × 105 M−1s−1 and 3.7 × 105 M−1s−1, respectively) suggests a lack of preference for either 10a or 10b. Single-substrate kinetic analysis with 10c revealed non-Michaelis Menten kinetics with apparent inhibition at increasing substrate concentration (Figure 3E); however, the data did not fit well to a kinetic equation describing simple substrate inhibition. Consequently, the specific activities at the observed maximal velocity were compared, which revealed ≥ 15-fold lower activity with 10c compared to the other substrates. Thus, the kinetic results suggest that methylation of ADR occurs following the Mur16-catalyzed reaction.
Mur17, a PLP-dependent l-Thr:11a transaldolase.
The mur17 gene was cloned and expressed in Streptomyces lividans TK24 to yield soluble protein (Figure 4A). UV-VIS spectroscopic analysis of recombinant Mur17 revealed a detectable UV maximum at 415 nm (Figure S5A), suggesting that a minor fraction of the protein copurified with PLP as the internal aldimine. Using HPLC for detection, activity tests with exogenously supplied PLP and potential substrates l-Thr and 11a revealed a new peak (Figure 4B). The formation of the new peak was significantly reduced without the addition of PLP (Figure S5B), which is consistent with the UV-VIS spectroscopic analysis and the necessity of PLP in catalysis as previously reported for LipK.17 LCMS analysis of the purified, new peak yielded an (M + H)+ ion at m/z = 318.1 (Figure 4B), which is consistent with the molecular formula for GlyU (12) [expected (M + H)+ ion at m/z = 318.1 for C11H15N3O8]. As expected, the new peak co-eluted with the product of LipK as well as with authentic 5′S,6′S-12,24–26 which is the relative stereochemistry that is observed in the ADR-GlyU disaccharide core of muraymycins.12 Mur17 did not utilize 11b or 11c as substrates, results of which are consistent with the structure of all known muraymycin congeners that have the identical, ‘hydroxylated’ 12 nucleoside. To provide evidence for the stereochemical assignment of 12, authentic 5′S,6′S-12 as well as its two 5′- and 6′-epimers (5′R,6′S-12 and 5′S,6′R-12) were synthesized,24,25,27 and each isomer was modified with 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (AQC) to generate the 6-quinolinylaminocarbonyl amines without alteration of the stereochemistry. LCMS comparison of the AQC-modified isomers in comparison to the AQC-modified Mur17 product was consistent with stereospecific formation of 5′S,6′S-12 (Figure 4C).
Figure 4.

Characterization of Mur17. (A) Reaction catalyzed by Mur17. (B) HPLC analysis of 12-h incubation of 11a with (i) no enzyme and (ii) Mur17. The inset depicts the mass spectrum for the ion peak eluting at t = 5.6 min corresponding to 12. (C) HPLC analysis of 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (AQC)-modified (5′S,6′S)-12 after 12-h incubation of 11a with (i) no enzyme and (ii) Mur17. The inset depicts the mass spectrum for the ion peak eluting at t = 16.2 min corresponding to AQC-modified (5′S,6′S)-12 [expected (M + H)+ ion at m/z = 488.1 for C21H21N5O9]. Under these HPLC conditions, AQC-modified (5′S,6′R)-12 and (5′R,6′S)-12 elute at t = 19.5 min and t = 25.1 min, respectively, and have similar mass spectra as shown for AQC-modified (5′S,6′S)-12. A260, absorbance at 260 nm.
Functional assignment of Mur20 as an l-Tyr:11-aminotransferase.
The mur20 gene was cloned and expressed in E. coli BL21(DE3) to yield soluble protein (Figure 5A). The successful production of Mur20 in E. coli is in contrast to prior results with the homologous protein LipO, which was only soluble upon production in Streptomyces lividans TK64.18 Using HPLC for detection, activity tests with potential substrates 11a and l-Met revealed a new peak with an identical retention time to the LipO product as well as synthetic 5′-amino-5′-deoxyuridine (13a) standard (Figure 5B).18 The identity of the product was further supported by LCMS analysis (Figure S6A), yielding an (M + H)+ ion at m/z = 244.0902, which is consistent with the molecular formula for 13a [expected (M + H)+ ion at m/z = 244.0855 for C9H13N3O5]. The expected co-product—α-keto-γ-methylthiol butyrate—was also detected by LCMS following derivatization of the Mur20-reaction components with 3-methyl-2-benzothiazolinone hydrazone hydrochloride (MBTH) to give a mixture of cis and trans imine adducts (Figure 5C).28
Figure 5.

Characterization of Mur20. (A) Reaction catalyzed by Mur20. (B) HPLC analysis of 6-h incubation of l-Met and 11a with (i) no enzyme, (ii) Mur20, and (iii) LipO. (C) HPLC analysis of MBTH-modified components of 6-h incubation of l-Met and 11a with (i) no enzyme, (ii) Mur20, and (iii) LipO. A260, absorbance at 260 nm.
In contrast to Mur17 (Figure S5A) and LipO, the latter of which co-purified with near stoichiometric amounts of PLP based on UV-VIS spectroscopic analysis,18 recombinant Mur20 did not have clear a diagnostic absorption feature above 300 nm that would suggest copurification with PLP (Figure S7A). Instead, a shoulder was evident at 363 nm within a long tail that proceeded to around 500 nm. Given the established PLP-dependency of LipO and other transaminases along with this inconclusive UV-VIS profile, we initially added PLP to the Mur20-catalyzed reactions. Upon further examination, however, 13a was shown to be generated in reactions without exogenously supplied PLP (Figure S7B). The activity under these conditions was 1.5- to 3-fold decreased depending upon the batch of purified Mur20. Sequential addition of substrates to Mur20 did not alter the UV-VIS spectrum (Figure S7A). As a consequence of these results, which are similar to that previously reported for glutamate-glycine transaminase,29 we can only conclude that PLP stimulates transamination at this time. Further experimental analysis will be needed to ascertain a definitive role for PLP in Mur20-mediated catalysis.
The substrate specificity of Mur20 toward different amine acceptors and donors was next examined. Activity tests with 11b or 11c, the latter generated in situ by a Mur16-catalyzed reaction starting from 10c, revealed both were amine acceptors (Figure S6B, C). Therefore, the timing of O-methylation during muraymycin biosynthesis could not be discerned based on the characterization of Mur20 along with consideration of the aforementioned Mur16 results. With respect to amine donors, l-Met was initially utilized with Mur20 since LipO exhibited the highest specific activity with this donor.18 Nonetheless, other potential amine donors—including the remaining proteinogenic amino acids—were tested with Mur20. HPLC analysis revealed several amine donors were readily utilized by Mur20 with 11a as the amine acceptor (Table S1). A comparable broad selectivity toward the amine donor was previously noted for LipO. In contrast to LipO, however, the specific activity for Mur20 was highest using l-Tyr as an amine donor, followed closely by l-Trp, l-Arg, l-Met, and AdoMet (Table S1). Comparison of the specific activities revealed a ~2.6-fold higher activity for LipO with l-Met compared to Mur20 with l-Tyr.
Sugar activation by Mur26 and Mur18.
The mur26 and mur18 genes were cloned and expressed in E. coli BL21(DE3) to yield soluble proteins (Figure 6 and 7). The putative phosphorylase activity of Mur26 was initially tested (Figure 6A). Using conditions that were employed during the functional assignment of the orthologous protein LipP,18 HPLC analysis revealed that Mur26 catalyzed phosphotransfer using orthophosphate with co-substrate uridine, 13a, or 13b to generate uracil (14) and presumably the sugar-1-phosphates: α-D-ribose-1-phosphate, 5-amino-5-deoxy- α-D-ribose-1-phosphate, and 5-amino-2,5-dideoxy-α-D-ribose-1-phosphate, respectively (Figure 6B and C). Contrastingly, Mur26 did not utilize 13c, providing the first, direct evidence to support that O-methylation occurs after formation of the sugar-1-phosphate.
Figure 6.

Characterization of Mur26. (A) Reaction catalyzed by Mur26. (B) HPLC analysis of 6-h incubations of 13a with (i) no enzyme, (ii) Mur26, and (iii) LipP. (C) HPLC analysis of 6-h incubations of 13b with (i) no enzyme, (ii) Mur26, and (iii) LipP. A260, absorbance at 260 nm.
Figure 7.

Characterization of Mur18. (A) Reaction catalyzed by Mur26 (or LipP) and Mur18. (B) HPLC analysis of 6-h incubations of 13b with (i) no enzymes, (ii) Mur26 or LipP, and (iii) LipP and Mur18. (C) Mass spectrum for the ion peak eluting at t = 5.9 min corresponding to 15b. A260, absorbance at 260 nm.
The activity of the putative nucleotidyltransferase Mur18 was next assessed using sugar-1-phosphates generated in situ by Mur26. Our previous analysis of the orthogous proteins LipP and LipM revealed that the product of the tandem catalyzed reaction starting from 13a was unstable, degrading to 10a and 5-amino-5-deoxy-α-d-ribose-1,2-cyclophosphate (Figure 2).18 Contrastingly, the UDP-sugar was attainable when starting with the deoxy variant 13b, which was utilized here with Mur26 and Mur18 (Figure 7A). Using HPLC for detection, analysis of reactions containing both Mur26 and Mur18 revealed the formation of a new peak that was not detected in the controls (Figure 7B). A peak with the same retention time was observed when either enzyme was substituted with the respective orthologous proteins LipP or LipM. LCMS analysis of the new peak revealed an (M − H)− ion at m/z = 518.1 (Figure 7C), which is consistent with the molecular formula for UDP-5-amino-2,5-dideoxy-α-d-ribose (15b) [expected (M − H)− ion at m/z = 518.1 for C14H23N3O14P2]. Thus, the data are consistent with Mur26 producing 5-amino-2,5-dideoxy-α-D-ribose-1-phosphate, which serves as the substrate for Mur18-catalyzed nucleotidyltransfer to yield 15b. Similar to the prior results reported for LipM,17 no Mur18 activity was observed when starting from uridine, which is converted to α-d-ribose-1-phosphate by Mur26. Therefore, Mur18 and LipM constitute an unusual group of nucleotidyltransferases that absolutely require an aminated sugar-1-phosphate for catalysis in contrast to the standard hydroxylated ribose.
Functional assignment of Mur19 as a 15:12 ribosyltransferase.
LipN was previously assigned as a ribosyltransferase by utilizing uridine as a surrogate sugar acceptor and 15a—enzymatically generated in situ starting from 13a—as a sugar donor.18 Here we aimed to identify the genuine sugar acceptor for LipN and Mur19, in combination with either 15a or 15b as sugar donors. The mur19 gene was cloned and expressed in S. lividans TK24 to yield soluble protein (Figure 8A).
Figure 8.

Characterization of Mur19. (A) Reaction catalyzed by Mur19 with in situ-generated 15a. (B) HPLC analysis of 6-h incubations of 12 and 13a with (i) Mur26; (ii) Mur26 and Mur18; and (iii) Mur26, Mur18, and Mur19. Identical HPLC chromatograms were obtained upon substitution of Mur26 and Mur18 with the respective ortholog involved in 4 biosynthesis. ADR-GlyU (17a) was prepared by chemical synthesis and used as a control. (C) Mass spectrum for the ion peak eluting at t = 2.6 min corresponding to 17a. A260, absorbance at 260 nm.
In contrast to the results with LipN, Mur19 incubated with uridine and in situ-generated 15a did not yield a new product based on HPLC analysis. When uridine was substituted with authentic 5′S,6′S-12, which was established here as the product of Mur16 and Mur17 catalysis, HPLC analysis of the reaction revealed a new peak (Figure 8B). The formation of the new peak was dependent upon the inclusion of 5′S,6′S-12, 13a, Mur18, Mur26, and Mur19. LCMS analysis of the new peak yielded an (M + H)+ ion at m/z = 449.1463 (Figure 8C), which is consistent with the molecular formula for ADR-GlyU disaccharide (17a) [expected (M + H)+ ion at m/z = 449.1442 for C16H24N4O11]. To simplify the analytical identification of the product, 17a was prepared by chemical synthesis (Figures S8-S12);30 analysis of the Mur19-catalyzed reaction revealed the new peak co-eluted with, and had identical UV and MS spectroscopic properties to, authentic 17a. The data are therefore consistent with the functional assignment of Mur19 as a 12:15a 5-amino-5-deoxyribosyltransferase.
The substrate selectivity of Mur19 was examined with respect to both sugar donor and acceptor. For the former, Mur19 was incubated with 12 and in situ-generated 15b (Figure S13A), and HPLC analysis revealed a new peak that was not present in the controls (Figure S13B). To identify the product, the reaction components were first modified with AQC, and the peak corresponding to the AQC-derivatized product was purified. Both HRMS (Figure S13C) and NMR spectroscopy (Figures S14 and S15) were consistent with the structure of AQC-modified 17b. The ability of Mur19 to transfer 2-deoxyribose to 12 is consistent with the structures of some muraymycin congeners, exemplified by 9 (Figure 1). Likewise the inability of LipN to transfer the 2-deoxyribose variant is consistent with the structure of known 4 congeners, all of which contain the standard 2-hydroxylated aminoribosyl component.9
The substrate selectivity for Mur19 with respect to the sugar acceptor was next interrogated, primarily to decipher whether ribose attachment occurs prior to or after N-alkylation of 12. Notably, muraymycins lacking the ADR component have been isolated from the producing strain as minor metabolites,12,13 suggesting that ribosyltransfer could possibly occur even later in the biosynthetic pathway, i.e., after peptide attachment. To entertain this possibility, muraymycin D4, which lacks the ADR component, was isolated following standard fermentation of the producing strain (Figure S16). However, incubation of Mur19 with in situ generated 15a and muraymycin D4 did not yield a new product. We also explored the potential reversibility of the Mur19-catalyzed reaction with the assumption that ribosylation occurs post peptidation. Muraymycins D1, D2, and D3, which contain the ADR component with the same modifications found in C1, C2, and C3, respectively, were isolated,14 and each congener was incubated with excess UDP and Mur19. However, HPLC analysis of the reactions did not reveal a new peak (Figure S17). Although negative results, the data with muraymycins D1-D4 are consistent with ribosylation happening before peptide attachment. To narrow down the timing of the Mur19-catalyzed reaction, the hypothetical pathway intermediate aminopropyl-12 (18), which lacks the ADR and peptide components, was synthesized and confirmed by HRMS and NMR spectroscopic analyses (Figures S18-S23). Similar to the results with muraymycin D4, incubation of Mur19 with in situ-generated 15a and 18 did not yield a new product. Therefore, the totality of the data suggest that Mur19-catalyzed ribosylation occurs prior to N-alkylation and that 12 is the most likely substrate in vivo.
Glycosyltransferases are mechanistically classified as either inverting or retaining enzymes based on the stereochemistry of the anomeric bond of the sugar before and after formation of the glycoside product.31–33 Inverting glycosyltransferases are generally accepted to follow a single displacement mechanism, thereby requiring the formation of a ternary complex during the reaction coordinate. Mur19 falls within the inverting glycosyltransferase classification, suggesting that the donor 15 and acceptor 12 are bound simultaneously at the active site preceding catalysis. Unlike typical inverting glycosyltransferases, which have structurally distinct donors and acceptors,34 Mur19 utilizes two structurally similar, uridine-containing substrates, raising the question of how this ribosyltransferase differentially binds and orients the two substrates for glycosidic bond formation. Mechanistic and structural investigations for this enzyme are ongoing to interrogate the molecular details behind this unusual substrate selection.
Total Enzymatic Synthesis of 17a and 17b.
We finally aimed to reconstitute the biosynthesis of 17a in vitro starting from the primary metabolite 10a using six enzymes: Mur16, Mur17, LipO, LipP, Mur18 and Mur19. The enzymes were selected based upon their desirable properties such as solubility when produced in E. coli (Mur18), substrate flexibility (Mur16 and Mur19), or superior catalytic activity (LipO). With the addition of each enzyme, a new peak corresponding to the expected product was detected by HPLC, including a small, new peak corresponding to 17a when all enzymes were present (Figure 9A). Yields of 17a were low under these conditions (5% with respect to 10a) with a significant amount of the enzymatically generated sugar acceptor 12 remaining after termination of the reaction. Similarly, using 10b and 12 as starting material, the production of 17b was attempted using five enzymes: Mur16, LipO, LipP, Mur18 and Mur19. In this case, the exogenous supply of 12 was essential given that Mur17 cannot utilize 11b, the product of Mur16. HPLC analysis of the five-enzyme reaction revealed complete consumption of 12 with the formation of the expected product, 17b (Figure 9B).
Figure 9.

Biosynthesis of ADR-GlyU. (A) HPLC analysis of one-pot reaction starting with 10a and UTP (*impurity in commercial UTP) after 12-h incubation with (i) no enzymes; (ii) Mur16; (iii) Mur16 and Mur17; (iv) Mur16, Mur17, and LipO; (v) Mur16, Mur17, LipO, and LipP; (vi) Mur16, Mur17, LipO, LipP, and Mur18; (vii) Mur16, Mur17, LipO, LipP, Mur18, and Mur19; and (viii) 17a standard. (B) HPLC analysis of one-pot reaction starting with 10b, UTP, and 12 after 12-h incubation with (i) no enzymes; (ii) Mur16; (iii) Mur16 and LipO; (iv) Mur16, LipO, and LipP; (v) Mur16, LipO, LipP, and Mur18; (vi) Mur16, LipO, LipP, Mur18, and Mur19. A260, absorbance at 260 nm.
The chemical synthesis of ADR-GlyU has previously been established starting from uridine (acceptor) and d-ribose (donor).30,35,36 This procedure involves eight linear steps to the final product with respect to either uridine or d-ribose, with convergence of the acceptor and donor halfway through the sequence. An overall yield of ~3.5% with respect to the starting reagent uridine has been achieved in our hands following this synthetic scheme.30 Here the synthesis of 17a starting from 10a was accomplished in a single pot, six-enzyme reaction with comparable yields. Thus, without any optimization, the enzymatic synthesis appears to be on par with the chemical route.
A considerable challenge for multiple enzyme-mediated in vitro synthesis is the tendency for enzyme inhibition to occur by components within the complete reaction mixture.37–39 An inspection of representative HPLC traces for the synthesis of 17a suggests this is indeed likely the case, as several enzymatic conversions including the final Mur19-catalyzed ribosyltransfer are incomplete (Figure 9A). As a preliminary strategy to overcome this issue and improve the yield, we aimed to increase the flux of the pathway by removal of the final coproduct UDP by adding phosphoenolpyruvate and pyruvate kinase, thus enzymatically converting UDP to UTP.40 This strategy is also appealing as it would concurrently provide more starting reagent. Unfortunately, however, no improvement was observed. Nevertheless, with the assays for each enzyme now in hand, the potential kinetic liabilities for every enzyme catalyst—along with the many documented factors that can contribute to the flux—can now be closely examined to optimize the yield.
CONCLUSION
The biosynthetic mechanism leading to the ADR-GlyU disaccharide core of 7-9 has now been defined (Figure 10). Six enzymes were functionally assigned and characterized: Mur16, a non-heme, Fe(II)-dependent αKG:10 dioxygenase; Mur17, a PLP-dependent l-Thr:11a transaldolase; Mur20, an l-Tyr:11 aminotransferase stimulated by PLP; Mur26, a 13 phosphorylase; Mur18, a UTP:5-amino-5-deoxy-α-d-ribose-1-phosphate uridylyltransferase; and Mur19, a 15:12 ribosyltransferase. Several discoveries were uncovered that are consistent with the structural variations of 7-9 in comparison to 1-6. Notably Mur16, in contrast to the ortholog LipL involved in 4 biosynthesis, is able to catalyze the hydroxylation of 10b along with 10a, thereby initiating the biosynthesis of the 2-deoxy-ADR-containing muraymycins exemplified by 9. Mur20, with 37% sequence identity with LipO, catalyzes transamination of 11 like LipO yet prefers l-Tyr instead of l-Met as the amine donor. Following the Mur20-catalyzed reaction, the phosphorylase Mur26 and nucleotidylyltransferase Mur18 work in tandem to generate an activated sugar. Mur26 initiates this activation by generating the ribose-1-phosphate. Importantly, Mur26 cannot utilize the methylether-containing 13c, thus suggesting that O-methylation—catalyzed by an unidentified enzyme—occurs following this step. Mur18 completes the sugar activation by generating the UDP-sugar. Unlike the typical nucletodiylyltransferase, Mur18 absolutely requires an amine functionality to form the activated sugar, thus joining LipN as a unique group of nucleotidylyltransferase enzymes with respect to this functional group specificity. Finally, the ribosyltransferases Mur19 and LipN were characterized with the bona fide biosynthetic pathway intermediates for the first time to reveal that 5′S,6′S-12 is the likely in vivo sugar acceptor and N-alkylation follows ribosylation. Based on the in vitro characterization of these recombinant proteins, an enzymatic synthesis of 17a (or 17b) starting from 10a (or 10b) was achieved, providing the opportunity to explore downstream enzymatic conversions, highlighted by N-alkylation, that form the basic scaffold that is shared among several promising nucleoside antibiotics.
Figure 10.

Biosynthesis of the ADR-GlyU disaccharide core of muraymycins. The results are consistent with O-methylation occurring downstream of ADR-GlyU formation. αKG, alpha-ketoglutarate; 4-HPP, 4-hydroxyphenylpyruvate.
EXPERIMENTAL SECTION
General Experimental Methods:
UV spectra were recorded on an Ultraspec 8000 spectrometer (GE, Pittsburgh, PA, USA). NMR data were recorded at 400 MHz for 1H and 100 MHz for 13C with Varian Inova NMR spectrometers (Agilent, Santa Clara, CA). HRMS spectra were recorded on an AB SCIEX Q-TOF 5600 System (AB Sciex, Framingham, MA, USA). Analytic HPLC was performed with Dionex Ultimate 3000 or Agilent 1200. Semipreparative HPLC was performed with a Waters 600 controller and pump (Milford, MA) equipped with a 996 diode array detector, 717 plus autosampler, and an Apollo C18 column (250 × 10 mm, 5 µm) purchased from Grace (Deerfield, IL). All solvents used were of HPLC grade and purchased from Fisher Scientific. Muraymycins D1-D4 were isolated from Streptomyces sp. NRRL 30475 as described.29 The synthesis and analytical characterization of 11a,16 12,17,24,26 13a,18 and 13b,18 have been previously reported (Figure S24-S27).
Synthesis of 10c.
The synthesis of 10c followed a previously described procedure.16 Briefly, pyrophosphoryl chloride (0.1 mL) was added to a cooled (4 °C) solution of 2′-O-methyluridine (52 mg) in m-cresol (2 mL). The mixture was stirred for 2 h at 4 °C, diluted with ice-cold water (7 mL), and extracted with diethyl ether (3 mL). The aqueous layer was adjusted to pH = 2 with 4 M sodium hydroxide. After lyophilization, the product was purified by HPLC equipped with a semipreparative Apollo C18 column (250 mm × 10 mm, 5 µm). A series of linear gradients was developed from 0.025% trifluoroacetic acid in water (A) to 0.025% trifluoroacetic acid in acetonitrile (B) in the following manner (beginning time and ending time with linear increase to % B): 0 min, 3% B; 0–8 min, 10% B; 8–9 min, 100% B; 9–13 min, 100% B; and 13–14 min, 3% B. The flow rate was kept constant at 3.5 mL/min, and elution was monitored at 260 nm. 1H NMR (400 MHz, D2O) δ 7.83 (d, J = 8.1 Hz, 1H), 5.92 (d, J = 4.1 Hz, 1H), 5.83 (d, J = 8.2 Hz, 1H), 4.35 (t, J = 5.3 Hz, 1H), 4.21 – 4.16 (m, 1H), 4.15 (q, J = 2.8 Hz, 1H), 4.11 – 4.04 (m, 1H), 3.96 (dd, J = 5.2, 4.2 Hz, 1H), 3.43 (s, 3H). 13C NMR (100 MHz, D2O) δ 166.0 151.3, 141.2, 102.2, 87.0, 82.8, 82.5, 67.8, 64.2, 58.0. HRMS (ESI/Q-TOF) m/z: [M – H]− Calcd for C10H14N2O9P 337.0437; Found 337.0438.
Synthesis of 17a.
The stereoselective synthesis of the disaccharide 17a followed a previously described procedure,30 which is based on the methodology first reported by Hirano et al.4,34,35 1H NMR (500 MHz, D2O) δ 7.74 (d, J = 8.0 Hz, 1H), 5.87 (d, J = 8.0 Hz, 1H), 5.81 (d, J = 2.7 Hz, 1H), 5.17 (s, 1H), 4.50 (br s, 1H), 4.35 (dd, J = 5.0, 2.7 Hz, 1H), 4.18–4.26 (m, 2H), 4.08–4.17 (m, 3H), 3.88 (br s, 1H), 3.25–3.38 (m, 1H), 3.06–3.15 (m, 1H); 13C NMR (126 MHz, D2O) δ 167.4, 152.3, 141.7, 109.0, 102.0, 91.0, 84.7, 79.1, 77.2, 75.0, 73.1, 72.0, 69.3, 57.2, 42.5; HRMS (ESI/Q-TOF) m/z: [M + H]+ Calcd for C16H25N4O11 449.1520; Found 449.1462; IR (ATR) ν 3139, 2935, 2365, 2336, 1703, 1684, 1627, 1510, 1470, 1395, 1272, 1197, 1121, 1051, 1005, 819, 796, 720; UV (H2O) λmax (log ε) 202 (2.13), 262 (2.10); mp 172 °C (decomposition); TLC Rf 0.05–0.16 (5:2:1 i-PrOH-H2O-AcOH as saturated NaCl solution); [α]D20 +3.8 (c 0.90, H2O).
Synthesis of 18.
Protected (5′S,6′S)-12 (previously named S3) was synthesized as described.29 The synthesis of 18 starting from protected (5′S,6′S)-12 followed a previously described method with minor modifications.36 Protected GlyU (100 mg, 0.19 mmol) and 10% Pd/C (10 mg) in MeOH (5 mL) was vigorously stirred under H2 atmosphere at rt for 4 h. The reaction was filtered through a Celite pad and dried at room temperative. A solution of 3-[(benzyloxycarbonyl)amino]propionaldehyde (78 mg, 0.3 mmol) and NaBH3CN (80 μL, 1.3 mmol) in THF (2 mL) was added to the product and stirred at room temperature for 3 h, quenched by adding 500 µL water, and dried. Without purification, the final deprotections were carried out according to the described protocol36 by sequential treatment with LiOH, THF-aq, TFA-aq, and Pd/C H2 to successfully afford 18. 1H NMR (400 MHz, D2O) δ 8.02 (d, J = 8.1 Hz, 1H), 5.92 (d, J = 3.7 Hz, 1H), 5.89 (d, J = 8.1 Hz, 1H), 4.38 (d, 1H), 4.32 (d, J = 5.4 Hz, 1H), 4.27 (d, J = 5.7 Hz, 1H), 4.24 (d, J = 5.7 Hz, 1H), 3.84 (d, J = 6.3 Hz, 1H), 3.22 (m, 2H), 3.10 (t, J = 7.5 Hz, 2H), 2.12 (dt, J = 7.6, 3.6 Hz, 2H). 13C NMR (100 MHz, D2O) δ 173.77, 169.22, 154.03, 145.40, 105.25, 93.07, 87.08, 76.11, 72.82, 70.72, 51.37, 47.34, 39.50, 26.62. HRMS (ESI/Q-TOF) m/z: [M + H]+ Calcd for C14H23N4O8 375.1516; Found 375.1510.
Cloning, Overexpression and Purification of Recombinant Proteins.
The genes mur16, 17, 18, 19, 20 and 26 were amplified by PCR from genomic DNA extracted from Streptomyces sp. NRRL 30473 using Phusion Hot Start II DNA Polymerase from Thermo Scientific with supplied HF buffer and 10 mM each of the following primer pairs: mur16 (forward) 5′- GGTATTGAGGGTCGCGTGGTCCGCGCTGAC −3′ / (reverse) 5′- AGAGGAGAGTTAGAGCCTCAGGGGCTCTCCAG −3′; mur17 (forward) 5′- GATAGGCATATGACCTCTTCGGACGACTGC −3′ / (reverse) 5′- CGAGTTGGATCCTCAGCCATGGAAGAGTCCGG −3′; mur18 (forward) 5′- GGTATTGAGGGTCGCATGGCTGACTTCGCCGAACC −3′ / (reverse) 5′- AGAGGAGAGTTAGAGCCTCATGACCAGCTCCCCGGA −3′; mur19 (forward) 5′- AAAAAACATATGAGCCGCCCGACAAGAGT −3′ / (reverse) 5′- AAAAAAGGATCCTCACAGGGTCGTAGTTCTCAG −3′; mur20 (forward) 5′- GGTATTGAGGGTCGCGTGAGCCCCCAGAGCG −3′ / (reverse) 5′- AGAGGAGAGTTAGAGCCTCAGGCCGTCGCCTCG −3′; and mur26 (forward) 5′- GGTATTGAGGGTCGCATGAGCACCTCCCTCGCG −3′ / (reverse) 5′- AGAGGAGAGTTAGAGCCTCACAGGACGGAGTGCACC −3′. Purified PCR products were inserted into pET-30 Xa/LIC (Novagen) or digested with NdeI/BamHI and ligated into pXY200 following standard prodcedure. PCR integrity was confirmed by DNA sequencing (ACGT, INC).
Plasmids pET30-mur16, pET30-mur18, pET30-mur20 and pET30-mur26 were introduced into E. coli BL21(DE3) cells, and the transformed strains were grown in LB supplemented with 50 μg/mL kanamycin. Following inoculation of 500 mL of LB with 50 μg/mL kanamycin, the cultures were grown at 37 °C until the cell density reached an OD600 ~ 0.5 when expression was induced with 0.1 mM IPTG. Cells were harvested after an overnight incubation at 18 °C and lysed in 100 mM KH2PO4, 300 mM NaCl, and 10 mM imidazole (pH 8.3) using a Qsonica sonicator (Qsonica LLC, Newtown, CT) for sonication for a total of 2 min at 40% amplitude with 2 s pulses separated by 8 s rest periods. Following centrifugation the protein was purified using affinity chromatography with HisPurTM Ni-NTA agarose (Thermo Scientific, Rockford, IL), and proteins were eluted with increasing concentrations of imidazole in Buffer A. Purified proteins were concentrated and buffer exchanged into 25 mM KH2PO4 and 100 mM NaCl (pH 8.3) using Amicon Ultra 10,000 MWCO centrifugal filter (Millipore) and stored as glycerol stocks (40%) at −20 °C. Protein purity was assessed as by 12% acrylamide SDS-PAGE; His6-tagged proteins were utilized without further modifications. Protein concentration was determined using UV/Vis spectroscopy, and the extension coefficients were calculated using the ProtParam tool available from ExPASY.
Plasmids pXY200-mur17 and pXY200-mur19 were transformed into S. lividans TK24 using PEG-mediated protoplast transformation and plated on R2YE supplemented with 50 μg/mL apramycin. After 6 days at 28 °C, positive transformants were confirmed by colony PCR using InstaGene Matrix from Bio-Rad (Hercules, CA) and LA-Taq polymerase with GC buffer I. The recombinant strains were utilized to inoculate 50 mL R2YE containing 50 μg/mL apramycin, grown for 3 days at 28 °C at 250 rpm, and 2 mL transferred to fresh 100 mL containing 50 μg/mL apramycin. Following growth for 3 days at 28 °C at 250 rpm, protein expression was induced by addition of thiostrepton (5 μg/mL) and the culture was incubated for another 24 h before harvesting. The cells from 400 mL of culture were collected by centrifugation. The pellet was thoroughly resuspended in ice-cold lysis buffer and supplemented with 4 mg/mL of lysozyme after suspension. After incubation at 30 °C for 30 min, the cell suspension was mixed by pipetting and lysed using a Qsonica sonicator (Qsonica LLC, Newtown, CT) for sonication for a total of 4 min at 40% amplitude with 2 s pulses separated by 8 s rest periods. The remaining steps were performed as described above.
Activity Assay for Mur16.
Reactions consisted of 50 mM HEPES (pH 7.5), 2.5 mM αKG, 2 mM ascorbic acid, 0.2 mM FeCl2, 1 mM 10a, 10b, or 10c, and 100 nM Mur16 at 30 °C. Reactions were terminated by ultrafiltration using Amicon® Ultra centrifugal filter units. Following centrifugation, the filtrate was analyzed by HPLC or LCMS equipped with an analytical Apollo C18 column (250 mm × 4.6 mm, 5 μm). A series of linear gradients was developed from 0.1% formic acid in water (C) to 0.1% formic acid in acetonitrile (D) in the following manner (beginning time and ending time with linear increase to % D): 0 min, 1% D; 0–16 min, 20% D; 16–28 min, 100% D; 28–37 min, 100% D; and 37–38 min, 1% D. The flow rate was kept constant at 0.5 mL/min, and elution was monitored at 260 nm.
Kinetics of Mur16.
To determine the kinetic constants, reactions were carried out in 50 mM Tris-HCl (pH 7.5), 2 mM αKG, 2 mM ascorbic acid, 0.4 mM FeCl2 with variable concentration of 10a, 10b or 10c (1 µM-500 or 1000 µM), and 40 nM Mur16 at 30 °C for 4 min. Phosphate formation was monitored using the malachite green binding assay and the formation of aldehyde 11a, 11b or 11c was monitored by HPLC. Each data point represents three replicate end point assays. Kinetic constants were obtained by nonlinear regression analysis using GraphPad Prism 7 (GraphPad Software, La Jolla, CA).
Activity Assay for Mur17.
Reactions consisted of 50 mM HEPES (pH 7.5), 2 mM 11a, 11b, or 11c, 5 mM l-Thr, 0.1 mM PLP and 1 μM Mur17 at 30 °C. Following removal of the protein by ultrafiltration, the reaction was analyzed by LCMS using the conditions described for Mur16. Alternatively, the reaction components or 12 standards were modified with 6-aminoquinolyl-N-hydroxysuccinimidyl carbamate (AQC) prior to analysis in a reaction consisting of 20 μL sample, 60 μL of 0.2 M sodium borate buffer (pH 8.8) and 20 μL of 3 mg/mL AQC acetonitrile solution. Samples were incubated at 55 °C for 10 min and then cooled to room temperature. The AQC-derivatized samples (50 μL) were applied to LCMS equipped with an analytical Acclaim™ 120 C18 column (100 mm × 4.6 mm, 5 µm). A series of linear gradients was developed in the following manner (beginning time and ending time with linear increase to % D): 0 min, 1% D; 0–40 min, 20% D; 40–41 min, 100% D; 41–44 min, 100% D; and 44–45 min, 1% D. The flow rate was kept constant at 0.4 mL/min, and elution was monitored at 260 nm.
Activity Assay of Mur20.
Reactions consisted of 50 mM potassium phosphate (pH 7.5), 1 mM 11a, 11b, or 11c, 1 mM l-amino acid, and 1 μM Mur20 at 30 °C. Following removal of the protein by ultrafiltration, the reaction was analyzed by LCMS using the conditions described for Mur16 or equipped with an analytical Apollo C18 column (250 mm × 4.6 mm, 5 μm). A series of linear gradients was developed from 40 mM triethylamine-acetic acid (pH 6.5) in water (E) to 40 mM triethylamine-acetic acid (pH 6.5) in 20% methanol (F) in the following manner (beginning time and ending time with linear increase to % F): 0 min, 0% F; 0–8 min, 100% F; 8–18 min, 60% F; and 18–19 min, 0% F. The flow rate was kept constant at 1.0 mL/min, and elution was monitored at 260 nm.
To identify the production of α-keto-γ-methylthiol butyric acid, reaction mixtures or the positive control (α-keto-γ-methylthiol butyric acid) were treated with 3-methyl-2-benzothiazolinone hydrazone hydrochloride (MBTH) according to the method described by Tanaka.28 Following removal of the protein by ultrafiltration, the filtrate (50 μL) was mixed with 50 μL of 1 M sodium acetate (pH 5.0) and 50 μL of 8 mM MBTH aqueous solution. The mixtures were incubated at 50 °C for 30 min and analyzed by LCMS equipped with an analytical Acclaim™ 120 C18 column (100 mm × 4.6 mm, 5 µm). A series of linear gradients was developed in the following manner (beginning time and ending time with linear increase to % D): 0 min, 10% D; 0–10 min, 40% D; 10–20 min, 100% D; 20–27 min, 100% D; and 27–28 min, 10% D. The flow rate was kept constant at 0.4 mL/min, and elution was monitored at 350 nm.
Activity Assay of Mur26.
Reactions consisted of 50 mM potassium phosphate (pH 7.5), 2 mM 13a or 13b (13c was generated in situ with Mur16 and Mur20) and 1 μM Mur26 at 30 °C. Following removal of the protein by ultrafiltration, the filtrate was analyzed by LCMS using the conditions described for Mur16.
Activity Assay of Mur18.
Reactions of Mur18 consisted of 50 mM potassium phosphate (pH 7.5), 2 mM 13a or 13b, 5 mM MgCl2, 2 mM UTP, 1 μM Mur26 (or LipP) and 1 μM Mur18. Following removal of the protein by ultrafiltration, the filtrate was analyzed by LCMS using the conditions described for Mur20 with a triethylamine-acetic acid mobile phase or with an analytical Apollo C18 column (250 mm × 4.6 mm, 5 μm). A series of linear gradients was developed in the following manner (beginning time and ending time with linear increase to % D): 0 min, 80% D; 0–12 min, 50% D; 12–26 min, 50% D; 26–27 min, 80% D; and 27–35 min, 80% D. The flow rate was kept constant at 0.4 mL/min, and elution was monitored at 260 nm.
Activity Assay of Mur19.
Reactions consisted of 50 mM potassium phosphate (pH 7.5), 2 mM 13a or 13b, 2 mM 12, 5 mM MgCl2, 2 mM UTP, 1 μM Mur26 (or LipP), 1 μM Mur18, and 1 μM Mur19. Following removal of the protein by ultrafiltration, the filtrate was analyzed by LCMS using the conditions described for Mur20 with a triethylamine-acetic acid mobile phase. Large-scale production of 17b (30 mL reaction) was identical to reactions described above using 13b, 12 and UTP with pre-column AQC derivatization. The samples were applied to an HPLC equipped with a semipreparative Apollo C18 column (250 mm × 10 mm, 5 µm). A series of linear gradients was developed in the following manner (beginning time and ending time with linear increase to % B): 0 min, 12% B; 0–15 min, 12% B; 15–16 min, 100% B; 16–19 min, 100% B; and 19–20 min, 12% B. The flow rate was kept constant at 4.0 mL/min, and elution was monitored at 260 nm.
Enzymatic Synthesis of 17a.
Reactions consisted of 50 mM HEPES (pH 7.5), 2.5 mM αKG, 2 mM ascorbic acid, 0.2 mM FeCl2, 4 mM 10a, 2 mM l-Thr, 0.1 mM PLP, 2 mM l-Met, 1 mM MgCl2, 2 mM UTP, and 1 μM each of Mur16, Mur17, LipO, LipP, Mur18 and Mur19. Following removal of the protein by ultrafiltration, the filtrate was analyzed by LCMS using the conditions described for Mur20 with a triethylamine-acetic acid mobile phase.
Enzymatic Synthesis of 17b.
Reactions consisted of 50 mM HEPES (pH 7.5), 2.5 mM αKG, 2 mM ascorbic acid, 0.2 mM FeCl2, 4 mM 10b, 100 μM 12, 0.1 mM PLP, 2 mM l-Met, 1 mM MgCl2, 2 mM UTP, and 1 μM each of Mur16, LipO, LipP, Mur18 and Mur19. Following removal of the protein by ultrafiltration, the filtrate was analyzed by LCMS using the conditions described for Mur20 with a triethylamine-acetic acid mobile phase.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health (NIH) grants R01 AI087849 (SVL), R01 GM115261 (JST), the University of Kentucky College of Pharmacy, the University of Kentucky Markey Cancer Center, the National Center for Advancing Translational Sciences (UL1TR001998), the Deutsche Forschungsgemeinschaft (DFG, grant DU 1095/5–1), the state of Lower Saxony [Lichtenberg doctoral fellowship (CaSuS program) for A.L.], the Konrad-Adenauer-Stiftung (doctoral fellowship for D.W.), and the Fonds der Chemischen Industrie (doctoral fellowship for G.N.).
Footnotes
Supporting Information Available: Supporting Information containing Table S1 and Figures S1-S27 is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Kimura K; Bugg TD Recent advances in antimicrobial nucleoside antibiotics targeting cell wall biosynthesis. Nat. Prod. Rep 2003, 20, 252–273. [DOI] [PubMed] [Google Scholar]
- (2).Winn M; Goss RJ; Kimura K; Bugg TD Antimicrobial nucleoside antibiotics targeting cell wall assembly: recent advances in structure-function studies and nucleoside biosynthesis. Nat. Prod. Rep 2010, 27, 279–304. [DOI] [PubMed] [Google Scholar]
- (3).Wiegmann D; Koppermann S; Wirth M; Niro G; Leyerer K; Ducho C Muraymycin nucleoside-peptide antibiotics: uridine-derived natural products as lead structures for the development of novel antibacterial agents. Beilstein J. Org. Chem 2016, 12, 769–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hirano S; Ichikawa S; Matsuda A Total synthesis of (+)-FR-900493 and establishment of its absolute stereochemistry. Tetrahedron 2007, 63, 2798–2804. [Google Scholar]
- (5).Isono K; Uramoto M; Kusakabe H; Kimura K; Izaki K; Nelson CC; McCloskey JA Liposidomycins: novel nucleoside antibiotics which inhibit bacterial peptidoglycan synthesis. J. Antibiot 1985, 38, 1617–1620. [DOI] [PubMed] [Google Scholar]
- (6).Kimura K; Ikeda Y; Kagami S; Yoshihama M Selective inhibition of the bacterial peptidoglycan biosynthesis by the new types of liposidomycins. J. Antibiot 1998, 51, 1099–1104. [DOI] [PubMed] [Google Scholar]
- (7).Igarashi M; Nakagawa N; Doi N; Hattori S; Naganawa H; Hamada M Caprazamycin B, a novel anti-tuberculosis antibiotic, from Streptomyces sp. J. Antibiot 2003, 56, 580–583. [DOI] [PubMed] [Google Scholar]
- (8).Igarashi M; Takahashi Y; Shitara T; Nakamura H; Naganawa H; Miyake T; Akamatsu Y Caprazamycins, novel lipo-nucleoside antibiotics, from Streptomyces sp. II. Structure elucidation of caprazamycins. J. Antibiot 2005, 58, 327–337. [DOI] [PubMed] [Google Scholar]
- (9).Fujita Y; Kizuka M; Funabashi M; Ogawa Y; Ishikawa T; Nonaka K; Takatsu T A-90289 A and B, new inhibitors of bacterial translocase I, produced by Streptomyces sp. SANK 60405. J. Antibiot 2011, 64, 495–501. [DOI] [PubMed] [Google Scholar]
- (10).Chi X; Baba S; Tibrewal N; Funabashi M; Nonaka K; Van Lanen SG The muraminomicin biosynthetic gene cluster and enzymatic formation of the 2-deoxyaminoribosyl appendage. MedChemComm 2013, 4, 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Funabashi M; Baba S; Takatsu T; Kizuka M; Ohata Y; Tanaka M; Nonaka K; Spork AP; Ducho C; Chen WC; Van Lanen SG Structure-based gene targeting discovery of sphaerimicin, a bacterial translocase I inhibitor. Angew. Chem. Int. Ed 2013, 52, 11607–11611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).McDonald LA; Barbieri LR; Carter GT; Lenoy E; Lotvin J; Petersen PJ; Siegel MM; Singh G; Williamson RT Structures of the muraymycins, novel peptidoglycan biosynthesis inhibitors. J. Am. Chem. Soc 2002, 124, 10260–10261. [DOI] [PubMed] [Google Scholar]
- (13).McDonald LA; Barbieri LR; Carter GT; Kruppa G; Feng X; Lotvin JA; Siegel MM FTMS structure elucidation of natural products: application to muraymycin antibiotics using ESI multi-CHEF SORI-CID FTMS(n), the top-down/bottom-up approach, and HPLC ESI capillary-skimmer CID FTMS. Anal Chem. 2003, 75, 2730–2739. [DOI] [PubMed] [Google Scholar]
- (14).Cui Z; Wang X; Koppermann S; Thorson JS; Ducho C; Van Lanen SG J. Nat. Prod 2018, 81, 942–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Funabashi M; Baba S; Nonaka K; Hosobuchi M; Fujita Y; Shibata T; Van Lanen SG The biosynthesis of liposidomycin-like A-90289 antibiotics featuring a new type of sulfotransferase. ChemBioChem 2010, 11, 184–190. [DOI] [PubMed] [Google Scholar]
- (16).Yang Z; Chi X; Funabashi M; Baba S; Nonaka K; Pahari P; Unrine J; Jacobsen JM; Elliott GI; Rohr J; Van Lanen SG Characterization of LipL as a non-heme, Fe (II)-dependent α-ketoglutarate: UMP dioxygenase that generates uridine-5′-aldehyde during A-90289 biosynthesis. J. Biol. Chem 2011, 286, 7885–7892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Barnard-Britson S; Chi X; Nonaka K; Spork AP; Tibrewal N; Goswami A; Pahari P; Ducho C; Rohr J; Van Lanen SG Amalgamation of nucleosides and amino acids in antibiotic biosynthesis: discovery of an l-threonine: uridine-5′-aldehyde transaldolase. J. Am. Chem. Soc 2012, 134, 18514–18517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chi X; Pahari P; Nonaka K; Van Lanen SG Biosynthetic origin and mechanism of formation of the aminoribosyl moiety of peptidyl nucleoside antibiotics. J. Am. Chem. Soc 2011, 133, 14452–14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Goswami A; Liu X; Cai W; Wyche TP; Bugni TS; Meurillon M; Peyrottes S; Perigaud C; Nonaka K; Rohr J; Van Lanen SG Evidence that oxidative dephosphorylation by the nonheme Fe(II), α-ketoglutarate:UMP oxygenase occurs by stereospecific hydroxylation. FEBS Lett 2017, 591, 468–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kaysser L; Lutsch L; Siebenberg S; Wemakor E; Kammerer B; Gust B Identification and manipulation of the caprazamycin gene cluster lead to new simplified liponucleoside antibiotics and give insights into the biosynthetic pathway. J. Biol. Chem 2009, 284, 14987–14996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kaysser L; Siebenberg S; Kammerer B; Gust B Analysis of the liposidomycin gene cluster leads to the identification of new caprazamycin derivatives. ChemBioChem 2010, 11, 191–196. [DOI] [PubMed] [Google Scholar]
- (22).Cheng L; Chen W; Zhai L; Xu D; Huang T; Lin S; Zhou X; Deng Z Identification of the gene cluster involved in muraymycin biosynthesis from Streptomyces sp. NRRL 30471. Mol. BioSyst 2011, 7, 920–927. [DOI] [PubMed] [Google Scholar]
- (23).Imai K; Fujii S; Takanohashi K; Furukawa Y; Masuda T; Honjo M Phosphorylation. IV. Selective phosphorylation of the primary hydroxyl group in nucleosides. J. Org. Chem 1969, 34, 1547–1550. [Google Scholar]
- (24).Spork AP; Koppermann S; Dittrich B; Herbst-Irmer R; Ducho C Efficient synthesis of the core structure of muraymycin and caprazamycin nucleoside antibiotics based on a stereochemically revised sulfur ylide reaction. Tetrahedron: Asymmetry 2010, 21, 763–766. [Google Scholar]
- (25).Spork AP; Büschleb M; Ries O; Wiegmann D; Boettcher S; Mihalyi A; Bugg TDH; Ducho C Lead structures for new antibacterials: stereocontrolled synthesis of a bioactive muraymycin analogue. Chem. Eur. J 2014, 20, 15292–15297. [DOI] [PubMed] [Google Scholar]
- (26).Ying H; Liu X; Cui Z; Wiegmann D; Niro G; Ducho C; Song Y; Yang Z; Van Lanen SG Pyridoxal-5′-phosphate as an oxygenase cofactor: Discovery of a carboxamide-forming, α-amino acid monooxygenase-decarboxylase. Proc. Natl. Acad. Sci. USA, 2018, 115, 974–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Spork AP; Ducho C Stereocontrolled synthesis of 5’- and 6’-epimeric analogues of muraymycin nucleoside antibiotics. Synlett 2013, 24, 343–346. [Google Scholar]
- (28).Tanaka H; Yamamoto A; Ishida T; Horiike K Simultaneous measurement of d-serine dehydratase and d-amino acid oxidase activities by the detection of 2-oxo-acid formation with reverse-phase high-performance liquid chromatography. Anal. Biochem 2007, 362, 83–88. [DOI] [PubMed] [Google Scholar]
- (29).Nakada HI Glutamic-glycine transaminase from rat liver. J. Biol. Chem 1964, 239, 468–471. [PubMed] [Google Scholar]
- (30).Cui Z; Wang X-C; Liu X; Lemke A; Koppermann S; Ducho C; Rohr J; Thorson JS; Van Lanen SG Self-resistance during muraymycin biosynthesis: A complementary nucleotidyltransferase and phosphotransferase with identical modification sites and distinct temperal order. Antimicrob. Agents. Chemother 2018, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lairson LL; Henrissat B; Davies GJ; Withers SG Glycosyltransferases: structures, functions, and mechanisms. Ann. Rev. Biochem 2008, 77, 521–555. [DOI] [PubMed] [Google Scholar]
- (32).Liang D-M; Liu J-H; Wu H; Wang B-B; Zhu H-J; Qiao J-J Glycosyltransferases: mechanisms and applications in natural product development. Chem. Soc. Rev 2015, 44, 8350–8374. [DOI] [PubMed] [Google Scholar]
- (33).Williams GJ; Thorson JS Natural product glycosyltransferases: properties and applications. Adv. Enzymol. Relat. Areas Mol. Biol 2009, 76, 55–119. [DOI] [PubMed] [Google Scholar]
- (34).Elshahawi SI; Shaaban KA; Kharel MK; Thorson JS A comprehensive review of glycosylated bacterial natural products. Chem. Soc. Rev 2015, 44, 7591–7697. [DOI] [PMC free article] [PubMed] [Google Scholar]; (34) Hirano S; Ichikawa S; Matsuda A Development of a highly β-selective ribosylation reaction without using neighboring group participation: Total synthesis of (+)-caprazol, a core structure of caprazamycins. J. Org. Chem 2007, 72, 9936–9946. [DOI] [PubMed] [Google Scholar]
- (35).Hirano S; Ichikawa S; Matsuda A Total synthesis of caprazol, a core structure of the caprazamycin antituberculosis antibiotics. Angew. Chem. Int. Ed 2005, 44, 1854–1856. [DOI] [PubMed] [Google Scholar]
- (36).Tanino T; Ichikawa S; Shiro M; Matsuda A Total synthesis of (−)-muraymycin D2 and its epimer. J. Org. Chem 2010, 75, 1366–1377. [DOI] [PubMed] [Google Scholar]
- (37).Schmid A; Dordick JS; Hauer B; Kiener A; Wubbolts M; Witholt B Industrial biocatalysis today and tomorrow. Nature 2001, 409, 258–268. [DOI] [PubMed] [Google Scholar]
- (38).Johannes T; Simurdiak MR; Zhao H Biocatalysis In Encyclopedia of Chemical Processing; Lee S, Ed.; CRC Press: Boca Raton, 2005; pp 101–109. [Google Scholar]
- (39).Bornscheuer U Biocatalysis In Catalysis: From Principles to Applications; Beller M, Renken A, van Santen R, Eds.; Wiley-VCH Verlag GmbH & Co, 2012; pp 171–200. [Google Scholar]
- (40).Kayne FJ Adenylate Kinase In The Enzymes; Boyer PD, Ed.; Academic Press: New York, NY, 1973; 3rd ed., Vol. 8, pt. A, pp 353–382. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.