

Abstract
Childhood-onset schizophrenia (COS) is a rare and severe form of schizophrenia defined as onset before age of 13. Here we report on two unrelated cases diagnosed with both COS and alternating hemiplegia of childhood (AHC), and for whom two distinct pathogenic de novo variants were identified in the ATP1A3 gene. ATP1A3 encodes the α-subunit of a neuron-specific ATP-dependent transmembrane sodium–potassium pump. Using whole exome sequencing (WES) data derived from a cohort of 17 unrelated COS cases, we also examined ATP1A3 and all of its interactors known to be expressed in the brain to establish if variants could be identified. This led to the identification of a third case with a possibly damaging missense mutation in ATP1A3 and three others cases with predicted pathogenic missense variants in the FXYD gene family (FXYD1, FXYD6 and FXYD6-FXYD2 readthrough). FXYD genes encode proteins that modulate the ATP-dependant pump function. This report is the first to identify variants in the same pathway for COS. Our COS study illustrates the interest of stratifying a complex condition according to the age of onset for the identification of deleterious variants. Whereas ATP1A3 is a replicated gene in rare neuropediatric diseases, this gene has previously been linked with COS in only one case report. The association with rare variants in FXYD gene family is novel and highlights the interest of exploring these genes in COS as well as in pediatric neurodevelopmental disorders.
Keywords: psychosis, genetic, early-onset schizophrenia, phospholemman, phosphohippolin
Introduction
Schizophrenia is a major mental disorder characterized by a spectrum of symptoms, including delusions, hallucinations, disorganisation of speech and behaviour, negative symptoms, and cognitive deficits. The age of onset of schizophrenia typically ranges from 15 to 25 years old, but rarely can begin before age 13. This early presentation is referred to as childhood-onset schizophrenia (COS) and has a similar presentation in the DSM compared to poor outcome adult-onset schizophrenia (AOS)1. The rate of comorbidity of developmental disorders such as autism spectrum disorder (ASD), motor developmental disorders and learning disabilities are higher than in the later onset forms of schizophrenia. Also, the rate of comorbid medical conditions is increased2. The presence of prominent delusions or hallucinations for at least one month defines COS and helps to differentiate it from ASD or pervasive developmental disorder (PDD). The prevalence of COS is estimated to be 0.03%3 compared to 1% for AOS.
Understanding the causes of this rare but severe form of schizophrenia improves our knowledge of the genetic architecture of schizophrenia. COS variants are observed as more penetrant whereas AOS seems to be more driven by genetic and environmental interactions4. Stratifying by age of onset has been useful in medicine, in particular for the identification of causal genetic variants. Several publications have shown that some polymorphisms are associated with COS. Identification of rare variants in COS is just starting, including Copy Number Variants5,6, truncating variants7 and de novo mutations8. Rare missense variants are more difficult to interpret due to their incomplete penetrance and the limitations of working with a relatively small cohort.
Here we report two COS cases who also have Alternating Hemiplegia of childhood (AHC), a rare disease with a prevalence below 1/100,000. Onset of AHC is typically before the age of 18 months and the clinical presentation is characterised by repeated episodes of hemiplegia that alternately affects one side of the body9. Some paroxysmal symptoms are associated, such as seizures, dystonic episodes, visuomotor disorders, dyspnea, dysautonomia signs. Other neurological symptoms include choreoathetosis and ataxia10. Alternating hemiplegia of childhood also causes mild to severe cognitive impairment. Most of the cases are associated with ATP1A3 mutation and very rarely, with a mutation in the ATP1A2 gene. In this current study, two de novo deleterious missense variants were identified in the ATP1A3 gene in two individuals experiencing comorbidities between COS and AHC. This gene has been previously associated with COS in one case with a history of PDD and selective mutism11. Very recently, de novo mutations in this gene have been found in ASD12. It encodes the catalytic α-subunit of a neuron-specific ATP-dependent transmembrane sodium–potassium pump. Then, we looked for variants in this gene and its interactors in a large COS cohort. One missense mutation was identified in the same exon in the ATP1A3 gene in one affected individual. Missense variants were found in genes that interact with ATP1A3 in four additional COS cases.
Population and methods
1) Participants
The two cases of COS who also have AHC were identified in the child and adolescent psychiatric unit in the Centre Hospitalier Spécialisé du Rouvray (Sotteville-lès-Rouen, France). This is an inpatient intensive care unit specialized in severe forms of child psychiatric disease. The psychiatric clinical assessment was performed by a specialized expert psychiatrist (VF) using the DSM-V criteria. A specialized geneticist (AG) conducted the general clinical examination including neurological examination. The parents gave their written informed consent for the study. The two probands and their families are Caucasian.
An American COS cohort was recruited by Dr. Rapoport’s group at the National Institute of Mental Health (NIMH) as part of their childhood onset schizophrenia research study. This study was approved by the Institutional Review Board of The National Institute of Mental Health. All participants provided written informed consent from a parent or legal guardian for minors. A total of 361 patients were screened. Seventeen sporadic COS cases (11 males and 6 females) meeting DSM-IIIR/DSM-IV criteria for schizophrenia with onset of psychosis before age 13 and their unaffected parents were selected for this study. Diagnosis was confirmed with inpatient medication-free observation according previously published recommendations13. To address the concern of false positives resulting from inclusion of language disorders, we included only patients with clear positive symptoms (delusions or hallucinations). Medical or neurological disorders were criteria of exclusion. Patients and their available first-degree relatives were interviewed for lifetime and current psychiatric disorders using structured psychiatric interviews and Autism Symptom Questionnaire. The mean age of onset was 9.8 years (range: 6–12 years old). Six of the patients were also diagnosed with ASD. CNV and de novo variants have been previously explored and published in this cohort5,8.
2) Genetic study
The molecular analysis of the two French cases was performed by targeted Sanger sequencing in the affected individuals, their parents, and their siblings.
In the American cohort, exome capture of all individuals in the COS trios was performed using SureSelectXT Human All Exon V4 kit (Agilent Technologies Inc. Mississauga, ON, Canada) in two different batches. The first batch consisting of 13 COS trios (39 samples) was captured and sequenced using Illumina HiSeq 2000 at the McGill University and Genome Quebec Innovation Centre (Montreal, QC, Canada). The second batch of 4 COS trios (12 samples) was sequenced using the Illumina HiSeq 2000 platform at the Université de Montréal's Beaulieu-Saucier Pharmacogenomics Centre at the Montreal Heart Institute (Montreal, Canada).
The sequenced reads of all the samples from Illumina HiSeq2000 were aligned to the reference genome (GRCh37/hg19) using Burrow-Wheeler Aligner14. The aligned reads were converted to binary format for the convenience of further analysis using SAMtools15. Samples had an average coverage of over 90% target covered at a depth of 20 ×. The quality of coverage was assessed by the total number of reads mapped to corresponding regions in the reference genome, over the total number of uniquely mapped reads. Next, variant calling was performed using Genome Analysis Tool Kit (GATK)16. The variants were called for the sequenced reads available within the coverage region for each of the samples. This process identified single-nucleotide variants and small insertions or deletions at different levels of stringency based on their quality scores.
The identified variants were annotated with ANNOVAR tool17, including minor allele frequencies from publicly available databases (1,000 Genomes project and ExAC database), pathogenicity scores based on Polyphen-2, SIFT, LRT, C-PAP and MutationTaster, phylogenetic conservation using GERP and PhyloP scores. Segregation analyses and extraction of variants located in genes of interest were performed using an in-house script. Only variants in exonic positions, with a frequency <0.01 in the 1000 Genome project and ExAC database, identified as possibly damaging by at-least three algorithms, and in a phylogenetically-conserved position, were retained.
3) Interactome analysis
In the American cohort, we looked for pathogenic mutations in genetic interactors of ATP1A3. The protein-protein interaction network was identified using the STRING software18 (http://string-db.org/) with data settings as follow: all active interaction sources, no more than 50 interactions and highest level of confidence (0.9). The pathway was secondarily explored using the curated database Reactome (http://reactome.org/)19. Then, all the interactors were retained for further analysis if they are expressed in any part of the brain, based on GTEx database (https://gtexportal.org/home/)20. The final list of candidate interactors is given in the Supplementary Table. We also looked for expression of the more interesting interactors across the lifespan using BrainCloud application. BrainCloud allows the query of genome-wide gene expression data in the normal human postmortem dorsolateral prefrontal cortex at different ages21.
4) Visualisation
To visualize the localisation of the predicted pathogenic missense variants, we constructed the 3D picture of the ATP1A3 gene and FXYD gene family using the UCSF ChimeraX software22 (http://www.rbvi.ucsf.edu/chimera/). Molecular data were obtained from the PHYRE2 Protein Fold Recognition Server23 (www.sbg.bio.ic.ac.uk/~phyre2/) with the following Uniprot entries: P13637 (ATP1A3), Q9H0Q3 (FXYD6), O00168 (FXYD1), and A0A0A6YYL5 (FXYD6-FXYD2 readthrough). Color markers have been manually placed in ChimeraX.
Results
Our study identified three variants in the ATP1A3 gene in three unrelated individuals with COS (Table 1). Mutations were found in the first two individuals because they also had AHC, which is caused by ATP1A3 mutations in 74% of the patients24.
Table 1.
summary of the molecular findings and the clinical presentation of the ATP1A3 mutation carriers
| Case 1 | Case 2 | NSB1251 | |
|---|---|---|---|
| Mutation in ATP1A3 gene | NM_152296.4(ATP1A3):c.2401G>A (p.Asp801Asn) | NM_152296.4(ATP1A3):c.2443G>A (p.Glu815Lys) | NM_152296.4(ATP1A3):c.2438C>T (p.Ala813Val) |
| Pathogenicity | Reported in ClinVar (ID:37108) | Reported in ClinVar (ID:37107) | Not reported, predicted damaging |
| Inheritance | De novo | De novo | Inherited from the mother |
| Age of onset for psychiatric symptoms | 10 | 12 | 10 |
| Sex | Male | Male | Male |
| Main psychiatric symptoms | Fluctuant visual and auditory hallucinations, delusions of persecution, psychomotor agitation and aggressiveness | Visual hallucinations (distortion of lights and shadows) followed by auditory and tactile hallucinations, delusion with persecutory and mystic ideas Negative symptoms with major social withdrawal |
Positive symptoms: delusions and hallucinations |
| Dysmorphic features | Short philtrum, large ears with low implantation, gum hypertrophy, exotropia and macrocephaly | Macroglossy, nystagmus, esotropia and short philtrum and cleft palate | ? |
| Neurodevelopmental delays | Moderate intellectual disability, developmental delays, reading and writing skills are not acquired | Walk at 25 months, first word around 4 years old, severe hypotonia | Intellectual disability with verbal intelligence quotient (IQ) of 75 and performance IQ of 57 |
| Response to treatment | Poor | Poor | ? |
| Associated phenotype | Recurrent major depressive disorder | Autism Spectrum Disorder | Autism Spectrum Disorder |
In Case 1, we found a de novo mutation in ATP1A3 gene: c.2401G>A. The patient presented at 3-month-old with seizures and repeated episodes of hemiplegia. Diagnosis of AHC was made at the age of 14 months old. He had severe developmental delays in early childhood and moderate intellectual disability without acquisition of reading and writing skills. He walked at 30 months, spoke his first words at 24 months, and he still has urinary incontinence. He had a failure to thrive, a gait disorder, and global hypotonia. He is the first child of unrelated and unaffected parents and has three siblings who do not carry the mutation, have no neuropsychiatric symptoms and have a normal development. The first psychotic features appeared at the age of 10 when he reported fluctuant symptoms such as visual and auditory hallucinations, delusions of persecution followed by behavioral disorders including psychomotor agitation and aggressiveness (Scale for Assessment of Positive Symptoms25 (SAPS): patient’s maximal score = 35). He also had depressive symptoms with suicidal ideation and psychomotor slowdown. Hallucinations and delusional ideation seemed to worsen when hemiplegic episodes occurred.
In the unrelated Case 2, we found a de novo missense mutation in ATP1A3: c.2443G>A in a boy. The diagnosis of AHC was made at the age of 3 months based on nystagmus episodes, major hypotonia, tonic and myoclonic limb movements and a hemiplegic episode complicated with recurrent seizures. He had a developmental delay with walking acquired at the age of 25 months, and first words around 4 years old. He had impaired social skills compatible with a diagnosis of autism-spectrum disorder (ASD). The first psychotic symptoms appeared at the age of 12 years with self-reported isolated visual hallucinations described as distortion of lights and shadows, auditory and tactile hallucinations followed by delusion with persecutory and mystic ideas and bizarre behavior. SAPS scored 40 and he also had negative symptoms (Scale for Assessment of Negative Symptoms (SANS): patient’s maximal score = 35).
No other potentially causative mutations were found for both patients. Neither had obstetrical complications. Their de novo variants have been reported as deleterious in ClinVar (Table 1). Following an approach developed for interpretation of de novo mutation in human disease and especially in autism26, we estimate that these variants are disease-relevant mutations. The constraint metric for missense variant in ExAC browser27 is very high (z=7.38) indicating an intolerance to variation in the ATP1A3 gene.
Response to treatment was evaluated by the May & Dencker scale28 and the score of 4 for both the patients indicated that they had a poor response to treatment. In the case 1 the patient did not respond to aripiprazole or antidepressant medication but respond to a combination of risperidone (1.5 mg/day) and lithium 800 mg/day. Antidepressant and lithium were introduced as he presented recurrence of major depressive episodes. In the case 2, he did not respond to risperidone but benefited from treatment with aripiprazole. After a follow-up of two years, the pharmacological treatments have not been modified. The tolerance is acceptable and no psychotic relapses have been noticed or reported until now. They also received psychotherapy, cognitive rehabilitation, as well as institutional care including psychomotor-training, physiotherapy and special needs education.
We looked for mutations in ATP1A3 in our American cohort. A nonsynonymous variant c.2438T>C was found in an affected male child. This variant has never been reported in any database, it is very conserved across species and all the tested algorithms suggest it was possibly damaging (Table 2). The carrier (NSB1251) was diagnosed with the symptoms of schizophrenia at the age of 10, after an initial diagnosis of ASD. The ethnicity of the trio was Caucasian. None of the parents has a history of psychiatric or neurological diseases. The variant was inherited from the mother. There were no de novo single nucleotide variants identified in the proband in our COS whole exome sequencing study.
Table 2.
list of mutations in ATP1A3 and its interactors in the American cohort annotated with their predicted pathogenicity and their conservative score. Scores in bold are considered as pathogenic.
| Family ID |
Chr | Position | Reference Allele |
Mutant Allele |
Gene symbol |
Detailed annotation of the variant |
Frequency in the 1000 Genome project |
Frequency in the ExAC database |
SIFT | Poly Phen |
LRT | Mutation Taster |
M- CAP |
PhyloP vertebrate |
GERP++ score |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NSB1251 | 19 | 42474441 | G | A | ATP1A3 | exon18:c.C2438T:p.A813V | not reported | not reported | 0 | 0.999 | 0.000 | 1.000 | 0.773 | 9.531 | 3.69 |
| NSB1949 | 9 | 139906315 | G | C | ABCA2 | exon35:c.C5606G:p.A1869G | not reported | 0.00002364 | 0.01 | 0.955 | 0.043 | 0.998 | . | 9.195 | 2.95 |
| NSB1814 | 19 | 35633635 | C | T | FXYD1 | exon7:c.C268T:p.R90C | 0.0014 | 0.007666 | 0 | 1.0 | . | 1.000 | . | 3.554 | 4.12 |
| NSB2720 | 11 | 117693403 | A | G | FXYD6-FXYD2 | exon7:c.T302C:p.V101A | 0.0009 | 0.0004984 | 0 | 0.999 | 0.000 | 0.967 | 0.143 | 4.289 | 4.35 |
| NSB1553 | 11 | 117711076 | C | G | FXYD6 | exon6:c.G217C:p.G73R | 0.0009 | 0.001911 | 0.04 | 0.175 | . | 1.000 | 0.133 | 4.818 | 5.44 |
The ATP1A3 gene encodes the alpha-3 catalytic subunit of the Na+/K(+)-ATPase transmembrane ion pump, which is exclusively expressed in neurons of various brain regions. The ATP1A3 Na,K-ATPase is heteromeric so we systematically looked for mutations in its known interactors (Supplementary Figure 1 and Supplementary Table). The interactome centered on ATP1A3 identified sixteen genes, essentially the genes coding for Na+/K+-ATPases and their interacting FXYD proteins (Supplementary Figure 2). Among them, twelve genes are expressed in the brain according to GTEx database (Supplementary Table). Possibly damaging variants in phylogenetically-conserved positions were identified in ABCA2, FXYD1, FXYD6 and FXYD6-FXYD2 readthrough (Table 2). FXYD6-FXYD2 readthrough is a conjoined gene that generates transcripts by combining exons from FXYD6 and FXYD2, which are on the same chromosome and in the same orientation. None of the variants were de novo. In total, we found 4 cases with rare damaging variants in ATP1A3 and FXYD gene family in the American cohort.
We have represented the amino-acid (AA) changes in Figure 1. The ATP1A3 variants are very close to each other (less than 15 AA between them) and are part of the transmembrane region of the protein. This region seems critical for the function of the pump. Amino-acid changes in FXYD proteins are only found in the N-terminal region. How this FXYD region interacts with ATP1A3 remains unknown. Visualization of the AA changes in ATP1A3 in other phenotypes has been previously reported using the same tools11.
Figure 1.
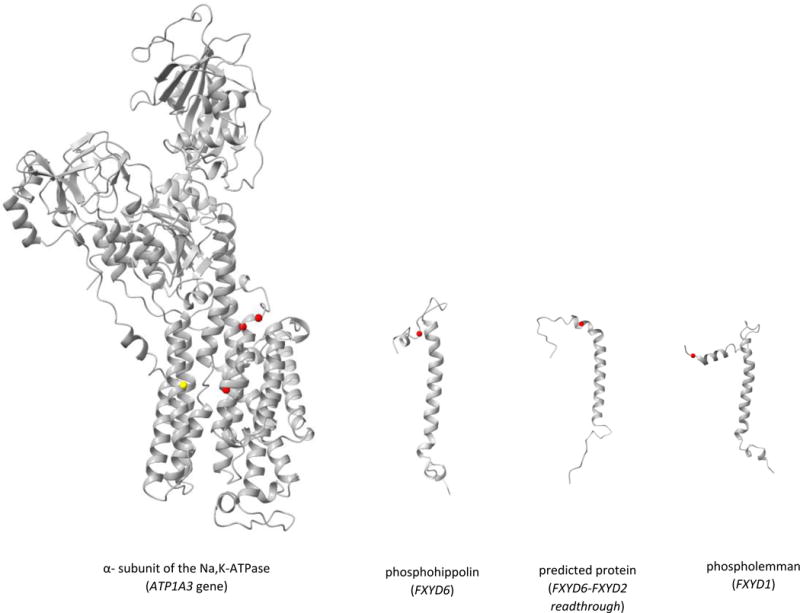
Model of the mutations observed in our cohort (red) and previously reported in COS38 (yellow) in ATP1A3 and its brain-expressed interactors (FXYD gene family).
As we focused on schizophrenia with an age of onset below 13 years-old, we looked for expression of these genes during childhood. Brain Cloud provides data for gene expression in normal postmortem dorsolateral prefrontal cortex during lifespan (Supplementary Figure 3). ATP1A3, FXYD1 and FXYD6 were expressed in the brain during childhood, but information is not provided for FXYD6-FXYD2. However, this transcript has been experimentally-validated and reported in the human brain in another study29. The expression of ATP1A3 and FXYD1 are quite stable during the lifespan, whereas the expression of FXYD6 decreased with age.
Discussion
We have identified three rare pathogenic variants in the ATP1A3 gene and three rare possibly damaging variants in FXYD gene family. ATP1A3 is a replicated gene in rare neuropediatric diseases and here we strengthen the evidence linking it to COS. The association with FXYD gene family is novel. These genes are closely related as they participate to the same heteromeric transmembrane protein complex. Indeed, the transmembrane Na,K-ATPase complex is composed of an essential α- and β-subunit30, and an auxiliary third subunit belonging to the FXYD proteins (sometimes named as the γ-subunit)31. The α-subunit is the catalytic subunit responsible for transport activities of the enzyme. The FXYD family has been identified as a modulator subunit of Na,K-ATPase by stabilizing the complex, altering its kinetic activity, regulating its affinity for Na+, K+, and ATP32,33. The subunits are tissue specific33 and ATP1A3 is selectively expressed in neurons of the central nervous system34. FXYD1 encodes the phospholemman protein, a transmembrane phosphoprotein expressed in the cerebellum and the frontal cortex35. Phospholemman integrates signals of many different kinases36 and modulates the neuronal excitability via its effect on the NA,K-ATPase37. FXYD6 encodes the phosphohippolin, which plays an important role in neuronal excitability during postnatal development and in adult brain38. The heterotrimer ATP1A:ATP1B:FXYD catalyzes the hydrolysis of ATP coupled with the exchange of sodium and potassium ions across the plasma membrane, creating the electrochemical gradient, critical for the neuronal excitability. Interestingly, the expression of ATP1A3, FXYD1 and FXYD6 in the prefrontal cortex is present since birth consistent with the precocious onset of the phenotype.
ATP1A3 has been previously involved in various severe neurological disorders such as rapid-onset dystonia-parkinsonism (RDP), AHC, and CAPOS syndrome (CAPOS=cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss)39. Most of the pathogenic ATP1A3 mutations are located in the conserved transmembrane or N-terminus domains40. Two rare damaging missense mutations in ATP1A3 have been reported in schizophrenia with neither clinical description nor mention of the age of onset41. Overall the frequency of ATP1A3 deleterious variants in adult-onset schizophrenia seems to be very low by comparison to the frequency we reported in COS. A unique COS case with de novo ATP1A3 has been previously reported in the literature11; the proband presented with psychotic symptoms at 6 years of age but also PDD and selective mutism. Interestingly this patient did not have motor phenotypes except decreased muscle tone at 2 months of age for which he received physical therapy. This presentation seems closer to the third case we reported from NIMH cohort, suggesting that ATP1A3 mutations can lead to isolated psychiatric symptoms such as delusions and hallucinations associated with severe behavioural changes. We screened the literature to identify the cognitive deficits and behavioural problems associated with ATP1A3 mutations. In AHC, impairment is nearly constant but variable, ranging from mild to moderate with a mean IQ estimation of 62.5 ± 14.042. The early development ranges from very slow to slight depending on the mutations43. However, detailed behavioural phenotypes are not often reported. One study has detailed the behaviour of a girl with AHC and reports deficits in sustained attention, in self-control, in regulation of her emotions and difficulties in inhibition capability44. Another case with Attention Deficit Hyperactivity Disorder has been reported45. Very recently, de novo mutations in the ATP-binding pathway have been found in ASD12. COS is preceded by and comorbid with ASD (or PDD) in 30% to 50% of cases and a large number of genetic variants are shared by these conditions46. Consequently, autistic features reported in ATP1A3 carriers could be considered as an early and prodromal expression of COS.
Interestingly, in RDP, psychiatric conditions (e.g. bipolar disorder) have been reported with a high frequency47. Brashear et al have systematically assessed psychiatric comorbidities in RDP, reporting psychotic symptoms emerging before or at the same time as motor symptom onset48. The prevalence of psychotic symptoms was 26% in ATP1A3 mutation carriers with RDP, which is significantly different from the prevalence in RDP-affected non-carrier individuals. Beyond these psychotic features, the carriers suffering from RDP also exhibited depressive symptoms10 and cognitive impairments specifically in memory and learning, attention, and executive functions49. Finally, quantifying protein levels using targeted mass spectrometry showed that ATP1A3 was reduced in auditory cortex gray matter of patients with schizophrenia compared to controls50. ATP1A3 was also upregulated by both clozapine and haloperidol in cerebral cortex tissue of antipsychotic-treated monkeys50. A heterozygous knock-in mouse model harboring a pathogenic mutation in position 801 (same position as case 1) has been generated and it displayed behavioural abnormalities such as hyperactivity and cognitive deficits51.
FXYD6 was found to be expressed in glutamatergic synapses52, one major component and actor in psychosis. Therefore, FXYD6 gene may be highly regulated with a peak of expression around birth in neurons of certain layers from the frontal cortex53, which represent crucial period and brain region for COS. Ito et al have looked at FXYD6 expression in the post-mortem brain collection of schizophrenia and bipolar disorder called the Stanley Brain Collection (http://www.stanleyresearch.org/brain/); they found that the expression of FXYD6 in the dorsolateral prefrontal cortex (Brodmann area 46) tended to be decreased compared with healthy subjects54. A linkage analysis followed by fine mapping has suggested an association of FXYD6 with AOS55. A candidate SNP association study has also identified a SNP and a haplotype associated with AOS in this gene56. Two SNPs in this gene have also been associated with schizophrenia in a family-based association study57. However, a meta-analysis did not confirm this association concluding that polymorphisms may not have a major influence on susceptibility to schizophrenia58. The expression level of FXYD6 in the normal prefrontal cortex decreases during the lifespan and may suggest that variants in this gene are more susceptible to be associated with COS than AOS.
The post-mortem levels of FXYD1 messenger RNA and corresponding protein are decreased in the entorhinal cortex of individuals with schizophrenia compared to controls59. FXYD1 has been proposed to regulate the genesis of the neuroepithelium during brain development60. The expression of FXYD1 is specifically-regulated in the frontal cortex by the nuclear protein methyl-CpG binding protein 2 (MECP2)61. Mutations in MECP2 cause Rett syndrome, a syndromic form of autism in girls, and are associated with an overexpression of FXYD1 in the brain35. A case of COS has been reported in a boy carrying a missense mutation in MECP262.
Brain expressed FXYD genes (including FXYD6 and FXYD1) localized in dendrites. The loss of the mRNA localization affects the function of the ATPase in dendrites. Variants in these genes could impact the synaptic functions63. It has been proposed that these ATPase regulator genes control the synaptic and perisynaptic membrane potential52.
Due to the devastating neurologic presentations of ATP1A3 mutations, an international task force has been created to standardize the clinical examination and to provide recommendations64. Our data support the idea that there is a purely psychiatric form associated with mutations in ATP1A3. Moreover, there is no previous report of rare mutations in the FXYD gene family in the neurological forms of the disease. It might be worthwhile screening for FXYD6 mutations in AHC, RDP or CAPOS patients where no ATP1A3 mutation has been identified. Given that we report recurrent missense variants in the same genes in COS, perhaps these genes may be routinely examined in COS.
Response to treatment was poor in our ATP1A3 carriers, as it is frequently the case in COS65. However, identification of a molecular target could be helpful in a personalized approach. New therapeutic strategies are currently being explored for ATP1A3 mutation related-pathologies. For example, oral supplementation with adenosine-5′-triphosphate has been reported to improve motor and cognitive skills in one case66 and could be considered for our patients.
In conclusion, we wish to highlight the interest of studying extreme phenotypes in psychiatric genetics. By studying COS, which is rare but may result from more penetrant mutations, we are increasing our chances to identify new genes. From a clinical point of view, psychotic symptoms can occur in various medical, neurological and genetic diseases of children and adolescents. Atypical clinical signs such as early-onset, visual hallucinations, a catatonic syndrome, fluctuation of symptoms, a cognitive regression, or a paradoxical reaction to psychotropic drugs are red flags67,68 that should urge psychiatrists to look for an underlying organic condition, including genetic ones.
Supplementary Material
Acknowledgments
We thank the patients and the family members who participate in the study as well as the involved medical teams. We thank Daniel Rochefort and Sylvia Dobrezenicka for the technical support; also Edouard Henrion and Ousmane Diallo for their bioinformatics support. We also thank Dr Maryam Soleimani and Dr Laure Bera for their comments, as well as Dr Elodie Hainque for her advice.
Funding/Support:
The American cohort was supported by the National institute of Mental health (NIMH). Its genetic assessment was supported by the Genome Canada and Genome Quebec (Grant No. RMF_92086). Bioinformatics analysis was supported by the Canadian Institutes of Health Research (CIHR). Boris Chaumette receives a postdoctoral fellowship from the Healthy Brains for Healthy Lives project (Talent program)
Footnotes
The authors declare no conflict of interest.
References
- 1.Driver DI, Gogtay N, Rapoport JL. Childhood Onset Schizophrenia and Early Onset Schizophrenia spectrum disorders. Child Adolesc Psychiatr Clin N Am. 2013;22:539–555. doi: 10.1016/j.chc.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giannitelli M, Consoli A, Raffin M, Jardri R, Levinson DF, Cohen D, et al. An overview of medical risk factors for childhood psychosis: Implications for research and treatment. Schizophr Res. 2017 doi: 10.1016/j.schres.2017.05.011. [DOI] [PubMed] [Google Scholar]
- 3.McKenna K, Gordon CT, Lenane M, Kaysen D, Fahey K, Rapoport JL. Looking for childhood-onset schizophrenia: the first 71 cases screened. J Am Acad Child Adolesc Psychiatry. 1994;33:636–644. doi: 10.1097/00004583-199406000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Asarnow RF, Forsyth JK. Genetics of childhood-onset schizophrenia. Child Adolesc Psychiatr Clin N Am. 2013;22:675–687. doi: 10.1016/j.chc.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahn K, Gotay N, Andersen TM, Anvari AA, Gochman P, Lee Y, et al. High rate of disease-related copy number variations in childhood onset schizophrenia. Mol Psychiatry. 2014;19:568–572. doi: 10.1038/mp.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou D, Gochman P, Broadnax DD, Rapoport JL, Ahn K. 15q13.3 duplication in two patients with childhood-onset schizophrenia. Am J Med Genet Part B Neuropsychiatr Genet Off Publ Int Soc Psychiatr Genet. 2016;171:777–783. doi: 10.1002/ajmg.b.32439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Addington AM, Gauthier J, Piton A, Hamdan FF, Raymond A, Gogtay N, et al. A novel frameshift mutation in UPF3B identified in brothers affected with childhood onset schizophrenia and autism spectrum disorders. Mol Psychiatry. 2011;16:238–239. doi: 10.1038/mp.2010.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ambalavanan A, Girard SL, Ahn K, Zhou S, Dionne-Laporte A, Spiegelman D, et al. De novo variants in sporadic cases of childhood onset schizophrenia. Eur J Hum Genet EJHG. 2015 doi: 10.1038/ejhg.2015.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tenney JR, Schapiro MB. Child neurology: alternating hemiplegia of childhood. Neurology. 2010;74:e57–59. doi: 10.1212/WNL.0b013e3181d7d85b. [DOI] [PubMed] [Google Scholar]
- 10.Brashear A, Sweadner KJ, Cook JF, Swoboda KJ, Ozelius L. ATP1A3-Related Neurologic Disorders. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJ, et al., editors. GeneReviews(®) University of Washington, Seattle; Seattle (WA): 1993. http://www.ncbi.nlm.nih.gov/books/NBK1115/ (accessed 10 May 2017). [PubMed] [Google Scholar]
- 11.Smedemark-Margulies N, Brownstein CA, Vargas S, Tembulkar SK, Towne MC, Shi J, et al. A novel de novo mutation in ATP1A3 and childhood-onset schizophrenia. Cold Spring Harb Mol Case Stud. 2016;2 doi: 10.1101/mcs.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takata A, Miyake N, Tsurusaki Y, Fukai R, Miyatake S, Koshimizu E, et al. Integrative Analyses of De Novo Mutations Provide Deeper Biological Insights into Autism Spectrum Disorder. Cell Rep. 2018;22:734–747. doi: 10.1016/j.celrep.2017.12.074. [DOI] [PubMed] [Google Scholar]
- 13.Gochman P, Miller R, Rapoport JL. Childhood-onset schizophrenia: the challenge of diagnosis. Curr Psychiatry Rep. 2011;13:321–322. doi: 10.1007/s11920-011-0212-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinforma Oxf Engl. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinforma. 2013;43:11.10.1–33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164–e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fabregat A, Sidiropoulos K, Garapati P, Gillespie M, Hausmann K, Haw R, et al. The Reactome pathway Knowledgebase. Nucleic Acids Res. 2016;44:D481–487. doi: 10.1093/nar/gkv1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Melé M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, et al. The human transcriptome across tissues and individuals. Human genomics. Science. 2015;348:660–665. doi: 10.1126/science.aaa0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT, et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature. 2011;478:519–523. doi: 10.1038/nature10524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 23.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heinzen EL, Swoboda KJ, Hitomi Y, Gurrieri F, Nicole S, de Vries B, et al. De novo mutations in ATP1A3 cause alternating hemiplegia of childhood. Nat Genet. 2012;44:1030–1034. doi: 10.1038/ng.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andreasen NC. Methods for Assessing Positive and Negative Symptoms1. 1990;24:73–88. doi: 10.1159/000418013. [DOI] [PubMed] [Google Scholar]
- 26.Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet. 2014;46:944–950. doi: 10.1038/ng.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.May P, Dencker S, Hubbard J. A systematic approach to treatment resistance in schizophrenic disorders. In: Dencker SJ, Kulhanek F, editors. Treatment Resistance in Schizophrenia. Braunschweig/Wiesbaden: Viewag Verlag; 1988. pp. 22–3. [Google Scholar]
- 29.Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics MCP. 2014;13:397–406. doi: 10.1074/mcp.M113.035600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lingrel JB, Kuntzweiler T. Na+,K(+)-ATPase. J Biol Chem. 1994;269:19659–19662. [PubMed] [Google Scholar]
- 31.Geering K. FXYD proteins: new regulators of Na-K-ATPase. Am J Physiol - Ren Physiol. 2006;290:F241–F250. doi: 10.1152/ajprenal.00126.2005. [DOI] [PubMed] [Google Scholar]
- 32.Garty H, Karlish SJD. Role of FXYD proteins in ion transport. Annu Rev Physiol. 2006;68:431–459. doi: 10.1146/annurev.physiol.68.040104.131852. [DOI] [PubMed] [Google Scholar]
- 33.Geering K, Béguin P, Garty H, Karlish S, Füzesi M, Horisberger J-D, et al. FXYD proteins: new tissue- and isoform-specific regulators of Na,K-ATPase. Ann N Y Acad Sci. 2003;986:388–394. doi: 10.1111/j.1749-6632.2003.tb07219.x. [DOI] [PubMed] [Google Scholar]
- 34.Li Z, Langhans SA. Transcriptional regulators of Na, K-ATPase subunits. Front Cell Dev Biol. 2015;3 doi: 10.3389/fcell.2015.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deng V, Matagne V, Banine F, Frerking M, Ohliger P, Budden S, et al. FXYD1 is an MeCP2 target gene overexpressed in the brains of Rett syndrome patients and Mecp2-null mice. Hum Mol Genet. 2007;16:640–650. doi: 10.1093/hmg/ddm007. [DOI] [PubMed] [Google Scholar]
- 36.Mounsey JP, Lu KP, Patel MK, Chen ZH, Horne LT, John JE, et al. Modulation of Xenopus oocyte-expressed phospholemman-induced ion currents by co-expression of protein kinases. Biochim Biophys Acta. 1999;1451:305–318. doi: 10.1016/s0167-4889(99)00102-0. [DOI] [PubMed] [Google Scholar]
- 37.Crambert G, Fuzesi M, Garty H, Karlish S, Geering K. Phospholemman (FXYD1) associates with Na,K-ATPase and regulates its transport properties. Proc Natl Acad Sci U S A. 2002;99:11476–11481. doi: 10.1073/pnas.182267299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kadowaki K, Sugimoto K, Yamaguchi F, Song T, Watanabe Y, Singh K, et al. Phosphohippolin expression in the rat central nervous system. Mol Brain Res. 2004;125:105–112. doi: 10.1016/j.molbrainres.2004.03.021. [DOI] [PubMed] [Google Scholar]
- 39.Sweney MT, Newcomb TM, Swoboda KJ. The expanding spectrum of neurological phenotypes in children with ATP1A3 mutations, Alternating Hemiplegia of Childhood, Rapid-onset Dystonia-Parkinsonism, CAPOS and beyond. Pediatr Neurol. 2015;52:56–64. doi: 10.1016/j.pediatrneurol.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Panagiotakaki E, De Grandis E, Stagnaro M, Heinzen EL, Fons C, Sisodiya S, et al. Clinical profile of patients with ATP1A3 mutations in Alternating Hemiplegia of Childhood—a study of 155 patients. Orphanet J Rare Dis. 2015;10:123. doi: 10.1186/s13023-015-0335-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sweney MT, Silver K, Gerard-Blanluet M, Pedespan J-M, Renault F, Arzimanoglou A, et al. Alternating Hemiplegia of Childhood: Early Characteristics and Evolution of a Neurodevelopmental Syndrome. Pediatrics. 2009;123:e534–e541. doi: 10.1542/peds.2008-2027. [DOI] [PubMed] [Google Scholar]
- 43.Sasaki M, Ishii A, Saito Y, Morisada N, Iijima K, Takada S, et al. Genotype-phenotype correlations in alternating hemiplegia of childhood. Neurology. 2014;82:482–490. doi: 10.1212/WNL.0000000000000102. [DOI] [PubMed] [Google Scholar]
- 44.Muriel V, Garcia-Molina A, Aparicio-Lopez C, Ensenat A, Roig-Rovira T. Neuropsychological deficits in alternating hemiplegia of childhood: a case study. Rev Neurol. 2015;61:25–28. [PubMed] [Google Scholar]
- 45.Hoei-Hansen CE, Dali C, Lyngbye TJB, Duno M, Uldall P. Alternating hemiplegia of childhood in Denmark: Clinical manifestations and ATP1A3 mutation status. Eur J Paediatr Neurol. 2014;18:50–54. doi: 10.1016/j.ejpn.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 46.Rapoport J, Chavez A, Greenstein D, Addington A, Gogtay N. Autism Spectrum Disorders and Childhood-Onset Schizophrenia: Clinical and Biological Contributions to a Relation Revisited. J Am Acad Child Adolesc Psychiatry. 2009;48:10–18. doi: 10.1097/CHI.0b013e31818b1c63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barbano RL, Hill DF, Snively BM, Light LS, Boggs N, McCall WV, et al. New triggers and non-motor findings in a family with rapid-onset dystonia-parkinsonism. Parkinsonism Relat Disord. 2012;18:737–741. doi: 10.1016/j.parkreldis.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brashear A, Cook JF, Hill DF, Amponsah A, Snively BM, Light L, et al. Psychiatric disorders in rapid-onset dystonia-parkinsonism. Neurology. 2012;79:1168–1173. doi: 10.1212/WNL.0b013e3182698d6c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cook JF, Hill DF, Snively BM, Boggs N, Suerken CK, Haq I, et al. Cognitive impairment in rapid-onset dystonia-parkinsonism. Mov Disord Off J Mov Disord Soc. 2014;29:344–350. doi: 10.1002/mds.25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.MacDonald ML, Ding Y, Newman J, Hemby S, Penzes P, Lewis DA, et al. Altered Glutamate Protein Co-Expression Network Topology Linked to Spine Loss in the Auditory Cortex of Schizophrenia. Biol Psychiatry. 2015;77:959–968. doi: 10.1016/j.biopsych.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holm TH, Isaksen TJ, Glerup S, Heuck A, Bøttger P, Füchtbauer E-M, et al. Cognitive deficits caused by a disease-mutation in the α3 Na+/K+-ATPase isoform. Sci Rep. 2016;6 doi: 10.1038/srep31972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Biesemann C, Grønborg M, Luquet E, Wichert SP, Bernard V, Bungers SR, et al. Proteomic screening of glutamatergic mouse brain synaptosomes isolated by fluorescence activated sorting. EMBO J. 2014;33:157–170. doi: 10.1002/embj.201386120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stansberg C, Ersland KM, van der Valk P, Steen VM. Gene expression in the rat brain: High similarity but unique differences between frontomedial-, temporal- and occipital cortex. BMC Neurosci. 2011;12:15. doi: 10.1186/1471-2202-12-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ito Y, Nakamura Y, Takahashi N, Saito S, Aleksic B, Iwata N, et al. A genetic association study of the FXYD domain containing ion transport regulator 6 (FXYD6) gene, encoding phosphohippolin, in susceptibility to schizophrenia in a Japanese population. Neurosci Lett. 2008;438:70–75. doi: 10.1016/j.neulet.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 55.Choudhury K, McQuillin A, Puri V, Pimm J, Datta S, Thirumalai S, et al. A Genetic Association Study of Chromosome 11q22-24 in Two Different Samples Implicates the FXYD6 Gene, Encoding Phosphohippolin, in Susceptibility to Schizophrenia. Am J Hum Genet. 2007;80:664–672. doi: 10.1086/513475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhong N, Zhang R, Qiu C, Yan H, Valenzuela RK, Zhang H, et al. A novel replicated association between FXYD6 gene and schizophrenia. Biochem Biophys Res Commun. 2011;405:118–121. doi: 10.1016/j.bbrc.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 57.Jiao L, Wang B, Niu X, Ma X, Li J, Shen B, et al. A family-based association study of FXYD6 gene polymorphisms and schizophrenia. Zhonghua Yi Xue Yi Chuan Xue Za Zhi Zhonghua Yixue Yichuanxue Zazhi Chin J Med Genet. 2011;28:539–542. doi: 10.3760/cma.j.issn.1003-9406.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 58.Iwata Y, Yamada K, Iwayama Y, Anitha A, Thanseem I, Toyota T, et al. Failure to confirm genetic association of the FXYD6 gene with schizophrenia: the Japanese population and meta-analysis. Am J Med Genet Part B Neuropsychiatr Genet Off Publ Int Soc Psychiatr Genet. 2010;153B:1221–1227. doi: 10.1002/ajmg.b.31095. [DOI] [PubMed] [Google Scholar]
- 59.Hemby SE, Ginsberg SD, Brunk B, Arnold SE, Trojanowski JQ, Eberwine JH. Gene expression profile for schizophrenia: discrete neuron transcription patterns in the entorhinal cortex. Arch Gen Psychiatry. 2002;59:631–640. doi: 10.1001/archpsyc.59.7.631. [DOI] [PubMed] [Google Scholar]
- 60.Chang JT, Lowery LA, Sive H. Multiple roles for the Na,K-ATPase subunits, Atp1a1 and Fxyd1, during brain ventricle development. Dev Biol. 2012;368:312–322. doi: 10.1016/j.ydbio.2012.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Banine F, Matagne V, Sherman LS, Ojeda SR. Brain region-specific expression of Fxyd1, an Mecp2 target gene, is regulated by epigenetic mechanisms. J Neurosci Res. 2011;89:840–851. doi: 10.1002/jnr.22608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cohen D, Lazar G, Couvert P, Desportes V, Lippe D, Mazet P, et al. MECP2 mutation in a boy with language disorder and schizophrenia. Am J Psychiatry. 2002;159:148–149. doi: 10.1176/appi.ajp.159.1.148-a. [DOI] [PubMed] [Google Scholar]
- 63.Shiina N, Yamaguchi K, Tokunaga M. RNG105 deficiency impairs the dendritic localization of mRNAs for Na+/K+ ATPase subunit isoforms and leads to the degeneration of neuronal networks. J Neurosci Off J Soc Neurosci. 2010;30:12816–12830. doi: 10.1523/JNEUROSCI.6386-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rosewich H, Sweney MT, DeBrosse S, Ess K, Ozelius L, Andermann E, et al. Research conference summary from the 2014 International Task Force on ATP1A3-Related Disorders. Neurol Genet. 2017;3:e139. doi: 10.1212/NXG.0000000000000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kumra S, Oberstar JV, Sikich L, Findling RL, McClellan JM, Vinogradov S, et al. Efficacy and tolerability of second-generation antipsychotics in children and adolescents with schizophrenia. Schizophr Bull. 2008;34:60–71. doi: 10.1093/schbul/sbm109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ju J, Hirose S, Shi X-Y, Ishii A, Hu L-Y, Zou L-P. Treatment with Oral ATP decreases alternating hemiplegia of childhood with de novo ATP1A3 Mutation. Orphanet J Rare Dis. 2016;11:55. doi: 10.1186/s13023-016-0438-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Consoli A, Raffin M, Laurent C, Bodeau N, Campion D, Amoura Z, et al. Medical and developmental risk factors of catatonia in children and adolescents: a prospective case-control study. Schizophr Res. 2012;137:151–158. doi: 10.1016/j.schres.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 68.Bonnot O, Klünemann HH, Sedel F, Tordjman S, Cohen D, Walterfang M. Diagnostic and treatment implications of psychosis secondary to treatable metabolic disorders in adults: a systematic review. Orphanet J Rare Dis. 2014;9:65. doi: 10.1186/1750-1172-9-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.