

Supplemental Digital Content is available in the text.
Keywords: liposomes; macrophages; myocardial infarction; receptors, mineralocorticoid; wound healing
Abstract
Myocardial infarction (MI) is a major cause of death worldwide. Here, we identify the macrophage MR (mineralocorticoid receptor) as a crucial pathogenic player in cardiac wound repair after MI. Seven days after left coronary artery ligation, mice with myeloid cell–restricted MR deficiency compared with WT (wild type) controls displayed improved cardiac function and remodeling associated with enhanced infarct neovascularization and scar maturation. Gene expression profiling of heart-resident and infarct macrophages revealed that MR deletion drives macrophage differentiation in the ischemic microenvironment toward a phenotype outside the M1/M2 paradigm, with regulation of multiple interrelated factors controlling wound healing and tissue repair. Mechanistic and functional data suggest that inactivation of the macrophage MR promotes myocardial infarct healing through enhanced efferocytosis of neutrophils, the suppression of free radical formation, and the modulation of fibroblast activation state. Crucially, targeted delivery of MR antagonists to macrophages, with a single administration of RU28318 or eplerenone-containing liposomes at the onset of MI, improved the healing response and protected against cardiac remodeling and functional deterioration, offering an effective and unique therapeutic strategy for cardiac repair.
Myocardial infarction (MI) and ensuing heart failure are leading causes of death worldwide. Ischemic cell death triggers a cascade of cellular and molecular events that promote wound healing and structural cardiac remodeling. Immune mechanisms/mediators regulating ischemic injury and tissue repair are considered important therapeutic targets to prevent cardiac functional decline and are the focus of intensive research efforts.1–4
MR (mineralocorticoid receptor) blockade reduces morbidity and mortality in the setting of myocardial infarction. Emerging evidence suggests that administration of MR antagonists early in the course of acute MI offers the greatest benefit in clinical outcome.5,6 Modulation of the inflammatory response, improvement of cardiac structural and electrical remodeling, and prevention of life-threatening arrhythmia appear to play an important role.7–10
Monocytes/macrophages act as key players in the wound repair process through the clearance of apoptotic cells and the release of cytokines/chemokines, proteases, growth factors, and oxygen-derived free radicals.2,11,12 Experimental studies using cell-specific MR knockout highlighted the cell-specific effects of MR activation in the cardiovascular system and identified the myeloid MR as a critical regulator of cardiovascular inflammation, fibrosis, and hypertrophy.7,13,14
However, it remains unknown whether the benefits of early MR blockade post-MI can be attributed to inhibition of the MR signaling in macrophages. Using mice with myeloid cell–restricted MR deficiency, we investigated the relevance of the macrophage MR for cardiac repair and remodeling, dissecting the pathogenic mechanisms associated with MR activation after acute MI. Moreover, we explored whether the targeted delivery of MR antagonists to macrophages at the onset of MI improved the healing response and protected against cardiac remodeling and functional deterioration.
Materials and Methods
The authors declare that all supporting data are available within the article and its online-only Data Supplement.
Detailed Materials and Methods are available in the online-only Data Supplement.
Study Protocol
All animal experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (Publication No. 85–23, revised 1985). All procedures were approved by the Regierung von Unterfranken (Würzburg, Germany; permit No. 54–2531.01-15/07) and by the Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit (Oldenburg, Germany; permit No. 33.12-42502-04-11/0644 and 33.9-42502-04-13/1124). Adult Nr3c2tm2Gsc (MRflox, WT controls), Nr3c2tm2GscLyz2tm1(cre)lfo/J (MRLysMCre), and C57Bl/6 mice of both sexes were used in this study. Liposome-encapsulated RU28318 (2 µmol/kg) or liposome-encapsulated eplerenone (20 µmol/kg) were administered intraperitoneally (100 μmol lipid per kg) in C57BL/6 mice at the onset of myocardial infarction. Control C57BL/6 mice were injected with equal volumes of plain (MR antagonists lacking) liposome. In additional experimental groups, starting immediately after coronary ligation, C57BL/6 mice were randomly selected for eplerenone (100 mg/kg of body weight) or placebo treatment (5% arabic gum) administered by gavage once daily. Mice subjected to coronary artery ligation were sacrificed at days 1, 3, or 7 after surgery. Mice were excluded from the analyses for 2 reasons: perioperative death (within the first 12 hours after surgery) and MI size <40.
Statistical Analysis
Results are reported as mean±SEM. One-way ANOVA with Tukey post hoc test or with the unpaired t test was performed using GraphPad Prism 6.01 (GraphPad Software, Inc). Values of P <0.05 were considered statistically significant.
Results
The schematic representation of the MR deletion strategy is shown in Figure S1A through S1C in the online-only Data Supplement. Quantitative reverse-transcriptase polymerase chain reaction revealed downregulation of MR expression (Figure S1D) in cardiac macrophages from mice with myeloid cell–restricted MR deficiency (hereafter referred to as MRLysMCre) compared with WT (wild type) controls (hereafter referred to as MRflox).
Mice With Myeloid Cell–Restricted MR Deficiency Displayed Improved Cardiac Function and Remodeling After MI
Infarct size was similar among MRflox and MRLysMCre mice (Figure 1A). We did not detect differences between MRflox and MRLysMCre sham-operated mice regarding left ventricular (LV) systolic or diastolic pressure, cardiac volume, and function (Figure 1B; Figure S1E). Myeloid cell–restricted MR deficiency prevented the rightward shift of the pressure-volume curve 7 days after left coronary artery ligation (Figure 1B). LV end-diastolic pressure, LV end-diastolic volume, and LV end-systolic volume were significantly decreased compared with MRflox (Figure 1C). Amelioration of LV remodeling in infarcted MRLysMCre mice was associated with a significant improvement in LV ejection fraction (Figure 1C). Correspondingly, MRLysMCre mice exhibited enhanced LV dP/dtmin, LV dP/dtmax, and LV dP/dtmax divided by instantaneous pressure—a load-independent measure of contractile function. Moreover, the time constant of LV pressure isovolumic decay (τ)—a relatively load-independent index of LV relaxation—was significantly shortened in MRLysMCre mice compared with control animals (Figure 1C). These results indicate that myeloid cell–restricted MR deficiency prevents early post-MI cardiac dilation, functional deterioration, and failure.
Figure 1.
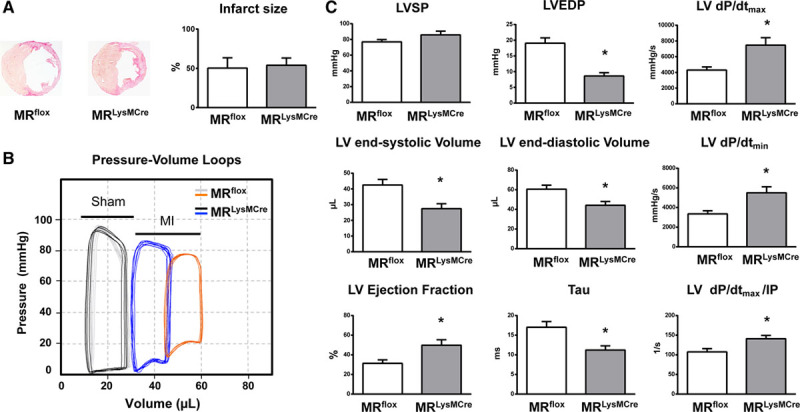
Mice with myeloid cell–restricted MR (mineralocorticoid receptor) deficiency display improved cardiac function and remodeling after myocardial infarction (MI). A, Representative sections from MRflox and MRLysMCre infarcted hearts and infarct size. B, Representative left ventricular (LV) pressure-volume loops measured in vivo with conductance catheter in sham-operated MRflox (gray) and MRLysMCre (black) mice and in MRflox (orange) and MRLysMCre (blue) mice with MI. C, LV systolic pressure (LVSP), LV end-diastolic pressure (LVEDP), LV end-systolic and end-diastolic volumes; LV ejection fraction, LV maximal rate of pressure rise (LV dP/dtmax), maximal rate of pressure decline (LV dP/dtmin), and LV dP/dtmax divided by instantaneous pressure (IP) and the time constant of LV pressure isovolumic decay (Tau). Mean±SEM (n=14–16). *P<0.01 vs MRflox.
Enhanced Infarct Neovascularization and Scar Maturation in MRLysMCre Mice After Ischemic Injury
Neovascularization of the ischemic myocardium plays an essential role in fibrous tissue formation and infarct healing. Seven days post-infarction, MRLysMCre mice displayed an enhanced angiogenic response to ischemic injury (Figure 2A through 2C). Immunofluorescence analysis showed an increased number of capillaries, identified as small lumen vessels positively staining for CD (cluster of differentiation) 31 (Figure 2C). As the scar matures, many vessels acquire a pericyte coat that stabilizes the infarct vasculature. Many more coated vessels, identified as thin-walled α-SMA (α-smooth muscle actin)–positive vascular structures, were observed in the infarcted myocardium of MRLysMCre mice compared with MRflox animals (Figure 2A and 2B).
Figure 2.
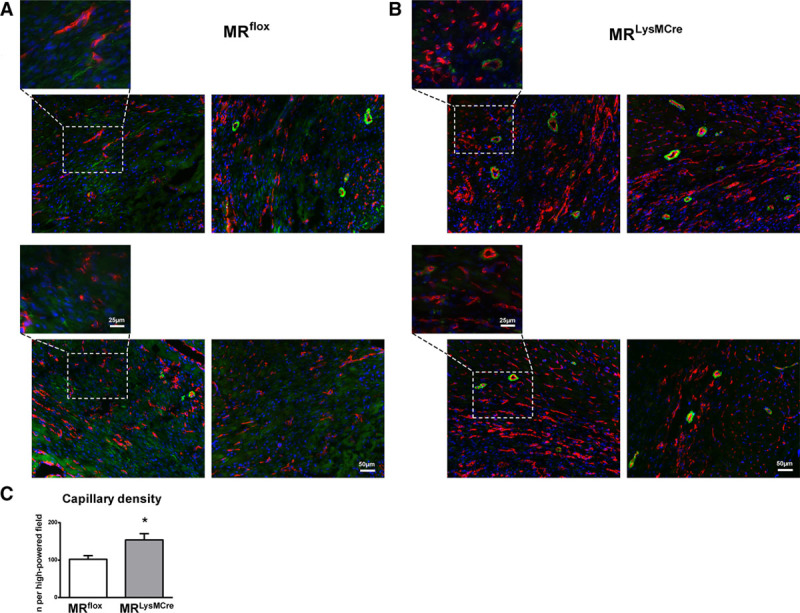
Enhanced cardiac neovascularization after ischemic injury in MRLysMCre mice. A and B, Immunofluorescence double staining (CD [cluster of differentiation] 31, red; α-SMA [α-smooth muscle actin], green) showing capillaries, coated vessels, and (myo)fibroblasts in the healing myocardium of MRflox and MRLysMCre mice, 7 d after myocardial infarction. In MRLysMCre infarcts, the expression of α-SMA was mostly restricted to pericytes and arterioles. C, Capillary density quantification in MRflox and MRLysMCre mice. Mean±SEM (n=6). *P<0.01 vs MRflox.
Immunofluorescence staining for α-SMA also demonstrated the presence of myofibroblasts, identified as extramural spindle-shaped α-SMA–positive cells, within the infarct region 7 days post-MI (Figures 2A and 3A; enlarged insets). Recent data indicated that in the infarcted myocardium, fibroblasts become activated, differentiate to a α-SMA–positive myofibroblast phenotype, and after the first 7 days, when the infarct scar is mature, begin to lose their α-SMA expression.15 It is noteworthy that in MRLysMCre infarcts, the expression of α-SMA was reduced in fibroblasts (Figures 2B and 3B; enlarged insets) and α-SMA–positive cells were mostly restricted to vessels.
Figure 3.
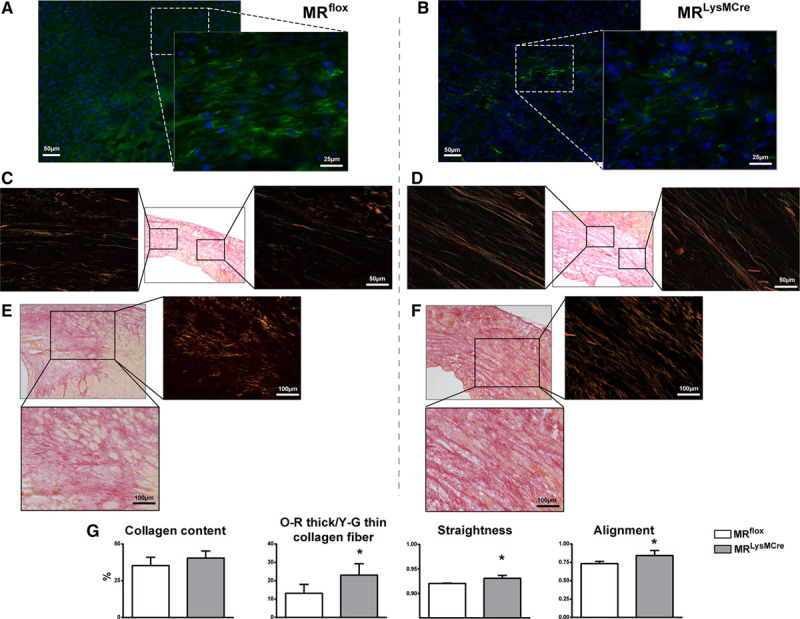
Enhanced collagen scar formation in MRLysMCre mice. Immunofluorescence staining for α-SMA (α-smooth muscle actin; green) showing the presence of myofibroblasts (spindle-shaped α-SMA–positive cells) within the infarct region of (A) MRflox and (B) MRLysMCre mice. MRLysMCre infarcts exhibited a loss of α-SMA expression in (myo)fibroblasts. C–F, Sirius red polarization microscopy of scar sections revealed a matrix with a predominance of thin and loosely assembled collagen fibers in MRflox mice (C and E) and well-aligned and tightly packed collagen fibers in MRLysMCre infarcts (D and F), 7 d after myocardial infarction. G, Infarct collagen content, ratio of orange-red (O-R) thick to yellow-green (Y-G) thin collagen fibers, and fiber straightness and alignment. Mean±SEM (n=6). *P<0.05 vs MRflox.
Scar collagen content was similar among MRflox and MRLysMCre mice, yet collagen maturation was improved in MRLysMCre infarcts (Figure 3C through 3G). Sirius red polarization microscopy revealed a mostly well-aligned collagen matrix with thick, tightly assembled mature collagen fibers in MRLysMCre mice (Figure 3D and 3F) in contrast to the matrix with a predominance of thin, loosely packed immature fibers observed in control-MI mice (Figure 3C and 3E). The ratio of thick (orange-red)-to-thin (yellow-green) birefringent collagen fibers and fiber straightness and alignment were significantly increased in MRLysMCre compared with MRflox infarcts (Figure 3G). Taken together, these findings show MR in myeloid cells to be a pathogenic player and important therapeutic target in cardiac wound repair.
MR Inactivation Drives Macrophage Differentiation in the Ischemic Microenvironment Toward a Phenotype Outside the M1/M2 Activation Paradigm
To delineate the macrophage-specific mechanisms underlying the improved healing response to ischemic injury by myeloid cell–restricted MR deficiency, we investigated phenotypic differences between macrophages isolated from MRflox and MRLysMCre hearts. Importantly, using a modified Langendorff perfusion system, the hearts were perfused for 6 minutes to remove blood cells and subsequently digested for 8 minutes to preserve cell surface antigens along with gene expression profile.2 Infarct macrophages were identified by flow cytometry as CD45+/CD11b+/Ly6G−/F4/80+ cells and then further stratified by Ly6C expression (Figure 4A). As shown in Figure S2A, infarct macrophages were Mertk+ and CD64+, whereas infiltrating monocytes (CD45+/CD11b+/Ly6G−/F4/80−) were Mertk−. At day 3 post-MI, corresponding to transition from the inflammatory to the reparative phase of cardiac wound repair, we showed that macrophages in the infarcted myocardium of MRLysMCre mice almost exclusively consisted of Ly6Clow cells (Figure 4A; Figure S2B). Interestingly, the number of infarct macrophages was similar in MRLysMCre versus MRflox mice, but the amount of CD45+/CD11b+/F4/80−/Ly6G+ cells was significantly lower, indicative of reduced neutrophil accumulation in the ischemic myocardium of MRLysMCre mice (Figure 4A).
Figure 4.
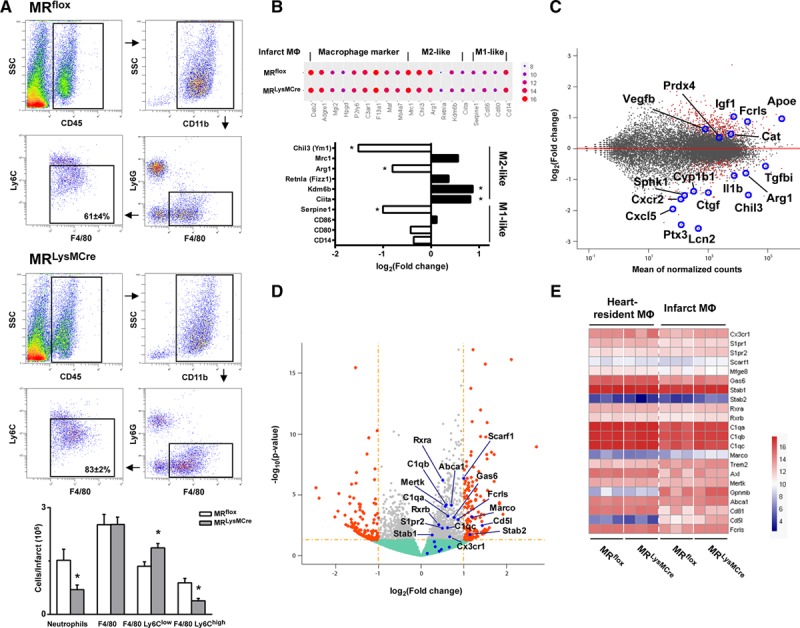
MR (mineralocorticoid receptor) inactivation drives macrophage differentiation in the ischemic microenvironment toward a phenotype outside the M1/M2 activation paradigm. A, Flow cytometry and gating strategy identifying macrophages (CD [cluster of differentiation] 45+/CD11b+/Ly6G−/F4/80+ cells −Ly6Clow and Ly6Chigh) and neutrophils (CD45+/CD11b+/F4/80−/Ly6G+ cells) in MRflox and MRLysMCre infarcts, 3 d after coronary artery ligation. SSC indicates side scatter. Mean±SEM (n=5). *P<0.05 vs MRflox. B–E, Comparison of gene expression profiles of infarct macrophages from MRLysMCre and MRflox mice (n=3). B, Dot plot showing gene expression of macrophage markers; fold changes of M1/M2-like markers are reported (*Padj<0.1). C, MA plot showing significantly regulated genes with Padj <0.1 (red). D, Volcano plot; significantly regulated genes with P<0.05 (gray) and with fold change >2 (orange) are shown; upregulated efferocytic genes are highlighted (blue). E, Heatmap showing efferocytic gene expression.
Next, we performed RNA sequencing of fluorescence-activated cell sorting (FACS)–isolated resident macrophages at steady state and of infarct macrophages sorted from the ischemic region at day 3. As shown in the Figure S3A principal component analysis comparing transcriptional profile determined by RNA-seq of heart-resident macrophages at steady state, infarct macrophages and blood monocytes revealed strong dissimilarity between circulating monocytes and cardiac macrophages in steady state and after infarction. Moreover, MA plot of gene expression from infarct macrophages versus resident macrophages at steady state (Figure S3B) showed significant regulation of genes detected by single-cell RNA-seq in macrophages present in the infarct region (from published single-cell RNA-seq data GSE10647316).
Comprehensive transcriptome analysis highlighted that MR inactivation induces macrophage differentiation toward a phenotype outside the M1/M2 classification (Figure 4B; Figures S4A through S4D, S5, and S6).17,18
Bioinformatic analysis revealed that several genes regulated by MR deletion in infarct macrophages contain at least 1 consensus mineralocorticoid response element motif19 (Figure S5). Notable, among the most downregulated genes containing a mineralocorticoid response element motif located close to the transcription-starting site, we found 2 key molecules (lipocain-2 [Lcn2], amphiregulin [Areg]) involved in fibroblast activation and fibrosis post-MI (Figure S5B). Gene set enrichment analysis, able to individuate though small but cumulatively significant changes on sets of functionally related genes, revealed Tnfα signaling via NF-κB (nuclear factor-kB; Figure S6A through S6C), one of the most significantly enriched pathways (Normalized enrichment score, −2.81; Padj=0.009). Among genes of leading edge mainly contributing to the enrichment score, Ptx3, Cxcl5, Il1b, Sphk1, Areg, and Tnc were significantly downregulated in infarct macrophages from MRLysMCre versus MRflox infarct macrophages (Figure S6A through S6C).
Overall, gene expression profiling of infarct macrophages revealed that multiple factors known to mediate tissue repair and wound healing20 were differently regulated in MR-deficient versus WT macrophages (Figure 4C). Noteworthy, we found that several of these genes were similarly downregulated/upregulated in the infarcted myocardium by eplerenone treatment (Figure S7A through S7C), thereby establishing a relationship between MR signaling in macrophages and the protective effects of MR antagonism after MI.
Transcriptome profiling of infarct and heart-resident macrophages (Figure 4D and 4E) also showed the upregulation of receptors and molecules involved in the phagocytosis of apoptotic cells.21 The enrichment of efferocytosis-related transcripts included receptors that are able to recognize the chemotactic find-me signals (Stab1, Cx3cr1, and S1pr1-2), receptors able to recognize eat-me signals (Mertk and Scarf1), and their associated opsonins (Gas6 and C1q). Also regulated were the scavenger receptor that mediates opsonin-independent phagocytosis (Marco), the ATP-binding cassette transporter (Abca1), which plays a critical function in the postengulfment response, and the RXR (retinoid X receptor), an obligatory heterodimeric partner for the activity of the peroxisome proliferator activated receptor. Other important prophagocytic genes (Axl, Mefge8, and Trem2) showed a trend toward upregulation contributing to the positive enrichment of efferocytic pathway (Figure 4D and 4E). Of note, Ingenuity pathway analysis identified role of pattern recognition receptors, that includes several genes involved in the clearance of apoptotic cells, among the significantly regulated pathways (Figure S6D).
The regulation of key molecules involved in efferocytosis (Mertk, C1qa, and C1qb), inflammation resolution (Gpnmb [glycoprotein nmb] and Apoe [apolipoprotein E]), oxidative stress (peroxiredoxin 4 and catalase), angiogenesis/wounding responses (Vegfb and Igf1), and fibroblast homeostasis (lipocalin-2) was confirmed by quantitative reverse-transcriptase polymerase chain reaction (Figure 5A).
Figure 5.
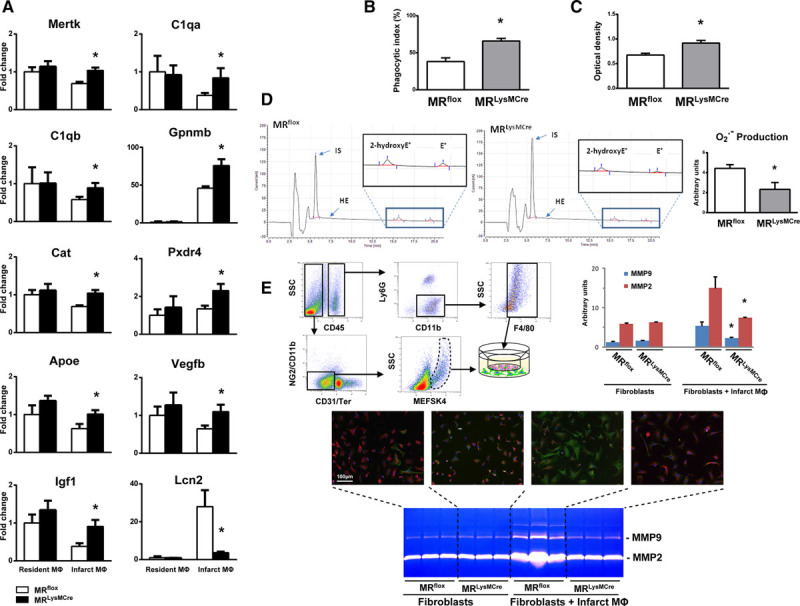
Macrophage MR (mineralocorticoid receptor) deficiency is associated with enhanced neutrophil efferocytosis, suppression of free radical formation and the modulation of fibroblast activation. A, Quantitative reverse-transcriptase polymerase chain reaction was used to detect the relative gene expression of Mertk, C1qa, C1qb, Gpnmb, Cat, Pxdr4, ApoE, Vegfb, IGF1, and Lcn2. B, Phagocytic index for apoptotic neutrophils and (C) phagocytic capacity for zymosan particles of macrophages (CD [cluster of differentiation] 45+/CD11b+/Ly6G−/F4/80+ cells) isolated by cell sorting from MRflox and MRLysMCre infarcts, 3 d after coronary artery ligation. D, Superoxide production by CD45+/CD11b+/Ly6G−/F4/80+ macrophages fluorescence-activated cell sorting–isolated from MRflox and MRLysMCre infarcts, assessed using a highly sensitive isocratic ion-pair high performance liquid chromatography–electrochemical method. E, Immunocytochemical staining showing fibroblasts and differentiated α-SMA (α-smooth muscle actin)–positive myofibroblasts (vimentin, red; α-SMA, green) and zymography of conditioned media. Macrophages (CD45+/CD11b+/Ly6G−/F4/80+ cells) and fibroblasts (CD45−/CD11b−/CD31−/TER-119−/NG2−/MEFSK4+ cells) were isolated by cell sorting from the infarct myocardium of MRflox and MRLysMCre mice and cocultured using the Boyden chamber system. Mean±SEM (n=4–5). MMP indicates matrix metalloproteinase. *P<0.05 vs MRflox. HE indicates hydroethidine; IS, internal standard; NG2, chondroitin sulfate proteoglycan; MEFSK4, anti-feeder antibody, clone mEF-SK4; and SSC, side scatter.
Macrophage MR Deficiency Is Associated With Enhanced Neutrophil Efferocytosis
To define the functional role of the MR in macrophage phagocytic activity in the ischemic microenvironment, we isolated CD45+/CD11b+/Ly6G−/F4/80+ macrophages from MRflox and MRLysMCre infarcts. Because the FACS data showed a significantly lower amount of neutrophils in the ischemic myocardium of MRLysMCre mice, we examined the potential involvement of the MR in modulating neutrophil efferocytosis. As shown in Figure 5B, the phagocytosis of apoptotic neutrophils (isolated from C57BL/6 mice) was significantly increased in macrophages sorted from MRLysMCre infarcts. Furthermore, MR deficiency significantly enhanced the phagocytic capacity of infarct macrophages for zymosan particles (Figure 5C). Collectively, these findings highlight a novel role for the macrophage MR in orchestrating phagocytosis after MI.
Reduced Superoxide Production in Infarct Macrophages Lacking the MR
Oxidative stress triggers a cascade of molecular alterations, leading to impaired wound repair. We assessed superoxide production by CD45+/CD11b+/Ly6G−/F4/80+ macrophages using a highly sensitive isocratic ion-pair high-performance liquid chromatography-electrochemical method (Figure 5D). FACS-isolated macrophages from MRLysMCre infarcts showed significantly decreased O2·− production (Figure 5D). It is noteworthy that transcriptional profiling revealed upregulation of the antioxidant enzymes, catalase and peroxiredoxin 4, in macrophages lacking the MR (Figures 4C and 5A).
Macrophage MR Affects Interactions Between Fibroblasts and Macrophages
Prompted by the histological and immunofluorescence results that showed enhanced scar collagen maturation and loss of α-SMA expression in infarct fibroblasts from MRLysMCre mice, we investigated whether the macrophage MR affects the interactions between fibroblasts and macrophages in the early phase of healing.
Recent studies have highlighted that infarct fibroblasts are derived from proliferation and migration of resident cardiac fibroblasts.15,22 In the ischemic wound environment, fibroblasts differentiate into myofibroblasts mostly driven by paracrine mediators produced by cardiac M(IL [interleukin]-4)-like macrophages.23 To consider extrinsic factors (released by infarct macrophages) and intrinsic changes in the fibroblast itself, macrophages and fibroblasts were isolated from the infarct myocardium of MRflox and MRLysMCre mice 3 days post-MI. We observed that fibroblasts in coculture with macrophages differentiate into α-SMA–positive myofibroblasts. Interestingly, the coculture of macrophages and fibroblasts led to an increase in MMP (matrix metalloproteinase) secretion into the media, as assessed by zymography (Figure 5E). In contrast, an increase in MMP secretion was not induced by the coculture of macrophages and fibroblasts isolated from MRLysMCre ischemic myocardium. (Figure 5E). These data suggest that dynamic interactions between macrophages and fibroblasts seem to be critically regulated by the macrophage MR
Targeted Delivery of MR Antagonists to Macrophages Improved Postischemic Wound Healing and Protected Against Cardiac Dysfunction and Remodeling
Next, we examined whether the inhibition of MR signaling in macrophages by targeted delivery of MR antagonists (RU28318/eplerenone encapsulated within liposomes) promotes cardiac wound repair. A single dose of liposomal RU28318 or liposomal eplerenone or empty (MR antagonist lacking) liposomes was injected into mice at the onset of MI.
The targeted delivery to macrophages in the infarcted myocardium was investigated with liposomes containing fluorescein isothiocyanate (FITC)-BSA.11 The liposomes were injected intraperitoneally at the onset of coronary ligation, and the in vivo uptake of liposomes encapsulating FITC-BSA was analyzed by FACS analysis and immunofluorescence (Figure S8). We observed that the overwhelming majority of infarct macrophages (Figure S8A) had uptaken FITC liposomes. In contrast, FACS analysis showed that almost all neutrophils, endothelial cells, fibroblasts, and vascular cells were FITC negative (Figure S8B). Moreover, immunofluorescence revealed that FITC-labeled cells in the infarcted heart were CD68-positive macrophages (Figure S8C), demonstrating the selectivity of the approach.
At 7 days post-MI, mice receiving empty liposomes developed elevated LV filling pressure, LV end-systolic and end-diastolic volumes, and marked cardiac dysfunction, as assessed by LV ejection performance, dP/dtmax, dP/dtmin, and LV dP/dtmax divided by instantaneous pressure (Figure 6B and 6C). Intriguingly, the administration of RU28318 or eplerenone-containing liposomes significantly decreased LV end-diastolic pressure, LV end-systolic volume, and end-diastolic volume, associated with a downward and leftward shift of the pressure-volume curve (Figure 6B and 6C). LV contractile function and relaxation were also significantly improved after targeted delivery of MR antagonists (Figure 6C). In addition, the time constant of LV pressure isovolumic decay was significantly shortened by administration of RU28318/eplerenone-loaded liposomes compared with mice receiving empty liposomes (Figure 6C). Of note, nearly identical hemodynamic data were obtained in infarcted MRLysMCre mice (Figure 1B and 1C).
Figure 6.
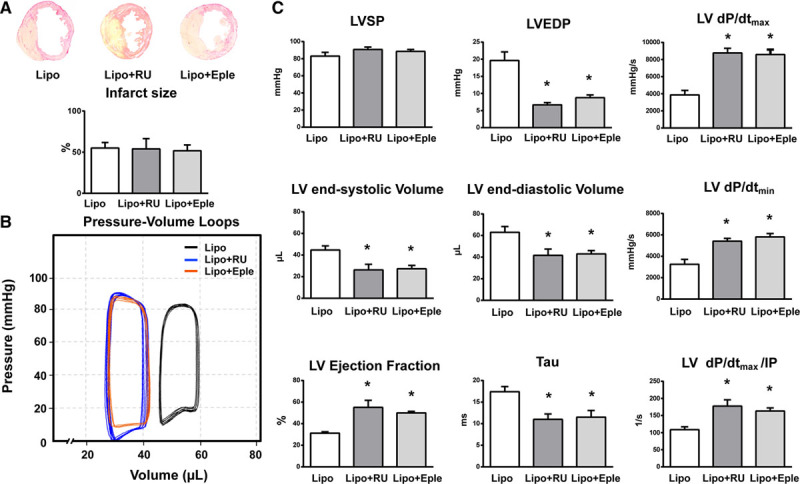
Targeted delivery of MR (mineralocorticoid receptor) antagonists to macrophages protects against cardiac dysfunction and remodeling after myocardial infarction. A single dose of liposomal RU28318 (Lipo+RU) or liposomal eplerenone (Lipo+Eple) or empty (MR antagonists lacking) liposomes (Lipo) were injected intraperitoneally into mice at the onset of myocardial infarction. A, Sections of infarcted hearts and infarct size. B, Representative left ventricular (LV) pressure-volume loops measured in vivo with conductance catheter, 7 d after myocardial infarction. C, LV systolic pressure (LVSP), LV end-diastolic pressure (LVEDP), LV maximal rate of pressure rise (LV dP/dtmax), maximal rate of pressure decline (LV dP/dtmin), LV end-systolic and end-diastolic volumes, LV ejection fraction, LV dP/dtmax divided by instantaneous pressure (IP), and the time constant of LV pressure isovolumic decay (Tau). Mean±SEM (n=6). *P<0.05 vs Lipo.
In addition, the administration of RU28318-loaded liposomes enhanced infarct neovascularization (Figure S9A). Similar to the effects observed in MRLysMCre infarcts, mice receiving RU28318-loaded liposomes exhibited more α-SMA–positive cells restricted to pericytes/vascular smooth muscle cells and a loss of α-SMA expression in fibroblasts within the infarcted myocardium (Figure S9A). Further, we performed flow cytometry analysis of fibroblasts isolated from the infarct region of mice receiving empty liposomes and liposomal RU28318 (Figure S9B). Quantitative analysis showed that liposomal RU28318 did not affect the number of fibroblasts present in the infarct scar (Figure S9B).
Taken together, these results suggest that inhibition of MR activation in macrophages by a targeted delivery of MR antagonists could represent a unique therapeutic strategy to promote healing and infarct scar maturation and to prevent cardiac dysfunction and remodeling after MI.
Discussion
This study highlights the pathogenic role of the macrophage MR in tissue repair mechanisms after acute MI. Our data strongly suggest that MR deficiency in macrophages improves healing and cardiac remodeling after ischemic injury through multiple mechanisms, including the promotion of neutrophil efferocytosis, the suppression of free radical formation, and the modulation of fibroblast activation state.
Previous studies in isolated peritoneal thioglycollate-elicited macrophages showed that loss of MR activity promotes an alternatively activated M2 macrophage phenotype and identified the myeloid MR as a regulator of macrophage polarization.14 Macrophages are sentinel cells that can respond to microenvironmental cues and polarize to different phenotypes. A combination of transcription factors and signaling pathway modulators dictate distinct functional responses.24 Classically activated M1 and alternatively activated M2 macrophages represent the extreme states of macrophage polarization, and this classification unmistakably belies the spectrum of macrophage activation.24–26 Our transcriptome analysis revealed that MR inactivation drives macrophage differentiation in the ischemic microenvironment toward a phenotype unrelated to the M1/M2 paradigm with regulation of factors promoting wound healing and tissue repair.
Monocytes/macrophages are essential for myocardial infarct healing.2,11,12,20 Depletion of monocytes/macrophages or defective macrophage recruitment impairs repair and promotes adverse remodeling post-MI.1,20,27 However, emerging evidence suggests that the number of infarct macrophages is not as important as the dynamic changes in macrophage differentiation state for wound healing and functional outcomes after ischemic injury.2,12,20 Modulation of macrophage activation/function and consequently of their trophic factor secretion could be an important therapeutic target for prevention of progressive functional deterioration and heart failure.
In addition to inducing an optimal pattern of pro-wound healing mediators, macrophage MR deficiency also regulates the expression of receptors and molecules involved in the clearance of apoptotic cells. We report that critical components of the phagocytosis machinery were upregulated in infarct macrophages from MRLysMCre hearts and that MR deficiency in macrophages was associated with enhanced neutrophil efferocytosis in the infarct area. In line with this, MR loss in macrophages was shown to decrease the number of apoptotic cells in atherosclerotic lesions by affecting efferocytosis.28,29 Moreover, Montes-Cobos et al30 recently showed that phagocytic activity of bone marrow-derived macrophages and peritoneal macrophages is increased in the absence of the MR. It is important to note that efficient phagocytic clearance of apoptotic/necrotic cells is crucial for the resolution of inflammation and for favorable cardiac repair.11,21 Increased expression of cell surface receptors (Cx3cr1 and S1pr1-2) probably rendered macrophages more sensitive to soluble find-me signals, such as fractalkine and sphingosine-1-phosphate released by apoptotic cells. In addition, the augmented expression of specific receptors (Mertk, Scarf1, and stabilin) and bridging molecules (Gas6 and C1q) likely promoted apoptotic cell clearance by an improved recognition of apoptotic cell–associated molecular pattern, like externalized phosphatidylserine. Also noteworthy is the upregulation of type I surface receptor stabilin-2, which triggers efferocytosis through a direct interaction with phosphatidylserine. Moreover, enhanced expression of the innate immune protein C1q may have promoted M2-like macrophage activation and clearance of apoptotic cells31 triggering macrophages to produce factors that positively regulate inflammation resolution.
Oxidative stress caused by the excessive formation of reactive oxygen species by inflammatory cells impairs wound healing.4 Aldosterone/MR activation has been shown to stimulate macrophage oxygen radical generation in mouse peritoneal macrophages from ApoE knockout mice.32 We found reduced superoxide production in infarct macrophages lacking the MR, most likely because of an increased expression of catalase and peroxiredoxin 4, antioxidant enzymes involved in maintaining cellular redox balance.
Our in vitro studies suggest that the macrophage MR could also play an active role in scar remodeling by controlling the secretion of matrix-degrading MMPs and fibroblast activation state. After infarction injury, cardiac fibroblasts become activated upon sensing microenvironmental factors and critically regulate the wound healing response.33 However, unrestrained myofibroblast differentiation can lead to fibrotic remodeling and impaired heart function. MR signaling in macrophages regulates phenotypic changes of fibroblasts in the infarcted heart most probably through mechanisms involving oxidative stress and lipocalin-2. Indeed, MR knockout prevented the extensive upregulation of lipocalin-2 in infarct macrophages. This is of particular relevance considering that lipocalin-2—a downstream MR activation target—is a key mediator of aldosterone/MR profibrotic effects in cardiac fibroblasts and that lipocalin-2 inactivation improves cardiac function and remodeling after MI.34
In an exciting recent study, Fu et al15 investigating the dynamics of fibroblast activation post-MI disclosed that myofibroblasts do not disappear but rather persist within the scar and lose α-SMA expression as the extracellular matrix and scar matures (7–10 days after MI). Therefore, the loss of the α-SMA–positive myofibroblast phenotype within the infarcted myocardium of MRLysMCre mice may be also indicative of a mature scar and an accelerated wound healing process. Interestingly, interventions impairing scar formation are associated with the persistence of α-SMA–expressing myofibroblasts within the infarct region and adverse cardiac remodeling.15,35
Despite strong clinical evidence that MR blockade improves outcome in patients with acute MI, MR antagonists are still underused.5,6 Individual participant data analysis of the REMINDER and ALBATROSS (Aldosterone Lethal Effects Blocked in Acute MI Treated With or Without Reperfusion to Improve Outcome and Survival at Six Months Follow-Up) trials suggests a significant reduction of death or resuscitated sudden death by MR antagonists given early (within 72 hours) after low-risk ST-segment–elevation myocardial infarction.5 Moreover, a recent meta-analysis of data from 10 randomized placebo-controlled trials (including patients with ST-segment–elevation myocardial infarction without evidence of heart failure or severe LV dysfunction) showed that the use of MR antagonists was associated with a 38% reduction in mortality and a significant increase in LV ejection fraction during follow-up.36,37
Decreased blood volume secondary to diuresis may have contributed to beneficial effects of MR antagonists in patients with acute MI. However, Rossignol et al38 showed that the benefit of MR antagonism on cardiovascular outcomes was independent from the early diuretic and K-sparing effects. Extrarenal effects on myocardial structural and electrical remodeling, on sympathoadrenergic stimulation, platelet activation, and endothelial dysfunction, appear to be important mechanisms underlying the benefits of MR antagonists in the post-MI setting.5–10,39 The present study identifies the macrophage MR as a pleiotropic modulator of myocardial infarct healing.
Our data also show that targeted delivery of MR antagonists to macrophages promotes wound healing, thus identifying a promising translational strategy for cardiac repair. Remarkably, a single administration of RU28318 or eplerenone-containing liposomes at the onset of MI was able to improve the postischemic angiogenic response and to protect against cardiac dysfunction and remodeling. Recent data indicate that myeloid MR activation contributes to progressive kidney disease and that macrophage deficiency of MR provides protection against declining renal function without affecting tubular regulation of salt balance.40,41 Thus, targeting of MR antagonists to macrophages after acute MI could be a strategic approach to avoid the side effects on regulation of salt homeostasis, particularly in patients with compromised renal function.
Perspectives
We provide important new insights into the mechanisms underlying the clinical benefits of early MR antagonist administration in patients with ST-segment–elevation myocardial infarction and demonstrate that MR signaling in macrophages may be a novel therapeutic target to prevent cardiac dysfunction and failure after ischemic injury.
Acknowledgments
We thank Margarete Göbel from the Core Unit SysMed (University of Würzburg) for excellent technical assistance. We are grateful for the support of Dr Matthias Ballmaier from the Research Facility Cell Sorting of Hannover Medical School.
Sources of Funding
D. Fraccarollo and J. Bauersachs received support from the Deutsche Forschungsgemeinschaft (BA 1742/8-1).
Disclosures
None.
Supplementary Material
Footnotes
These authors contributed equally to this work.
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/HYPERTENSIONAHA.118.12162.
Novelty and Significance
What Is New?
MR (mineralocorticoid receptor) inactivation drives macrophage differentiation in the ischemic microenvironment toward a phenotype outside the M1/M2 paradigm, with regulation of multiple interrelated factors controlling inflammation resolution and tissue repair.
Macrophage MR deficiency promotes wound healing through enhanced efferocytosis of neutrophils, the suppression of free radical formation, and the modulation of fibroblast activation state.
Targeted delivery of MR antagonists to macrophages protects against cardiac dysfunction and remodeling after myocardial infarction.
What Is Relevant?
The MR in macrophages is a pathogenic player and important therapeutic target in cardiac repair after myocardial infarction.
Summary
This study identifies the MR in macrophages as a pleiotropic modulator of myocardial infarct healing. Macrophage MR deficiency or targeted liposomal delivery of MR antagonists to macrophages improves cardiac repair and remodeling after ischemic injury.
References
- 1.Fraccarollo D, Galuppo P, Bauersachs J. Novel therapeutic approaches to post-infarction remodelling. Cardiovasc Res. 2012;94:293–303. doi: 10.1093/cvr/cvs109. doi: 10.1093/cvr/cvs109. [DOI] [PubMed] [Google Scholar]
- 2.Galuppo P, Vettorazzi S, Hövelmann J, Scholz CJ, Tuckermann JP, Bauersachs J, Fraccarollo D. The glucocorticoid receptor in monocyte-derived macrophages is critical for cardiac infarct repair and remodeling. FASEB J. 2017;31:5122–5132. doi: 10.1096/fj.201700317R. doi: 10.1096/fj.201700317R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pfeffer MA, Rutherford JD. Therapeutic attenuation of cardiac remodeling after acute myocardial infarction: a conversation with Marc A. Pfeffer, MD, PhD. Circulation. 2018;137:2430–2434. doi: 10.1161/CIRCULATIONAHA.118.033665. doi: 10.1161/CIRCULATIONAHA.118.033665. [DOI] [PubMed] [Google Scholar]
- 4.Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res. 2016;119:91–112. doi: 10.1161/CIRCRESAHA.116.303577. doi: 10.1161/CIRCRESAHA.116.303577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beygui F, Van Belle E, Ecollan P, Machecourt J, Hamm CW, Lopez De Sa E, Flather M, Verheugt FWA, Vicaut E, Zannad F, Pitt B, Montalescot G. Individual participant data analysis of two trials on aldosterone blockade in myocardial infarction. Heart. 2018;104:1843–1849. doi: 10.1136/heartjnl-2018-312950. doi: 10.1136/heartjnl-2018–312950. [DOI] [PubMed] [Google Scholar]
- 6.Docherty KF, Jhund PS. Early use of mineralocorticoid receptor antagonists in st-elevation myocardial infarction: is it ever too early?. Heart. 2018;104:1812–1813. doi: 10.1136/heartjnl-2018-313371. doi: 10.1136/heartjnl-2018–313371. [DOI] [PubMed] [Google Scholar]
- 7.Fraccarollo D, Berger S, Galuppo P, Kneitz S, Hein L, Schütz G, Frantz S, Ertl G, Bauersachs J. Deletion of cardiomyocyte mineralocorticoid receptor ameliorates adverse remodeling after myocardial infarction. Circulation. 2011;123:400–408. doi: 10.1161/CIRCULATIONAHA.110.983023. doi: 10.1161/CIRCULATIONAHA.110.983023. [DOI] [PubMed] [Google Scholar]
- 8.Fraccarollo D, Galuppo P, Schraut S, Kneitz S, van Rooijen N, Ertl G, Bauersachs J. Immediate mineralocorticoid receptor blockade improves myocardial infarct healing by modulation of the inflammatory response. Hypertension. 2008;51:905–914. doi: 10.1161/HYPERTENSIONAHA.107.100941. doi: 10.1161/HYPERTENSIONAHA.107.100941. [DOI] [PubMed] [Google Scholar]
- 9.Ouvrard-Pascaud A, Sainte-Marie Y, Bénitah JP, et al. Conditional mineralocorticoid receptor expression in the heart leads to life-threatening arrhythmias. Circulation. 2005;111:3025–3033. doi: 10.1161/CIRCULATIONAHA.104.503706. doi: 10.1161/CIRCULATIONAHA.104.503706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perrier E, Kerfant BG, Lalevee N, Bideaux P, Rossier MF, Richard S, Gómez AM, Benitah JP. Mineralocorticoid receptor antagonism prevents the electrical remodeling that precedes cellular hypertrophy after myocardial infarction. Circulation. 2004;110:776–783. doi: 10.1161/01.CIR.0000138973.55605.38. doi: 10.1161/01.CIR.0000138973.55605.38. [DOI] [PubMed] [Google Scholar]
- 11.Harel-Adar T, Ben Mordechai T, Amsalem Y, Feinberg MS, Leor J, Cohen S. Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proc Natl Acad Sci USA. 2011;108:1827–1832. doi: 10.1073/pnas.1015623108. doi: 10.1073/pnas.1015623108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weinberger T, Schulz C. Myocardial infarction: a critical role of macrophages in cardiac remodeling. Front Physiol. 2015;6:107. doi: 10.3389/fphys.2015.00107. doi: 10.3389/fphys.2015.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rickard AJ, Morgan J, Tesch G, Funder JW, Fuller PJ, Young MJ. Deletion of mineralocorticoid receptors from macrophages protects against deoxycorticosterone/salt-induced cardiac fibrosis and increased blood pressure. Hypertension. 2009;54:537–543. doi: 10.1161/HYPERTENSIONAHA.109.131110. doi: 10.1161/HYPERTENSIONAHA.109.131110. [DOI] [PubMed] [Google Scholar]
- 14.Usher MG, Duan SZ, Ivaschenko CY, Frieler RA, Berger S, Schütz G, Lumeng CN, Mortensen RM. Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest. 2010;120:3350–3364. doi: 10.1172/JCI41080. doi: 10.1172/JCI41080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, Sargent MA, Prasad V, Valiente-Alandi I, Blaxall BC, Molkentin JD. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Invest. 2018;128:2127–2143. doi: 10.1172/JCI98215. doi: 10.1172/JCI98215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.King KR, Aguirre AD, Ye YX, et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat Med. 2017;23:1481–1487. doi: 10.1038/nm.4428. doi: 10.1038/nm.4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walter W, Alonso-Herranz L, Trappetti V, Crespo I, Ibberson M, Cedenilla M, Karaszewska A, Núñez V, Xenarios I, Arroyo AG, Sánchez-Cabo F, Ricote M. Deciphering the dynamic transcriptional and post-transcriptional networks of macrophages in the healthy heart and after myocardial injury. Cell Rep. 2018;23:622–636. doi: 10.1016/j.celrep.2018.03.029. doi: 10.1016/j.celrep.2018.03.029. [DOI] [PubMed] [Google Scholar]
- 18.Skelly DA, Squiers GT, McLellan MA, Bolisetty MT, Robson P, Rosenthal NA, Pinto AR. Single-cell transcriptional profiling reveals cellular diversity and intercommunication in the mouse heart. Cell Rep. 2018;22:600–610. doi: 10.1016/j.celrep.2017.12.072. doi: 10.1016/j.celrep.2017.12.072. [DOI] [PubMed] [Google Scholar]
- 19.Le Billan F, Khan JA, Lamribet K, Viengchareun S, Bouligand J, Fagart J, Lombès M. Cistrome of the aldosterone-activated mineralocorticoid receptor in human renal cells. FASEB J. 2015;29:3977–3989. doi: 10.1096/fj.15-274266. doi: 10.1096/fj.15-274266. [DOI] [PubMed] [Google Scholar]
- 20.Ben-Mordechai T, Palevski D, Glucksam-Galnoy Y, Elron-Gross I, Margalit R, Leor J. Targeting macrophage subsets for infarct repair. J Cardiovasc Pharmacol Ther. 2015;20:36–51. doi: 10.1177/1074248414534916. doi: 10.1177/1074248414534916. [DOI] [PubMed] [Google Scholar]
- 21.Elliott MR, Koster KM, Murphy PS. Efferocytosis signaling in the regulation of macrophage inflammatory responses. J Immunol. 2017;198:1387–1394. doi: 10.4049/jimmunol.1601520. doi: 10.4049/jimmunol.1601520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, J Lin SC, Aronow BJ, Tallquist MD, Molkentin JD. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;7:12260. doi: 10.1038/ncomms12260. doi: 10.1038/ncomms12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shiraishi M, Shintani Y, Shintani Y, Ishida H, Saba R, Yamaguchi A, Adachi H, Yashiro K, Suzuki K. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest. 2016;126:2151–2166. doi: 10.1172/JCI85782. doi: 10.1172/JCI85782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi: 10.12703/P6-13. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duffield JS. Macrophages in kidney repair and regeneration. J Am Soc Nephrol. 2011;22:199–201. doi: 10.1681/ASN.2010121301. doi: 10.1681/ASN.2010121301. [DOI] [PubMed] [Google Scholar]
- 26.Xue J, Schmidt SV, Sander J, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40:274–288. doi: 10.1016/j.immuni.2014.01.006. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frantz S, Hofmann U, Fraccarollo D, et al. Monocytes/macrophages prevent healing defects and left ventricular thrombus formation after myocardial infarction. FASEB J. 2013;27:871–881. doi: 10.1096/fj.12-214049. doi: 10.1096/fj.12-214049. [DOI] [PubMed] [Google Scholar]
- 28.Shen ZX, Chen XQ, Sun XN, Sun JY, Zhang WC, Zheng XJ, Zhang YY, Shi HJ, Zhang JW, Li C, Wang J, Liu X, Duan SZ. Mineralocorticoid receptor deficiency in macrophages inhibits atherosclerosis by affecting foam cell formation and efferocytosis. J Biol Chem. 2017;292:925–935. doi: 10.1074/jbc.M116.739243. doi: 10.1074/jbc.M116.739243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van der Heijden CDCC, Deinum J, Joosten LAB, Netea MG, Riksen NP. The mineralocorticoid receptor as a modulator of innate immunity and atherosclerosis. Cardiovasc Res. 2018;114:944–953. doi: 10.1093/cvr/cvy092. doi: 10.1093/cvr/cvy092. [DOI] [PubMed] [Google Scholar]
- 30.Montes-Cobos E, Schweingruber N, Li X, Fischer HJ, Reichardt HM, Lühder F. Deletion of the mineralocorticoid receptor in myeloid cells attenuates central nervous system autoimmunity. Front Immunol. 2017;8:1319. doi: 10.3389/fimmu.2017.01319. doi: 10.3389/fimmu.2017.01319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bohlson SS, O’Conner SD, Hulsebus HJ, Ho MM, Fraser DA. Complement, c1q, and c1q-related molecules regulate macrophage polarization. Front Immunol. 2014;5:402. doi: 10.3389/fimmu.2014.00402. doi: 10.3389/fimmu.2014.00402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keidar S, Kaplan M, Pavlotzky E, Coleman R, Hayek T, Hamoud S, Aviram M. Aldosterone administration to mice stimulates macrophage NADPH oxidase and increases atherosclerosis development: a possible role for angiotensin-converting enzyme and the receptors for angiotensin II and aldosterone. Circulation. 2004;109:2213–2220. doi: 10.1161/01.CIR.0000127949.05756.9D. doi: 10.1161/01.CIR.0000127949.05756.9D. [DOI] [PubMed] [Google Scholar]
- 33.Turner NA, Porter KE. Function and fate of myofibroblasts after myocardial infarction. Fibrogenesis Tissue Repair. 2013;6:5. doi: 10.1186/1755-1536-6-5. doi: 10.1186/1755-1536-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martínez-Martínez E, Buonafine M, Boukhalfa I, Ibarrola J, Fernández-Celis A, Kolkhof P, Rossignol P, Girerd N, Mulder P, López-Andrés N, Ouvrard-Pascaud A, Jaisser F. Aldosterone target NGAL (Neutrophil Gelatinase-Associated Lipocalin) is involved in cardiac remodeling after myocardial infarction through NFκB pathway. Hypertension. 2017;70:1148–1156. doi: 10.1161/HYPERTENSIONAHA.117.09791. doi: 10.1161/HYPERTENSIONAHA.117.09791. [DOI] [PubMed] [Google Scholar]
- 35.Eschenhagen T. A new concept of fibroblast dynamics in post-myocardial infarction remodeling. J Clin Invest. 2018;128:1731–1733. doi: 10.1172/JCI121079. doi: 10.1172/JCI121079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dahal K, Hendrani A, Sharma SP, Singireddy S, Mina G, Reddy P, Dominic P, Modi K. Aldosterone antagonist therapy and mortality in patients with ST-segment elevation myocardial infarction without heart failure: a systematic review and meta-analysis. JAMA Intern Med. 2018;178:913–920. doi: 10.1001/jamainternmed.2018.0850. doi: 10.1001/jamainternmed.2018.0850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pitt B, Zannad F. Mineralocorticoid receptor antagonists in ST-segment elevation myocardial infarction. JAMA Intern Med. 2018;178:920–921. doi: 10.1001/jamainternmed.2018.1940. doi: 10.1001/jamainternmed.2018.1940. [DOI] [PubMed] [Google Scholar]
- 38.Rossignol P, Ménard J, Fay R, Gustafsson F, Pitt B, Zannad F. Eplerenone survival benefits in heart failure patients post-myocardial infarction are independent from its diuretic and potassium-sparing effects. Insights from an EPHESUS (Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study) substudy. J Am Coll Cardiol. 2011;58:1958–1966. doi: 10.1016/j.jacc.2011.04.049. doi: 10.1016/j.jacc.2011.04.049. [DOI] [PubMed] [Google Scholar]
- 39.Belden Z, Deiuliis JA, Dobre M, Rajagopalan S. The role of the mineralocorticoid receptor in inflammation: focus on kidney and vasculature. Am J Nephrol. 2017;46:298–314. doi: 10.1159/000480652. doi: 10.1159/000480652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barrera-Chimal J, Estrela GR, Lechner SM, Giraud S, El Moghrabi S, Kaaki S, Kolkhof P, Hauet T, Jaisser F. The myeloid mineralocorticoid receptor controls inflammatory and fibrotic responses after renal injury via macrophage interleukin-4 receptor signaling. Kidney Int. 2018;93:1344–1355. doi: 10.1016/j.kint.2017.12.016. doi: 10.1016/j.kint.2017.12.016. [DOI] [PubMed] [Google Scholar]
- 41.Huang LL, Nikolic-Paterson DJ, Han Y, Ozols E, Ma FY, Young MJ, Tesch GH. Myeloid mineralocorticoid receptor activation contributes to progressive kidney disease. J Am Soc Nephrol. 2014;25:2231–2240. doi: 10.1681/ASN.2012111094. doi: 10.1681/ASN.2012111094. [DOI] [PMC free article] [PubMed] [Google Scholar]