

Abstract
Resolution of inflammation is essential for tissue homeostasis and a promising approach to inflammatory disorders. Here we found that DEL-1, a secreted protein inhibiting leukocyte-endothelial adhesion and inflammation initiation, also functions as a non-redundant downstream effector in inflammation clearance. In human and murine periodontitis, waning of inflammation correlated with DEL-1 upregulation, whereas resolution of experimental periodontitis failed in DEL-1 deficiency. This concept was mechanistically substantiated in acute monosodium urate crystal-induced inflammation, where the pro-resolution function of DEL-1 was attributed to effective apoptotic neutrophil clearance (efferocytosis). DEL-1-mediated efferocytosis induced liver-X-receptor-dependent macrophage reprogramming to pro-resolving phenotype and was required for optimal production of at least certain specific pro-resolving mediators. Experiments in transgenic mice with cell-specific overexpression of DEL-1 linked its anti-leukocyte recruitment action to endothelial-derived DEL-1 and its efferocytic/pro-resolving action to macrophage-derived DEL-1. Thus, the compartmentalized expression of DEL-1 facilitates distinct homeostatic functions in an appropriate context that can be harnessed therapeutically.
Introduction
Timely termination of an inflammatory response is of major importance to prevent unwarranted tissue damage and promote reconstitution of tissue integrity after sterile or microbial inflammatory insults1. Resolution of acute neutrophilic inflammation1,2 requires a well-orchestrated series of processes, including down-regulation of proinflammatory mediators, termination of further neutrophil recruitment, and clearance of neutrophils1,2. In this regard, after performing acute proinflammatory actions, recruited neutrophils in the inflamed site undergo apoptosis and are cleared by macrophage phagocytosis through a process known as efferocytosis3,4. Efficient engulfment of apoptotic neutrophils is mediated by specific macrophage receptors that directly or indirectly recognize the major ‘eat-me’ signal phosphatidylserine (PS) on the apoptotic cell surface5,6. Resolving macrophages upregulate efferocytic receptors and opsonins that bridge them to apoptotic cells6,7. Efferocytosis serves more than waste disposal as this process promotes a ‘pro-resolving’ phenotype in macrophages; defective efferocytic activity has been associated with persistence and chronicity of inflammation1. Consistently, efferocytic macrophages release immunomodulatory molecules, such as transforming growth factor-β (TGF-β)6,7.
Developmental endothelial locus-1 (DEL-1) is a secreted multifunctional protein that interacts with integrins8. DEL-1 comprises three N-terminal epidermal growth factor (EGF)-like repeats and two C-terminal discoidin I-like domains, hence also known as EDIL3 (EGF-like repeats and discoidin I-like domains 3). An RGD (Arg–Gly–Asp) motif in the second EGF-like repeat (E2) mediates DEL-1 binding to αv integrins9,10. Moreover, we have demonstrated that DEL-1 interacts with αLβ2 (CD11a/CD18; LFA-1) and αMβ2 (CD11b/CD18; Mac-1) integrin11,12. The interaction of DEL-1 with LFA-1 blocks the binding of the latter to its endothelial counter-receptor, intercellular cell adhesion molecule-1 (ICAM-1), thereby inhibiting leukocyte adhesion and recruitment to inflamed sites11. By virtue of its anti-inflammatory properties, DEL-1 protects against several inflammation-associated pathologies, such as experimental autoimmune encephalitis/multiple sclerosis13, inflammatory bone loss9,14, pulmonary inflammation and fibrosis11,15 and inflammation associated with islet transplantation16. Pro-inflammatory cytokines, such as interleukin 17 (IL-17), inhibit endothelial DEL-1 expression, thereby promoting leukocyte infiltration and tissue inflammation14,17. Interestingly, the IL-17–dependent downregulation of DEL-1 is reversed by the pro-resolving lipid mediator resolvin-D1, which in turn depends on DEL-1 to mediate protection in a model of inflammatory bone loss17. The connection of DEL-1 with resolvins raised the possibility that DEL-1 does not simply inhibit the initiation of inflammation but might also promote its resolution, a hypothesis that has not been addressed thus far.
By using two different inflammation models and multiple complementary genetic and pharmacologic approaches, we demonstrate here that DEL-1 contributes to resolution of inflammation by promoting αvβ3 integrin-dependent apoptotic neutrophil efferocytosis by macrophages and the emergence of a pro-resolving macrophage phenotype. Consistently, inflammation clearance correlated with resurgence of DEL-1 expression in humans and mice. DEL-1-dependent efferocytosis triggered a pro-resolving macrophage phenotype in a manner dependent on the liver X receptor (LXR). Importantly, by generating different transgenic mice with cell-specific overexpression of DEL-1, we showed that the cellular source of DEL-1 is a critical determinant of its inflammation-modulatory functions. Specifically, we found that the anti-leukocyte recruitment action (and hence inhibitory action on initiation of inflammation) was associated with endothelial cell-derived DEL-1, whereas the efferocytic/pro-resolving action with macrophage-derived DEL-1. Therefore, we demonstrate here a new tissue homeostatic concept: The same molecule, DEL-1, not only regulates initiation of inflammation but also promotes inflammation resolution and this functional versatility is facilitated by its compartmentalized expression. Our present findings on the pro-resolution function of DEL-1 underscore its therapeutic potential against inflammatory diseases.
Results
DEL-1 promotes inflammation resolution in periodontitis
DEL-1 expression is downregulated by inflammation, and humans with inflammatory diseases, including multiple sclerosis or the oral mucosal inflammatory disease periodontitis, express significantly less EDIL3 mRNA in active diseased sites than in inactive or healthy sites13,14,18. Periodontitis, in which neutrophils play a major pathogenic role19, is a pathology amenable to studies of resolution of inflammation20. Resolution of inflammation in human periodontitis correlates with reduction in the amount of inflammatory mediators (e.g., IL-6 and IL-17) in the fluid exudate adjacent to periodontitis inflamed lesions (gingival crevicular fluid; GCF)21. We found that DEL-1 protein is significantly elevated in the GCF upon treatment of periodontitis by mechanical debridement (scaling and root planing, SRP), which leads to clinical inflammatory resolution22, as also confirmed here by assessing probing pocket depth (PPD), a parameter that measures inflammatory tissue destruction (Supplementary Fig. 1a). Specifically, DEL-1 protein concentration was compared per patient in GCF samples from a single or multiple sites of periodontal inflammation, before and after clinical inflammatory resolution, and found to be increased after treatment (Fig. 1). Thus, DEL-1 is upregulated during clearance of human periodontal inflammation. To establish a potential cause-and-effect relationship between DEL-1 and resolution, we used the murine ligature-induced periodontitis (LIP) model, which has been used extensively to study mechanisms of inflammation and bone loss23. To study clearance of inflammation, we employed a modified version of the standard LIP model, in which we removed the ligature at day 5 and monitored the course of the inflammation for another 5 days. As control, we used mice where the ligatures were not removed. In wild-type mice, high mRNA expression of the pro-inflammatory cytokines Il6 and Il17 was induced in the gingival tissue during the ‘initiation phase’ of inflammation (days 1–5). However, during the ‘resolution phase’ (days 6–10), Il6 and Il17 mRNA expression declined sharply to baseline (Fig. 2a), consistent with the reduction of IL-6 and IL-17 protein in the GCF of periodontitis patients after treatment21. Conversely, Edil3 (hereafter referred to as Del1) mRNA and DEL-1 protein (as assessed by specific ELISA; Supplementary Fig. 1b), declined during the inflammation initiation phase but were swiftly upregulated in the resolution phase (Fig. 2a,b). Interestingly, the re-emergence of Del1 in the resolution phase was associated with elevated expression of Tgfb1 and Tgfb2, which are involved in inflammation resolution6,24 (Fig. 2a). In wild-type mice where the ligatures were not removed, the expression of Del1, Tgfb1, or Tgfb2 failed to increase after day 5, whereas high expression of Il6 and Il17 persisted through day 10 (Fig. 2a).
Fig. 1. DEL-1 is elevated in the gingival crevicular fluid (GCF) following resolution of periodontitis due to treatment with scaling and root planing (SRP).
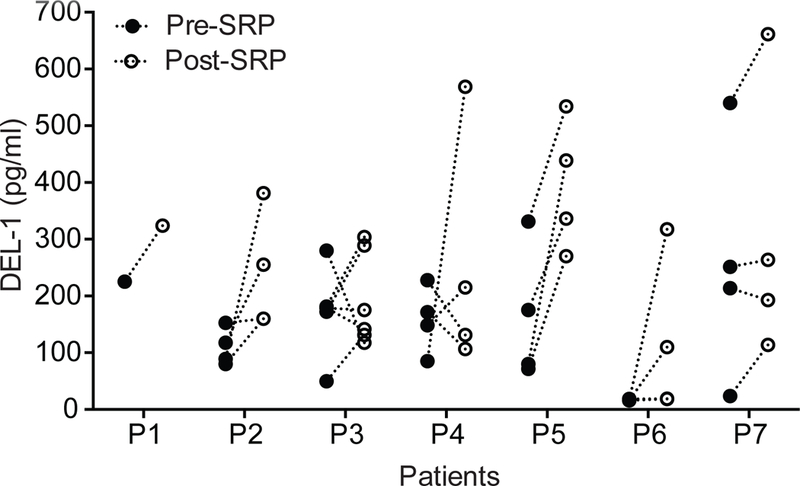
GCF was collected using PerioPaper strips from distinct periodontitis-involved sites from 7 patients before and after SRP. After elution in 0.1 ml buffer, the GCF was analyzed by ELISA for DEL-1 content. Data represent the concentration of DEL-1 in the eluted GCF samples. P = 0.0041 (Pre-post paired 2-way ANOVA with repeated measures by patient).
Fig. 2. The pro-resolution effect of DEL-1 in a resolving model of murine ligature-induced periodontitis (LIP).
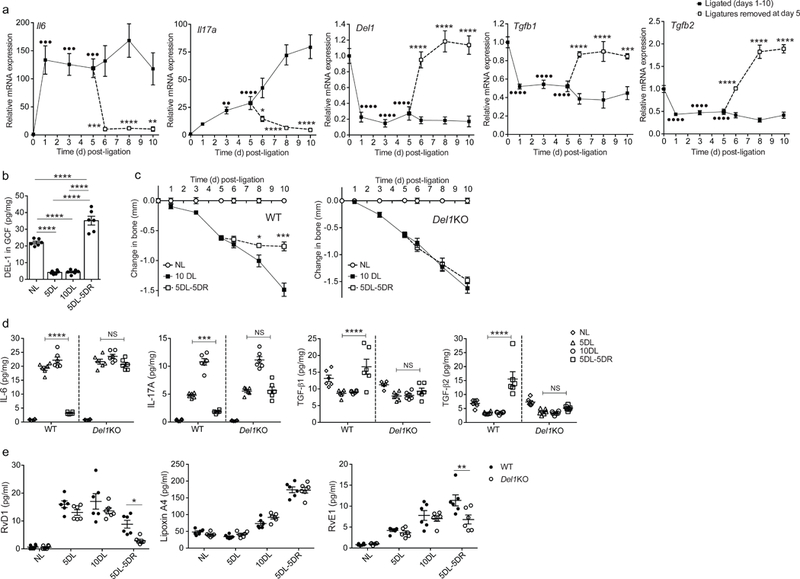
(a) Wild-type mice were subjected to LIP for up to 10 days and ligatures were removed or not at day 5 (n = 5 mice per group per time point). Relative mRNA expression of the indicated molecules in gingival tissue was assayed by real-time PCR. Data were normalized to Gapdh mRNA and are presented as fold change relative to baseline, set as 1. (b) Wild-type mice were subjected to LIP for up to 10 days and ligatures were removed or not at day 5 (n = 6 mice per group per time point). Protein concentration of DEL-1 was measured by ELISA in GCF that was recovered from ligatures and data were expressed as pg per mg of total protein (NL, not ligated; 5DL, 5 days ligated; 10DL, 10 days ligated; 5DL-5DR, 5 days ligated and 5 days with ligatures removed). (c) Bone loss was measured in littermate wild-type (WT) or Del1KO mice that were subjected to LIP for up to 10 days with or without ligature removal at day 5 (n = 5 mice per group). (d,e) Wild-type (WT) or Del1KO littermate mice were subjected to LIP for up to 10 days and ligatures were removed or not at day 5 (n = 6 mice per group) (NL, not ligated; 5DL, 5 days ligated; 10DL, 10 days ligated; 5DL-5DR, 5 days ligated and 5 days with ligatures removed). (d) Protein concentrations of IL-6, IL-17A, TGF-β1 and TGF- β2 were assessed in gingival tissue by ELISA (pg/mg of total protein are shown). (e) Concentrations of RvD1, Lipoxin A4 and RvE1 were measured in GCF recovered from ligatures (pg/ml are shown). ••P < 0.01, •••P < 0.001, ••••P < 0.0001 vs. day 0 (baseline) (a), *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. corresponding time-point without ligature removal (a, c) or between indicated groups (b, d, e). NS, non-significant. One-way ANOVA with Dunnett’s multiple comparisons test for comparison with baseline and two-way ANOVA with Sidak’s multiple comparisons test for comparisons with corresponding time-point without ligature removal (a); one-way ANOVA with Tukey’s multiple comparisons test (b); two-tailed Student’s t-test (c); two-way ANOVA and Tukey’s multiple comparisons test (d,e). Data are means ±SEM and are derived from one experiment with independent mice at each time point (a, c) or pooled from two experiments (b, d, e).
The removal or not of the ligatures after day 5 defines resolving versus non-resolving inflammation. Consistently, wild-type mice subjected to LIP developed progressive bone loss through day 10, unless the ligatures were removed at day 5, in which case bone loss was arrested (Fig. 2c). Notably, in the same model, DEL-1–deficient (Del1KO) mice exhibited persistent bone loss through day 10 even when the ligatures were removed at day 5 (Fig. 2c). In line with their failure to resolve bone loss even when the ligatures were removed, Del1KO mice maintained high expression of IL-6 and IL-17 and did not upregulate TGF-β1 and TGF-β2 expression, at both mRNA (Supplementary Fig. 1c) and protein level (Fig. 2d), despite ligature removal. Conversely, bone loss arrest upon ligature removal in wild-type littermates correlated with downregulation of IL-6 and IL-17 and upregulation of TGF-β1 and TGF-β2 protein and mRNA (Fig. 2d and Supplementary Fig. 1c). Histological analysis of the gingival tissue in the course of LIP and its resolution revealed that neutrophils in Del1KO samples were significantly increased compared with wild-type controls, especially in the resolution phase (Supplementary Fig. 1d). To further examine the role of DEL-1 in the regulation of resolution in the LIP model, we determined the concentrations of specialized pro-resolving mediators (SPMs). The amounts of resolvin D1 (RvD1) and resolvin E1 (RvE1) were decreased in the GCF from Del1KO mice compared with samples from wild-type controls collected five days after ligature removal (5DL-5DR; Fig. 2e). Lipoxin A4 was comparable between the two groups (Fig. 2e). Additionally, we tested the ability of exogenous RvD1 administration during the resolution phase of LIP to promote inflammation resolution in Del-1 deficiency. Whereas exogenously added RvD1 further promoted inflammation resolution in wild-type mice, it failed to promote resolution in Del1KO mice (Supplementary Fig. 1e-g). Together, DEL-1 deficiency is associated with defective resolution of inflammation.
DEL-1 clears inflammation through efferocytosis
We next investigated mechanisms of DEL-1 involvement in inflammation resolution. To obtain mechanistic insights and to investigate whether the observed novel action of DEL-1 represents a more general principle in resolution of inflammation, we engaged established models of peritoneal inflammation and resolution25–27. Analysis of the peritoneal exudate (Supplementary Fig. 2a) following monosodium urate (MSU) crystal injection revealed higher neutrophil numbers in Del1KO mice as compared to wild-type controls (Fig. 3a-c). DEL-1 deficiency also enhanced monocyte/macrophage recruitment only at the 8 h time point (Supplementary Fig. 2b). Given our previous reports that DEL-1 can inhibit both αLβ2 and αMβ2 integrins on leukocytes11,12, we first studied the involvement of these recruitment receptors in initiation of MSU-induced inflammation. Absence of αLβ2 integrin resulted in decreased neutrophil recruitment at 8 h (Supplementary Fig. 2c), whereas the infiltration of monocytes/macrophages remained unaffected (Supplementary Fig. 2d). On the other hand, antibody blockade of αΜβ2 integrin led to decreased neutrophil (Supplementary Fig. 2e) and monocyte/macrophage accumulation at the 8 h time point (Supplementary Fig. 2f). Thus, DEL-1 likely inhibits initial αLβ2- and αΜβ2-dependent recruitment of neutrophils and αΜβ2-dependent recruitment of monocytes in MSU-induced inflammation.
Fig. 3. DEL-1 modulates resolution of acute inflammation.
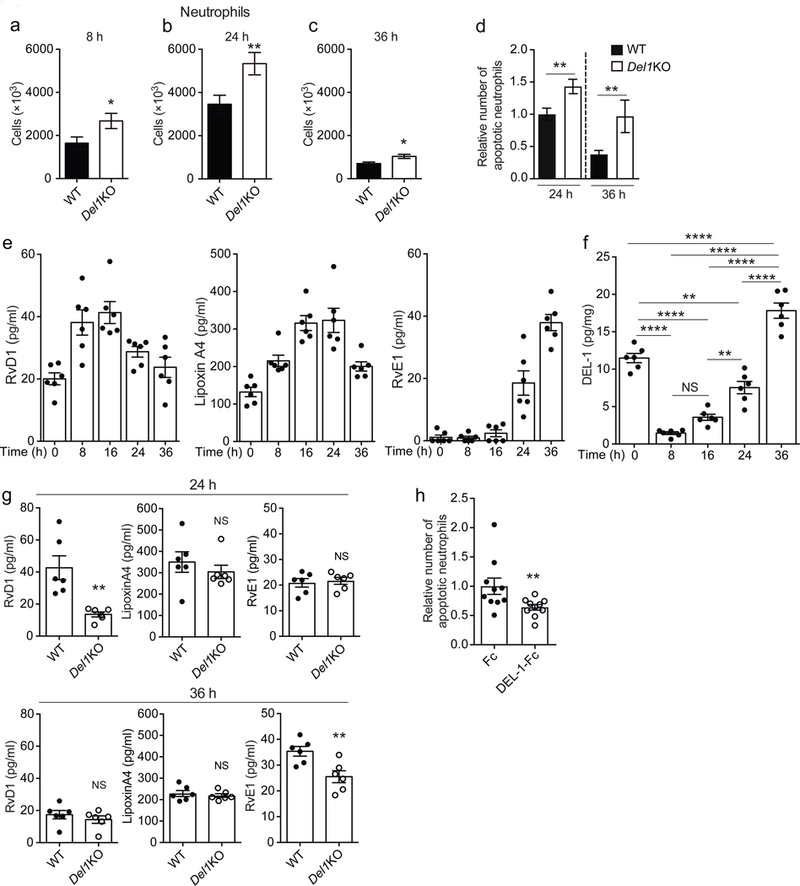
(a-c) Numbers of neutrophils in peritoneal exudates obtained from Del1KO and their respective wild-type (WT) mice at 8 h (a, n = 11 WT and 13 Del1KO mice), 24 h (b, n = 18 WT and 16 Del1KO mice) and 36 h (c, n = 8 mice per group) after MSU crystal-induced inflammation. (d) Relative number of apoptotic neutrophils detected in peritoneal exudates 24 h and 36 h after MSU crystal injection in wild-type (WT) or Del1KO mice. Number of apoptotic neutrophils was measured and expressed relative to the wild-type/24 h group, set as 1 (24 h, n = 18 WT and 16 Del1KO mice and 36 h, n = 8 mice per group). (e-f) Concentrations of RvD1, Lipoxin A4 and RvE1 (e) and protein concentration of DEL-1 (pg/mg of total protein) (f) were measured in the supernatant of peritoneal fluid obtained from untreated (0 h) wild-type mice or wild-type mice at 8 h, 16 h, 24 h and 36 h after MSU crystal-induced inflammation (n = 6 mice per group). (g) Concentrations of RvD1, Lipoxin A4 and RvE1 were measured in the supernatant of peritoneal exudates obtained from wild-type (WT) or Del1KO mice at 24 h and 36 h after MSU crystal-induced inflammation (n = 6 mice per group). (h) MSU crystals were injected in wild-type mice and, after 20 h, 5 μg of DEL-1-Fc or Fc control protein were administered intraperitoneally (i.p.); 4 h after the latter injection, the relative amount of apoptotic neutrophils in peritoneal exudates was assessed. Numbers of apoptotic neutrophils are expressed relative to the Fc control group, set as 1 (n = 10 mice per group). *P < 0.05, **P < 0.01, ****P < 0.0001, NS, non-significant ((a, b) two-tailed Student’s t-test; (c-d, g, h) two-tailed Mann-Whitney U-test; (f) One-way ANOVA with Tukey’s multiple comparisons test). Data are means ±SEM and are pooled from two experiments (a, c, d – 36 h time point, e-h) or three experiments (b, d – 24 h time point).
To address the role of DEL-1 in inflammation clearance in this model, we determined the abundance of apoptotic neutrophils at 24 h and 36 h post-MSU injection. We found increased numbers of apoptotic neutrophils in Del1KO mice as compared to wild-type mice (Fig. 3d), which might be attributed to non-redundant DEL-1-mediated regulation of apoptotic cell clearance. Given the importance of SPMs in mediating inflammation resolution28, we next performed kinetic analysis of SPMs and DEL-1 in the supernatant of peritoneal exudates in the course of MSU crystal-induced inflammation (Fig. 3e,f). The kinetic SPM data in the MSU peritonitis model (Fig. 3e) were comparable to previous SPM kinetic analyses in acute self-limited inflammation29–31. Consistent with findings from the LIP model, in the course of MSU crystal-induced inflammation, RvE1 followed a similar pattern with that of DEL-1; the concentrations of both molecules in the peritoneal exudate progressively increased from the 16 h time point onward (Fig. 3e,f). To further investigate the pro-resolving role of DEL-1, SPM concentrations were assessed in peritoneal exudates from wild-type and Del1KO mice collected during inflammation resolution. Notably, and in line with findings from the LIP model, DEL-1 deficiency resulted in decreased concentrations of RvD1 and RvE1 at 24 h and 36 h post-MSU injection, respectively (Fig. 3g). Conversely, expression analysis of Gpr18, Chemr23 and Fpr2, which encode SPM receptors, in peritoneal monocytes/macrophages sorted 24 h after MSU crystal-induced inflammation revealed comparable expression between wild-type and Del1KO mice (Supplementary Fig. 2g).
To assess whether DEL-1 is a non-redundant effector of SPMs, we examined the effect of exogenous RvD1 administration on inflammation resolution in the presence or absence of endogenous DEL-1 using the MSU crystal-induced inflammation model. Simultaneous administration of RvD1 and MSU crystals led to enhanced clearance of apoptotic neutrophils in wild-type but not in Del1KO mice 24 h after induction of inflammation (Supplementary Fig. 2h). That RvD1 failed to restore defective clearance of apoptotic neutrophils in Del1KO mice suggests that DEL-1 mediates at least a part of the SPM-dependent pro-resolution program.
To further confirm the pro-resolving function of DEL-1, we administered soluble recombinant DEL-1 (expressed as an Fc fusion protein; DEL-1-Fc) 20 h after treatment with MSU crystals (i.e., after a large portion of neutrophil recruitment has already taken place26,27) and analyzed the cellular exudate 4 h thereafter. Treatment with DEL-1-Fc reduced numbers of apoptotic neutrophils in the exudate as compared to treatment with Fc control protein (Fig. 3h), suggesting that DEL-1 enhances the clearance of apoptotic neutrophils.
We therefore set out to understand the mechanism whereby DEL-1 enhances the phagocytosis of apoptotic neutrophils by macrophages. At its C-terminus, DEL-1 has two discoidin I-like domains that are involved in the binding of phospholipid moieties32. Consistent with this property, DEL-1 bound with high affinity (Kd ≈ 26 nM) to liposomes containing PS (Fig. 4a), a major ‘eat-me’ signal on the surface of apoptotic cells. The binding of DEL-1 to PS was attributed to the discoidin I-like domains since a truncated version of DEL-1 lacking these domains (DEL-1[E1-E3]-Fc) failed to interact with PS (Fig. 4b). Accordingly, DEL-1-Fc but not DEL-1[E1-E3]-Fc or Fc control protein promoted efferocytosis of apoptotic neutrophils by macrophages in vitro both in the murine and human system (Fig. 4c,d).
Fig. 4. DEL-1 enhances efferocytic activity through interaction with phosphatidylserine and αvβ3 integrin.
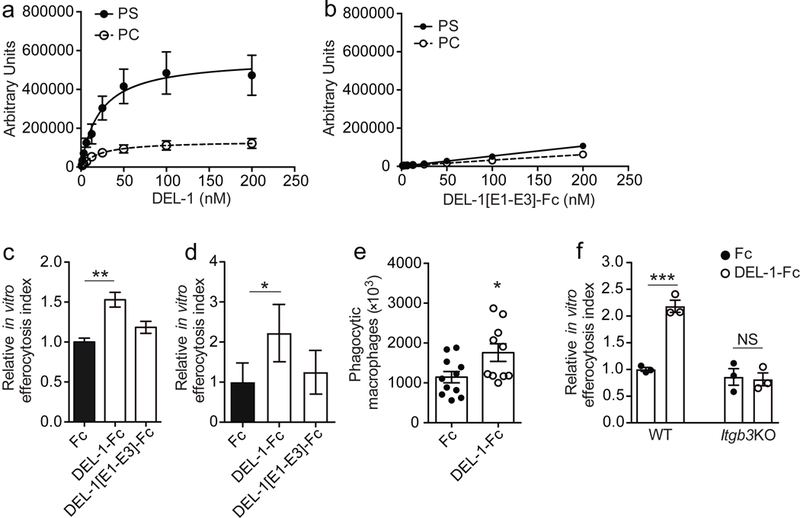
(a, b) Saturation binding of (a) full-length DEL-1, or (b) DEL-1 protein that lacks the discoidin I-like domains expressed as an Fc fusion protein (DEL-1[E1-E3]-Fc) to liposomes composed of PC/Chol. (PC) or PC/Chol./PS (PS) (n = 3 preparations in (a) and n = 2 preparations in (b)). (c, d) Primary BMDMs from wild-type mice (n = 3 separate cell isolations) (c), or human monocyte-derived macrophages (MDMs) (n = 5 separate cell isolations) (d) were co-cultured for 30 min with apoptotic neutrophils (mouse or human, respectively) that were pre-stained with BCECF-AM ((2’,7’-Bis-(2-Carboxyethyl)-5-(and-6)-Carboxyfluorescein, Acetoxymethyl Ester) in the presence of Fc control, full-length DEL-1-Fc or DEL-1 protein lacking the discoidin I-like domains (DEL-1[E1-E3]-Fc) (each 500 ng/ml). Relative efferocytosis index is shown and was calculated as the ratio of macrophages that have phagocytosed apoptotic material to the total number of macrophages. Data are expressed relative to the Fc control group set as 1. (e) Wild-type mice were injected i.p. with thioglycollate and 72 h thereafter they received a second i.p. injection of 5 × 106 pre-stained apoptotic neutrophils together with 5 μg of either Fc control or DEL-1-Fc; 3 h after the latter injection, mice were sacrificed and the number of phagocytic macrophages was analyzed (n = 11 mice for Fc control and 10 mice for Del-1-Fc). (f) In vitro phagocytosis assay was performed as described in (c). Relative efferocytosis index of BMDMs from mice deficient in β3 integrin (Itgb3KO) and of BMDMs from respective control mice (WT) in the presence of Fc control or DEL-1-Fc is shown (n = 3 separate cell isolations per genotype). *P < 0.05, **P < 0.01, ***P < 0.001, NS, non-significant (One-way ANOVA with Dunnett’s (c, d) or Tukey’s (f) multiple comparisons test; two-tailed Student’s t-test (e)). Data are means ± SEM and are derived from three experiments (a), from two experiments (b, e), from one experiment (c, f), or are pooled from five experiments (d).
To further establish the role of DEL-1-Fc in promoting efferocytosis using an independent in vivo model, we performed thioglycollate-induced peritonitis, which is conducive for studying specific aspects of efferocytosis. Specifically, wild-type mice were injected intraperitoneally with thioglycollate and after 72 h, when the peritoneal exudate has few neutrophils33, they received a second i.p. injection of 5 × 106 fluorescently (BCECF)-prestained apoptotic neutrophils together with DEL-1-Fc or Fc control. Mice were sacrificed 3 h later and the numbers of efferocytic (BCECF+F4/80+) macrophages in the peritoneal exudate were determined by flow cytometry. Treatment with DEL-1-Fc significantly increased efferocytic macrophages as compared to Fc control protein (Fig. 4e).
To address which of the two DEL-1 receptors on macrophages that are involved in macrophage efferocytosis10,12,34,35, αvβ3 integrin or αΜβ2 integrin, was the receptor mediating the DEL-1–dependent efferocytic activity, we engaged macrophages from β3 integrin- and αM integrin-deficient mice. DEL-1-Fc failed to promote efferocytosis of apoptotic neutrophils by β3 integrin-deficient macrophages (Fig. 4f). However, DEL-1-Fc significantly enhanced efferocytosis of apoptotic neutrophils by αM integrin-deficient macrophages (data not shown). Taken together, DEL-1 enhances resolution of peritoneal inflammation by promoting αvβ3 integrin-mediated efferocytosis.
DEL-1 triggers a pro-resolving macrophage phenotype
We next investigated the consequences of DEL-1–mediated clearance of apoptotic neutrophils on the efferocytic macrophages. Wild-type and Del1KO mice were subjected to MSU crystal-induced peritoneal inflammation for 24 h (a time point by which significant efferocytosis occurs) and monocytes/macrophages were then sorted and analyzed for the expression of genes serving as markers of the resolving macrophage phenotype7,36. We found that DEL-1 deficiency caused reduction in the expression of genes encoding TGF-β1, the nuclear receptors/transcription factors LXRα and LXRβ, retinoid X receptor α (RXRα), the molecules downstream of LXR signaling ATP-binding cassette transporter 1 (ABCA1), and transglutaminase 2 (TGM2), as well as the efferocytosis-associated molecules AXL, CD36 and uncoupling protein 2 (UCP2) (Fig. 5a). Importantly, the absence of DEL-1 had no effect on the expression of factors associated with the resolving macrophage phenotype at steady-state (Supplementary Fig. 3a), suggesting that DEL-1 regulates these factors only in the context of inflammation resolution.
Fig. 5. DEL-1 promotes phenotypic alterations in macrophages during inflammation resolution through LXR.
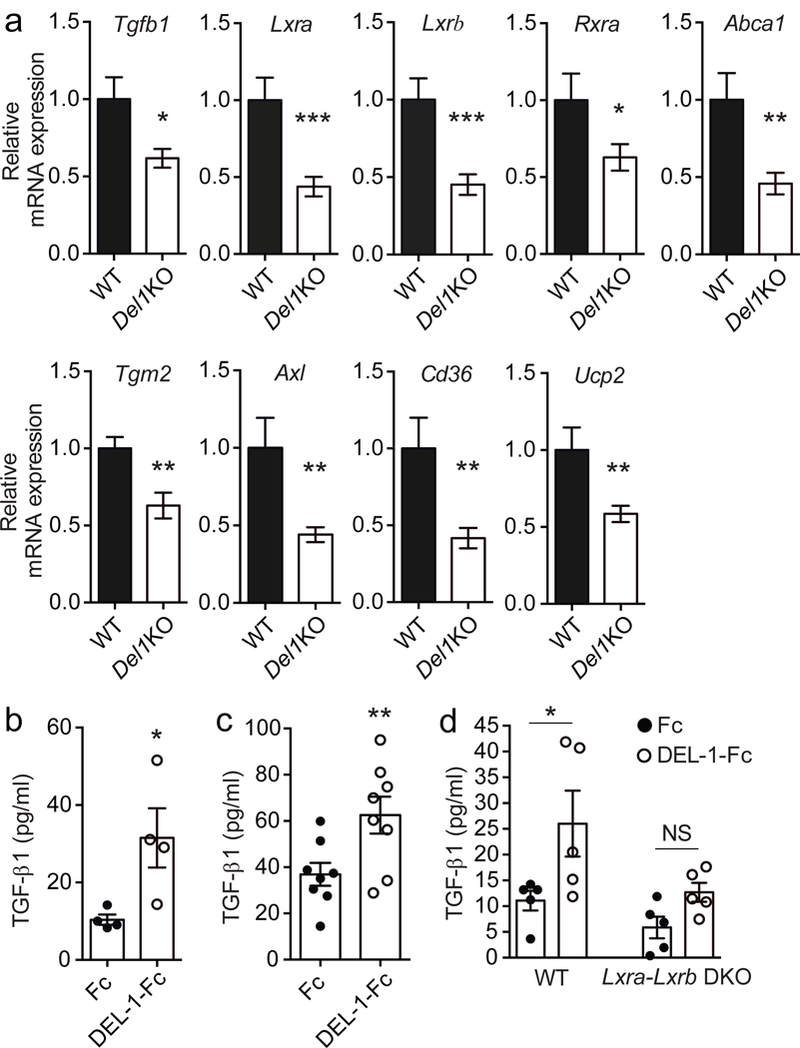
(a) Relative mRNA expression of Tgfb1, Lxra, Lxrb, Rxra, Abca1, Tgm2, Axl, Cd36 and Ucp2 in peritoneal monocytes/macrophages sorted from Del1KO and their littermate wild-type (WT) mice 24 h after MSU-induced inflammation. Relative mRNA expression was normalized against 18s and was set as 1 in wild-type monocytes/macrophages (n = 7 WT and 12 Del1KO mice). (b,c) Protein concentrations of TGF-β1 were measured in efferocytosis culture supernatants of BMDMs (n = 4 separate cell isolations) (b) or isolated peritoneal macrophages (n = 8 separate cell isolations) (c) incubated for 3 h with mouse apoptotic neutrophils in the presence of Fc control or DEL-1-Fc (500 ng/ml each). (d) Protein concentrations of TGF-β1 in the supernatants of co-cultures of apoptotic neutrophils with BMDMs from mice deficient in both LXRα and LXRβ (Lxra-LxrbDKO) or BMDMs from their respective control mice (WT) for 3 h in the presence of Fc control or DEL-1-Fc (each 500 ng/ml) (n = 5 separate cell isolations per genotype). *P < 0.05, **P < 0.01, ***P < 0.001, NS, non-significant ((a,b,c) two-tailed Student’s t-test, (d) One way ANOVA with Tukey’s multiple comparisons test). Data are means ± SEM and are pooled from two experiments (a, c), are from one experiment representative of two experiments (b) or are from one experiment (d).
In further in vitro experiments, we studied the role of DEL-1-dependent efferocytosis in the expression of factors associated with resolution of inflammation. Addition of DEL-1-Fc to efferocytosis cultures (macrophages, either bone marrow-derived macrophages [BMDMs] or isolated peritoneal macrophages, incubated with apoptotic neutrophils) stimulated the up-regulation of TGF-β1 protein secretion in the cultures (Fig. 5b,c). Moreover, DEL-1-Fc enhanced mRNA expression of Tgfb1 and additional markers of resolution (Lxra, Lxrb, Ucp2, Abca1, Tgm2 and Rxra) in macrophages from the efferocytosis cultures (Supplementary Fig. 3b–i). Macrophages treated with DEL-1-Fc in the absence of apoptotic neutrophils did not demonstrate any phenotypic alterations (Supplementary Fig. 3j), confirming that these upregulatory effects of DEL-1 are exerted only in the context of efferocytosis. Both the point mutant DEL-1[RGE]-Fc, which lacks the functional RGD motif and hence does not interact with the αvβ3 integrin, and the truncated protein DEL-1[E1-E3]-Fc, which lacks the discoidin I-like domains and does not bind PS, failed to induce the resolving phenotype in macrophages (Supplementary Fig. 4a–c), thus confirming that the pro-resolving effects of DEL-1 require its concomitant interactions with β3 integrin and PS. Inactivation of β3 integrin conclusively demonstrated that this integrin is required for the DEL-1–mediated switch of efferocytic macrophages towards a resolving phenotype (Supplementary Fig. 4d).
The in vivo and ex vivo experiments described thus far showed that DEL-1 regulates the expression of Lxra, Lxrb and their downstream effector Tgm2, which enhances the efferocytic activity of macrophages37. We therefore determined the role of LXRs as possible mediators of the DEL-1-dependent resolving macrophage phenotype. Genetic deficiency of both Lxra and Lxrb or treatment of macrophages with an LXR antagonist significantly attenuated the DEL-1-dependent up-regulation of TGF-β1 in macrophages from efferocytosis cultures (Fig. 5d, Supplementary Fig. 4e), but did not affect Tgfb1 expression in macrophages that were not exposed to apoptotic neutrophils (Supplementary Fig. 4f,g). In conclusion, DEL-1 promotes a resolving phenotype in macrophages by enhancing efferocytosis and production of pro-resolving markers in a manner that is – at least in part – dependent on LXR signaling.
DEL-1 localization determines its pro-resolving activity
The anti-leukocyte recruitment activity of DEL-1 was assumed to be mediated by endothelial cell-secreted DEL-111,14. In the present study, although DEL-1 deficiency resulted in defective resolution, the endogenous cellular source of DEL-1 promoting efferocytosis in wild-type mice was uncertain. We thus next investigated the cellular sources of DEL-1 predominantly involved in these distinct functions under the hypothesis that macrophage-derived DEL-1 regulates efferocytosis whereas endothelial cell-derived DEL-1 regulates leukocyte recruitment. Interestingly, DEL-1 expression was previously identified in resident macrophages and Del1 was identified as a peritoneal resident macrophage-selective gene38,39. We verified that Del1 was expressed by resident peritoneal macrophages, and, in particular, by GATA-6+ macrophages (Fig. 6a), which were previously ascribed an essential role in acute inflammation resolution39.
Fig. 6. Compartmentalized expression of DEL-1 regulates resolution of acute inflammation.
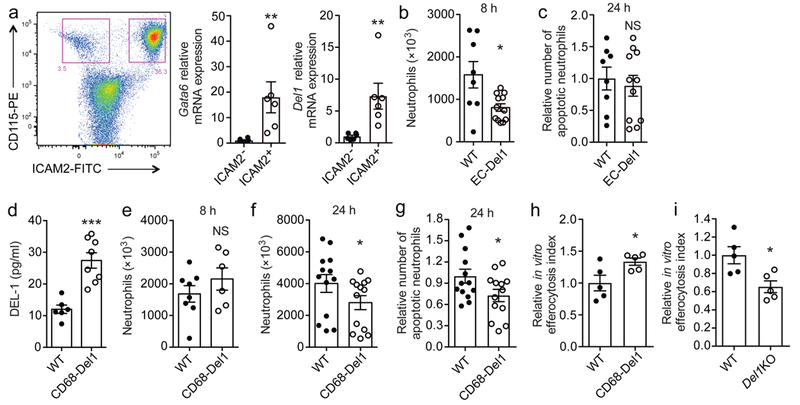
(a) Representative FACS plot and relative mRNA expression of Gata6 and Del1 in CD115+ICAM2- macrophages and CD115+ICAM2+ resident macrophages sorted from peritoneum of wild-type mice at steady-state (n = 6 mice) are shown. Relative mRNA expression was normalized against 18s and was set as 1 in CD115+ICAM2- macrophages. (b) Neutrophil numbers in peritoneal exudates collected 8 h after MSU crystal injection in mice overexpressing DEL-1 in the endothelium (EC-Del1 mice) or their littermate wild-type (WT) mice (n = 8 WT and 12 EC-Del1 mice). (c) Relative number of apoptotic neutrophils detected in peritoneal exudates 24 h after MSU crystal injection in littermate wild-type (WT) or EC-Del1 mice. Number of apoptotic neutrophils was measured and expressed relative to the WT group, set as 1 (n = 8 wild-type and 11 EC-Del1 mice). (d) DEL-1 protein concentration in the supernatant of isolated peritoneal macrophages derived from untreated (not subjected to MSU crystal-induced inflammation) mice that overexpress DEL-1 under control of the CD68 promoter (CD68-Del1) or their littermate wild-type (WT) mice (n = 6 separate cell isolations for WT mice and 8 for CD68-Del1 mice). (e, f) Numbers of neutrophils in peritoneal exudates obtained from CD68-Del1 mice or littermate wild-type (WT) mice at 8 h (e) and 24 h (f) after MSU crystal-induced inflammation ((e) n = 8 WT and 6 CD68-Del1 mice, (f) n = 13 mice per group). (g) Relative numbers of apoptotic neutrophils in peritoneal exudates obtained from wild-type (WT) and CD68-Del1 mice 24 h after MSU crystal-induced inflammation. Numbers of apoptotic neutrophils are expressed relative to the WT group, set as 1 (n = 13 mice per group). (h, i) Isolated peritoneal macrophages from untreated CD68-Del1 mice (h), or untreated Del1KO mice (i), and from their untreated respective wild-type (WT) mice were incubated for 30 min with apoptotic neutrophils that were pre-stained with BCECF-AM. Relative efferocytosis index is shown and was calculated as the ratio of macrophages that have phagocytosed apoptotic material to the total number of macrophages. Data are expressed relative to the respective WT group, set as 1 (n = 5 separate cell isolations per genotype). *P < 0.05, **P < 0.01, ***P < 0.001. NS, non-significant ((b, c, g) two-tailed Student’s t-test, (a, d, e, f, h, i) two-tailed Mann-Whitney U-test). Data are means ± SEM and are from one experiment representative of two (a) or are pooled from two experiments (b-g) or from three experiments (h, i).
We next assessed whether the two distinct functions of DEL-1, inhibition of leukocyte recruitment and promotion of efferocytosis, correlate with its spatial cellular distribution in endothelial cells and macrophages, respectively. Using mice with endothelial-specific overexpression of DEL-1 (EC-Del1) in the acute MSU-induced inflammation model, we found that neutrophil recruitment during the initial phase of MSU crystal-induced inflammation was significantly decreased in peritoneal exudates from EC-Del1 mice as compared to littermate controls (Fig. 6b). In contrast, endothelial-specific DEL-1 overexpression failed to promote the clearance of apoptotic neutrophils assessed at 24 h post-MSU injection (Fig. 6c). Conversely, in the same assays, mice with macrophage-specific DEL-1 overexpression (under control of the CD68 promoter; Fig. 6d, Supplementary Fig. 5a) did not display any difference in initial neutrophil recruitment (Fig. 6e), but demonstrated a significant decrease in total neutrophils and apoptotic neutrophils at 24 h (Fig. 6f,g). The impact of macrophage-specific overexpression of DEL-1 on inflammation resolution was corroborated using our in vitro efferocytosis assay. DEL-1–overexpressing peritoneal macrophages exhibited enhanced efferocytosis compared to wild-type cells (Fig. 6h). Conversely, but consistently, peritoneal macrophages from Del1KO mice exhibited decreased efferocytosis index as compared to macrophages derived from wild-type controls (Fig. 6i). Macrophage-specific overexpression of truncated DEL-1 that lacks the discoidin I-like domains (DEL-1[E1-E3]; Supplementary Fig. 5b) in mice failed to influence the numbers of total neutrophils and of apoptotic neutrophils, as compared to their respective wild-type controls (Supplementary Fig. 5c,d), confirming the specificity of the overexpression effect and the requirement for PS binding.
Having shown the importance of macrophage αvβ3 integrin in DEL-1–dependent efferocytosis, we next examined whether macrophage-derived DEL-1 might interfere with further receptor pairs potentially involved in the formation of the phagocytic synapse. To this end, we performed in vitro efferocytosis assays with macrophages over-expressing full-length DEL-1, in the presence or absence of antibody-mediated inhibition of the receptors LFA-1 and Mac-1 or their counter-receptor ICAM-1. None of the tested inhibitors influenced DEL-1–dependent efferocytosis (Supplementary Fig. 6a,b). Together, these data unveil that the efferocytosis and inflammation resolution functions are primarily associated with macrophage-derived DEL-1, whereas endothelial-derived DEL-1 is primarily involved in restraining leukocyte recruitment and initiation of inflammation.
Discussion
Inflammation resolution does not represent passive termination of inflammatory responses but rather a well-coordinated active process to reinstate tissue integrity and function1,2. Although SPMs play important roles in resolution28, the network of molecules and mechanisms that coordinate this complex process is incompletely understood. DEL-1 was previously implicated as an inhibitor of inflammatory cell recruitment and initiation of inflammation11,14,16. We now demonstrate that DEL-1 pro-actively modulates inflammation resolution by facilitating efferocytosis and by inducing phenotypic alterations in macrophages, driving them toward a resolving phenotype. Moreover, the present study indicates that DEL-1 is a non-redundant effector of SPM-dependent inflammation resolution and is required for optimal production of at least certain important SPMs (RvD1 and RvE1). Our findings, therefore, establish DEL-1 as an endogenous regulator of functional immune plasticity.
In line with its upregulation during resolution of human or experimental mouse periodontitis, DEL-1 had a non-redundant pro-resolving role in two murine inflammatory models. First, DEL-1 deficiency resulted in non-resolving inflammation and persistent bone loss, associated with reduced amounts of RvD1 and RvE1, in a modified model of LIP that allowed the study of inflammation resolution. These findings are therapeutically important for periodontitis, an oral inflammatory disease associated with increased risk for systemic disorders40.
To determine if the pro-resolving action of DEL-1 in periodontitis can be generalized to different settings and to elucidate potential mechanisms of inflammation resolution involving DEL-1, we employed a model of self-limited MSU crystal–induced peritoneal inflammation. In this model, endogenous DEL-1 deficiency caused defective clearance of apoptotic neutrophils and attenuated expression of resolving markers in macrophages; conversely, administration of exogenous DEL-1 resulted in improved inflammation resolution. In terms of structure-function relationships, we showed that DEL-1 binds to PS through its discoidin I-like domains, thereby enabling the recognition of apoptotic neutrophils. Moreover, the additional interaction of DEL-1 with αvβ3 integrin on macrophages through the RGD site in its second EGF-repeat promotes the uptake of apoptotic neutrophils.
Intriguingly, the pro-resolving effect of exogenously added RvD1 failed in Del1KO mice and, furthermore, the defective efferocytosis in Del1KO mice resulted in decreased production of endogenous SPMs, an effect, which would further aggravate defective resolution associated with DEL-1 deficiency. Thus, DEL-1 is not only an essential effector of inflammation resolution but also promotes SPM production, therefore possibly generating a positive-feedback loop that reinforces resolution. These mechanistic findings reveal a new connection between DEL-1 and resolvins and might in part explain the successful use of resolvins in preclinical models of inflammatory diseases41,42.
The nuclear receptor LXR regulates important aspects of immunity and inflammation43,44. The ability of DEL-1 to upregulate TGF-β1 in efferocytic macrophages was, at least in part, dependent on Lxra and Lxrb. Moreover, DEL-1 up-regulated both Lxr isoforms and dependent downstream molecules, such as Tgm2, which enhances apoptotic cell clearance by macrophages37, suggesting that DEL-1 promotes LXR activation. In this regard, given its lipid-binding capacity, DEL-1 might increase the local concentration or availability of lipid agonists derived from engulfed apoptotic material, thereby promoting LXR activation in efferocytic macrophages43,45. LXR activation in macrophages was also shown to inhibit LPS-induced expression of Th17-inducing cytokines such as IL-1β and IL-646. This mechanism might in part explain the persistence of the gingival IL-17 response in Del1KO mice despite ligature removal, which in DEL-1-proficient mice leads to resolution. It should be noted that IL-17 has been implicated as the driver of destructive inflammation associated with DEL-1 deficiency in preclinical models of periodontitis and multiple sclerosis13,14.
DEL-1 associates with the cell surface and extracellular matrix and is a highly adhesive protein, which might substantially prevent its diffusion upon secretion11,47. Therefore, the expression of DEL-1 at a site where it is specifically needed becomes a critical regulatory issue for homeostatic immunity. This notion is consistent with our experimental evidence herein showing that mice overexpressing DEL-1 in their endothelium displayed decreased neutrophil recruitment upon MSU crystal-induced inflammation while maintaining unaltered efferocytic/pro-resolving activity. The latter activity was enhanced by macrophage-specific overexpression of DEL-1, lending support to this novel location-dependent homeostatic principle. This ‘location principle’ might constitute a critical determinant of the immune-modulatory functions of additional homeostatic molecules. A future study using mice with a conditional Del1 allele and a myeloid cell-specific deletion of DEL-1 could fully address the physiological relevance of the pro-resolving action of macrophage-derived DEL-1.
We additionally confirmed here that Del1 was predominantly expressed by Gata6+ macrophages38,39, which represent resident macrophages. This finding, together with the local endothelial expression of DEL-1, is consistent with the emerging understanding that local tissues are not passive recipients of recruited immune surveillance but rather have a ‘regulatory say’ over inflammatory responses8,48,49.
Taken together, our present findings support that the versatile functionality of DEL-1 is dependent upon distinct structural domains and the differential temporospatial expression of this molecule. DEL-1 is a determinant of functional immune plasticity at the tissue level and has a dual mode of action in the course of inflammation. It can both regulate inflammatory cell recruitment and shape macrophage plasticity toward a resolving phenotype, each of which properties could be therapeutically exploited to inhibit destructive inflammation and mediate the reconstitution of tissue integrity in inflammatory diseases.
METHODS
Human subjects and gingival crevicular fluid (GCF) sample collection
Research procedures were performed in compliance with all relevant ethical regulations under a protocol approved by the Institutional Review Board of the University of Pennsylvania. Signed informed consent was obtained to procure GCF from individuals treated in the postgraduate clinic of the Department of Periodontics, University of Pennsylvania School of Dental Medicine. All subjects were 28 years of age or older with no serious medical illness contraindicative of periodontal treatment. The inclusion criteria for chronic periodontitis were diseased sites with probing pocket depth ≥ 5 mm associated with clinical attachment loss ≥ 4 mm, presence of bleeding on probing, and radiographic evidence of bone loss. GCF was collected from 7 patients (3 males and 4 females) prior to initiation of scaling and root planing (SRP) and at a reevaluation appointment 6 weeks later. To collect GCF, teeth were dried and isolated with cotton rolls and samples were collected from distinct periodontitis-involved sites using PerioPaper strips (OraFlow Inc.). The strips were placed in the selected sites until mild resistance was felt and kept in place for 30 s, as previously described50,51. Samples contaminated with blood or saliva were discarded. Each PerioPaper strip was placed into a coded sealed polypropelene tube containing 0.1 ml phosphate-buffered saline with proteinase inhibitors. The samples were placed at 4 °C for 2 h, and then stored at −80 °C until analysis.
Mice
Mice were housed under specific pathogen-free conditions on a standard 12 / 12 h light/dark cycle. 8–10 weeks old sex-matched mice were used. Food and water were provided ad libitum. In experiments where only wild-type C57BL/6 mice were used, these were purchased from Janvier Labs or the Jackson Laboratory. Del1KO mice and mice overexpressing DEL-1 in the endothelium (EC-Del1; generated by using a Tie2 promoter/enhancer construct) have been previously described11,16,52,53. Mice overexpressing in macrophages full-length DEL-1 (CD68-Del1) or the truncated version of DEL-1 that lacks the discoidin I-like domains (CD68-Del1-[E1-E3]) were generated in C57BL/6N background, using the human CD68 promoter54,55. The CD68-IVS1 promoter construct was subcloned into the pUC19 vector backbone. Mouse Edil3 (encoding full length DEL-1 and E1-E3 derivative) were amplified by PCR and Edil3 cDNA (full length or E1-E3) was cloned into the pUC19-CD68 vector and subsequently amplified in Escherichia coli strain DH5α. For generation of transgenic mice, the constructs were linearized and microinjected into pronuclei of mouse oocytes (C57BL/6NCrl). Offspring were genotyped by PCR and transgene-bearing founders were further crossed with C57BL/6N mice (Charles River). Following screening for transgene transmission to the offspring and Del1 overexpression by qPCR, two founders from each strain (CD68-Del1 and CD68-Del1-[E1-E3]) were selected. Data are presented from one founder per strain; key experiments were repeated using the second founder in each case and similar results were obtained. Mice deficient in αL integrin (ItgalKO), or αM integrin (ItgamKO) were previously described11,12,56,57. Mice deficient in β3 integrin (Itgb3KO) and their respective wild-type control mice were from the Jackson Laboratory. Mice deficient in both LXR isoforms (Nr1h3KONr1h2KO; here referred to as Lxra-LxrbDKO) in a C57BL/6 background43,58 were originally provided from D. Mangelsdorf (University Texas Southwestern). All animal experiments were performed in compliance with all relevant ethical regulations. Specifically, Lxra-LxrbDKO mice and their respective wild-type control mice were housed and used in accordance with the CSIC and ULPGC Animal Research Committees; all other animal procedures were approved by the Landesdirektion Sachsen, Germany or the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania.
MSU crystals
MSU crystals were prepared as described59. Briefly, uric acid (Sigma) was added to H2O containing NaOH and the solution was boiled until dissolution was reached. Afterwards, the solution was passed through a 0.2-μM filter and NaCl was added. After seven days of storage at 26 °C, crystals were washed with ethanol and their shape was examined using microscopy.
MSU peritonitis and treatment of mice with antibodies and proteins
MSU crystals (3 mg) were injected intraperitoneally (i.p.) in mice and peritoneal exudates were collected at different time points (8, 24, 36 h) for the analysis of inflammatory cell composition. To examine the role of αΜβ2 integrin in MSU crystal-induced leukocyte recruitment, 50 μg of a blocking antibody against αM integrin (clone M1/70; BioLegend) or an isotype control (clone RTK4530; BioLegend) were injected intravenously in mice 10 min prior to i.p. administration of the MSU-crystals. Mice were sacrificed and peritoneal exudates were analyzed 8 h after the MSU crystal injection.
The effect of DEL-1 on the regulation of resolution of MSU crystal-induced inflammation was investigated by injecting 5 μg of either DEL-1-Fc or Fc control i.p. 20 h after the treatment of mice with MSU crystals. 4 h after DEL-1-Fc injection, mice were sacrificed and clearance of apoptotic neutrophils was evaluated in peritoneal exudates.
In other experiments, 300 ng of RvD1 or vehicle control (PBS) were administered i.p. together with MSU crystals in wild-type and Del1KO mice. 24 h after injection, the numbers of apoptotic neutrophils were measured in peritoneal exudates.
Resolving & non-resolving model of ligature-induced periodontitis in mice
The placement of ligatures accelerates bacteria-mediated inflammation and bone loss60. To induce ligature-induced periodontitis, a 5–0 silk ligature was tied around the maxillary left second molar of mice, as previously described61. The contralateral molar tooth in each mouse was left unligated to serve as baseline control for bone loss measurements and gingival gene expression. The ligatures remained in place for 5 or 10 days (5DL and 10DL groups, respectively). In another group, ligatures were removed at day 5 to allow transition to inflammation resolution and the mice were monitored for another 5 days (5DL-5DR group). The animals were euthanized at various timepoints up to day 10 as specified in figure legends. Defleshed maxillae were used to measure bone heights (i.e., the distances from the cementoenamel junction [CEJ] to the alveolar bone crest [ABC]) at 6 predetermined points on the ligated site as previously described61. To calculate bone loss, the 6-site total CEJ-ABC distance for the ligated site of each mouse was subtracted from the 6-site total CEJ-ABC distance of the contralateral unligated site. The results were presented in mm and negative values indicate bone loss relative to the baseline (unligated control). Excised gingival tissue was used to extract total RNA using the RNeasy Mini Kit (Qiagen). The RNA was quantified by spectrometry at 260 and 280 nm and was reverse-transcribed using the High Capacity RNA-to-cDNA Kit (Life Technologies). Quantitative real-time PCR with cDNA was performed using the Applied Biosystems 7500 Fast Real-Time PCR System according to the manufacturer’s protocol (Life Technologies). Data were analyzed using the comparative (ΔΔCt) method. TaqMan probes, sense primers, and antisense primers for detection and quantification of genes investigated in Fig. 2a and Supplementary Fig. 1c were purchased from Life Technologies. Mouse GCF was collected based on a previously described method62. Briefly, a fresh ligature was placed in the gingival crevice for 10 min and the collected GCF was eluted in a solution of PBS containing 1% Triton-X-100. Coronal sections of maxillae and intact surrounding gingival tissue from wild-type and Del1KO mice were prepared as previously described63 and stained with a primary monoclonal antibody against Ly6G (clone RB6–8C5, eBioscience) followed by a secondary goat anti-rat IgG antibody conjugated to Alexa Fluor-488. Ly6G+ cells were enumerated from three random sections from each of six mice per time point per genotype.
Isolation and culture of primary human and mouse cells
Human monocyte-derived macrophages (MDMs):
Peripheral blood mononuclear cells were isolated from healthy donors by density gradient separation using Histopaque 1077 (Sigma). To induce differentiation to macrophages, cells were plated and cultured in the presence of 20 ng/ml human macrophage colony stimulating factor (M-CSF) (eBioscience) for 4 days.
Human neutrophils:
Neutrophils were isolated from peripheral blood of healthy donors using double gradient separation (Histopaque 1119 and 1077; Sigma). To induce apoptosis, purified cells were incubated in complete culture medium overnight at 37 °C. Apoptotic rate was confirmed with annexin V staining (eBioscience).
Peripheral blood was collected from healthy volunteers in compliance with relevant ethical regulations (after written informed consent; the procedure was approved by the Ethics Committee of the Technische Universität Dresden).
Mouse bone marrow-derived macrophages (BMDMs) and neutrophils:
Bone marrow cells were flushed from femurs and tibiae of wild-type, CD68-Del1, CD68-Del1-[E1-E3] or different KO mice. After lysis of erythrocytes using RBC lysis buffer (eBioscience), cells were plated and cultured in the presence of recombinant murine granulocyte macrophage colony-stimulating factor (GM-CSF; 20 ng/ml, eBioscience). Culture medium was replaced every two days, and after seven days differentiated BMDMs were used for further experiments25,56. To isolate neutrophils, bone marrow cells, after erythrocyte lysis, were centrifuged in 62% Percoll gradient (GE Healthcare). Induction of apoptosis was performed after overnight culture of the obtained neutrophil population in HBSS containing 1% FBS.
Isolated peritoneal macrophages:
For peritoneal macrophage isolation, peritoneal fluid was obtained from untreated Del1KO, CD68-Del1 or CD68-Del1-[E1-E3] mice and their respective wild-type controls. Isolated peritoneal macrophages were plated and cultured overnight. Culture medium was then removed, cells were washed in PBS and the adherent cells population was used in in vitro efferocytosis assays and gene expression analyses.
Efferocytosis assays
In vitro:
In vitro analysis of efferocytosis as well as subsequent gene and protein expression analysis in efferocytic macrophages were performed as described43,64–66. Human MDMs or mouse wild-type BMDMs were pretreated with DEL-1-Fc, DEL-1[E1–E3]-Fc, DEL-1[RGE]-Fc or Fc control protein for 15 min in 96-well plates. Apoptotic neutrophils were stained with 1 μg/ml of 2’,7’-Bis-(2-Carboxyethyl)-5-(and-6)-Carboxyfluorescein, Acetoxymethyl Ester (BCECF AM; ThermoFisher Scientific). Afterwards, human MDMs or mouse wild-type BMDMs were co-cultured with apoptotic neutrophils of human or mouse origin, respectively, in a 5:1 ratio of apoptotic neutrophils to macrophages (unless otherwise indicated) for 30 min. In other experiments, isolated peritoneal macrophages (see ‘Isolation and culture of primary human and mouse cells’) from Del1KO or CD68-Del1 mice, or from their respective wild-type mice, were incubated with apoptotic neutrophils as described above for BMDMs. Macrophages were then washed three times with PBS and fixed with 4% paraformaldehyde for 30 min. Efferocytosis index was evaluated using microscopy (Axio Observer Z1 microscope, Carl Zeiss). Zeiss ZEN 2012 (blue edition) software was used for collection and analysis of images. In some cases, ImageJ software was also used for image analysis. At least three different fields per sample were analyzed and at least 30 macrophages per field were evaluated.
For gene expression analysis, macrophages were incubated with apoptotic neutrophils for 3 h; thereafter, macrophages were washed three times with PBS prior to RNA isolation. For protein analysis, macrophages were incubated with apoptotic neutrophils for 3 h in serum-free medium; thereafter, the cell-free supernatant of the co-culture was collected and analyzed by ELISA.
Intervention studies were performed by treating BMDMs with a selective antagonist for LXR (1 μM; Axon) for 15 min prior to the addition of DEL-1 proteins.
In order to examine the effect of LFA-1, Mac-1 or ICAM-1 on DEL-1–dependent efferocytosis, isolated peritoneal macrophages from CD68-Del1 and littermate wild-type mice were incubated with apoptotic neutrophils in the presence of isotype controls or a neutralizing antibody (10 μg/ml each) against LFA-1 integrin (clone 17/4), Mac-1 integrin (clone M1/70) or ICAM-1 (clone YN1/1.7.4). Apoptotic neutrophils were added to the macrophage culture for 30 min. Macrophages were then washed and efferocytosis index was evaluated, as described above.
In vivo:
Wild-type mice were injected intraperitoneally (i.p.) with 2% w/v thioglycollate (Sigma). After 72 h, animals received i.p. 5 × 106 apoptotic neutrophils, that were pre-stained with BCECF-AM, together with 5 μg of either Fc control or DEL-1-Fc; 3 h after the latter injection, mice were sacrificed and the efferocytosis index was analyzed in the peritoneal exudates with flow cytometry. Phagocytic macrophages were identified as BCECF+F4/80+ cells.
DEL-1 proteins
Full-length human DEL-1 was generated and purified as a fusion protein with the human IgG1-Fc fragment (DEL-1–Fc) as described9,10. DEL-1 that lacks the discoidin I-like domains (DEL-1[E1–E3]-Fc) and a point mutant protein in which Glu [E] was substituted for Asp [D] in the RGD motif of the second EGF repeat (DEL-1[RGE]-Fc) were prepared as previously described9. Recombinant human DEL-1 and IgG Fc protein were purchased from R&D Systems.
Liposome preparation and quality control
Chloroform (Roth) / methanol (VWR) (10/1) solutions of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC, Avanti Polar Lipids Inc.), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS, Avanti Polar Lipids Inc.) and cholesterol (Chol; Sigma Aldrich), were mixed in a molar ratio of 60:40 (POPC:Chol) or 55:40:5 (POPC:Chol:POPS) with a total lipid content of 1 mg. The solvent was evaporated under a gentle nitrogen stream to form a uniform lipid film and further dried under vacuum for at least 1 h. The lipid film was rehydrated in 1 ml of HBS (25 mM HEPES, 150 mM NaCl, pH 7.4), subjected to 11 freeze-thaw cycles and extruded 21 times through a 100 nm membrane (Whatman).
The quality of liposomes was checked by thin layer chromatography (TLC, Supplementary Fig. 6c); therefore 30 μl of liposomes were extracted with 90 μl chloroform:methanol (1:1). The lower organic phase was collected and loaded on a silica plate (Merck KGaA). Lipids were separated in a solvent mixture of chloroform : ethanol:water:triethylamine (30:35:7:35 v/v). The dried plate was stained with primuline (Sigma Aldrich; 5 mg in 100 ml acetone:water (80:20)) and the fluorescence was recorded on a Typhoon 9410 (Amersham Bioscience).
For Zeta potential measurements (Supplementary Fig. 6d), liposomes were diluted 10 times in water (ZetaSizer - Malvern).
In-vitro lipid binding assay
The binding assay was performed using a platform of Mesoscale Discoveries. Liposomes were passively adsorbed on the electrode surface for 1 h at 23°C, the residual sites on the surface were blocked for 1 h with 0.2% (w/v) porcine gelatin (Sigma) in HBS. After three wash steps with HBS a serial dilution of recombinant DEL-1 in blocking buffer, covering the desired range, was applied and incubated for 2 h. Unbound protein was removed and rabbit-anti-DEL-1 serum (1:2000 in blocking buffer) was applied for 1 h followed by three wash steps with HBS. For detection a goat-anti-rabbit secondary antibody conjugated with Sulfo-Tag (Mesoscale Discoveries) was used at 1.25 μg/ml in blocking buffer for 1 h in the dark. Free secondary antibody was washed off and reading buffer (surfactant-free reading buffer from Mesoscale Discoveries) was added. The readout was performed on a SECTOR Imager 6000 chemiluminescence reader. The resulting data was analyzed with GraphPad Prism6. A non-linear curve fitting was applied and the binding kinetics calculated using one-sited total and nonspecific binding; the binding of DEL-1 to PC vesicles was defined as background.
Flow cytometry and sorting
Antibodies against CD11b (clone M1/70, BD Biosciences), Ly-6G (clone 1A8, BD Biosciences), CD115 (clone AFS98, BioLegend), ICAM2 (CD102; clone 3C4 (mIC2/4), BD Biosciences) and F4/80 (clone BM8, eBioscience) were used for flow cytometry67. Antibody against mouse CD16/32 was used to block Fcγ receptors (clone 2.4G2, BD Biosciences). Peritoneal neutrophils were identified as Ly-6G+CD11b+ cells. Peritoneal monocytes/macrophages from MSU crystal-treated mice were defined as Ly-6G–CD11b+ cells after excluding the SSC high eosinophil population68 (gate R3 in Supplementary Fig. 2a; cells in R3 were identified as eosinophils by additional Siglec-F staining (not shown)). Apoptotic neutrophils were detected using an annexin V apoptosis detection kit (eBioscience) and cell viability was determined after staining with Hoechst 33258 (Thermo Fisher Scientific). Flow cytometric analysis of stained cells was performed on a FACS Canto II flow cytometer (BD Biosciences). After the removal of erythrocytes with RBC lysis buffer (eBioscience), cells from the peritoneal exudates were pre-incubated with anti-mouse CD16/CD32 antibody and then incubated with antibodies against specific surface receptors in Phosphate-Buffered Saline (PBS) supplemented with 5% fetal bovine serum (FBS; Life Technologies).
Cell sorting was performed using a FACS Aria cell sorter (BD Biosciences) to >95% purity. Peritoneal monocytes/macrophages from MSU crystal-treated mice, defined as above, were sorted at 24 h after the induction of MSU crystal-induced inflammation. In other experiments, macrophages were sorted as CD11b+F4/80+ cells from peritoneal fluids of untreated mice (not subjected to MSU crystal-induced inflammation). GATA-6+ resident macrophages in peritoneal fluids of untreated mice were characterized and sorted as the CD115+ICAM2+ cells, as previously described69. In addition, CD115+ICAM2- macrophages were sorted from peritoneum of untreated mice. Data analysis was performed using FlowJo software (Tree Star).
Quantitative real-time PCR
Total RNA was extracted from cultured or sorted cells and was quantified by spectrometry at 260 and 280 nm. Complementary DNA was synthesized using the iScript cDNA Synthesis Kit (Bio-Rad) or the High Capacity RNA-to-cDNA Kit (Life Technologies). Real-time PCR was performed using the SsoFast EvaGreen Supermix (BioRad) and gene-specific primers (Supplementary Table 1) in a CFX384 Real time PCR detection system (BioRad). 18s was used as an internal control for normalization56. Data were analyzed using the comparative (ΔΔCt) method.
Enzyme-linked immunosorbent assays (ELISAs)
Mouse IL-6, IL-17A, TGF-β1 and TGF-β2 proteins were measured in gingival tissue lysates using kits from eBioscience or R&D Systems. TGF-β1 protein in cell culture supernatants was assessed with ELISA using a commercially available kit (BioLegend). The DEL-1 concentration in murine GCF, supernatants of peritoneal exudates and isolated peritoneal macrophage cultures was determined using a sandwich ELISA, as previously described9. Specificity of this ELISA was confirmed as shown in Supplementary Fig. 1b. Concentrations of SPMs were measured in murine GCF and supernatant of peritoneal exudates using commercially available ELISA kits (Cayman Chemical for RvD1, MyBioSource for RvE1 and Neogen for Lipoxin A4). According to the manufacturers, the ELISA for Lipoxin A4 exhibits 24% cross-reactivity for 15-epi-Lipoxin A4, 5% for 5(S),6(R)-DiHete and 1% for Lipoxin B4; the ELISA for RvD1 has 20% cross-reactivity for 5(S),6(R)-Lipoxin A4, 4.2% cross-reactivity for 17(R)-Resolvin D1 and 0.7% cross-reactivity for 10(S),17(S)-DiHDoHE. Minor cross-reactivities (≤ 0.1%) of both Lipoxin A4 and RvD1 also exist for other SPMs or analogues thereof. According to the manufacturer, no significant cross-reactivity between RvE1 and analogues has been observed, although cross-reactivities cannot be formally excluded. At the time of analysis for human DEL-1 content of collected GCF from periodontitis patients, 1% Triton X-100 was added to the buffer in each tube sample and incubated at 4 °C for 2 h to elute the GCF contents. After centrifugation (500g for 5 min), the concentration of human DEL-1 in the eluted GCF samples was measured using an ELISA kit according to the instructions of the manufacturer (R&D Systems).
Statistics
Results are presented as means ± standard error of means (SEM). Data were analyzed by two-tailed Student’s t-test or two-tailed Mann-Whitney U-test, as appropriate (after testing for normality). Multiple-group comparisons were performed using analysis of variance (ANOVA) and the Dunnett’s, Tukey’s or Sidak’s multiple comparison tests. Analysis of human GCF samples (Fig. 1) and PPD (Supplementary Fig. 1a) was performed by pre-post paired 2-way ANOVA with repeated measures by patient. All statistical analyses were performed on GraphPad Prism software (GraphPad Inc). P values <0.05 were considered to be statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the European Research Council (DEMETINL to T.C.), the Deutsche Forschungsgemeinschaft (Transregio-SFB 83 to Ü.C. as well as Transregio-SFB 127 and Transregio-SFB 205 to T.C.) and the NIH (AI068730, DE024153, DE024716, DE026152 and DE015254 to G.H.). I.K. was supported by the MeDDriveGrant (60.400) from the Technische Universität (TU) Dresden Medical Faculty. A.Ca was supported by Spanish MINECO SAF2014–56819R and SAF2015–71878-REDT. This work was supported in part by intramural NIDCR, NIH funding. We thank R. Naumann (Transgenic Core Facility, MPI-CBG Dresden, Germany) for microinjections and B. Gercken and M. Schuster for technical assistance.
Parts of this work are included in the Master’s theses of C. Doreth at the TU Dresden, Dresden, Germany and of A. Ferreira at the University of Porto, Porto, Portugal.
Footnotes
COMPETING FINANCIAL INTERESTS
All authors declare that they have no competing interests.
Life Sciences Reporting Summary
Further information on experimental design and reagents is available in the Life Sciences Reporting Summary.
Data availability
The data that support the findings of this study are available from the corresponding authors upon request.
REFERENCES
- 1.Fullerton JN & Gilroy DW Resolution of inflammation: a new therapeutic frontier. Nat Rev Drug Discov 15, 551–567 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Serhan CN Pro-resolving lipid mediators are leads for resolution physiology. Nature 510, 92–101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Serhan CN, Chiang N & Dalli J The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Semin. Immunol 27, 200–215 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenlee-Wacker MC Clearance of apoptotic neutrophils and resolution of inflammation. Immunol. Rev 273, 357–370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Birge RB et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ 23, 962–978 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ortega-Gómez A, Perretti M & Soehnlein O Resolution of inflammation: an integrated view. EMBO Mol Med 5, 661–674 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.A-Gonzalez N & Hidalgo A Nuclear Receptors and Clearance of Apoptotic Cells: Stimulating the Macrophage’s Appetite. Front Immunol 5, 211 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hajishengallis G & Chavakis T Endogenous modulators of inflammatory cell recruitment. Trends Immunol 34, 1–6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shin J et al. DEL-1 restrains osteoclastogenesis and inhibits inflammatory bone loss in nonhuman primates. Sci Transl Med 7, 307ra155 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mitroulis I et al. Secreted protein Del-1 regulates myelopoiesis in the hematopoietic stem cell niche. J. Clin. Invest 127, 3624–3639 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi EY et al. Del-1, an endogenous leukocyte-endothelial adhesion inhibitor, limits inflammatory cell recruitment. Science 322, 1101–1104 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitroulis I et al. Developmental endothelial locus-1 attenuates complement-dependent phagocytosis through inhibition of Mac-1-integrin. Thromb. Haemost 111, 1004–1006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi EY et al. Developmental endothelial locus-1 is a homeostatic factor in the central nervous system limiting neuroinflammation and demyelination. Mol. Psychiatry 20, 880–88 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eskan MA et al. The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat. Immunol 13, 465–473 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang Y-Y, Kim D-Y, Lee S-H & Choi EY Deficiency of developmental endothelial locus-1 (Del-1) aggravates bleomycin-induced pulmonary fibrosis in mice. Biochem. Biophys. Res. Commun 445, 369–374 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Kourtzelis I et al. Developmental endothelial locus-1 modulates platelet-monocyte interactions and instant blood-mediated inflammatory reaction in islet transplantation. Thromb. Haemost 115, 781–788 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maekawa T et al. Antagonistic effects of IL-17 and D-resolvins on endothelial Del-1 expression through a GSK-3β-C/EBPβ pathway. Nat Commun 6, 8272 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Folwaczny M, Karnesi E, Berger T & Paschos E Clinical association between chronic periodontitis and the leukocyte extravasation inhibitors developmental endothelial locus-1 and pentraxin-3. Eur. J. Oral Sci 125, 258–264 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Hajishengallis G, Moutsopoulos NM, Hajishengallis E & Chavakis T Immune and regulatory functions of neutrophils in inflammatory bone loss. Semin. Immunol 28, 146–158 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kantarci A & Van Dyke TE Resolution of inflammation in periodontitis. J. Periodontol 76, 2168–2174 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kajikawa T et al. Milk fat globule epidermal growth factor 8 inhibits periodontitis in non-human primates and its gingival crevicular fluid levels can differentiate periodontal health from disease in humans. J. Clin. Periodontol 44, 472–483 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohen RE & Research, Science and Therapy Committee, American Academy of Periodontology Position paper: periodontal maintenance. J. Periodontol 74, 1395–1401 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Hajishengallis G, Lamont RJ & Graves DT The enduring importance of animal models in understanding periodontal disease. Virulence 6, 229–235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ariel A & Timor O Hanging in the balance: endogenous anti-inflammatory mechanisms in tissue repair and fibrosis. J. Pathol 229, 250–263 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Choi EY et al. Regulation of LFA-1-dependent inflammatory cell recruitment by Cbl-b and 14–3-3 proteins. Blood 111, 3607–3614 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Getting SJ et al. Molecular determinants of monosodium urate crystal-induced murine peritonitis: a role for endogenous mast cells and a distinct requirement for endothelial-derived selectins. J. Pharmacol. Exp. Ther 283, 123–130 (1997). [PubMed] [Google Scholar]
- 27.Martin WJ, Shaw O, Liu X, Steiger S & Harper JL Monosodium urate monohydrate crystal-recruited noninflammatory monocytes differentiate into M1-like proinflammatory macrophages in a peritoneal murine model of gout. Arthritis Rheum 63, 1322–1332 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Serhan CN, Chiang N & Van Dyke TE Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol 8, 349–361 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dalli J et al. Resolvin D3 and aspirin-triggered resolvin D3 are potent immunoresolvents. Chem. Biol 20, 188–201 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bannenberg GL et al. Molecular circuits of resolution: formation and actions of resolvins and protectins. J. Immunol 174, 4345–4355 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Hong S et al. Resolvin E1 metabolome in local inactivation during inflammation-resolution. J. Immunol 180, 3512–3519 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Villoutreix BO & Miteva MA Discoidin Domains as Emerging Therapeutic Targets. Trends Pharmacol. Sci 37, 641–659 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Lastrucci C et al. Molecular and cellular profiles of the resolution phase in a damage-associated molecular pattern (DAMP)-mediated peritonitis model and revelation of leukocyte persistence in peritoneal tissues. FASEB J 29, 1914–1929 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Savill J, Dransfield I, Hogg N & Haslett C Vitronectin receptor-mediated phagocytosis of cells undergoing apoptosis. Nature 343, 170–173 (1990). [DOI] [PubMed] [Google Scholar]
- 35.Mevorach D, Mascarenhas JO, Gershov D & Elkon KB Complement-dependent clearance of apoptotic cells by human macrophages. J. Exp. Med 188, 2313–2320 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stables MJ et al. Transcriptomic analyses of murine resolution-phase macrophages. Blood 118, e192–208 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rébé C et al. Induction of transglutaminase 2 by a liver X receptor/retinoic acid receptor alpha pathway increases the clearance of apoptotic cells by human macrophages. Circ. Res 105, 393–401 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Hanayama R, Tanaka M, Miwa K & Nagata S Expression of developmental endothelial locus-1 in a subset of macrophages for engulfment of apoptotic cells. J. Immunol 172, 3876–3882 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Rosas M et al. The transcription factor Gata6 links tissue macrophage phenotype and proliferative renewal. Science 344, 645–648 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hajishengallis G Periodontitis: from microbial immune subversion to systemic inflammation. Nat. Rev. Immunol 15, 30–44 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chiang N et al. Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 484, 524–528 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hasturk H et al. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J. Immunol 179, 7021–7029 (2007). [DOI] [PubMed] [Google Scholar]
- 43.A-Gonzalez N et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 31, 245–258 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kidani Y & Bensinger SJ Liver X receptor and peroxisome proliferator-activated receptor as integrators of lipid homeostasis and immunity. Immunol. Rev 249, 72–83 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choi J-Y et al. Mer signaling increases the abundance of the transcription factor LXR to promote the resolution of acute sterile inflammation. Sci Signal 8, ra21 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Hong C et al. Constitutive activation of LXR in macrophages regulates metabolic and inflammatory gene expression: identification of ARL7 as a direct target. J Lipid Res 52, 531–539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hidai C, Kawana M, Kitano H & Kokubun S Discoidin domain of Del1 protein contributes to its deposition in the extracellular matrix. Cell Tissue Res 330, 83–95 (2007). [DOI] [PubMed] [Google Scholar]
- 48.Galli SJ, Borregaard N & Wynn TA Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat. Immunol 12, 1035–1044 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matzinger P & Kamala T Tissue-based class control: the other side of tolerance. Nat. Rev. Immunol 11, 221–230 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Griffiths GS Formation, collection and significance of gingival crevice fluid. Periodontol 2000 31, 32–42 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Bostanci N et al. Gingival crevicular fluid levels of RANKL and OPG in periodontal diseases: implications of their relative ratio. J. Clin. Periodontol 34, 370–376 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Subramanian P et al. Endothelial cell-specific overexpression of developmental endothelial locus-1 does not influence atherosclerosis development in ApoE−/− mice. Thromb. Haemost 117, 2003–2005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen LS et al. Endothelial cell-specific overexpression of Del-1 drives expansion of haematopoietic progenitor cells in the bone marrow. Thromb. Haemost doi: 10.1055/s-0038-1624582. [Epub ahead of print] (2018) [DOI] [PMC free article] [PubMed]
- 54.Lang R, Rutschman RL, Greaves DR & Murray PJ Autocrine deactivation of macrophages in transgenic mice constitutively overexpressing IL-10 under control of the human CD68 promoter. J. Immunol 168, 3402–3411 (2002). [DOI] [PubMed] [Google Scholar]
- 55.Gough PJ, Gordon S & Greaves DR The use of human CD68 transcriptional regulatory sequences to direct high-level expression of class A scavenger receptor in macrophages in vitro and in vivo. Immunology 103, 351–361 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chung K-J et al. A self-sustained loop of inflammation-driven inhibition of beige adipogenesis in obesity. Nat. Immunol 18, 654–664 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Coxon A et al. A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity 5, 653–666 (1996). [DOI] [PubMed] [Google Scholar]
- 58.A-Gonzalez N et al. The nuclear receptor LXRα controls the functional specialization of splenic macrophages. Nat. Immunol 14, 831–839 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Joosten LAB et al. Engagement of fatty acids with Toll-like receptor 2 drives interleukin-1β production via the ASC/caspase 1 pathway in monosodium urate monohydrate crystal-induced gouty arthritis. Arthritis Rheum 62, 3237–3248 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Graves DT, Fine D, Teng Y-TA, Van Dyke TE & Hajishengallis G The use of rodent models to investigate host-bacteria interactions related to periodontal diseases. J. Clin. Periodontol 35, 89–105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abe T & Hajishengallis G Optimization of the ligature-induced periodontitis model in mice. J. Immunol. Methods 394, 49–54 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matsuda S et al. A novel method of sampling gingival crevicular fluid from a mouse model of periodontitis. J. Immunol. Methods 438, 21–25 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abe T et al. The B Cell-Stimulatory Cytokines BLyS and APRIL Are Elevated in Human Periodontitis and Are Required for B Cell-Dependent Bone Loss in Experimental Murine Periodontitis. J. Immunol 195, 1427–1435 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mukundan L et al. PPAR-δ senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med 15, 1266–1272 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xiong W, Frasch SC, Thomas SM, Bratton DL & Henson PM Induction of TGF-β1 synthesis by macrophages in response to apoptotic cells requires activation of the scavenger receptor CD36. PLoS ONE 8, e72772 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fadok VA et al. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest 101, 890–898 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mitroulis I et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 172, 147–161.e12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Davies LC et al. A quantifiable proliferative burst of tissue macrophages restores homeostatic macrophage populations after acute inflammation. Eur. J. Immunol 41, 2155–2164 (2011). [DOI] [PubMed] [Google Scholar]
- 69.Gautier EL et al. Gata6 regulates aspartoacylase expression in resident peritoneal macrophages and controls their survival. J. Exp. Med 211, 1525–1531 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.