

The choice of a primary endpoint is a matter of ongoing debate in the design of clinical oncology trials testing new treatment regimens. Overall survival (OS) time of patients serves as a gold-standard endpoint in phase III clinical trials testing first-line chemotherapy for cancers, because this outcome variable ultimately represents survival benefits from chemotherapy regimens and has minimal measurement errors. Using validated surrogate endpoints that can be determined prior to a patient’s death would facilitate early completion of clinical trials, thereby accelerating regulatory approval of effective chemotherapeutic agents and reducing costs associated with the drug approval. In the setting of phase II clinical trials, surrogate end points for OS have been used to detect the potential effectiveness of new treatment strategies and to make a decision to proceed to phase III trials. Among potential surrogate endpoints (1), progression-free survival (PFS) and objective response rate (ORR) have been well validated as surrogate endpoints for OS in various tumor types including colorectal cancer (2-5): i.e., PFS and ORR are positively correlated with OS at the treatment-arm level, and a reduction in the hazard of PFS or an increase in the ORR is associated with a reduced hazard of OS at the trial level. In particular, PFS has been successfully utilized as a study endpoint in several phase III trials testing first-line chemotherapy (6), leading to accelerated drug approval. In contrast to OS as a study endpoint, intermediate endpoints are not affected by variations in chemotherapy strategies following the initial treatment failure (6). This advantage is of particular importance for tumor types for which multiple effective second-line or subsequent chemotherapy regimens are available and cross-over study designs are commonly adopted (7).
Immune checkpoint inhibitors have become an attractive treatment modality for chemorefractory solid neoplasms. These agents can reactivate T lymphocyte-mediated immune response against the tumor in the microenvironment through blocking the immune checkpoint molecules, including PDCD1 (programmed cell death 1, PD-1), CD274 (PDCD1 ligand 1, PD-L1), and CTLA4 (cytotoxic T-lymphocyte associated protein 4) (8-10). Remarkable clinical response associated with those monoclonal antibodies has been documented in various cancer types; however, in the current clinical practice, survival benefits from the immune checkpoint blockade therapy have been confined to a subset of patients. High-level microsatellite instability (MSI) or mismatch repair deficiency has been the most validated tumor biomarker for survival benefits from the immune checkpoint inhibitors (9-11). Indeed, the anti-PDCD1 (PD-1) monoclonal antibodies, pembrolizumab and nivolumab, have been approved by the U.S. Food and Drug Administration (FDA) for solid tumors with high-level MSI or mismatch repair deficiency (pembrolizumab approved for all MSI-high tumors and nivolumab for MSI-high colorectal cancer). Host and tumor factors predictive for clinical response to the immune checkpoint blockade beyond high-level MSI status have been extensively investigated [e.g., tumor mutational burden, tumor neoantigen loads, tumor CD274 (PD-L1) expression status] (11-15). With unprecedented survival benefits reported in a selected group of patients, the immune checkpoint blockade therapy is now indicated not only for refractory tumors but also for treatment-naïve tumors. Therefore, it is of considerable importance to approve promising immune checkpoint inhibitors in a timely manner, potentially through the use of surrogate study endpoints. When validating surrogate endpoints in trials testing the immune checkpoint inhibitors, we should encounter several specific challenges. Clinical response observed in sensitive patients receiving an immune checkpoint inhibitor is characterized by considerably durable tumor suppression. In addition, patterns of tumor response and progression associated with the immune checkpoint blockade therapy have been reported to be different from those observed in patients receiving a conventional chemotherapeutic agent and/or a molecular-targeted agent (14,16). Therefore, from the perspective of clinical trials and subsequent drug approval, surrogate endpoints for OS should be evaluated specifically for trials testing the immune checkpoint inhibitors (Figure 1).
Figure 1.
Surrogate study endpoints for overall survival in clinical trials testing immune checkpoint inhibitors.
In a recent issue of JAMA Oncology, Ritchie and colleagues reported a literature-based meta-analysis of published clinical trials that had investigated the effectiveness of the immune checkpoint inhibitors, and provided evidence on possible surrogate endpoints for OS among patients treated with this promising treatment modality (17). Among 87 phase II trials identified through the systematic electrical review of the articles published between 2000 and 2017, ORR was most commonly used as the primary endpoint (in 60% of the studies), followed by PFS (13%). Notably, the Response Evaluation Criteria In Solid Tumors (RECIST) criteria (18) rather than the immune-related response criteria were used to assess tumor response and progression in a vast majority (94%) of the trials identified. Subsequently, the researchers identified 20 phase II and III randomized controlled trials of the immune checkpoint inhibitors involving a total of 10,828 patients, and primarily examined the surrogacy of ORR for PFS and OS. The most predominant primary disease was non-small cell lung cancer (in 45% of the trials), followed by melanoma (20%). Experimental treatment regimens included PDCD1 (PD-1) inhibitor monotherapy, CD274 (PD-L1) inhibitor monotherapy, CTLA4 inhibitor monotherapy, and combination of an immune checkpoint inhibitor and chemotherapy. In their primary analysis of 24 randomized treatment comparisons (Figure 2), a between-arm difference in ORR was only moderately correlated with that in PFS or OS. The correlation coefficients were 0.63 [95% confidence interval (CI), 0.35–0.89)] between the odds ratio (OR) for ORR and the hazard ratio (HR) for PFS, and 0.57 (95% CI, 0.23–0.89) between the OR for ORR and the HR for OS. In the secondary analysis, a difference in PFS was also moderately correlated with that in OS with a correlation coefficient of 0.42 (95% CI, 0.04–0.81). In exploratory analyses limited to 24 treatment arms including the immune checkpoint inhibitors (Figure 2), the correlation between ORR and 6-month PFS or 12-month OS was shown to be weak with correlation coefficients of 0.37 (95% CI, −0.06 to 0.95) and 0.08 (95% CI, −0.17 to 0.70), respectively. Of note, the 6-month PFS rate was strongly correlated with the 12-month OS rate with a correlation coefficient of 0.74 (95% CI, 0.57–0.92). Based on these findings, the authors generated a linear regression model for prediction of the 12-month OS rate using the 6-month PFS rate. When the prediction model was validated using 19 single-arm or multi-arm phase II trials with an immune checkpoint inhibitor arm, a good calibration between the actual and predicted 12-month OS rates was noted. In contract, when ORR was used to predict the 6-month PFS or 12-month OS rate, the calibration between the actual and predicted rates was not satisfactory. The authors concluded that ORR might not be a robust surrogate endpoint for OS when examining the immune checkpoint inhibitors and that the 6-month PFS rate might be a more suitable surrogate for patient survival.
Figure 2.
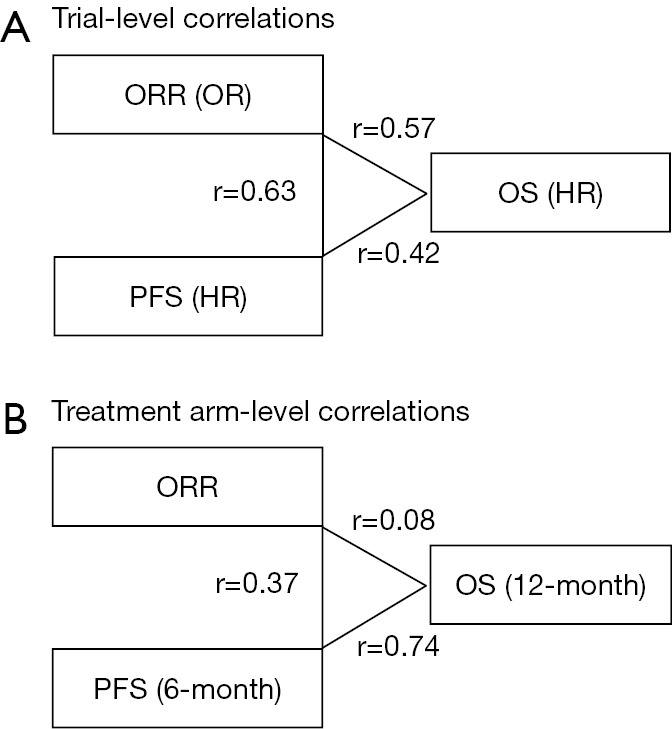
Results of correlation tests between study endpoints (17). (A) Correlations of differences in endpoints between treatment arms (analyses at the trial level); (B) correlations of endpoints within treatment arms (analyses at the treatment-arm level). HR, hazard ratio; OR, odds ratio; ORR, objective response rate; OS, overall survival; PFS, progression-free survival; r, correlation coefficient.
Given the great promise of the immune checkpoint inhibitors for advanced cancers, the findings of the current study would have substantial impact on future studies in clinical oncology. The main finding of this study was a moderate correlation between the 6-month PFS and 12-month OS rates. In contrast, the correlation between the HR of PFS and that of OS was weak. As the authors described, a major limitation of this study was unavailability of individual patient data from the studies included. For time-to-event outcome variables (i.e., PFS and OS), some investigators advocate that the HR comparing an experimental arm to a control arm can represent the between-arm difference in the pattern of event occurrence along the entire follow-up time (19). In the current study, survival rates at specific time-points rather than the HRs of the corresponding clinical event during the entire period of follow-up were analyzed based on the premise that these statistics might surrogate clinical benefits from the immune checkpoint inhibitors (“milestone analyses”) (20). It would be interesting to examine how the surrogacy of PFS for OS differed by the time-points analyzed; however, individual patient data would be required to fully address this point. Individual patient data would also allow us to assess other potential surrogate endpoints (e.g., disease control rate, time to progression, time to treatment failure; Figure 1), to define outcome variables consistently across the studies, thereby increasing the number of studies analyzed, and to adjust for potential confounding factors consistently. In this regard, the recent trend for data sharing may increase opportunities of individual patient data analyses and help examine alternative outcome variables to OS in a more comprehensive fashion, potentially improving generalizability of findings of meta-analyses.
In analyses of surrogate endpoints in clinical oncology trials, both definitions of outcome variables and evaluation criteria for treatment response require discussions (1,21). Inconsistency in definitions of outcome variables may result in exclusion of a part of identified studies due to unavailability of the data in published articles and thereby hinder a robust meta-analysis. Tumor response and progression in patients receiving an immune checkpoint inhibitor may need to be evaluated by a different algorithm from the conventional guidelines for patients receiving chemotherapy and/or a molecular-targeted agent. The RECIST criteria (18) and the World Health Organization criteria (WHO) (22) have been traditionally utilized to evaluate radiological findings of tumor status after non-surgical anti-tumor treatment. In particular, the RECIST criteria have been widely utilized as a simple and pragmatic scheme for evaluation of the activity of new cancer therapeutics in solid tumors based on validated and consistent criteria to assess temporal changes in tumor burden. However, a fraction of patients receiving an immune checkpoint inhibitor represent specific patterns of treatment responses. Those tumor behaviors that are atypical in patients receiving a conventional chemotherapeutic agent include pseudoprogression and hyperprogression, which are defined as a delayed response following a temporal apparent progression and a rapid progression after treatment administration, respectively (14,16). Pseudoprogression, which is thought to manifest due to T cell infiltrates enhanced by the therapy, poses a particular challenge for evaluation of tumor response after the treatment initiation. Namely, the effectiveness of the immune checkpoint inhibitors may be underestimated by the conventional WHO and RECIST criteria, which define tumor progression immediately at the time of documenting a new lesion or a predefined amount of increase in calculated tumor burden. Patients whose tumors are considered progressive based on those criteria might undergo delayed treatment response. To take into account distinctive patterns of tumor response after administration of the immune checkpoint inhibitors, the modified WHO criteria have been proposed as the immune-related response criteria (irRC) for evaluation of the efficacy of immunomodulatory anti-tumor agents (23). In contrast to the conventional WHO criteria, the irRC criteria require confirmation of progressive disease on two consecutive scans at least four weeks apart and define the overall tumor burden including measurements of new lesions. Similarly, iRECIST, which has been derived from the conventional RECIST criteria as an immunotherapy-specific evaluation scheme (24), requires confirmation of progressive disease on a subsequent imaging. Therefore, the findings of the current meta-analysis should be validated by including a larger number of trials that were conducted based on specific criteria for cancer immunotherapy or applying the criteria for individual patient data.
Considering other limitations of the current study may help us to obtain important implications for future research on the topic. The surrogacy of intermediate endpoints for OS might differ by the primary tumor location due to considerable variations in OS times across cancer types (25). The survival benefit from first-line treatment may be confounded and apparently eliminated by effective chemotherapy regimens administered subsequently. Indeed, in a subgroup analysis limited to the trials including non-small cell lung cancer, ORR appeared to be more strongly correlated with PFS or OS both at the trial and treatment-arm levels than in the current meta-analysis overall. Further subgroup analyses were not available due to a limited number of the trials included and the lack of relevant data. Analyses stratified by tumor MSI status would be intriguing because levels of this tumor phenotype have been a strong determinant of response to the immune checkpoint blockade. Since the immune checkpoint inhibitors are relatively new treatment modalities, the sample size in each arm of the reported studies might be small to obtain robust risk estimates for the endpoint variables. A correlation analysis at the trial level, which examines correlations of between-arm differences in surrogate endpoints and that in OS, may inform future trials through providing an estimated reduction in OS that could be achieved by a certain level of reduction in the surrogate endpoint (2-4). We should also acknowledge that the HRs for PFS and OS were not reported in all the trials and that the detailed definitions of tumor progression and censoring rules for each endpoint were unavailable in several trials. Again, individual patient data are required to address these limitations.
In summary, the current literature-based analysis by Ritchie et al. does not support the potential of ORR as a surrogate endpoint for OS in trials testing the immune checkpoint inhibitors. The data suggest the potential surrogacy of the 6-month PFS rate for the 12-month OS rate in this setting. The current study also provided us with important insights for future directions. Accumulating data and analyzing individual patient data would help us to validate surrogate endpoints more rigorously, to sophisticate designs of trials testing the immune checkpoint inhibitors, and to obtain early approval of promising anti-cancer treatment strategies to further improve clinical outcomes of cancer patients.
Acknowledgements
Funding: This work was supported in part by a Grant-in-Aid for Scientific Research from Japan Society for the Promotion of Science (grant number, 16K19941 to K Kosumi) and JSPS Fujita Memorial Fund for Medical Research (to K Kosumi). K Kosumi was supported by an Overseas Research Fellowship grant from Japan Society for the Promotion of Science (grant number, JP2017-775).
Provenance: This is an invited Editorial commissioned by Section Editor Jianrong Zhang (Candidate of Master of Public Health, George Warren Brown School of Social Work; Graduate Policy Scholar-in-training, Clark-Fox Policy Institute, Washington University in St. Louis, St. Louis, MO, USA).
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Bellera CA, Pulido M, Gourgou S, et al. Protocol of the Definition for the Assessment of Time-to-event Endpoints in CANcer trials (DATECAN) project: formal consensus method for the development of guidelines for standardised time-to-event endpoints' definitions in cancer clinical trials. Eur J Cancer 2013;49:769-81. 10.1016/j.ejca.2012.09.035 [DOI] [PubMed] [Google Scholar]
- 2.Hamada T, Nakai Y, Isayama H, et al. Progression-free survival as a surrogate for overall survival in first-line chemotherapy for advanced pancreatic cancer. Eur J Cancer 2016;65:11-20. 10.1016/j.ejca.2016.05.016 [DOI] [PubMed] [Google Scholar]
- 3.Tang PA, Bentzen SM, Chen EX, et al. Surrogate end points for median overall survival in metastatic colorectal cancer: literature-based analysis from 39 randomized controlled trials of first-line chemotherapy. J Clin Oncol 2007;25:4562-8. 10.1200/JCO.2006.08.1935 [DOI] [PubMed] [Google Scholar]
- 4.Buyse M, Burzykowski T, Carroll K, et al. Progression-free survival is a surrogate for survival in advanced colorectal cancer. J Clin Oncol 2007;25:5218-24. 10.1200/JCO.2007.11.8836 [DOI] [PubMed] [Google Scholar]
- 5.Johnson KR, Ringland C, Stokes BJ, et al. Response rate or time to progression as predictors of survival in trials of metastatic colorectal cancer or non-small-cell lung cancer: a meta-analysis. The Lancet Oncology 2006;7:741-6. 10.1016/S1470-2045(06)70800-2 [DOI] [PubMed] [Google Scholar]
- 6.Yothers G. Toward progression-free survival as a primary end point in advanced colorectal cancer. J Clin Oncol 2007;25:5153-4. 10.1200/JCO.2007.13.6796 [DOI] [PubMed] [Google Scholar]
- 7.Ishak KJ, Proskorovsky I, Korytowsky B, et al. Methods for adjusting for bias due to crossover in oncology trials. Pharmacoeconomics 2014;32:533-46. 10.1007/s40273-014-0145-y [DOI] [PubMed] [Google Scholar]
- 8.Boussiotis VA. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N Engl J Med 2016;375:1767-78. 10.1056/NEJMra1514296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409-13. 10.1126/science.aan6733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Overman MJ, McDermott R, Leach JL, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol 2017;18:1182-91. 10.1016/S1470-2045(17)30422-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jenkins RW, Thummalapalli R, Carter J, et al. Molecular and Genomic Determinants of Response to Immune Checkpoint Inhibition in Cancer. Annu Rev Med 2018;69:333-47. 10.1146/annurev-med-060116-022926 [DOI] [PubMed] [Google Scholar]
- 12.Yarchoan M, Hopkins A, Jaffee EM. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med 2017;377:2500-1. 10.1056/NEJMc1713444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamada T, Soong TR, Masugi Y, et al. TIME (Tumor Immunity in the MicroEnvironment) classification based on tumor CD274 (PD-L1) expression status and tumor-infiltrating lymphocytes in colorectal carcinomas. Oncoimmunology 2018;7:e1442999. 10.1080/2162402X.2018.1442999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishino M, Ramaiya NH, Hatabu H, et al. Monitoring immune-checkpoint blockade: response evaluation and biomarker development. Nat Rev Clin Oncol 2017;14:655-68. 10.1038/nrclinonc.2017.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124-8. 10.1126/science.aaa1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Champiat S, Dercle L, Ammari S, et al. Hyperprogressive Disease Is a New Pattern of Progression in Cancer Patients Treated by Anti-PD-1/PD-L1. Clin Cancer Res 2017;23:1920-8. 10.1158/1078-0432.CCR-16-1741 [DOI] [PubMed] [Google Scholar]
- 17.Ritchie G, Gasper H, Man J, et al. Defining the Most Appropriate Primary End Point in Phase 2 Trials of Immune Checkpoint Inhibitors for Advanced Solid Cancers: A Systematic Review and Meta-analysis. JAMA Oncol 2018;4:522-8. 10.1001/jamaoncol.2017.5236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228-47. 10.1016/j.ejca.2008.10.026 [DOI] [PubMed] [Google Scholar]
- 19.Buyse M, Molenberghs G, Burzykowski T, et al. The validation of surrogate endpoints in meta-analyses of randomized experiments. Biostatistics 2000;1:49-67. 10.1093/biostatistics/1.1.49 [DOI] [PubMed] [Google Scholar]
- 20.Hellmann MD, Kris MG, Rudin CM. Medians and Milestones in Describing the Path to Cancer Cures: Telling "Tails". JAMA Oncol 2016;2:167-8. 10.1001/jamaoncol.2015.4345 [DOI] [PubMed] [Google Scholar]
- 21.Punt CJ, Buyse M, Kohne CH, et al. Endpoints in adjuvant treatment trials: a systematic review of the literature in colon cancer and proposed definitions for future trials. J Natl Cancer Inst 2007;99:998-1003. 10.1093/jnci/djm024 [DOI] [PubMed] [Google Scholar]
- 22.Miller AB, Hoogstraten B, Staquet M, et al. Reporting results of cancer treatment. Cancer 1981;47:207-14. [DOI] [PubMed] [Google Scholar]
- 23.Wolchok JD, Hoos A, O'Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res 2009;15:7412-20. 10.1158/1078-0432.CCR-09-1624 [DOI] [PubMed] [Google Scholar]
- 24.Seymour L, Bogaerts J, Perrone A, et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol 2017;18:e143-e52. 10.1016/S1470-2045(17)30074-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin 2017;67:7-30. 10.3322/caac.21387 [DOI] [PubMed] [Google Scholar]