

ABSTRACT
Rozanolixizumab (UCB7665), a humanized high-affinity anti-human neonatal Fc receptor (FcRn) monoclonal antibody (IgG4P), has been developed to reduce pathogenic IgG in autoimmune and alloimmune diseases. We document the antibody isolation and compare rozanolixizumab with the same variable region expressed in various mono-, bi- and trivalent formats. We report activity data for rozanolixizumab and the different molecular formats in human cells, FcRn-transgenic mice, and cynomolgus monkeys. Rozanolixizumab, considered the most effective molecular format, dose-dependently and selectively reduced plasma IgG concentrations in an FcRn-transgenic mouse model (no effect on albumin). Intravenous (IV) rozanolixizumab dosing in cynomolgus monkeys demonstrated non-linear pharmacokinetics indicative of target-mediated drug disposition; single IV rozanolixizumab doses (30 mg/kg) in cynomolgus monkeys reduced plasma IgG concentration by 69% by Day 7 post-administration. Daily IV administration of rozanolixizumab (initial 30 mg/kg loading dose; 5 mg/kg daily thereafter) reduced plasma IgG concentrations in all cynomolgus monkeys, with low concentrations maintained throughout the treatment period (42 days). In a 13-week toxicology study in cynomolgus monkeys, supra-pharmacological subcutaneous and IV doses of rozanolixizumab (≤ 150 mg/kg every 3 days) were well tolerated, inducing sustained (but reversible) reductions in IgG concentrations by up to 85%, with no adverse events observed. We have demonstrated accelerated natural catabolism of IgG through inhibition of IgG:FcRn interactions in mice and cynomolgus monkeys. Inhibition of FcRn with rozanolixizumab may provide a novel therapeutic approach to reduce pathogenic IgG in human autoimmune disease. Rozanolixizumab is being investigated in patients with immune thrombocytopenia (NCT02718716) and myasthenia gravis (NCT03052751).
Keywords: FcRn, rozanolixizumab, UCB7665, autoantibody, autoimmunity, FcRn blockade, immune thrombocytopenia, myasthenia gravis, target-mediated drug disposition, IgG catabolism
Introduction
The presence of pathogenic autoantibodies is a fundamental feature of autoimmune and alloimmune diseases such as anti-glomerular basement membrane antibody disease, immune thrombocytopenia (ITP), myasthenia gravis, hemolytic anemia, and pemphigus vulgaris. Reduction of circulating pathogenic immunoglobulin G (IgG) concentrations can have therapeutic benefit, and is currently achieved by plasmapheresis, specifically immunoadsorption1 or by intravenous immunoglobulin (IVIg), although IVIg may have additional mechanisms of action beyond this.2,3 However, these current treatments occupy time in specialized units and can be associated with considerable side effects and health economic implications.1,4 An alternative, more specific approach to reduce plasma IgG in autoimmune conditions, such as accelerated catabolism of endogenous IgG, could benefit patients and address significant unmet clinical needs. Inhibition of the FcRn-IgG interaction has been shown to rapidly decrease IgG concentrations in a mouse model.5 We have generated rozanolixizumab, a novel anti-human neonatal Fc receptor (FcRn) monoclonal antibody (mAb). In a healthy volunteer study (n = 48), dose-dependent reductions in serum IgG were observed, and an acceptable safety profile was demonstrated, with single (subcutaneously [SC] and intravenously [IV] administered) doses of rozanolixizumab.6
The long half-life associated with IgG antibodies is attributed to the FcRn, a heterodimeric receptor composed of a major histocompatibility complex class I-like α-chain and a β2-microglobulin (β2m) chain. In endothelial and hematopoietic cells, FcRn is responsible for salvaging and recycling IgG (and albumin). FcRn binds IgG at endosomal acidic pH, and traffics it away from the degradative pathway that leads to the lysosome, and back to the cell membrane, where the IgG is released intact back into the plasma.7-10 Lack of FcRn function (and therefore lack of IgG salvage and recycling) in animal models is associated with low plasma IgG concentrations and resistance to autoimmune disease.11-14 Studies have shown that in humans, β2m mutation can lead to defective FcRn function, resulting in low plasma IgG concentrations, which is a symptom of familial hypercatabolic hypoproteinemia.15-17 In vivo studies in human FcRn-transgenic mouse models and non-human primates support the rationale of disrupting FcRn function in order to reduce IgG concentrations.18-20 Disease amelioration following administration of an anti-FcRn antibody has been demonstrated in a number of experimental autoimmune disease models in animals.5,21-25 Thus, inhibition of the FcRn-dependent IgG salvage pathway in autoimmune conditions could lead to lower plasma concentrations of pathogenic IgG and may result in therapeutic benefits.
Here, we present in vitro data functionally characterizing rozanolixizumab (UCB7665; 1519.g57 IgG4P) and its variable (V) region (1519.g57), expressed in various mono-, bi- and trivalent formats. We also present in vivo data in human FcRn-transgenic mice and in cynomolgus monkeys to investigate mechanism of action, and to allow characterization of pharmacokinetics (PK), pharmacodynamics (PD) and safety. The cynomolgus monkey was selected as a suitable species for investigation of the PK, PD and safety of human IgG, and mutants thereof, because human and cynomolgus monkey IgG4 bind comparably to cynomolgus monkey FcRn26,27 and have similar Fc effector functions.28
Results
Isolation and characterization of 1519.g57, the V-region of rozanolixizumab
Following an immunization campaign in both rats and mice, we identified a large panel of antibodies that bound to the human FcRn α-chain (but not to β2m) and that blocked human IgG binding to FcRn by both flow cytometry and surface plasmon resonance (SPR; data not shown). Using an affinity-based FcRn-binding selection criterion (KD ≤ 200 pM at pH 6), 156 unique antibody V region sequences were identified. This represented 95 unique clonotype families (based on VH complementarity-determining region (CDR) 3 homology). Eleven of these families were selected, on the basis of affinity for FcRn and ability to block IgG:FcRn interactions (and not albumin), for subsequent humanization. Representative V regions from the sequence families were each expressed in Escherichia coli (E.coli) as a histidine-tagged humanized Fab’ fragment, and numerous grafts of each antibody were engineered and prepared, so as to identify the optimal humanized sequence. To identify the antibody candidates with the highest activity, in vitro cell function inhibition assays and an in vivo study in the human FcRn-transgenic mouse model were undertaken. For this the candidates, in Fab’ format, were PEGylated with 2 × 20 kDa PEG polymer. Biophysical and biochemical characterizations were carried out to ensure that the selected anti-human FcRn molecules were suited to large-scale manufacture, formulation and long-term stability. V region 1519, graft 57 (1519.g57; bold text in Supplementary Figure 1) was selected as the development candidate due to its optimal combination of activity, biophysical and biochemical properties (Supplementary Table 1 and Supplementary Table 2). To enable further characterization, this V region was expressed in multiple antibody formats (Fab’, Fab’PEG, IgG1, IgG4P, triFab’, FabFv, Fab’human albumin conjugate and Fab’mouse albumin fusion; Supplementary Table 3). When subcloned as a humanized IgG4P (serine-to-proline mutation at residue 241 to reduce propensity for Fab-arm exchange31) isotype, this antibody is known as rozanolixizumab (Supplementary Figure 1; these sequence data have been submitted to the DDBJ/EMBL/GenBank databases under accession number MG976502 (light chain) and MG976503 (heavy chain)).
Rozanolixizumab was humanized using CDR-grafting.32 Under the 2014 INN/WHO method for assigning generic names, rozanolixizumab was given the -xizu- sub-stem, designating it a chimeric/humanized molecule; the chimeric designation was assigned due to sequence similarities with Macaque monkey IgG sequences, however, no Macaque monkey sequences were used in the humanization process. This designation has been criticized and used as an example in the literature to show some of the inaccuracies and inconsistencies with the INN/WHO system;33,34 the naming system has subsequently been revised and superseded.34
The binding epitope of 1519.g57 Fab’ overlaps with that of human IgG Fc on FcRn
X-ray crystallography and hydrogen-deuterium exchange (HDX) were both used to identify the binding epitopes of 1519.g57 Fab’ domain on human FcRn. Crystallization of human FcRn (deglycosylated extracellular domain) in complex with 1519.g57 Fab’ was achieved and the crystal structure of the complex solved (Figure 1A and Supplementary Table 4; protein data bank (PDB) entry 6FGB). The structure of FcRn was unchanged by binding of 1519.g57 Fab’ (structure of FcRn alone not shown). 1519.g57 Fab’ binds to human FcRn via an epitope in the α-chain (Figure 1A and 1C), which overlaps with the binding epitope of human IgG Fc domain on FcRn (Figure 1B and 1C). The 1519.g57 Fab’ binding site on human FcRn in solution was determined by HDX and was consistent with that determined by X-ray crystallography (data not shown).
Figure 1.
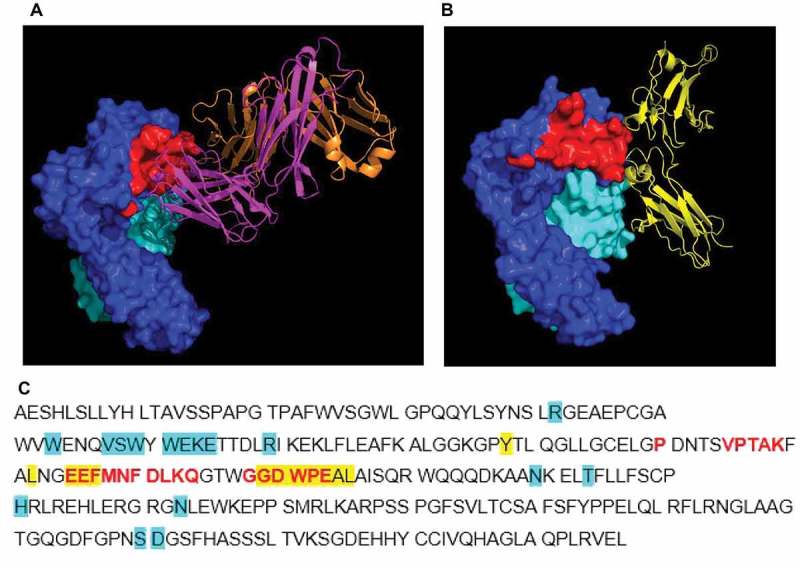
Crystal structure of complex of human FcRn (deglycosylated extracellular domain) and 1519.g57 Fab’. (A) The binding epitope of 1519.g57 Fab’ on FcRn α-chain is shown in red; (B) PDB 4N0U – human IgG Fc domain interacting with FcRn, demonstrating the overlapping epitope with that of 1519.g57Fab’,29 (C) FcRn α-chain sequence, showing residues involved in interaction with 1519.g57 Fab’ (in red). Residues involved in interaction between human FcRn and IgG (Fc domain) or albumin (as described by Oganesyan et al, 2014, highlighted in yellow or in blue, respectively) are also shown. The sequence of human FcRn α-chain ECD was taken from Swiss Prot (P55899-1); Residue F44, identified by Oganesyan et al.29 as interacting with albumin, is residue E44 in this sequence and in the sequence of Schmidt et al.30 Dark blue = FcRn α-chain; pale blue = β2 M; magenta = 1519.g57 heavy chain; orange = 1519.g57 light chain; yellow = IgG Fc domain; red = 1519.g57 Fab’ binding epitope on FcRn.
Rozanolixizumab has high affinity for FcRn at both pH 6.0 and pH 7.4, assessed by SPR and cell-based analyses
The binding affinity of rozanolixizumab and 1519.g57 expressed in various molecular formats (Fab’, Fab’PEG and IgG) was assessed by SPR analysis and a cell-based binding assay (Table 1). The estimates of KD, made by SPR analysis, were considered to be similar for the different formats (59, 40, 22, 23 pM for Fab’, Fab’PEG, IgG1 and rozanolixizumab, respectively; binding to human FcRn at pH 6.0), taking into consideration the range of intra- and inter-assay variation observed (~ 1.5- and 2-fold from the mean, respectively) and the numbers of replicates. The SPR assay was designed to avoid an avidity effect, with no reflection of bi-, as opposed to mono-valency. Rozanolixizumab bound to human FcRn and cynomolgus monkey FcRn with a similar affinity at both pH 6.0 (23 pM and 25 pM, human and cynomolgus monkey, respectively) and pH 7.4 (34 pM and 53 pM, human and cynomolgus monkey, respectively; Table 1) based on SPR analyses; the observed differences in binding to human and cynomolgus monkey at either pH 6.0 or pH 7.4 were not considered significant. The greater binding of rozanolixizumab to cynomolgus monkey FcRn at pH 6.0 versus that at pH 7.4 (25 pM vs 53 pM, respectively) was considered significant; no functional consequence of this difference was expected. Human IgG of any specificity may bind to human FcRn at acidic pH, via the Fc domain, but we believe that the binding described here occurred via the V region of the anti-FcRn antibody, and not its Fc region, for the following 3 reasons: 1) formats lacking the Fc domain also bound human FcRn with similar affinity; 2) binding occurred at high affinity at pH 7.4, whereas binding of the Fc domain is weak at neutral pH (the difference between 1519.g57 binding to human FcRn at pH 6.0 and at 7.4 was not significant); and 3) binding at pH 6.0 was ~ 104-fold stronger than non-specific (i.e., Fc-mediated) human IgG binding to FcRn (2.35 µM;35 2.52 µM;36 in a 2-site model, with an additional higher affinity binding, with the two KDs of 370 nM and 2.1 µM37).
Table 1.
Binding characteristics of rozanolixizumab and other 1519.g57 formats to different species of FcRn. The extracellular domain of FcRn was used in SPR analyses, whereas cell-based analyzes (MDCK cells) were conducted on membrane-bound FcRn. In SPR analyses, where kd results reached the limit of the instrument, a value of 1.0 × 10−5 was assigned. Results are given as geometric means.
| Species of FcRn | Format | System | pH | n | ka, M−1s−1 | kd, s−1 | KD, pM |
|---|---|---|---|---|---|---|---|
| Human | Rozanolixizumab | SPR | 6.0 | 8 | 4.4 × 105 | 1.0 × 10−5 | 23 |
| 7.4 | 8 | 3.7 × 105 | 1.3 × 10−5 | 34 | |||
| Cells | 6.0 | 3 | – | – | 3.7 × 102 | ||
| 7.4 | 3 | – | – | 4.3 × 102 | |||
| Fab’ | SPR | 6.0 | 6 | 3.7 × 105 | 2.2 × 10−5 | 59 | |
| 7.4 | 5 | 3.8 × 105 | 3.0 × 10−5 | 79 | |||
| Cells | 6.0 | 3 | – | – | 4.4 × 102 | ||
| 7.4 | 3 | – | – | 4.2 × 102 | |||
| Fab’PEG | SPR | 6.0 | 5 | 4.3 × 105 | 1.7 × 10−5 | 40 | |
| 7.4 | 5 | 3.3 × 105 | 1.8 × 10−5 | 56 | |||
| IgG1 | SPR | 6.0 | 3 | 4.6 × 105 | 1.0 × 10−5 | 22 | |
| 7.4 | 3 | 3.8 × 105 | 1.2 × 10−5 | 32 | |||
| Cynomolgus monkey | Rozanolixizumab | SPR | 6.0 | 5 | 4.3 × 105 | 1.2 × 10−5 | 25 |
| 7.4 | 5 | 3.3 × 105 | 1.8 × 10−5 | 53 | |||
| Cells | 6.0 | 3 | – | – | 1.1 × 103 | ||
| 7.4 | 3 | – | – | 1.0 × 103 | |||
| Fab’ | Cells | 6.0 | 3 | – | – | 9.6 × 102 | |
| 7.4 | 3 | – | – | 9.0 × 102 | |||
| Mouse | Rozanolixizumab | SPR | 6.0 | 3 | 1.2 × 105 | 1.3 × 10−2 | 1.1 × 105 |
| Rat | Rozanolixizumab | SPR | 6.0 | 3 | 8.5 × 104 | 8.4 × 10−3 | 9.9 × 104 |
| Rabbit | Rozanolixizumab | SPR | 6.0 | 3 | 0 | 0 | – |
FcRn, neonatal Fc receptor; IgG, immunoglobulin; KD, dissociation constant; ka, association rate constant; kd, dissociation rate constant; pM, picomolar; SPR, surface plasmon resonance.
Madin-Darby canine kidney (MDCK) cells transfected with either human or cynomolgus monkey FcRn (and β2m) were used to assess the binding of rozanolixizumab and 1519.g57 Fab’PEG to FcRn in the context of a cell membrane; comparable binding was observed at pH 6.0 and pH 7.4, consistent with the SPR data (Table 1, Supplementary Figure 2), though KD values were ~ 10-fold greater than those determined by SPR.
Rozanolixizumab had only very weak binding to mouse and rat FcRn at pH 6.0 (KD three orders of magnitude weaker), and showed no detectable binding to rabbit FcRn (Table 1). In support of these binding data, analysis of the sequence alignment of the FcRn α-chains of human, cynomolgus monkey, rat, mouse and rabbit FcRn showed sequence variability in the region of the 1519.g57 binding epitope (data not shown; Swiss Prot: P55899, Q8SPV9, P13599, Q61559, A9Z0W1, respectively). Thus, while cynomolgus monkey was potentially equivalent to human for reproducing the intended in vivo pharmacological effect of 1519.g57 antibodies, wild-type murine and rabbit species were not suitable.
Lack of 1519.g57 IgG1 binding to human β2m and lack of inhibition of human albumin binding to human FcRn by 1519.g57 IgG1 was confirmed by SPR analysis (Supplementary Table 2), and was consistent with screening and epitope-mapping results.
Rozanolixizumab inhibits FcRn-mediated IgG recycling in vitro
MDCK cells stably transfected with human FcRn (and β2m) are capable of recycling human IgG, while anti-FcRn antibodies can block this activity. Rozanolixizumab was observed to inhibit the recycling (IC50 0.41 nM; n = 4) of human IgG by human FcRn-transfected MDCK cells in a dose-dependent manner. Recycling of IgG was also inhibited in cynomolgus monkey FcRn-transfected MDCK cells (IC50 0.98nM; n = 6). In a separate experiment, differences in the potency of FcRn blockade on MDCK cells were observed depending on the molecular format of the antibody (Supplementary Table 5). The potencies of the monovalent formats, Fab’, Fab and Fab’PEG binding to FcRn, were not significantly different (n = 6). However, bivalent rozanolixizumab had a greater effect on IgG recycling in cells in vitro, suggesting that this format may have gained potency by avidity (due to the proximity of a second Fab domain).
To further understand the disruption of IgG transport in a system with endogenous human FcRn, we explored the trafficking of an AlexaFluor (AF) 647-conjugated IgG in human umbilical vein endothelial cells (HUVECs) after FcRn blockade. We observed an increase in intracellular IgG AF647 (Figure 2A and 2B), which would be consistent with disrupted recycling and accumulation of the AF647 label. Limited co-localization with Lysotracker was seen, suggesting the IgG could have been degraded (Figure 2C).
Figure 2.
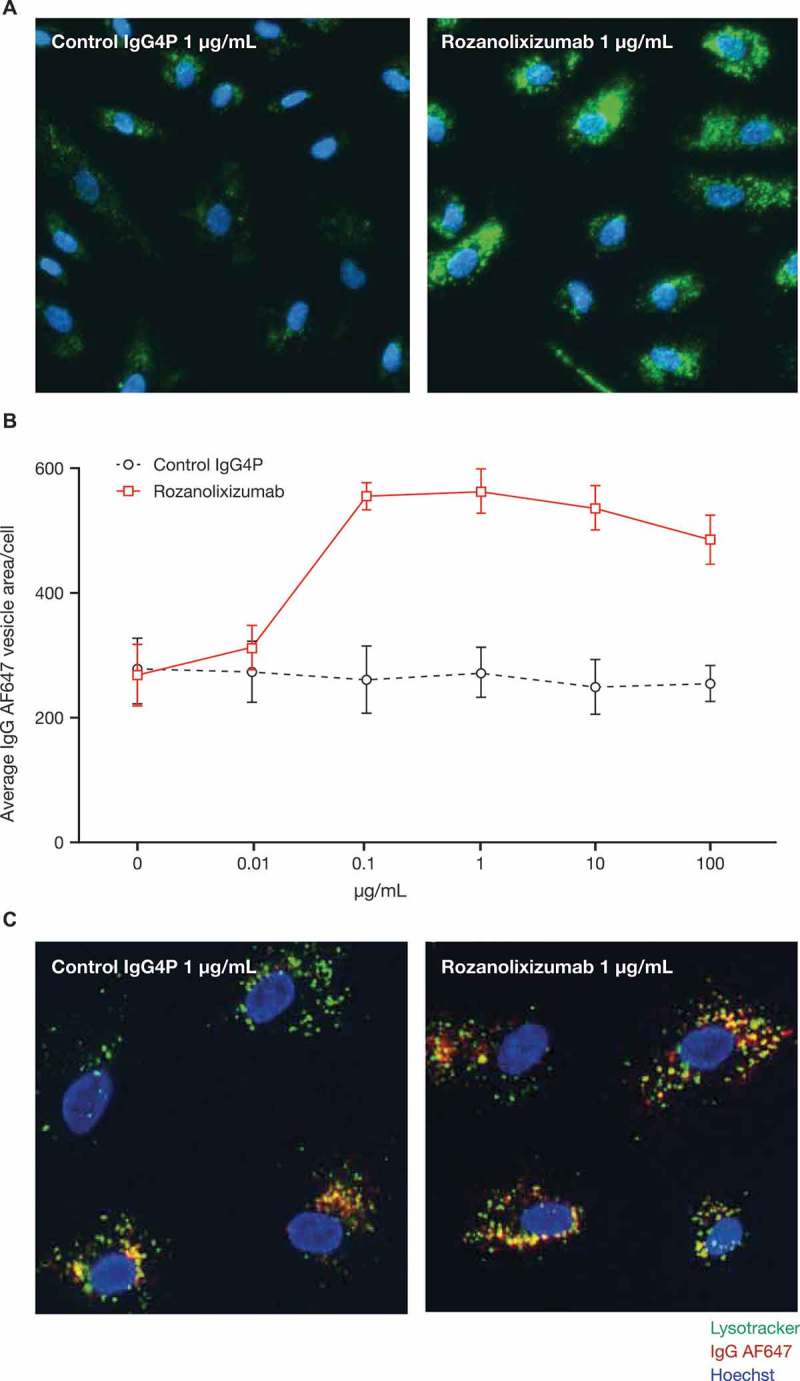
Rozanolixizumab causes an increase in intracellular IgG AF647. (A) HUVECs incubated for 24 hours with AF647-conjugated IgG (green) in the presence of either rozanolixizumab or a control IgG4P antibody. Nuclei labelled with Hoechst (blue). (B) Quantification of vesicular area per cell from three independent experiments. Graph shows arithmetic mean ± SD. (c) Co-localization of Lysotracker (green) with internalized AF647-conjugated IgG (red). Nuclei labelled with Hoechst (blue). Areas of co-localization shown in yellow.
To test if the increased trafficking of IgG to lysosomes disrupted their function, we assessed the lysosomal function in HUVECs using lysosomal associated membrane protein 1 (LAMP-1) staining or Lysotracker labelling. Rozanolixizumab treatment was compared with two other agents, chloroquine and gentamicin, which are known to disrupt lysosome function in vivo.38,39 Chloroquine caused swelling of lysosomes as judged by LAMP-1 staining and an initial increase in Lysotracker labelling followed by a reduction at higher concentrations. At these higher concentrations, we also observed a reduction in cell counts, thought to be due to the high concentrations of chloroquine. Gentamicin caused a modest reduction in the accumulation of Lysotracker label in lysosomes. Rozanolixizumab did not appear to affect cell viability as judged by cell counts (Supplementary Figure 3) or measures of nuclear staining (data not shown) consistent with increased degradative targeting.
Rozanolixizumab does not induce cytokine release in vitro
Using a variety of cell-based assay formats we investigated the potential of rozanolixizumab to affect the production of seven cytokines (interferon (IFN)γ, interleukin (IL)-12p70, IL-1β, IL-6, IL-8, IL-10 and tumor necrosis factor (TNF)). While the appropriate positive controls were able to induce cytokine release as expected, rozanolixizumab treatment did not cause cytokine release (nor did any other 1519.g57 molecular format tested). The induction of cytokine release in vivo is therefore considered to be very unlikely. Exemplary results of some assay formats tested are depicted in Supplementary Figure 4.
Rozanolixizumab has high specificity for FcRn with no cross-reactive protein or tissue binding
In order to exclude off-target binding by rozanolixizumab in humans, a blinded-cell surface receptor screen was conducted against over 4500 human plasma membrane proteins (Supplementary Table 6) expressed on the surface of human embryonic kidney (HEK)-293 cells. Rozanolixizumab was shown to bind only to the α-chain of human FcRn with no cross-reactive binding to any other proteins, confirming its high specificity.
To evaluate any species differences in the potential binding of rozanolixizumab to human and cynomolgus monkey tissues prior to the first-in-human study,6 a tissue cross-reactivity study was performed using immunohistochemical staining and microscopy of frozen tissue sections. Rozanolixizumab showed specific staining in vascular endothelium, epithelial cells, urothelium, connective tissue, glomerular mesangium, and smooth muscle (see Supplementary Figure 5 for examples of staining). Specific, predominantly cytoplasmic, staining with rozanolixizumab was also observed in cells in most tissues, including cells within lymphoid tissues, scattered perivascular and periglandular cells, the neuropil and peripheral nervous tissues, and within the connective tissue and cells between muscle myofibres. The stained cells were usually fusiform, stellar or cigar-shaped; based on their distribution (mostly within the connective tissue framework), the scattered specific positive cells are likely to be endothelial or endothelial precursors and were more likely to be tissue-resident cells rather than leucocytes. The expression of FcRn in multiple tissues was consistent with published data.40-44 There was good concordance of staining patterns between humans and cynomolgus monkeys (results not shown), providing further evidence (together with the comparable binding and pharmacology data) that the cynomolgus monkey was the most relevant species to assess the safety of rozanolixizumab prior to the first-in-human study.
Rozanolixizumab induces a rapid decrease in plasma IgG in vivo in human FcRn transgenic mice
To investigate the effect of molecular format on activity in vivo, human-FcRn transgenic mice were injected with human IgG, and then received a single dose of rozanolixizumab or another 1519.g57 molecular format (Fab’, Fab’PEG, IgG1, triFab’, FabFv, Fab’human albumin conjugate, Fab’mouse albumin fusion) at 10, 30, 100 or 250 mg/kg, or with vehicle. In this system, the concentration of human IgG in vehicle-treated mouse plasma decreases due to IgG turnover in the absence of synthesis.14 The effect of FcRn blockade was considered to be exhausted when the rate of decline of human plasma IgG concentration in treated animals was equal to that in vehicle-treated animals. The baseline clearance rate of human IgG was established using a mutated IgG4 antibody10 that was unable to bind human FcRn (Supplementary Figure 6); rates of clearance for the mutated IgG4 antibody were comparable to previously reported data.45 FcRn blockade led to an accelerated rate of decline in the concentration of human IgG in mouse plasma compared with vehicle. All molecular formats exhibited a dose-response relationship; the larger the dose, the longer the duration of effect, as exemplified in Supplementary Figure 6 for rozanolixizumab.
Differences were observed in the activity of the antibodies in the human FcRn transgenic mouse model according to molecular format (Figure 3A and 3B). At equivalent doses of 100 mg/kg, the activities of monovalent 1519.g57 formats (Figure 3A and 3B) could be ranked according to the differences in drug clearance and plasma concentration (Figure 4; Fab’ [without attached PEG] was undetectable by 8 hours after injection and so is not shown). Formats maintaining higher concentrations for a longer period of time demonstrated a prolonged effect on human IgG catabolism (Figures 3 and 4). The profiles of all formats except for the non-FcRn specific control Fab’PEG were typical of drugs that suffer target-mediated drug disposition (TMDD), where above a certain inflexion point the FcRn receptor is saturated, while below that inflexion point a varying degree of saturation is observed, with changes in the rate of elimination of the drug being concentration-dependent. Interestingly, the IgG1 format was less effective than the IgG4P format, and had an apparent faster elimination. This difference in PK might be explained by a difference in off-target binding, for instance to murine Fc receptors (other than FcRn).
Figure 3.
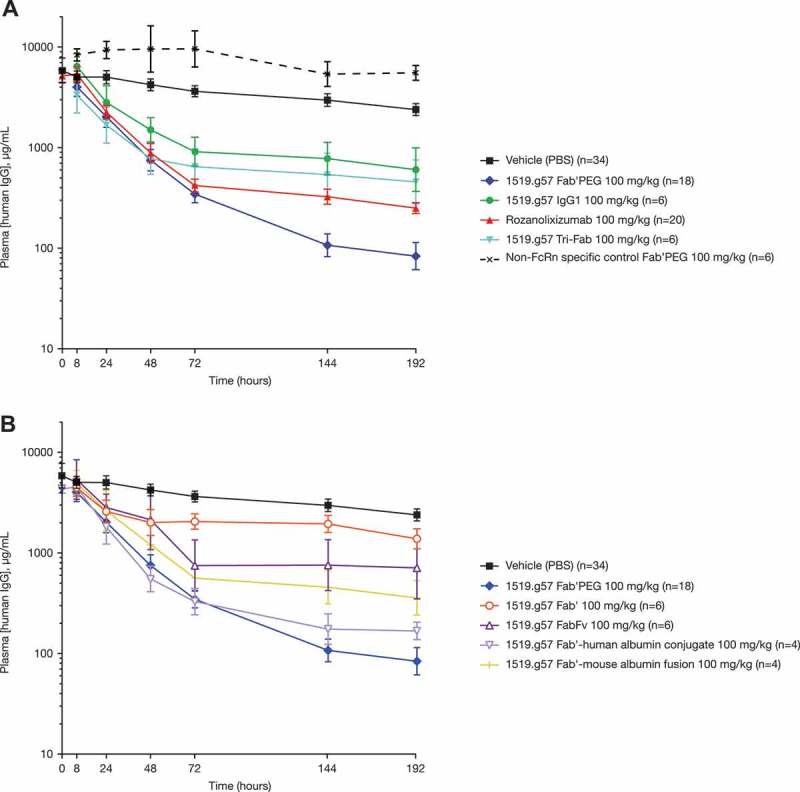
Effect of a single dose of various 1519.g57 formats (100 mg/kg) on human plasma IgG concentration in human FcRn-transgenic mice. Group sizes (n) as indicated in the legend, combined from multiple experiments. For clarity, data are divided between two graphs: (A) and (B). Geometric mean and 95% confidence interval are shown.
Figure 4.
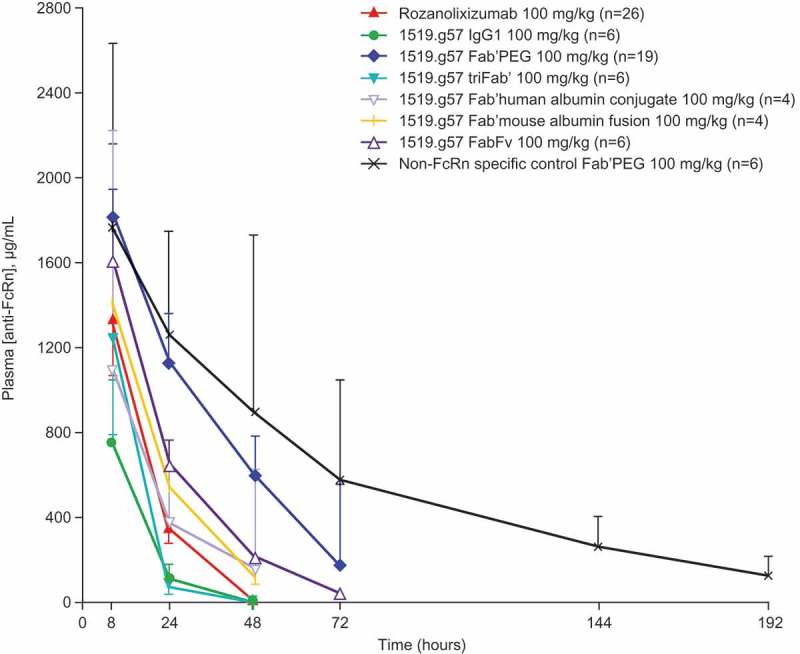
The PK of a single dose of various 1519.g57 formats in human FcRn-transgenic mice: the effect of molecular format on the concentration of anti-FcRn free in plasma. Group sizes (n) were as indicated in the legend, combined from multiple experiments. Geometric mean and 95% confidence interval are shown (one arm shown only, for greater clarity).
The valency of the various anti-FcRn molecules (mono- to tri-valent) had no clear effect on efficacy. At the same dose of 100 mg/kg, the monovalent Fab’PEG showed greater activity than either rozanolixizumab (bivalent format) or the triFab’ format (Figure 3A), indicating no increase of effect by multiple, crosslinking binding. However, in molar terms, the dose of Fab’PEG was about a third greater, which might explain its greater activity. In addition, the plasma concentrations of the Fab’PEG format were sustained for longer (Figure 4); this may be due not only to the higher molar dose, but also due to the presence of a PEG moiety. The PEG moiety is responsible for extending half-life in an FcRn-independent manner,46 and therefore could provide a PK advantage compared with the IgG formats that are blocking their own recycling mechanism. Rozanolixizumab and the triFab’ format were dosed approximately equally in molar terms, but the IgG4P format showed greater activity (Figure 3A), although this might, in part, be due to prolonged PK of rozanolixizumab (Figure 4).
Because mouse albumin binds to, and is recycled by, human FcRn,47 the effect of FcRn blockade on mouse plasma albumin concentration in the human FcRn transgenic mouse was measured. Neither rozanolixizumab, nor any other 1519.g57 format (with the exception of IgG1 for unknown reasons), was observed to have an effect on endogenous mouse plasma albumin concentration (Supplementary Figure 7A and 7B), indicating little or no blockade of albumin binding, even by those formats that included or bound mouse albumin (Fab-albumin fusion or conjugate, or FabFv). The maintenance of mouse plasma albumin concentration suggested no effect on albumin turnover (the sum of synthesis and degradation), and so possibly little or no change of expression of FcRn. No adverse events were noted in any mouse treated with rozanolixizumab or 1519.g57 in any molecular format.
Rozanolixizumab induces a rapid decrease in plasma IgG in vivo in cynomolgus monkeys
Single-dose administration
IV administration of a single dose of rozanolixizumab in cynomolgus monkeys caused a dose-dependent reduction in plasma IgG concentration (Table 2, Figure 5A); comparable results were observed with 1519.g57 Fab’PEG (Supplementary Table 7). The two formats appeared similar in efficacy, with no obvious difference in effect due to valency. The extent of reduction of plasma IgG concentration correlated with the presence of anti-FcRn antibody in the plasma. Doses of 5, 10 or 30 mg/kg rozanolixizumab (IV) gave 49%, 63% or 69% reduction in plasma IgG concentration in 3.5, 5 or 7 days, respectively (Table 2; Figure 5A). In other experiments (data not shown) larger doses of rozanolixizumab have generated only slightly greater reductions in plasma IgG concentration; the 30 mg/kg single dose of rozanolixizumab was considered to give close to maximum effect in this system.
Table 2.
Calculated PK parameters after single IV doses of rozanolixizumab in cynomolgus monkeys. Data shown are geometric mean (coefficient of variation).
| Pharmacodynamics |
||||||||
|---|---|---|---|---|---|---|---|---|
| Plasma [IgG] |
Pharmacokinetics, mean (%CV) |
|||||||
| Format | Dose, mg/kg | n | % change from baseline | Time to nadir, days | Cmax, µg/mL | Cmax/actual dose | AUClast, µg/mL/day |
AUClast/actual dose |
| Rozanolixizumab | 0 | 4 | 0 | n/a | n/a | n/a | n/a | n/a |
| 5 | 4 | 49 | 3.5 | 84.7 (17.6%) | 4.6 (19.2%) | 56.1 (18.4%) | 3.1 (27.8%) | |
| 10 | 4 | 63 | 5 | 266.2 (4.9%) | 7.4 (16.4%) | 216.1 (12.6%) | 6.1 (20.8%) | |
| 30 | 4 | 69 | 7 | 730.5 (11.5%) | 9.1 (14.5%) | 853.7 (7.6%) | 10.7 (10.4%) | |
AUClast, area under the concentration-time course until last quantifiable concentration; Cmax, maximum observed plasma concentration; CV, coefficient of variation; IgG, immunoglobulin G; n/a, not applicable.
Figure 5.
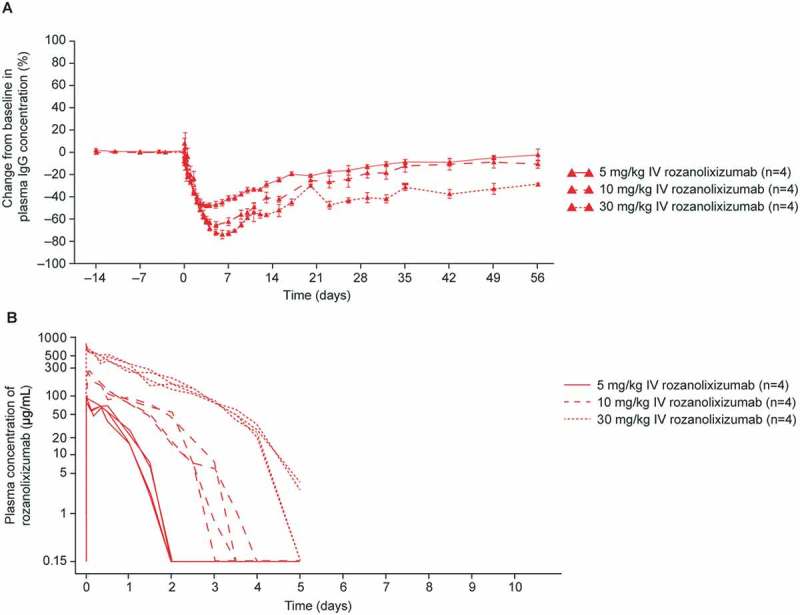
(A) The effect of single IV doses of rozanolixizumab (5, 10 and 30 mg/kg) on plasma IgG concentration in cynomolgus monkeys, relative to baseline. Data are arithmetic mean ± standard errors. (B) The PK of single doses (IV) of rozanolixizumab in cynomolgus monkey plasma.
After a single IV dose of rozanolixizumab, reduction of plasma IgG concentration continued after the disappearance of free anti-FcRn in plasma, suggesting a delayed effect; IgG returned to physiological ranges within 8 weeks. 1519.g57 Fab’PEG (20 mg/kg) resulted in a maximum reduction of about 70% following IV administration (Supplementary Table 7); dosing via the SC route was also effective, although to a lesser extent, with a maximum reduction of about 50% (data not shown).
The concentration-time profile for rozanolixizumab (Figure 5B) in cynomolgus monkeys showed marked non-linear PK behavior, indicating TMDD, consistent with the findings in the human FcRn-transgenic mouse model (Figure 4). Non-proportionality to dose was observed for Cmax (maximum plasma concentration) and AUClast (area under the concentration–time curve until last quantifiable concentration for single dosing) values in the derived non-compartmental PK parameters, exposure increased greater than proportionally with dose (Table 2), which may be related to the degree of saturation of the FcRn receptor with higher doses of rozanolixizumab. The non-linearity was accompanied by a rapid disappearance of rozanolixizumab from the plasma. At the highest dose (30 mg/kg), rozanolixizumab was undetectable in plasma beyond Day 5, demonstrating faster elimination compared with that previously reported for other IgG4 mAbs in cynomolgus monkeys,48 due to a combination of blockade of its own recycling process and TMDD. 1519.g57 Fab’PEG had comparable results (Supplementary Table 7), with non-linear increases in exposure with dose, a profile characteristic of TMDD, and rapid elimination (no drug detectable by Day 7 with a dose of 20 mg/kg). An advantage in terms of PK was expected for the 1519.g57 Fab’PEG format compared with rozanolixizumab because the pegylated portion is independent of the FcRn recycling system, and therefore could be expected to provide a longer exposure at comparable dosing. However, based on the PK results (Table 2 and Supplementary Table 7), little advantage was observed, which could indicate that the drug disappearance is predominantly driven by TMDD.
Plasma albumin concentration was monitored, but no clear dose-related trend was observed (Supplementary Figure 8). In separate experiments, treatment of cynomolgus monkeys with rozanolixizumab did not affect IgM or IgA concentrations (data not shown), consistent with these molecules not being salvaged and recycled by FcRn, and with data reported by others on elimination of FcRn function (by a peptide antagonist;19 in FcRn knockout mice14). Generation of anti-drug antibodies (ADA) was detected in the majority of animals during the dosing period; however, no impact to PK or PD was observed. No adverse events were noted in any cynomolgus monkey treated with rozanolixizumab.
Repeat dosing: infrequent administration
In order to explore the possibility of treatment with rozanolixizumab over an extended period, studies were made of repeated dosing with either long or short periods between doses. To evaluate the PK/PD of rozanolixizumab with infrequent dosing, the animals that received single IV doses of 30 mg/kg described above were then further treated with rozanolixizumab 30 mg/kg IV on Days 63 and 126. These time points were selected because in previous experiments ~ 60 days were required for plasma IgG concentrations to return to pre-dose levels. PK profiles for rozanolixizumab following the first and second administrations were similar, but exposure was reduced following the third administration (Supplementary Figure 9A). The magnitude of this can be seen in the different Cmax values achieved, with geometric means of 730, 764 and 569 μg/mL, after the first, second and third administrations, respectively.
The second IV dose of rozanolixizumab induced a reduction in plasma IgG concentration comparable to the first dose (~ 75%; mean value) with a similar PD profile. In contrast, the third IV dose on Day 126 achieved a lesser (~ 55%; mean value) reduction in plasma IgG, but recovery was faster than that of the first or second dose (Supplementary Figure 9B). Thus, PD correlated with PK, as observed in the human FcRn-transgenic mouse model (Figures 3 and 4). Generation of ADA was detected in the majority of animals during the dosing period, which may in part explain the reduction in drug effect over time and the faster recovery of IgG after the third dose. However, interpretation of the quantitative effect of ADA on IgG concentrations is limited because the IgG assay detected all IgG including ADA itself, so quantification of the margin in the reduction in IgG was not possible with this method.
Repeat dosing: frequent administration
The effects of frequent treatment with rozanolixizumab were evaluated by daily IV (initial 30 mg/kg loading dose on Day 0, followed by 41 daily doses of 5 mg/kg) and SC (5 mg/kg for 60 consecutive days) administration. IV administration of rozanolixizumab for 42 consecutive days showed that the highest peak drug concentration occurred after the loading dose, followed by diminishing peak values after each dose until ~ Day 12, when peak drug concentrations achieved a broadly steady level that was maintained for the duration of the study in most animals (Supplementary Figure 10A). The individual profiles showed a rapid clearance during the dose interval leading to pre-dose concentrations close to or below the limit of quantification; some animals appeared to suffer effects from ADA towards the end of the treatment period with decreases observed in the peak rozanolixizumab concentrations.
The PK of rozanolixizumab following daily SC dosing (with no loading dose) showed that plasma concentrations increased until ~ Day 10 as expected, but decreased thereafter (Supplementary Figure 10B). This decrease in exposure was attributed to the formation of ADA.
Plasma IgG concentrations fell by ~ 70–75% (mean values) from pre-dose levels by Day 10 with repeated dosing (Figure 6). Daily IV administration reduced IgG concentrations in all animals, and maintained low IgG concentrations throughout the treatment period. As with the single and intermittent IV doses, ADAs were detected during daily IV dosing, but with no discernible effect on PD. Following daily SC administration, the observed reduction in IgG concentrations was sustained in only two of four animals; the absence of a sustained IgG reduction in two animals may have been caused by the generation of ADAs. An overshoot in the IgG profile with respect to baseline was observed for one animal over the treatment period (baseline IgG concentration ~ 8 mg/mL, maximum IgG concentration during the treatment period 18.5 mg/mL), investigations showed no anomalies in this animal. There was no effect on plasma albumin concentration or any drug-related adverse events in cynomolgus monkeys dosed IV or SC, including those with detectable ADA.
Figure 6.
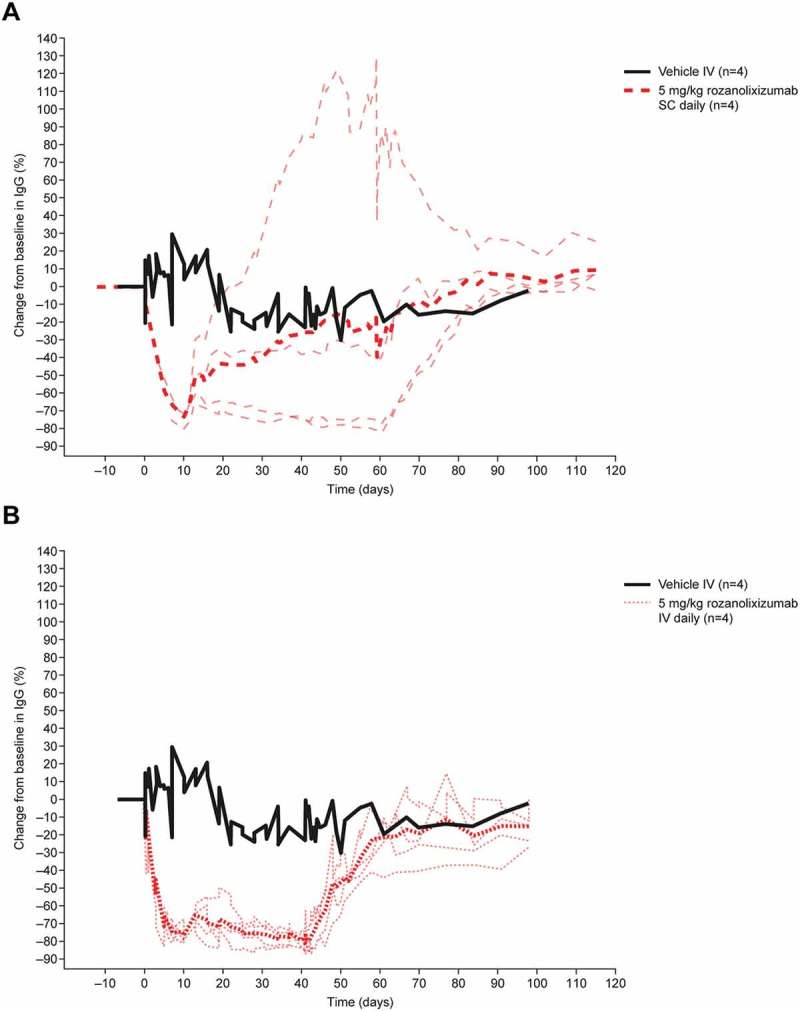
The effect of daily IV and SC doses of rozanolixizumab on plasma IgG concentrations in cynomolgus monkeys. For IV dosing, a loading dose of 30 mg/kg on Day 1 was followed by a daily dose of 5 mg/kg for 41 days. For SC dosing, 5 mg/kg was administered daily for 60 days. Thick lines represent mean values; thin lines represent individual animals. *Animals received a loading dose of 30 mg/kg on Day 1.
Rozanolixizumab induced marked sustained but reversible decreases in plasma IgG in the absence of adverse events in a cynomolgus monkey toxicology study
A 13-week toxicology study in cynomolgus monkeys was conducted to evaluate the potential toxicity, toxicokinetics and PD of high and repeated IV and SC doses of rozanolixizumab. Results from this study support the findings of the previously reported 4-week toxicology study.6 SC (50 and 150 mg/kg) or IV (150 mg/kg) administration of rozanolixizumab every 3 days for 13 weeks and intermittent SC dosing of rozanolixizumab (150 mg/kg) every 3 days in each of Weeks 1, 6 and 10 (Table 3) was well tolerated in cynomolgus monkeys. The no-observed-adverse-effect level (NOAEL) was 150 mg/kg dosed IV or SC every 3 days.
Table 3.
Study design and mean PK parameters of rozanolixizumab in the 13-week toxicity study in cynomolgus monkeys. Data shown are arithmetic mean (SD).
| Dosing regimen | Number of males/females | Necropsy at Day 91 | Necropsy at Day 144 | Cmax (µg/mL), mean (SD) |
AUC0–τ (µg/mL/day), mean (SD) |
||
|---|---|---|---|---|---|---|---|
| Day 1 | Day 64 or 88 | Day 1 | Day 64 or 88 | ||||
| Vehicle control | 6M/6F* | 3M/3F | 2M/2F | – | – | – | – |
| 50 mg/kg SC q3 days | 4M/4F | 4M/4F | n/a | 404 (84.2) |
157 (192) |
828 (244) |
327 (408) |
| 150 mg/kg SC q3 days | 6M/6F* | 4M/4F | 2M/2F | 1780 (639) |
1730 (998) |
3740 (1130) |
3780 (2290) |
| 150 mg/kg SC q3 days (Weeks 1, 6, 10) |
6M/6F* | 4M/4F | 2M/2F | 1700 (338) |
1160 (503) |
3570 (569) |
2410 (1220) |
| 150 mg/kg IV q3 days | 6M/6F* | 4M/4F | 2M/2F | 4230 (618) |
4870 (1030) |
5450 (841) |
5370 (2230) |
*includes 2M + 2F for recovery
AUC0–τ, area under the plasma concentration (for repeated dosing) versus time curve on Day 1 or Day 64 or 88; Cmax, maximum observed plasma concentration; D, day; F, female; M, male; W, week.
Plasma IgG concentrations were decreased in all rozanolixizumab-treated groups (150 mg/kg), maximal reductions of up to 85% were observed by Week 13; plasma IgG concentrations returned to baseline by the end of the 8-week recovery phase (Figure 7). Reduction of plasma IgG concentration was less pronounced when animals were treated only in Weeks 1, 6 and 10.
Figure 7.
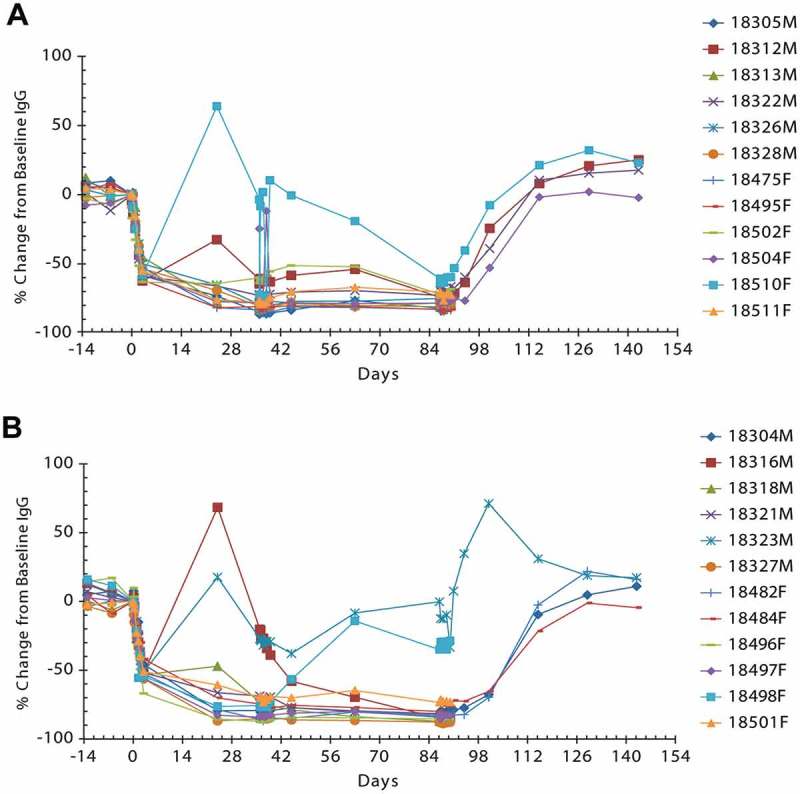
Individual percent change from baseline in plasma IgG concentration in cynomolgus monkeys following IV (A) or SC (B) rozanolixizumab dosing at 150 mg/kg every 3 days for 13 weeks. Lines represent individual animal data.
As previously observed in the 4-week study, there were no rozanolixizumab-related mortalities nor adverse effects on the standard toxicology, clinical pathology (apart from the large decreases in IgG and the related small decreases in globulin and total protein as expected with this mode of action), safety pharmacology, immunotoxicology and histopathology. No rozanolixizumab-related effects were observed on total IgM or IgA; there was no evidence of infection as a result of reduced total IgG concentrations. Small rozanolixizumab-related decreases in albumin were observed (generally in the range of a 1 to 13% decrease from baseline; Supplementary Figure 11); the decreases were all within acceptable ranges, not considered adverse and returned to baseline by the end of the dosing phase. Albumin concentrations recovered during the 8-week recovery period. No corresponding effect on serum calcium concentration was observed.
Rozanolixizumab treatment, both IV and SC, resulted in an expected decreased T-cell-dependent IgG antibody response (TDAR) to keyhole limpet hemocyanin (KLH) immunization. Both the primary and secondary responses were reduced (Supplementary Figure 12). Reduced titers upon treatment with rozanolixizumab reflect the pharmacologically intended effect, clearance of IgG antibodies due to blockade of FcRn and the associated effect this has on IgG recycling, they are not due to an impact on the cellular level of IgG antibody production. No effect was observed in the IgM-mediated anti-KLH response, this was as anticipated since IgM does not interact with FcRn. Hematological assessments showed no effects on immune cell population counts after rozanolixizumab treatment. Immunophenotyping did not reveal any rozanolixizumab-related effect on relative or absolute numbers of lymphocytes and lymphocyte subpopulations in blood samples and lymphoid organs (data not shown). Rozanolixizumab had no effect on a variety of factors released from immune cells or factors affecting the activity of immune cells (e.g., cytokines, C-reactive protein, fibrinogen, complement components).
Discussion
Here, we described the generation of a high-affinity anti-human FcRn antibody, in an optimal format, that works to inhibit IgG salvage and so reduces plasma IgG concentration in FcRn-transgenic mice and in cynomolgus monkeys. In a series of in vitro SPR and cell-based binding experiments using a panel of anti-FcRn antibody formats, rozanolixizumab (IgG4P) demonstrated comparable binding to human FcRn and cynomolgus monkey FcRn at both pH 6.0 and pH 7.4. Rozanolixizumab demonstrated significantly greater potency when blocking IgG recycling in MDCK cells (stably transfected with human FcRn and β2m), compared with other molecular formats, and increased intracellular trafficking of IgG to lysosomes.
Further in vivo characterizations in both a transgenic FcRn mouse model and cynomolgus monkeys demonstrated dose-dependent reductions in plasma IgG following rozanolixizumab treatment, and remarkably fast clearance of rozanolixizumab with non-linear PK, typical of drugs that suffer TMDD; no clinically relevant effect was observed on plasma albumin concentrations. These data support the clinical development of rozanolixizumab.6
We have demonstrated that rozanolixizumab acts as a competitive inhibitor for IgG, by physically overlapping with the IgG Fc binding site on the α-chain of FcRn. Greater KD values (10-fold lower binding affinity) were observed when the binding of rozanolixizumab and 1519.g57 Fab’PEG were measured on cells transfected with human or cynomolgus monkey FcRn compared with SPR analyses. It is proposed that this lower binding affinity could be due to steric hindrance by the nearby cell membrane or other neighboring molecules present thereon. The low binding affinity of rozanolixizumab for mouse FcRn was most likely due to Fc (and not Fab) binding, since the binding was comparable to that reported for human IgG Fc domain binding to mouse FcRn (60 – 270 × 103 pM).35–37 The low rodent cross-reactivity was insufficient to support the study of rozanolixizumab in wild-type mice or rats; hence transgenic mice and cynomolgus monkeys were selected as the test species for in vivo studies.
The in vitro data presented here supports the concept that, although rozanolixizumab is bivalent, it binds to FcRn in a monovalent manner. We have shown comparable efficacy (in molar terms) for the bivalent rozanolixizumab and monovalent 1519.g57 Fab’PEG in cynomolgus monkeys, corroborating the monovalent binding of rozanolixizumab to FcRn. From these data it is inferred that cross linking of FcRn molecules by rozanolixizumab in vivo is unlikely or infrequent. In vitro immunocytochemistry analyzes indicated that the mechanism of reduction in plasma IgG concentration was inhibition of IgG salvage, which causes trafficking of IgG to lysosomes for degradation instead of recycling, in agreement with earlier reports.49
Both transgenic mouse and cynomolgus monkey data indicated a dose-response relationship of anti-FcRn treatment to plasma IgG concentration; the longer free drug was present, the longer the observed reduction in plasma IgG concentration and the more pronounced the overall decrease.
Although the amount administered affected the extent of the decrease in IgG concentration, the format of the anti-FcRn was also seen to be important. A single dose of 1519.g57 Fab’ was weakly effective in transgenic mice, probably because of its rapid elimination. Formats with slower elimination appeared more effective at equivalent doses, presumably because they gave greater coverage (more extended period of anti-FcRn presence that translates to prolonged saturation of FcRn above a certain level). Marked TMDD was observed, consistent with the mechanism of action of FcRn and the high receptor expression, which can affect drug PK. In support of this, elimination of rozanolixizumab was faster than that reported by Haraya et al.50 for 11 non-anti-FcRn IgG1 and IgG2 antibodies in mice (half-lives ranging between 3.5 and 13.2 days), but consistent with the observations of Liu et al,22 who showed rapid disappearance of anti-rat FcRn from rat plasma. The expected PK exposure benefit from PEGylation was not observed in these studies; no obvious advantage was seen over the other IgG formats, Fab’PEG was similarly effective to IgG. These results suggest that pronounced TMDD prevails over the non-specific elimination of drugs as the main driver of clearance, with blockade of IgG’s own recycling system forming a secondary driver of clearance. This was particularly so since 1519.g57 antibody bound FcRn at both pH 6.0 and at neutral pH, leading to injected anti-FcRn rapidly encountering and binding its target at neutral pH on the cell surface before endocytosis had occurred.
FcRn is expressed in many tissues, so some loss of anti-FcRn given via the SC route might be expected; indeed, some loss would be expected with any mAb. It is unsurprising, therefore, that a small drop of 7% in bioavailability in cynomolgus monkeys was seen after SC injection of a 20 mg/kg dose of the 1519.g57 Fab’PEG format. Lower doses for the Fab’PEG format were not tested in this work; additional investigations would be required to better understand the potential non-linear behavior of the drug’s PK during the absorption phase. In this study, single and multiple administrations showed the SC route to be a viable drug delivery option. An IV dose of 100 mg/kg of 1519.g57 Fab’PEG caused an approximate 80% reduction in plasma IgG concentration in cynomolgus monkeys; 20–30 mg/kg doses of either format (rozanolixizumab or 1519.g57 Fab’PEG) approached this extent of effect and so are expected to be close to the maximally effective dose. In cynomolgus monkeys, normal IgG concentration was regained (with no rebound) by about 8 weeks after dosing, the effect of a single dose was prolonged in line with the prolonged mAb exposure, and was recoverable.
FcRn salvages and recycles IgG against a background of continued IgG synthesis, so it is unlikely that inhibition of FcRn could reduce plasma IgG concentration to zero. Very high and prolonged dosing with rozanolixizumab resulted in what we consider to be the maximum possible reduction of plasma IgG concentration in cynomolgus monkeys, of ~ 90%. A more practical single dose of 30 mg/kg, or repeated 5 mg/kg doses achieved ~ 70–75% reduction. These results are consistent with previously published data: two humans with β2m-deficiency (and consequently FcRn-deficient) had plasma IgG concentrations of about 11% and 25% of normal,51 and FcRn-deficient mice had plasma IgG concentrations of about 11.8% of normal.45 The extent of plasma IgG reduction achieved in these preclinical investigations and in the clinical study6 are comparable with the reduction (~ 50–60%) observed following a full course of repeated plasmapheresis procedures in humans, which is one of the current standards of care in autoimmune diseases.52,53 This suggests that FcRn blockade may not only have clinical efficacy in autoimmune disease, but also that it could be more simply and rapidly administered, and may be more convenient and more specific in its mechanism of action than current treatments. Importantly, when rozanolixizumab dosing was stopped, plasma IgG concentrations returned to pre-dose levels, demonstrating that there was no lasting effect on FcRn.
Rozanolixizumab was effective across both SC and IV dosing regimens, indicating flexibility in clinical use. While a single IV dose (30 mg/kg) in cynomolgus monkeys reduced plasma IgG concentrations immediately after infusion, and attained ~ 70% reduction within ~ 7 days, 5 mg/kg/day SC took ~ 10 days to reach a similar nadir. No other differences were observed between IV and SC administration. In either case, recovery after dosing was to the physiological range of plasma IgG concentrations, without rebound to concentrations greater than pre-dose levels, by ~ 8 weeks after dosing. There appeared to be no significant toxicity associated with these effects, although in humans, prolonged maintenance of very low IgG concentrations (< 4–7 mg/mL) might be expected to lead to an increased risk of infection, as observed in hypogammaglobulinemia.54 Reduced IgG concentrations persisted even after clearance of rozanolixizumab from the plasma, suggesting that rozanolixizumab remains bound to FcRn as it cycles from the cell surface to the endosome and retains the ability to block IgG recycling even after antibody is no longer detectable in the plasma, in line with FcRn turnover.
FcRn is responsible for binding and recycling albumin as well as IgG.55 Significant reductions in plasma albumin concentrations in patients would be undesirable, therefore during development, the anti-FcRn variable region (1519.g57) was selected for its lack of inhibition of the FcRn-albumin interaction. We have demonstrated that the binding epitope of 1519.g57 Fab’ was separate from that of albumin. Furthermore, the PK/PD studies reported here in transgenic mice and cynomolgus monkeys showed that rozanolixizumab caused no reduction in plasma albumin concentration outside the normal range; an observation also reported in healthy volunteers.6 The reductions in albumin observed in the 13-week cynomolgus monkey toxicology study were likely attributed to partial steric hindrance at high concentrations of rozanolixizumab; however, it is important to note that in this study, rozanolixizumab was administered at a concentration 100-fold in excess of the planned human exposure level. In cynomolgus monkeys there was no effect of rozanolixizumab or 1519.g57 Fab’PEG on concentrations of IgM and IgA (which are not recycled by FcRn), these data support the results observed in the clinical healthy volunteer study of rozanolixizumab.6 Overall, the effects of rozanolixizumab and 1519.g57 Fab’PEG were specific to IgG, in contrast to currently available therapies, which can affect a range of immune modulators (with the exception of immunoadsorption, in which IgG alone is removed by affinity chromatography).
The PK of rozanolixizumab was consistent with expectations for a human mAb, in cynomolgus monkeys, against a high abundance cell surface target that is widely distributed on the extracellular membrane, including vascular endothelium.10 Since FcRn is critical in salvaging IgG from degradation, FcRn inhibition would be expected to accelerate IgG clearance in a dose-dependent manner. Indeed, in these studies and in the clinical healthy volunteer study,6 rozanolixizumab exhibited pronounced TMDD, manifested as non-linearity in clearance with dose, and decreasing clearance with increased dose. Even at high doses, rozanolixizumab was cleared remarkably rapidly compared with other IgG-type mAbs, which typically demonstrate half-lives of ~ 6–10 days in monkeys.48
Administered human or humanized biologic drugs can be recognized as foreign by the immune system and, as such, ADA are commonly generated in animals and, usually to a lesser extent, in humans.56 Measurement of ADA in animal models is undertaken within the PK/PD studies to inform the relevant PK and IgG data (unaffected by ADA) that could be used to guide dose selection in the first in human study, and to ensure that a sufficient number of animals are adequately exposed to the test item, to enable derivation of toxicological effects; the frequency of ADA is not representative or translatable into humans. The ADAs identified in rozanolixizumab-treated cynomolgus monkeys possibly contributed to the gradual loss of PD observed in some repeat-dosed animals after an initial period of efficacy of ~ 10 days or longer during which the ADA response was mounted. As the drug tolerance of the ADA assay was not determined, the results are not definitive. However, the lack of detectable ADAs or loss of PD effect in normal mice dosed repeatedly with rabbit-mouse chimeric anti-mouse FcRn Ab indicate that blockade of FcRn per se did not cause or potentiate generation of ADA, even against an essentially foreign protein. The generation of ADA in cynomolgus monkeys was therefore possibly due to humanized rozanolixizumab being a heterologous protein in cynomolgus monkeys. In a Phase 1 clinical study in 48 healthy volunteers, ADAs were detected in subjects across all treatment groups following single SC and IV doses of rozanolixizumab 1–7 mg/kg, but concentrations were considered low, with quantifiable concentrations observed in only five subjects.6 ADA had no effect on IgG concentrations or on the PK of rozanolixizumab.6 Phase 2 studies will further explore the ADA profile and any impact on the pharmacological effect following repeated dosing of rozanolixizumab.
The safety of FcRn inhibition was further confirmed after dosing of rozanolixizumab at supra-pharmacological exposures (up to 6-fold above the predicted maximum human exposure) for 13 weeks in cynomolgus monkeys. Minor effects on albumin were observed, all within acceptable ranges. There were no adverse effects. A reduction in the TDAR was reported in the absence of any other immune changes, but this was due to the expected clearance of KLH-specific IgG and was not thought to be due to any direct effects on immune functions, such as the ability to mount antibody responses. As expected, no effects on KLH-specific IgM antibody titers were observed, since FcRn is not involved in the recycling of IgM. This is supported by in vitro safety analyses, which demonstrated that rozanolixizumab showed no effect on cytokine production. Although the FcRn C-terminal (intracellular) domain contains motifs that may be associated with endocytosis and trafficking,57,58 it lacks the immunoreceptor tyrosine-based activation motif (ITAM) and immunoreceptor tyrosine-based inhibition motif (ITIM) that are associated with the transmembrane signalling observed with other Fc receptors.59 Although Baker et al.60 reported that in dendritic cells, interaction between FcRn and immune complexes can lead to secretion of IL-12, to our knowledge there is no mention in the literature of cellular signalling that occurs as a result of FcRn binding its natural ligands, IgG or albumin. The induction of cytokine release by stimulation of FcRn-expressing cells directly was therefore considered to be low. Moreover, rozanolixizumab is an IgG4P isotype with reduced capacity to interact with Fcγ receptors, inability to activate the complement cascade and strongly reduced capacity to cause cytokine release by cells interacting with the Fc part of the antibody via Fcγ receptors.
In summary, we have described the generation and characterization of a humanized anti-human FcRn antibody, rozanolixizumab, as a potential therapeutic to reduce the concentration of plasma IgG (but not albumin, IgA or IgM). A Phase 1 study (NCT02220153) was recently published,6 providing the first clinical evidence in healthy volunteers of rozanolixizumab as a potential new treatment concept for autoimmune diseases. SC administration of rozanolixizumab up to 7 mg/kg and IV administration up to 4 mg/kg was well tolerated in healthy volunteers.6 Clinical development of rozanolixizumab continues with Phase 2 studies in myasthenia gravis (NCT03052751) and ITP (NCT02718716).
Materials and methods
Immunization
Rats and mice were both immunized with FcRn extracellular domain (ECD) or NIH3T3 mouse fibroblasts transiently co-transfected with a mutant surface-stabilized form of human full length FcRn (L320A and L321A)61 and mouse β2m. The NIH3T3 cells were transfected (using electroporation) with a 1:1 ratio of the human FcRn α-chain and mouse β2m DNA (75 µg of each vector was transfected per 3 × 107 cells). Rats and mice were immunized by SC injection of between 1 × 107 and 5 × 107 NIH3T3 mouse fibroblasts expressing mutant human FcRn and mouse β2m (three injections) in Weeks 1, 6 and 9, with a fourth final (IV) injection of 10 µg human FcRn ECD (in combination with mouse β2m) in Week 15. Rat and mouse B cells were harvested in Week 17.
Screening of antibody in B-cell cultures and V region recovery
Sera from immunized animals were screened by flow cytometry on a FACSCanto II flow cytometer (BD Biosciences, UK) for antibody binding (at neutral pH) to HEK-293 cells that were transiently transfected to express the mutant surface-stabilized form of human FcRn (L320A; L321A). Binding of IgG from mouse or rat sera was revealed with phycoerythrin (PE)-labelled anti-mouse or anti-rat Fc antibodies, respectively. Sera were also screened for the ability to block binding of AF 488-conjugated IgG to human FcRn at pH 6.0 on these cells, monitored by flow cytometry on a FACSCanto II flow cytometer (BD Biosciences, UK).
B-cell cultures were prepared from animals whose sera showed FcRn binding and blockade of IgG binding to FcRn, using a method similar to that described by Zubler et al.62 Further details are provided in Supplementary Methods, as are details of V region recovery63 and cloning.
Conversion to humanized Fab’ and other formats
Genes encoding humanized V-region heavy (VH) and light (kappa, VK) chain sequences were designed and constructed by Entelechon GmbH. VK and VH genes were cloned into proprietary expression vectors, containing DNA encoding the human κ-chain constant, the human gamma 1 heavy-chain constant (CH1) and truncated hinge regions with a C-terminal 6x Histidine (6HIS) tag, respectively. Transient co-transfection of heavy and light chain vectors enabled the expression of humanized Fab-6HIS in HEK-293F cells (11625–019; Life Technologies). Candidates were screened for retention of their FcRn binding (SPR analysis) and stability (biophysical characterization), and selected candidates were re-cloned for expression in E.coli as the Fab’ format to allow PEGylation and further study in mice.
The optimal candidate, antibody 1519, was humanized by grafting the CDRs from the parent rat antibody V-regions onto human germline antibody V-region frameworks. The CDRs grafted from the donor to the acceptor sequence were as defined by Kabat,64,65 with the exception of CDR-H1 where the combined Chothia/Kabat definition was used.32 Human V-region IGKV1-17 plus IGKJ2 J-region (IMGT, http://www.imgt.org/) was chosen as the acceptor for the light chain CDRs. Human V-region IGHV3-7 plus IGHJ4 J-region (IMGT, http://www.imgt.org/) was chosen as the acceptor for the heavy chain CDRs. The ultimate V-region pairing was named 1519.g57. Various formats of this antibody were generated as indicated in Supplementary Table 3.
Preparation of protein reagents
Human FcRn ECD (sequence AESH… to …AKSS; UniProt P55899)66 and the equivalent cynomolgus monkey FcRn ECD were each expressed in HEK-293 cells co-transfected with the respective α-chain and homologous β2m. The FcRn ECD was purified by affinity chromatography on IgG (CnBr-activated Sepharose 4 Fast Flow; GE Healthcare, UK), binding at pH 5.8 and eluting at pH 8.67,68 Rabbit, mouse and rat FcRn ECD were expressed, each with the homologous β2m, in proprietary Chinese Hamster Ovary (CHO) SXE69 cells and protein purified.
Humanized Fab’ was expressed in, and prepared from E. coli,46,70 and further modified as detailed in Supplementary Table 3. IgG molecules were prepared from CHO SXE69 cell culture medium by affinity chromatography on MabSelect™ SuRe™ (GE Healthcare, UK), followed by size-exclusion chromatography (SEC) on HiLoad XK 50/60 Superdex 200 PG (GE Healthcare, UK). The purity of the final product was confirmed by reducing and non-reducing SDS-PAGE, and by analytical scale SEC. Reagents were sterilized by filtration through a 0.22 µm filter and stored at 2 to 8°C, in the dark.
Biochemical and biophysical analysis
Candidates in Fab’6HIS format were analyzed to identify those with greater stability and suitability to manufacturing and formulation; detailed characterization was undertaken for rozanolixizumab (1519.g57 IgG4P; Supplementary Table 1; Supplementary Methods).
Crystallography was undertaken with deglycosylated human FcRn ECD complexed with 1519.g57 Fab’ (Supplementary Methods).
Characterization of binding affinity to human FcRn by SPR
Binding affinity measurements were conducted using SPR technology on a Biacore T200 system (GE Healthcare, UK). For screening of B-cell supernatants, IgG was captured out of the culture supernatant using an immobilized goat anti-rat IgG, Fc gamma (AP138; Chemicon International Inc.) antibody; human FcRn ECD (100 nM) was allowed to flow over the chip surface. Human IgG (009-000-003; Jackson ImmunoResearch Lab Inc, UK) was then passed over the surface to bind to captured FcRn. Failure of IgG to bind FcRn indicated blockade of FcRn by the anti-FcRn antibody.
For analysis of humanized anti-FcRn candidates binding to FcRn an Affinipure F(ab’)2 goat anti-human IgG Fc fragment-specific (109-006-098; Jackson ImmunoResearch Lab Inc., UK) antibody fragment was immobilized to the sensor chip and anti-FcRn antibody captured. Human FcRn ECD was titrated from 20 nM to 1.25 nM over the captured anti-FcRn. For analysis of binding of anti-human FcRn antibodies to rat, mouse or rabbit FcRn, the maximum concentration of FcRn was greater (200 nM). Estimates of KD were the means of 5 replicates. Where dissociation was very slow and kd values could not be accurately quantified, a value of 1 × 10–5 s–1 was assigned. Further details of SPR methods are provided in Supplementary Methods.
Characterization of binding affinity to FcRn on cell surfaces
MDCK cells were transfected with human FcRn α-chain and human β2m and a stable line was generated. Similarly, an MDCK stable cell line expressing cynomolgus FcRn α-chain with cynomolgus β2m was also generated. Cells were incubated in flow cytometry (FACS) buffer (phosphate-buffered saline (PBS) with 0.2% (w/v) bovine serum albumin (BSA), with 0.09% (w/v) sodium azide) for 30 mins prior to addition of AF 488-conjugated anti-FcRn antibody for 1 hour in FACS buffer (at either pH 7.4 or pH 5.9). The final antibody concentrations ranged from 400 nM to 0.003 nM. The cells were then washed in ice cold FACS buffer and analyzed by flow cytometry using a Guava flow cytometer (Millipore). The number of moles of bound antibody was calculated using interpolated values from a standard curve generated from beads comprised of differing amounts of fluorescent dye (Quantum FITC-5 Molecules of equivalent soluble fluorochrome beads, AbD Serotec FCSC555). Geometric mean fluorescence values were determined in the flow cytometric analyzes of cells and beads. Titration data sets were also produced for isotype-matched control (non-FcRn-specific) antibodies for each antibody format, in order to determine non-specific binding characteristics. Values of non-specific binding (of control antibody) were subtracted from the anti-FcRn antibody values and the generated specific binding curve was analyzed by non-linear regression using a one-site binding equation (Graphpad Prism®) to determine the KD. Three replicates experiments were conducted. KD values could be determined when binding of 1519.g57 Fab’PEG and rozanolixizumab was assessed in the context of a cell membrane; however, individual ka and kd values could not.
IgG recycling in cells
MDCK cells over-expressing human or cynomolgus monkey FcRn and β2m were plated in a 96-well plate and incubated overnight at 37°C, 5% CO2, to allow cells to adhere. The cells were washed with Hanks’ balanced salt solution with calcium and magnesium (HBSS + Ca2+/Mg2+) pH 7.2 + 1% BSA and incubated with a range of concentrations of anti-FcRn molecules for 1 hour. The supernatant was removed and 500 ng/ml of biotinylated human IgG (009-060-003; Jackson ImmunoResearch Lab Inc., UK) added to the cells in HBSS + Ca2+/Mg2+ pH 5.9 + 1% BSA (v/v) for 1 hour at 37°C, 5% CO2. After a pulse time of 60 minutes, the cells were washed and incubated in HBSS + Ca2+/Mg2+ pH 7.2 at 37°C, 5% CO2 for 2 hours. IgG released into the supernatant was measured using a meso scale discovery (MSD) assay with an anti-human IgG capture antibody (109-005-003; Jackson ImmunoResearch Lab Inc., UK) and a streptavidin-sulpho tag reveal antibody (MSD). The inhibition of IgG recycling was analyzed by non-linear regression to determine the IC50.
Intracellular IgG trafficking
HUVECs were maintained in M-200 media containing Large Vessel Endothelial Supplement (Life Technologies, USA) at 37°C in a humidified incubator and used until Passage 8. Cells were then incubated with 2.5 µg/mL AF 647-conjugated IgG for 24 hours in 96-well imaging plates (Corning, USA) in the presence of indicated concentrations of either rozanolixizumab or a control IgG4 Ab. Nuclei were labelled by the addition of Hoechst (Nucblue Dye; Life Technologies, USA) in media for 30 minutes. Cells were then washed twice in Live Cell Imaging Solution (Life Technologies, USA) and left in Live Cell Imaging Solution. Images were acquired using a Thermo Arrayscan with a 20x objective and appropriate excitation and emission parameters. Triplicate wells were used for each condition with at least 9 fields per well acquired. The spot detector algorithm was used to analyze AF 647-conjugated IgG vesicles and the Total Spot Area per Object parameter was obtained from the Cellomics Scan software (Life Technologies, USA). Replicate wells were then averaged and the data presented are the mean from three independent experiments.
For Lysotracker labelling, cells prepared as above were incubated with 100 nM Lysotracker Green (Life Technologies, USA) for 10 minutes at 37°C. Cells were then washed twice with Live Cell Imaging Solution and images acquired using a Molecular Devices ImageXpress with a 20x objective and appropriate excitation and emission parameters.
Assessment of lysosomes
Human serum was prepared from donors by allowing blood to clot in red-topped Vacutainers (BD Biosciences, UK) followed by centrifugation. HUVECs were grown in 50% human serum and 50% normal growth medium as described in the previous section. For LAMP-1 staining, cells were fixed using 4% paraformaldehyde (methanol-free; Life Technologies, USA), washed twice in PBS, and then permeabilized with −20°C methanol. Cells were then washed in PBS and blocked with PBS containing 5% goat serum for 30 minutes. Cells were incubated with anti-LAMP-1 (9091S; Cell Signaling Technology, USA) followed by goat AF 488-conjugated anti-rabbit IgG secondary antibody (A-11034, Life Technologies, USA) and DAPI (D1306; Life Technologies, USA). Cells were then left in PBS before imaging on a Thermo Arrayscan. Images were acquired as described previously, except the ‘Spot Average Area’ parameter of the Spot Detector Analysis, which was used to quantify lysosome area. Lysotracker labelling was carried out by incubating cells first with Hoechst and 100 nM Lysotracker Red DND99 (Life Technologies, USA) for 90 minutes at 37°C. Cells were then washed 3 times in Live Cell Imaging solution and images acquired on a Thermo Arrayscan and the ‘Spot Area per Object’ used to quantify Lysotracker staining. Data from independent experiments were normalized to control wells (with no drug addition) and then pooled.
Assessment of cytokine release in vitro
The potential of rozanolixizumab to affect cytokine production was investigated using four cell-based assay formats (peripheral blood mononuclear cells (PBMC); HUVEC confirmed to express FcRn; whole blood combined with HUVEC and PBMC combined with HUVEC). These cell types were used in combination with various rozanolixizumab delivery formats (soluble rozanolixizumab and rozanolixizumab cross-linked with an anti-human Fc F(ab’)2 [simulation of ADA-mediated additional cross-linking of the target]). See supplementary materials for detailed methodology.
Transgenic mouse model
Male and female muFcRn−/− huFcRn Tg32 transgenic mice (weighing 18–40g; B6.Cg-Fcgrttm1Dcr Tg(FCGRT)32Dcr; Jackson ImmunoResearch Lab Inc., UK [stock number 004919]) were used to study the effect of FcRn blockade on human IgG concentration, anti-FcRn antibody PK, and mouse albumin concentration in the plasma. All experiments were conducted in accordance with the UK Home Office Animals (Scientific Procedures) Act 1986, with local ethical approval and recently published guidelines.71
Effects of rozanolixizumab in a human FcRn-transgenic mouse model
Male and female Tg32 transgenic mice were treated with rozanolixizumab in PBS (variable volume according to dose size) at 10, 30, 100 and 250 mg/kg IV (n = 4–32 per group), human IgG4 mutated to abolish FcRn binding (200 mg/kg; n = 4) or with various 1519.g57 formats (100 mg/kg; n = 4–34 per group). Twenty-four hours prior to administration of rozanolixizumab, each mouse was injected IV with 500 mg/kg of a 10% w/v solution of polyclonal human IgG (726820; Gamunex®-C, Grifols, US) to give a plasma concentration of ~ 10 mg/mL. Serial tail bleeds were taken at regular intervals throughout the experiment and used to quantify the plasma concentration of human IgG, rozanolixizumab and mouse albumin by liquid chromatography tandem mass spectrometry (LC-MS/MS) following tryptic digestion (details of this method are shown in the supplementary information).
Cynomolgus monkey studies
Safety and PK/PD studies were conducted in cynomolgus monkeys. Studies were conducted by Covance Laboratories Ltd, UK, in accordance with the requirements of the United Kingdom Home Office Animals (Scientific Procedures) Act 1986, with local ethical approval and recently published guidelines.71 All procedures in the study were also in compliance with the German Animal Welfare Act (1972), were approved by the local Institutional Animal Care and Use Committee, and in consideration of European Commission Recommendation 2007/526/EC on guidelines for the accommodation and care of animals used for experimental and other scientific purposes (Appendix A of Convention ETS 123).
Tissue cross-reactivity study
Binding of rozanolixizumab to fresh frozen tissue samples from three unrelated human donors and three cynomolgus monkeys was assessed by immunohistochemistry. Rozanolixizumab was conjugated in house (without detectable loss of FcRn binding activity) with fluorescein isothiocyanate (FITC) at a molar labeling ratio of FITC to rozanolixizumab of 0.83:1; three concentrations of labelled rozanolixizumab were tested (0.2, 0.1 and 0.05 μg/mL). Binding was detected immunohistochemically using a biotinylated anti-FITC secondary Ab, followed by incubation with horseradish peroxidase-coupled streptavidin and a chromogene substrate (Covance, UK). Placental tissue was used as a positive control for FcRn-specific staining. A fully human IgG4 isotype control-FITC-conjugated antibody (ET904-FITC, Eureka Therapeutics) was used as an isotype control (molar labeling ratio FITC to ET904: 2.5), since it binds a hapten not existent in human cells.
Pharmacodynamics and PK analysis in cynomolgus monkeys
A PK/PD study was conducted to characterize the in vivo pharmacologic activity of rozanolixizumab, and to facilitate the translation of non-clinical pharmacology to expected clinical effect. A range of doses were tested to explore the dose-exposure-IgG response curve and to characterize PK. Each dosing group comprised four healthy, young adult males weighing a minimum of 2.3 kg; animals were ~ 135 to 185 weeks old at initiation of dosing. For single-dose studies, a 5, 10 or 30 mg/kg dose was administered IV on Day 0; for infrequent dosing a 30 mg/kg dose was administered IV on Days 0, 63 and 126; for frequent dosing, animals received either daily SC doses of 5 mg/kg (for 60 days), or a 30 mg/kg IV loading dose followed by daily IV doses of 5 mg/kg (for 41 days). Control animals received daily IV doses of vehicle (PBS). The concentration of samples for injection was adjusted with PBS to allow a dose volume of 2 mL/kg, based on individual body weight. Further details of the 1519.g57 Fab’PEG PK study are provided in the supplementary materials.
General health and body weight were monitored, and histopathology conducted at the end of the study. Blood samples were taken at regular intervals for analysis of total plasma IgG, albumin, and rozanolixizumab concentrations. Blood samples for IgG and rozanolixizumab analysis were collected from the cephalic/saphenous/femoral vein into tubes containing lithium heparin anticoagulant, gently mixed and kept on ice prior to centrifugation (10 min, 1500 g, 4°C). Plasma was immediately separated and stored at – 20°C. Derived PK variables (Cmax, AUC0–t, tmax [time to occurrence of Cmax, obtained directly from the observed plasma concentration–time curves], Cmax/Dose, AUC0–t/Dose) were calculated by non-compartmental analysis methods from individual plasma concentration vs actual time profiles after single dose administration using Phoenix® WinNonlin® v6.4. PK variables were calculated using actual blood sampling time points.
IgG bioanalysis
To quantify total IgG in monkey plasma, samples (10 µL) were mixed with an internal standard (isotopically labelled unique IgG peptide; Eurogentec), denatured and trypsin digested. After digestion, the samples were diluted and the resulting signature peptides (Analyte: H2N – TTPPVLDSDGSYFLYSK – COOH, internal standard: H2N – TTPP(U-13C5, 15N)VLDSDGSYFLYSK – COOH) were analyzed by reversed phase ultra-high-pressure liquid chromatography coupled with electrospray MS/MS detection (details in supplementary information). The assay had a lower limit of quantification (LLOQ) of 0.102 mg/mL and an upper limit of quantification of 15.345 mg/mL. The assay did not distinguish between endogenous IgG and administered rozanolixizumab, hence a transient peak of total IgG was observed shortly after dosing with rozanolixizumab.
Individual IgG-time profiles were plotted for absolute IgG and used to calculate the percentage change from baseline IgG. For baseline values, the mean of all IgG samples collected previous to the first dose administration was used. The arithmetic mean of the percentage change from baseline IgG at each time point was then calculated and plotted, stratified by dose regimen.
Albumin analysis
Albumin concentrations were determined by SPIFE electrophoresis as part of a standard clinical chemistry panel by Covance, UK.
Rozanolixizumab bioanalysis
A validated MSD assay72 with an LLOQ of 0.31 μg/mL was used to determine concentrations of rozanolixizumab in cynomolgus monkey plasma. Briefly, rozanolixizumab was captured by biotinylated FcRn immobilized on a streptavidin-coated MSD plate and revealed using commercially available goat anti-human kappa antibody (NB7463; Novus Biologicals) that had been conjugated to sulpho-TAG in-house (qualified assay developed by UCB). The assay diluent was PBS/1% BSA/1% bovine gamma globulin (BGG)/1% block B at neutral pH (1/100 minimum required dilution, MRD). The calibrator was prepared using pooled 1% cynomolgus monkey plasma (to reflect the MRD); the inclusion of a zero calibrator ensured no endogenous IgG bound to immobilized FcRn. The electrochemiluminescence signal was proportional to the amount of rozanolixizumab in the sample.
Rozanolixizumab ADA detection
A semiquantitative homogeneous qualified MSD assay72 (LLOQ 0.14 units/mL) with a pooled ADA calibrator harvested from cynomolgus monkeys who were dosed with 1519.g57 Fab’ was used to detect anti-rozanolixizumab antibody. This assay is similar in format to the mouse ADA assay with the exception of the reagents. For ADA bridging, rozanolixizumab conjugated to biotin and rozanolixizumab conjugated to sulpho-TAG were used. The positive control used in the calibrator and controls was purified from the plasma of a cynomolgus monkey previously immunized with a molecule of the same idiotype. Data were read from the calibrator curve to give a semi-quantitative measure of units/mL, as the pooled ADA calibrator was not identical to the ADA in the sample.
13-week toxicology study
Male and female cynomolgus monkeys were injected with rozanolixizumab 50 mg/kg or 150 mg/kg SC every 3 days for 13 weeks, or 150 mg/kg IV every 3 days for 13 weeks, or 150 mg/kg SC every 3 days in Weeks 1, 6 and 10 (Table 3), followed by an 8-week treatment-free recovery period. This study was conducted as detailed for the 4-week toxicology study.6 Additional investigations undertaken in the 13-week toxicology study included immune function assessment (TDAR to KLH immunization), and measurement of serum cytokines and complement activation markers. To assess the effect of rozanolixizumab on the TDAR to KLH, cynomolgus monkeys were immunized SC with 1 mg of KLH (1 mL in saline) on Day 41 and Day 69. A validated MSD assay was used to detect specific anti-KLH IgG and IgM. Complement activation markers (C3, C3a, C4 and C5a) were quantified in cynomolgus monkey plasma using Konelab Prime 60 or Tecan Sunrise (Thermo Scientific, US); cytokines (IFN-γ, TNF-α, lymphotoxin-alpha (also known as TNF-β), IL-1β, IL-2, IL-6, IL-8 and IL-10) were quantified in cynomolgus monkey serum using a validated flow cytometric assay (FACScalibur, Becton Dickinson).
Biography
BS, AK, RLG, KLD, MCC, PA, LED’H, HF, KG, HH, LK, DL, CM, RM, OQ, KS, SPS, RT, AT, KT, SW, SS, LC and FRB contributed to study conception or design; AK, RLG, KLD, MCC, PA, LED’H, HF, KG, HH, LK, DL, CM, RM, OQ, KS, SPS, RT, AT, KT, SW, LC and FRB contributed to the acquisition of data; BS, AK, RLG, KLD, MCC, PA, LED’H, HF, KG, HH, LK, DL, CM, RM, OQ, KS, SPS, RT, AT, KT, SW, LC and FRB were responsible for data analysis and interpretation. All authors critically reviewed the manuscript and approved the final version for submission.
Funding Statement
The study was funded by UCB Pharma.
Acknowledgments
The authors would like to acknowledge Helen Brand, Katharine Cain, Richard Davies, Jonathan Symmons and Bernadette Sweeney of UCB Pharma for their input into construct design, expression and purification of reagents; Remi Okoye of UCB Pharma for her input to the in vivo characterizations; Julia Steel of UCB Pharma for her input into the in vitro SPR characterizations; Andy Ventom, Mark Jairaj and Ludovicus Staelens of UCB Pharma for their input to the LC-MS/MS; Sarah Hamilton of UCB Pharma for her contribution to the aspartyl isomerization work; Tom Ceska of UCB Pharma for his contributions to the X-ray crystallography work; Gillian McCluskey and Rona Cutler of UCB for their input to the cytokine release work; Alastair Lawson of UCB Pharma for his invaluable guidance during the initial stages of the project, Andrew Popplewell, Alison Eddleston and Anthony Shock of UCB Pharma for their guidance during development of the manuscript and other UCB staff who have contributed throughout the project. The authors would like to acknowledge C. Marc Luetjens at Covance Preclinical Services GmbH, Muenster, Germany for conducting and reporting the 13-week toxicity study in cynomolgus monkey. Finally, the authors would also like to acknowledge Laura Griffin, PhD, of iMed Comms, Macclesfield, UK, an Ashfield Company, part of UDG Healthcare plc for medical writing support that was funded by UCB Pharma in accordance with Good Publications Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Disclosure of Potential Conflicts of interest
BS, AK, RLG, KLD, MCC, PA, LED’H, HF, KG, HH, LK, DL, CM, RM, OQ, KS, LC, SPS, RT, AT, KT, SW, SS and FRB are/were all employees of UCB Pharma and may hold stock and/or stock options.
Supplementary material
Supplemental data for this article can be accessed here.
Abbreviations
- ADA
anti-drug antibody
- AUC
area under curve
- β2m
β2-microglobulin
- CDR
complementarity-determining region
- CHO
Chinese hamster ovary
- Cmax
maximum plasma concentration
- E.coli
Escherichia coli
- ECD
extracellular domain
- FACS
fluorescence-activated cell sorting
- FcRn
neonatal Fc receptor
- HDX
hydrogen-deuterium exchange
- HEK cells
human embryonic kidney cells
- HUVEC
human umbilical vein endothelial cell
- IgG
immunoglobulin G
- IV
intravenous
- IVIg
intravenous immunoglobulin
- ITAM
immunoreceptor tyrosine-based activation motif
- ITIM
immunoreceptor tyrosine-based inhibition motif
- ITP
immune thrombocytopenia
- KD
dissociation constant
- ka
association rate constant
- kd
dissociation rate constant
- KLH
keyhole limpet hemocyanin
- LAMP-1
lysosomal-associated membrane protein 1
- LLOQ
lower limit of quantification
- MDCK
Madin-Darby canine kidney
- mAb
monoclonal antibody
- MRD
minimum required dilution
- MSD
mesoscale diagnostics
- PCR
polymerase chain reaction
- PD
pharmacodynamics
- PEG
polyethylene glycol
- pI
isoelectric point
- PK
pharmacokinetics
- RSE
relative standard error
- RU
response unit
- SC
subcutaneous
- SPR
surface plasmon resonance
- TDAR
T-cell-dependent IgG antibody response
- tmax
time to occurrence of Cmax
- TMDD
target-mediated drug distribution.
References
- 1.Schwartz J, Winters JL, Padmanabhan A, Balogun RA, Delaney M, Linenberger ML, Szczepiorkowski ZM, Williams ME, Wu Y, Shaz BH.. Guidelines on the use of therapeutic apheresis in clinical practice-evidence-based approach from the Writing Committee of the American Society for Apheresis: the sixth special issue. J Clin Apher. 2013;28:145–284. doi: 10.1002/jca.21276. [DOI] [PubMed] [Google Scholar]
- 2.Wang J, McQuilten ZK, Wood EM, Aubron C.. Intravenous immunoglobulin in critically ill adults: when and what is the evidence? J Crit Care. 2015;30:652.e9–16. doi: 10.1016/j.jcrc.2015.01.022. [DOI] [PubMed] [Google Scholar]
- 3.Matucci A, Maggi E, Vultaggio A. Mechanisms of action of Ig preparations: immunomodulatory and anti-inflammatory effects. Front Immunol. 2014;5:690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar R, Birinder SP, Gupta S, Singh G, Kaur A. Therapeutic plasma exchange in the treatment of myasthenia gravis. Indian J Crit Care Med. 2015;19:9–13. doi: 10.4103/0972-5229.148631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vaccaro C, Zhou J, Ober RJ, Ward ES. Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat Biotechnol. 2005;23:1283–1288. doi: 10.1038/nbt1143. [DOI] [PubMed] [Google Scholar]
- 6.Kiessling P, Lledo-Garcia R, Watanabe S, Langdon G, Tran D, Bari M, Christodoulou L, Jones E, Price G, Smith B, et al. The FcRn inhibitor rozanolixizumab reduces human serum IgG concentration: A randomized phase 1 study. Sci Transl Med. 2017;9:eaan1208. doi: 10.1126/scitranslmed.aan1208. [DOI] [PubMed] [Google Scholar]
- 7.Challa DK, Velmurugan R, Ober RJ, Sally Ward E. FcRn: from molecular interactions to regulation of IgG pharmacokinetics and functions. Curr Top Microbiol Immunol. 2014;382:249–272. doi: 10.1007/978-3-319-07911-0_12. [DOI] [PubMed] [Google Scholar]
- 8.Martins JP, Kennedy PJ, Santos HA, Barrias C, Sarmento B. A comprehensive review of the neonatal Fc receptor and its application in drug delivery. Pharmacol Ther. 2016;161:22–39. doi: 10.1016/j.pharmthera.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 9.Pyzik M, Rath T, Lencer WI, Baker K, Blumberg RS. FcRn: the architect behind the immune and nonimmune functions of IgG and albumin. J Immunol. 2015;194:4595–4603. doi: 10.4049/jimmunol.1403014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–725. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 11.Akilesh S, Petkova S, Sproule TJ, Shaffer DJ, Christianson GJ, Roopenian D. The MHC class I-like Fc receptor promotes humorally mediated autoimmune disease. J Clin Invest. 2004;113:1328–1333. doi: 10.1172/JCI18838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li N, Zhao M, Hilario-Vargas J, Prisayanh P, Warren S, Diaz LA, Roopenian DC, Liu Z. Complete FcRn dependence for intravenous Ig therapy in autoimmune skin blistering diseases. J Clin Invest. 2005;115:3440–3450. doi: 10.1172/JCI24394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Z, Roopenian DC, Zhou X, Christianson GJ, Diaz LA, Sedmak DD, Anderson CL. Beta2-microglobulin-deficient mice are resistant to bullous pemphigoid. J Exp Med. 1997;186:777–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roopenian DC, Christianson GJ, Sproule TJ, Brown AC, Akilesh S, Jung N, Petkova S, Avanessian L, Choi EY, Shaffer DJ, et al. The MHC class I-like IgG receptor controls perinatal IgG transport, IgG homeostasis, and fate of IgG-Fc-coupled drugs. J Immunol. 2003;170:3528–3533. [DOI] [PubMed] [Google Scholar]
- 15.Ardeniz O, Onay H, Gerceker B, Cianga P, Ikinciotullari A, Mete Gokmen N, Guloglu D, Fuchs I, Schlesier M, Ehl S, et al. Major histocompatibility complex class I deficiency-but not the sole abnormality caused by a different genetic defect. Eur J Allergy Clin Immunol. 2013;68:661–662. [Google Scholar]
- 16.Wani MA, Haynes LD, Kim J, Bronson CL, Chaudhury C, Mohanty S, Waldmann TA, Robinson JM, Anderson CL. Familial hypercatabolic hypoproteinemia caused by deficiency of the neonatal Fc receptor, FcRn, due to a mutant beta2-microglobulin gene. Proc Natl Acad Sci U S A. 2006;103:5084–5089. doi: 10.1073/pnas.0600548103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warren DS, Simpkins CE, Cooper M, Montgomery RA. Modulating alloimmune responses with plasmapheresis and IVIG. Curr Drug Targets Cardiovasc Haematol Disord. 2005;5:215–222. [DOI] [PubMed] [Google Scholar]
- 18.Christianson GJ, Sun VZ, Akilesh S, Pesavento E, Proetzel G, Roopenian DC. Monoclonal antibodies directed against human FcRn and their applications. MAbs. 2012;4:208–216. doi: 10.4161/mabs.4.2.19397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mezo AR, McDonnell KA, Hehir CA, Low SC, Palombella VJ, Stattel JM, Kamphaus GD, Fraley C, Zhang Y, Dumont JA, et al. Reduction of IgG in nonhuman primates by a peptide antagonist of the neonatal Fc receptor FcRn. Proc Natl Acad Sci U S A. 2008;105:2337–2342. doi: 10.1073/pnas.0708960105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nixon AE, Chen J, Sexton DJ, Muruganandam A, Bitonti AJ, Dumont J, Viswanathan M, Martik D, Wassaf D, Mezo A, et al. Fully human monoclonal antibody inhibitors of the neonatal fc receptor reduce circulating IgG in non-human primates. Front Immunol. 2015;6:176. doi: 10.3389/fimmu.2015.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Getman KE, Balthasar JP. Pharmacokinetic effects of 4C9, an anti-FcRn antibody, in rats: implications for the use of FcRn inhibitors for the treatment of humoral autoimmune and alloimmune conditions. J Pharm Sci. 2005;94:718–729. doi: 10.1002/jps.20297. [DOI] [PubMed] [Google Scholar]
- 22.Liu L, Garcia AM, Santoro H, Zhang Y, McDonnell K, Dumont J, Bitonti A. Amelioration of experimental autoimmune myasthenia gravis in rats by neonatal FcR blockade. J Immunol. 2007;178:5390–5398. [DOI] [PubMed] [Google Scholar]
- 23.Patel DA, Puig-Canto A, Challa DK, Perez Montoyo H, Ober RJ, Ward ES. Neonatal Fc receptor blockade by Fc engineering ameliorates arthritis in a murine model. J Immunol. 2011;187:1015–1022. doi: 10.4049/jimmunol.1003780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petkova SB, Akilesh S, Sproule TJ, Christianson GJ, Al Khabbaz H, Brown AC, Presta LG, Meng YG, Roopenian DC. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol. 2006;18:1759–1769. doi: 10.1093/intimm/dxl110. [DOI] [PubMed] [Google Scholar]
- 25.Raghavan M, Chen MY, Gastinel LN, Bjorkman PJ. Investigation of the interaction between the class I MHC-related Fc receptor and its immunoglobulin G ligand. Immunity. 1994;1:303–315. [DOI] [PubMed] [Google Scholar]
- 26.Dall’Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem. 2006;281:23514–23524. doi: 10.1074/jbc.M604292200. [DOI] [PubMed] [Google Scholar]
- 27.Neuber T, Frese K, Jaehrling J, Jager S, Daubert D, Felderer K, Linnemann M, Hohne A, Kaden S, Kolln J, et al. Characterization and screening of IgG binding to the neonatal Fc receptor. MAbs. 2014;6:928–942. doi: 10.4161/mabs.28744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warncke M, Calzascia T, Coulot M, Balke N, Touil R, Kolbinger F, Heusser C. Different adaptations of IgG effector function in human and nonhuman primates and implications for therapeutic antibody treatment. J Immunol. 2012;188:4405–4411. doi: 10.4049/jimmunol.1200090. [DOI] [PubMed] [Google Scholar]
- 29.Oganesyan V, Damschroder MM, Cook KE, Li Q, Gao C, Wu H, Dall’Acqua WF. Structural insights into neonatal Fc receptor-based recycling mechanisms. J Biol Chem. 2014;289:7812–7824. doi: 10.1074/jbc.M113.537563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmidt MM, Townson SA, Andreucci AJ, King BM, Schirmer EB, Murillo AJ, Dombrowski C, Tisdale AW, Lowden PA, Masci AL, et al. Crystal structure of an HSA/FcRn complex reveals recycling by competitive mimicry of HSA ligands at a pH-dependent hydrophobic interface. Structure. 1993;2013(21):1966–1978. [DOI] [PubMed] [Google Scholar]
- 31.Angal S, King DJ, Bodmer MW, Turner A, Lawson AD, Roberts G, Pedley B, Adair JR. A single amino acid substitution abolishes the heterogeneity of chimeric mouse/human (IgG4) antibody. Mol Immunol. 1993;30:105–108. [DOI] [PubMed] [Google Scholar]
- 32.Adair JRA, Athwal DS, Emtage JS. Humanised antibodies. WO91/09967 [accessed 2018. Aug]. https://patents.google.com/patent/WO1991009967A1.
- 33.Jones TD, Carter PJ, Pluckthun A, Vasquez M, Holgate RG, Hotzel I, Popplewell AG, Parren PW, Enzelberger M, Rademaker HJ, et al. The INNs and outs of antibody nonproprietary names. MAbs. 2016;8:1–9. doi: 10.1080/19420862.2015.1114320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parren P, Carter PJ, Pluckthun A. Changes to International Nonproprietary Names for antibody therapeutics 2017 and beyond: of mice, men and more. MAbs. 2017;9:898–906. doi: 10.1080/19420862.2017.1341029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Firan M, Bawdon R, Radu C, Ober RJ, Eaken D, Antohe F, Ghetie V, Ward ES. The MHC class I-related receptor, FcRn, plays an essential role in the maternofetal transfer of gamma-globulin in humans. Int Immunol. 2001;13:993–1002. [DOI] [PubMed] [Google Scholar]
- 36.Dall’Acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, Wu H, Kiener PA, Langermann S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J Immunol. 2002;169:5171–5180. [DOI] [PubMed] [Google Scholar]
- 37.Zhou J, Mateos F, Ober RJ, Ward ES. Conferring the binding properties of the mouse MHC class I-related receptor, FcRn, onto the human ortholog by sequential rounds of site-directed mutagenesis. J Mol Biol. 2005;345:1071–1081. doi: 10.1016/j.jmb.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 38.Giurgea-Marion L, Toubeau G, Laurent G, Heuson-Stiennon JA, Tulkens PM. Impairment of lysosome-pinocytic vesicle fusion in rat kidney proximal tubules after treatment with gentamicin at low doses. Toxicol Appl Pharmacol. 1986;86:271–285. [DOI] [PubMed] [Google Scholar]
- 39.Gonzalez-Noriega A, Grubb JH, Talkad V, Sly WS. Chloroquine inhibits lysosomal enzyme pinocytosis and enhances lysosomal enzyme secretion by impairing receptor recycling. J Cell Biol. 1980;85:839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.BioGPS Site FCGRT (Fc fragment of IgG receptor and transporter). [accessed 2018. Aug]. http://biogps.org/#goto=genereport&id=2217. [Google Scholar]
- 41.Borvak J, Richardson J, Medesan C, Antohe F, Radu C, Simionescu M, Ghetie V, Ward ES. Functional expression of the MHC class I-related receptor, FcRn, in endothelial cells of mice. Int Immunol. 1998;10:1289–1298. [DOI] [PubMed] [Google Scholar]
- 42.Akilesh S, Christianson GJ, Roopenian DC, Shaw AS. Neonatal FcR expression in bone marrow-derived cells functions to protect serum IgG from catabolism. J Immunol. 2007;179:4580–4588. [DOI] [PubMed] [Google Scholar]
- 43.Uno Y, Utoh M, Iwasaki K. Polymorphisms of neonatal Fc receptor in cynomolgus and rhesus macaques. Drug Metab Pharmacokinet. 2014;29:427–430. [DOI] [PubMed] [Google Scholar]
- 44.Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 2014;13:397–406. doi: 10.1074/mcp.M113.035600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Junghans RP, Anderson CL. The protection receptor for IgG catabolism is the beta2-microglobulin-containing neonatal intestinal transport receptor. Proc Natl Acad Sci U S A. 1996;93:5512–5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chapman AP, Antoniw P, Spitali M, West S, Stephens S. Therapeutic antibody fragments with prolonged in vivo half-lives. Nat Biotechnol. 1999;17:780–783. doi: 10.1038/11717. [DOI] [PubMed] [Google Scholar]
- 47.Andersen JT, Daba MB, Berntzen G, Michaelsen TE, Sandlie I. Cross-species binding analyses of mouse and human neonatal Fc receptor show dramatic differences in immunoglobulin G and albumin binding. J Biol Chem. 2010;285:4826–4836. doi: 10.1074/jbc.M109.081828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Datta-Mannan A, Witcher DR, Tang Y, Watkins J, Wroblewski VJ. Monoclonal antibody clearance. Impact of modulating the interaction of IgG with the neonatal Fc receptor. J Biol Chem. 2007;282:1709–1717. doi: 10.1074/jbc.M607161200. [DOI] [PubMed] [Google Scholar]
- 49.Ward ES, Velmurugan R, Ober RJ. Targeting FcRn for therapy: from live cell imaging to in vivo studies in mice. Immunol Lett. 2014;160:158–162. doi: 10.1016/j.imlet.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haraya K, Tachibana T, Nanami M, Ishigai M. Application of human FcRn transgenic mice as a pharmacokinetic screening tool of monoclonal antibody. Xenobiotica. 2014;44:1127–1134. doi: 10.3109/00498254.2014.941963. [DOI] [PubMed] [Google Scholar]
- 51.Waldmann TA, Terry WD. Familial hypercatabolic hypoproteinemia. A disorder of endogenous catabolism of albumin and immunoglobulin. J Clin Invest. 1990;86:2093–2098. doi: 10.1172/JCI114947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kaplan AA. Therapeutic plasma exchange: a technical and operational review. J Clin Apher. 2013;28:3–10. doi: 10.1002/jca.21257. [DOI] [PubMed] [Google Scholar]
- 53.Ohkubo A, Okado T, Kurashima N, Maeda T, Arai S, Miyamoto S, Itagaki A, Seshima H, Iimori S, Naito S, et al. Removal characteristics of immunoglobulin G subclasses by conventional plasma exchange and selective plasma exchange. Ther Apher Dial. 2015;19:361–366. doi: 10.1111/1744-9987.12346. [DOI] [PubMed] [Google Scholar]
- 54.Besada E. Low immunoglobulin levels increase the risk of severe hypogammaglobulinemia in granulomatosis with polyangiitis patients receiving rituximab. BMC Musculoskelet Disord. 2016;17:6. doi: 10.1186/s12891-016-1134-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anderson CL, Chaudhury C, Kim J, Bronson CL, Wani MA, Mohanty S. Perspective– FcRn transports albumin: relevance to immunology and medicine. Trends Immunol. 2006;27:343–348. doi: 10.1016/j.it.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 56.Krishna M, Nadler SG. Immunogenicity to biotherapeutics - the role of anti-drug immune complexes. Front Immunol. 2016;7:21. doi: 10.3389/fimmu.2016.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lizee G, Basha G, Tiong J, Julien JP, Tian M, Biron KE, Jefferies WA. Control of dendritic cell cross-presentation by the major histocompatibility complex class I cytoplasmic domain. Nat Immunol. 2003;4:1065–1073. doi: 10.1038/ni989. [DOI] [PubMed] [Google Scholar]
- 58.Wu Z, Simister NE. Tryptophan- and dileucine-based endocytosis signals in the neonatal Fc receptor. J Biol Chem. 2001;276:5240–5247. doi: 10.1074/jbc.M006684200. [DOI] [PubMed] [Google Scholar]
- 59.Getahun A, Cambier JC. Of ITIMs, ITAMs, and ITAMis: revisiting immunoglobulin Fc receptor signaling. Immunol Rev. 2015;268:66–73. doi: 10.1111/imr.12336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baker K, Rath T, Flak MB, Arthur JC, Chen Z, Glickman JN, Zlobec I, Karamitopoulou E, Stachler MD, Odze RD, et al. Neonatal Fc receptor expression in dendritic cells mediates protective immunity against colorectal cancer. Immunity. 2013;39:1095–1107. doi: 10.1016/j.immuni.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ober RJ, Radu CG, Ghetie V, Ward ES. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int Immunol. 2001;13:1551–1559. [DOI] [PubMed] [Google Scholar]
- 62.Zubler RH, Erard F, Lees RK, Van Laer M, Mingari C, Moretta L, MacDonald HR. Mutant EL-4 thymoma cells polyclonally activate murine and human B cells via direct cell interaction. J Immunol. 1985;134:3662–3668. [PubMed] [Google Scholar]
- 63.Lightwood D, O’Dowd V, Carrington B, Veverka V, Carr MD, Tservistas M, Henry AJ, Smith B, Tyson K, Lamour S, et al. The discovery, engineering and characterisation of a highly potent anti-human IL-13 fab fragment designed for administration by inhalation. J Mol Biol. 2013;425:577–593. doi: 10.1016/j.jmb.2012.11.036. [DOI] [PubMed] [Google Scholar]
- 64.Kabat EA. Sequences of proteins of immunological interest Bethesda (MD), Washington (DC): U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health; 1987. [Google Scholar]
- 65.Johnson G, Wu TT. Kabat database and its applications: 30 years after the first variability plot. Nucleic Acids Res. 2000;28:214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.UniProt UniProtKB - P55899 (FCGRN_HUMAN). [accessed 2018 Aug]. https://www.uniprot.org/uniprot/P55899. [Google Scholar]
- 67.Gastinel LN, Simister NE, Bjorkman PJ. Expression and crystallization of a soluble and functional form of an Fc receptor related to class I histocompatibility molecules. Proc Natl Acad Sci USA. 1992;89:638–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.West AP Jr., Bjorkman PJ. Crystal structure and immunoglobulin G binding properties of the human major histocompatibility complex-related Fc receptor(,). Biochemistry. 2000;39:9698–9708. [DOI] [PubMed] [Google Scholar]
- 69.Cain K, Peters S, Hailu H, Sweeney B, Stephens P, Heads J, Sarkar K, Ventom A, Page C, Dickson A. A CHO cell line engineered to express XBP1 and ERO1-Lalpha has increased levels of transient protein expression. Biotechnol Prog. 2013;29:697–706. doi: 10.1002/btpr.1693. [DOI] [PubMed] [Google Scholar]
- 70.Humphreys DP, Vetterlein OM, Chapman AP, King DJ, Antoniw P, Suitters AJ, Reeks DG, Parton TA, King LM, Smith BJ, Lang V, Stephens PE. F(ab’)2 molecules made from Escherichia coli produced Fab' with hinge sequences conferring increased serum survival in an animal model. J Immunol Methods. 1998;217:1–10. [DOI] [PubMed] [Google Scholar]
- 71.Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, Double JA, Everitt J, Farningham DA, Glennie MJ, et al. Guidelines for the welfare and use of animals in cancer research. Br J Cancer. 2010;102:1555–1577. doi: 10.1038/sj.bjc.6605642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chowdhury F, Williams A, Johnson P.. Validation and comparison of two multiplex technologies, Luminex and Mesoscale Discovery, for human cytokine profiling. J Immunol Methods. 2009;340:55–64. doi: 10.1016/j.jim.2008.10.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.