

Abstract
Hemoproteins have recently emerged as promising biocatalysts for promoting a variety of carbene transfer reactions including cyclopropanation and Y—H insertion (Y = N, S, Si, B). For these and synthetic carbene transfer catalysts alike, achieving high chemoselectivity toward cyclopropanation in olefin substrates bearing unprotected Y—H groups has proven remarkably challenging due to competition from the more facile carbene Y—H insertion reaction. In this report, we describe the development of a novel artificial metalloenzyme based on an engineered myoglobin incorporating a serine-ligated Co-porphyrin cofactor that is capable of offering high selectivity toward olefin cyclopropanation over N—H and Si—H insertion. Intramolecular competition experiments revealed a distinct and dramatically altered chemoselectivity of the Mb(H64V,V68A,H93S)[Co(ppIX)] variant in carbene transfer reactions compared to myoglobin based variants containing the native histidine-ligated heme cofactor or other metal/proximal ligand substitutions. These studies highlight the functional plasticity of myoglobin as a ‘carbene transferase’ and illustrate how modulation of the cofactor environment within this metalloprotein scaffold represents a valuable strategy for accessing carbene transfer reactivity not exhibited by naturally occurring hemoproteins or transition metal catalysts.
Graphical Abstract
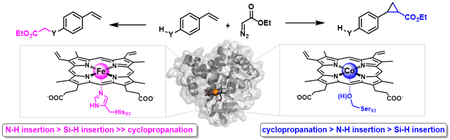
Introduction
The transition-metal catalyzed insertion of carbenoids into C=C, C—H, and Y—H bonds (Y = N, S, O, Si, B), represents an important class of transformations for the construction of new carbon-carbon and carbon-heteroatom bonds.1 A plethora of organometallic catalysts, including Rh-, Ir-, Ru-, Cu-, Co-, and Fe-based complexes, have been reported to promote these transformations.1 More recently, our group and others have demonstrated the ability of engineered hemoproteins derived from myoglobin,2 and cytochrome P450s,3, respectively, to provide viable biocatalysts for mediating olefin cyclopropanations as well as other types of carbene-mediated transformations, including N—H insertion,4 S—H insertion,5 and Si—H insertion.6 Artificial ‘carbene transferases’ based on these2c,7 or other protein scaffolds8 have also been reported.
Among carbene-mediated transformations, olefin cyclopropanations are particularly attractive owing to the relevance of cyclopropane rings as structural motifs in medicinal chemistry and drug discovery.9 Despite major progress made in transition metal-catalyzed carbene transfer chemistry,1 the development of catalytic systems capable of favoring olefin cyclopropanation over carbene Y—H insertion reactions has remained an unmet challenge. Indeed, studies involving different types of organometallic catalysts invariably showed that the more facile Y—H carbene insertion outcompetes olefin cyclopropanation in both inter- and intramolecular settings.10 A similar reactivity trend is exhibited by hemoprotein-based carbene transfer catalysts, as documented by our own studies (vide infra) and reports from other groups.4a,6 Indeed, Arnold and coworkers reported the exclusive occurrence of N—H insertion in the P450-catalyzed transformation of p-amino-styrene with ethyl α-diazoacetate4a and exclusive formation of the Si—H insertion product in the cytochrome c-catalyzed transformation of a silane group-containing styrene derivative with ethyl α-diazopropanoate.6 In this context, the development of carbene transfer catalysts capable of favoring cyclopropanation in substrates bearing unprotected Y—H groups is highly desirable as these systems would not only complement the chemoselectivity of (bio)catalysts currently available for these reactions, but also disclose new opportunities toward the selective cyclopropanation of densely functionalized olefin substrates. Herein, we report the development and characterization of a myoglobin-based biocatalyst that is able to display high chemoselectivity toward olefin cyclopropanation over N—H and Si—H insertion, a unique reactivity acquired through substitution of the native histidine-ligated heme cofactor with a non-native serine-ligated Co-porphyrin.
Results and Discussion
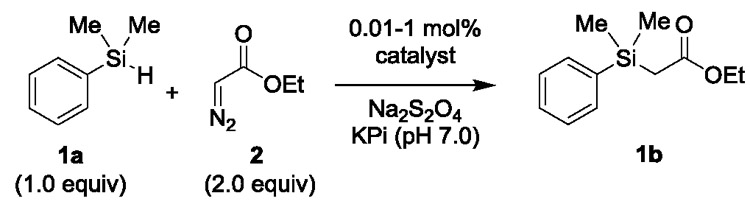
At the incipit of these studies, we selected the Mb(H64V,V68A) variant as the starting point for catalyst development in reason of its high catalytic activity (>10,000 turnovers (TON)) and stereoselectivity (95–99% de and ee) in the cyclopropanation of vinylarene substrates in the presence of ethyl α-diazoacetate (EDA) as the carbene donor.2a This variant also exhibits high activity toward the functionalization of arylamines4b and mercaptan substrates5 with diazoester reagents via carbene N—H and S—H insertion, respectively. Carbene insertion into the Si—H bonds of silanes is a facile transformation for carbene transfer catalysts.11 To assess the Si—H insertion activity of Mb(H64V,V68A), we examined the transformation of dimethylphenyl silane (1a) in presence of EDA (2) to give the functionalized product 1b (Table 1). As expected, this reaction proceeded efficiently resulting in the clean formation of the Si—H insertion product in 66% yield in the presence of 0.2 mol% catalyst (330 TON; Table 1, Entry 3). By comparison, negligible Si—H insertion activity was observed for free hemin (15 TON; Table 1, Entry 1), whereas wild-type sperm whale myoglobin (Mb) displayed a two-fold lower activity compared to the Mb(H64V,V68A) variant (175 TON; Table 1, Entry 2). Under catalyst-limited conditions, Mb(H64V,V68A) was found to support over 1,500 turnovers in this reaction (Table 1, Entry 4). This catalytic activity compares favorably with that recently reported for the same silane substrate and ethyl α-diazopropanoate by an engineered cytochrome c variant optimized by directed evolution for this reaction (2,520 TON)6 or that of other metallopeptide/protein complexes investigated in the context of related Si—H insertion reactions.12 Altogether, these results demonstrated that Si—H insertion reactions are readily catalyzed by the Mb-based ‘carbene transferase’, as expected based on its previously observed reactivity toward other electronrich Y—H substrates.4b,5
Table 1.
Myoglobin-catalyzed carbene Si—H insertion with dimethylphenyl silane 1a and EDA 2.a
 | ||||
|---|---|---|---|---|
| Entry | Catalyst | Cat. loading | % yieldb | TON |
| 1 | Hemin | 1 mol% | 15 | 15 |
| 2 | Mb | 0.2 mol% | 35 | 175 |
| 3 | Mb(H64V,V68A) | 0.2 mol% | 66 | 330 |
| 4 | Mb(H64V,V68A) | 0.01 mol% | 15 | 1545 |
Reaction conditions: 10 mM dimethylphenyl silane, 20 mM EDA, 10 mM Na2S2O4, 1-100 μM catalyst in 50 mM phosphate buffer (pH 7.0).
Based on GC conversion using calibration curves with isolated 1b.
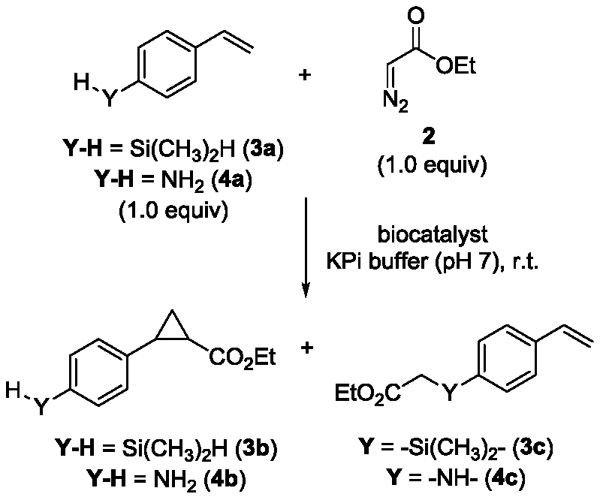
On the basis of these results, we were interested in assessing the chemoselectivity of Mb(H64V,V68A) in the cyclopropanation of 4-(dimethylsilyl)styrene (3a), in which the silane group (Si—H) can compete for carbene insertion with the olefinic group (Table 2). This reaction led to minimal formation (4%) of the desired cyclopropane product 3b, with the Si—H insertion adduct 3c representing the largely predominant product (96%; Table 2, Entry 2). Albeit with lower efficiency (1–10% vs. 13% conv.), a similar reactivity profile was observed for a variety of other hemoproteins, including horseradish peroxidase (HRP), catalase, cytochrome P450BM3 and its engineered variant FL#6213, and cytochrome c (Table 2, Entries 3–7), with which the cyclopropanation product 3b is formed only in minor amounts (<5–11%) compared to the Si—H insertion product. The results with wild-type equine heart cytochrome c are consistent with those recently reported in a related reaction with cytochrome c from Rhodothermus marinus.6
Table 2.
Chemoselectivity of myoglobin and other hemoproteins in the reaction with 4-(dimethylsilyl)styrene (3a) or para-amino-styrene (4a) and EDA.a
 |
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | % yield (3b+3c)b |
3b:3c ratio |
% yield (4b+4c) b |
4b:4c ratio |
| 1 | Mb | 6 | 3:97 | 65 | 1:99 |
| 2 | Mb(H64V,V68A) | 13 | 4:96 | 85 | 0:100 |
| 3 | HRP | 1 | 11:89 | 96 | 0:100 |
| 4 | Catalase | 1 | 9:91 | 6 | 2:98 |
| 5 | P450BM3 | <0.5 | 5:95 | 24 | 1:99 |
| 6 | FL#62 | 4 | 5:95 | 25 | 1:99 |
| 7 | cytochrome c | 10 | 5:95 | 57 | 1:99 |
Reaction conditions: 10 mM 3a or 4a, 10 mM EDA, 10 mM Na2S2O4, 20 μM catalyst in 50 mM phosphate buffer (pH 7.0).
Based on GC conversion using calibration curves with isolated 3b, 3c, 4b, and 4c.
To examine the relative reactivity toward olefin cyclopropanation vs. N—H insertion, similar experiments were conducted in the presence of para-amino-styrene (4a) and EDA as the carbene donor reagent. Interestingly, in addition to Mb(H64V,V68A), horseradish peroxidase and cytochrome c were found to be particularly active in the transformation of this substrate, leading to the N—H insertion product in 96% and 57% yield, respectively (Table 2, Entries 3 and 7). For these and all the other hemoproteins, however, the desired cyclopropanation product 3b was either not formed (HRP) or produced only in negligible amounts (<1–2%). Along with observations previously made with synthetic catalysts,10a-c these results corroborated the notion that preferential formation of the cyclopropanation product in substrates containing an unprotected Si—H or N—H group is not attainable using currently available carbene transfer catalysts and biocatalysts.
The carbene transfer activity of Mb hinges upon the formation of a carbene-heme intermediate at the level of the active site (i.e., distal heme pocket),2a whose reactivity is influenced by the nature of the porphyrin ligand and axial residue.14 Previously, we established that modification of the metalloporphyrin cofactor environment within this scaffold, i.e., through substitution of the metal,2c porphyrin ligand,7a or metal-ligating proximal residue,14 can modulate the carbene transfer reactivity of this metalloprotein. Based on these findings, we hypothesized that this approach could furnish a means to tune the chemoselectivity of myoglobin-based cyclopropanation catalysts and thus access Mb variants capable of favoring olefin cyclopropanation over the inherently more facile carbene Y—H insertions in functionalized substrates where both reactions are possible (e.g., 3a and 4a).
Toward this goal, we prepared a library of Mb(H64V,V68A) derived variants in which the native heme is substituted with Co(ppIX), Mn(ppIX), and Rh(mpIX)). In addition, the native metal-coordinating His residue (His93) was varied to include alternative nucleophilic residues (i.e., Cys, Ser, Tyr) as well as non-Lewis basic amino acids (i.e., Ala and Phe). Based on the available crystal structure of Mb,15 the His93Ala and His93Phe variants were expected to alter steric access at the proximal side of the metalloporphyrin cofactor for either enabling or disfavoring, respectively, axial coordination of the metal by a water molecule. Conveniently, these artificial metalloenzymes could be produced directly in E. coli cells arrayed in 96-well plates by exploiting our recently introduced protocol for the recombinant expression of cofactor-substituted myoglobins.2c,16
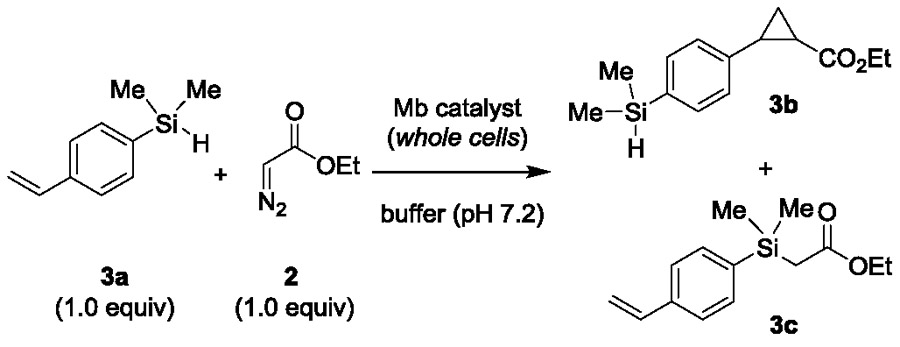
The catalytic activity and selectivity toward cyclopropanation vs. Si—H insertion of the cofactor-modified Mb variants was then assessed in whole-cell reactions2b containing 4-(dimethylsilyl)styrene 3a and EDA. The results from these experiments are graphically summarized in Figure 1a and the most representative data are provided in Table 3. Interestingly, these analyses revealed a significant impact of both the metal and proximal residues on the catalytic activity and chemoselectivity of the reaction. For example, in the presence of the native heme cofactor (Fe(ppIX)), only the His93Ser variant exhibited comparable activity to the parent protein Mb(H64V,V68A) (Table 3, Entries 1 and 2). In contrast, the most active catalysts among the Mn-containing Mb variants feature a non-nucleophilic residue (i.e., Ala or Phe) in place of the proximal histidine residue (Table 3, entries 3–5). More importantly, heme replacement with the non-native cofactor Co-protoporphyrin IX (Co(ppIX)) induced a dramatic shift in the chemoselectivity of the Mb catalyst to favor the formation of the cyclopropanation product, leading to 3b:3c ratios ranging from 40:60 to 74:26 (Figure 1a). This reactivity is in stark contrast with that of the Fe-, Mn-, and Rh-containing variants, which greatly favors the formation of the Si—H insertion product (3b:3c ratios from 4:96 to 11:89; Figure 1a).
Figure 1.

Screening data for the cofactor/proximal ligand substituted Mb(H64V,V68A) variants in the reactions with (a) 4-(dimethylsilyl)styrene 3a or (b) 4-amino-styrene 4a, and EDA. The pie charts indicate the product distribution (light blue for Si—H and N—H insertion products 3c and 4c, respectively; blue for cyclopropanation product 3b and 4b, respectively; dark blue for multiple insertion byproduct(s)). The activity (% GC yield) is represented as filling color.
Table 3.
Catalytic activity and selectivity of selected Mb(H64V,V68A) variants in the reaction with 4-(dimethylsilyl)styrene (3a) and EDA.a
 | |||||
|---|---|---|---|---|---|
| Entry | Protein | Cofactor | % yieldb |
% 3bc |
% 3cc |
| 1 | Mb(H64V,V68A) | Fe(ppIX) | 41 | 6 | 92 |
| 2 | Mb(H64V,V68A,H93S) | Fe(ppIX) | 36 | 4 | 96 |
| 3 | Mb(H64V,V68A) | Mn(ppIX) | 18 | 5 | 94 |
| 4 | Mb(H64V,V68A,H93A) | Mn(ppIX) | 36 | 3 | 96 |
| 5 | Mb(H64V,V68A,H93F) | Mn(ppIX) | 35 | 4 | 96 |
| 6 | Mb(H64V,V68A) | Co(ppIX) | 37 | 33 | 49 |
| 7 | Mb(H64V,V68A,H93S) | Co(ppIX) | 44 | 74 | 24 |
| 8 | Mb(H64V,V68A,H93F) | Co(ppIX) | 25 | 63 | 10 |
| 9 | Mb(H64V,V68A) | Rh(mpIX) | 33 | 6 | 94 |
Reaction conditions: 10 mM 3a, 10 mM EDA, E. coli whole cells expressing Mb variant (OD600 = 40) in 50 mM phosphate buffer (pH 7.2).
Based on GC conversion using calibration curves with isolated 3b and 3c.
The remainder to 100% corresponds to double insertion product(s).
Among the Co-substituted variants, Mb(H64V,V68A,H93S)[Co(ppIX)] displayed the most optimal combination of high chemoselectivity for cyclopropanation over Si—H insertion (74% selectivity) and good catalytic activity (Table 3, Entry 7). Attempts to further optimize the Mb(H64V,V68A,H93S)[Co(ppIX)]-catalyzed reaction by varying the substrate loading and/or 3a: EDA ratio, did not result in significant improvements in the yield of 3b, indicating that the reaction conditions applied during the catalyst screening were already optimal for this transformation.
From the systematic set of structure-activity data collected in these experiments, it became apparent that whereas the cobalt metal center is critical for inducing the desired selectivity toward cyclopropanation (6%→33%; Table 3, Entry 6 vs. 1), axial coordination by the serine residue is beneficial toward enhancing this reactivity (33%→74%; Table 3, Entry 7 vs. 6). Control experiments using free Co(ppIX) as the catalyst (2 mol%) under identical reaction conditions (10 mM 3a, 10 mM EDA, 10 mM Na2S2O4 in phosphate buffer, pH 7.2) resulted in negligible formation of product (<1% yield), most of which consisted of the Si—H insertion product (80%). These results further evidenced the critical role of the protein matrix in modulating the reactivity and selectivity of the Co-protoporphyrin IX cofactor.
Encouraged by these results, the panel of Mb(H64V,V68A)-based variants was screened against 4-amino-styrene (4a), in which carbene insertion into the N—H bond of the amino group was found to greatly outcompete cyclopropanation of the C=C double bond in the reactions catalyzed by Mb(H64V,V68A) and other carbene transfer (bio)catalysts (Table 2). As shown by the data summarized in Figure 1b, also in this case substitution of the metal and axial ligand was found to alter significantly the efficiency and/or chemoselectivity of the reaction. Specifically, all the Fe-containing variants maintain high preference for the N—H insertion reaction (95–96%) over the cyclopropanation reaction, regardless of the nature of the proximal residue. Most of these variants also display significantly reduced activity compared to Mb(H64V,V68A), with the only exception of the H93F variant, whose catalytic activity is only moderately affected (47% vs 63% yield; Table 4, Entries 2 vs. 1).
Table 4.
Catalytic activity and selectivity of selected Mb(H64V,V68A) variants in the reaction with 4-amino-styrene (4a) and EDA.a
 | |||||
|---|---|---|---|---|---|
| Entry | Protein | Cofactor | % yieldb |
% 4bc |
% 4cc |
| 1 | Mb(H64V,V68A) | Fe(ppIX) | 63 | 3 | 96 |
| 2 | Mb(H64V,V68A,H93F) | Fe(ppIX) | 47 | 2 | 98 |
| 3 | Mb(H64V,V68A) | Mn(ppIX) | 25 | 7 | 91 |
| 4 | Mb(H64V,V68A,H93Y) | Mn(ppIX) | 11 | 16 | 81 |
| 5 | Mb(H64V,V68A) | Co(ppIX) | 6 | 18 | 75 |
| 6 | Mb(H64V,V68A,H93C) | Co(ppIX) | 19 | 10 | 83 |
| 7 | Mb(H64V,V68A,H93S) | Co(ppIX) | 38 | 62 | 28 |
| 8d | Mb(H64V,V68A,H93S) | Co(ppIX) | 45 | 72 | 24 |
| 9 | Mb(H64V,V68A) | Rh(mpIX) | 4 | 56 | 28 |
| 10 | Mb(H64V,V68A,H93A) | Rh(mpIX) | 50 | 6 | 90 |
Reaction conditions: 10 mM 4a, 10 mM EDA, E. coli whole cells expressing Mb variants (OD600 = 40) in 50 mM phosphate buffer (pH 7.2).
Based on GC conversion using calibration curves with isolated 4b and 4c.
The remainder to 100% corresponds to double insertion product(s).
With 10 mM 4a, 7.5 mM EDA, and whole cells at OD600 = 80.
In comparison, metal substitution appeared to have a more profound impact on the chemoselectivity of the metalloprotein catalyst. Across the board, the Co-substituted variants displayed a more pronounced propensity to promote the formation of the cyclopropanation product 4b (10–62% selectivity; Figure 1b) when compared to the Mn-containing variants and most of the Rh-containing variants (5–15% selectivity).
As observed for the Si—H insertion vs. cyclopropanation reaction with 3a, the Mb variant combining the Co(ppIX) and His93Ser substitution emerged as the most efficient and chemoselective catalyst for promoting the cyclopropanation of 4a to give the desired product 4b (62% selectivity; Table 4, Entry 7). Again, coordination of the cobalt center by the serine residue appears to be critical for enhancing this chemoselectivity feature, as evidenced by the 3- to 6-fold higher selectivity of this variant toward favoring cyclopropanation over N—H insertion when compared to both the histidine-ligated and the structurally similar cysteine-ligated counterparts (Table 4, Entry 7 vs. 5 and 6). The Mb(H64V,V68A,H93S)[Co(ppIX)]-catalyzed conversion of 4a could be further optimized to yield the cyclopropanation product 4b with 72% selectivity in 45% yield (Table 3, Entry 8). Control reactions with free Co(ppIX) cofactor in buffer show minimal conversion of 4a to a mixture of 4b and 4c (<1% yield).
Interestingly, the Rh-based variant Mb(H64V,V68A)[Rh(mpIX)] also exhibited significantly improved selectivity toward cyclopropanation vs. N—H insertion compared to the parent variant Mb(H64V,V68A) (3%→56% selectiv.; Table 4, Entry 9 vs. 1). However, this change in chemoselectivity is accompanied by a drastic reduction in catalytic activity, making it an inferior catalyst compared to Mb(H64V,V68A,H93S)[Co(ppIX)]. Still, it is interesting to note the distinctive effect of the proximal histidine toward inducing the observed chemoselectivity shift in the Rh-based variant compared to the other residues introduced at this position (Figure 1b) or to the His93Ala counterpart, in which axial coordination of the metal is not available (Table 4, Entry 10 vs. 9). As for Mb(H64V,V68A,H93S)[Co(ppIX)], these results highlight the interplay of the effects resulting from metal and proximal ligand variation in tuning the reactivity and chemoselectivity of these biocatalysts.
Based on the results above, Mb(H64V,V68A,H93S)[Co(ppIX)] was selected for further characterization. This variant could be efficiently expressed and isolated from E. coli as holoprotein in good yields (~8 mg/L culture). Its UV-vis spectrum shows a strong absorption band at 425 nm and two weak absorption bands at 535 and 570 nm, corresponding to the Soret and Q-bands of the protein, respectively (Figure 2a). Upon addition of sodium dithionite (Na2S2O4), the Soret band shifts to 405 nm, which was assigned to the reduced, Co(II) form of the metalloprotein. Unlike the iron-based counterpart, only partial reduction (~40%) of Mb(H64V,V68A,H93S)[Co(ppIX)] was observed with excess sodium dithionite even after overnight incubation (Figure 2a, red line). More efficient conversion to the reduced form of this protein was achieved instead using the stronger reductant Ti(III) citrate in combination with the electron transfer mediator methyl viologen (Figure 2a, orange line).17
Figure 2.

Characterization of Mb(H64V,V68A,H93S)-[Co(ppIX)]. (a) Overlay of the electronic absorption spectra of the protein prior to (blue) and after reduction with sodium dithionite (red) or Ti(III) citrate (orange). (b) Thermal denaturation curves for Mb(H64V,V68A) (green) and Mb(H64V,V68A,H93S)[Co(ppIX)] (red) as determined by circular dichroism (θ222).
Control experiments involving reactions with 3a or 4a in the presence of either the oxidized or reduced form of Mb(H64V,V68A,H93S)[Co(ppIX)] revealed that the latter species is catalytically less active but more selective for cyclopropanation over Si—H (or N—H) insertion compared to the oxidized form of the protein. Moreover, both TON and the cyclopropanation vs. Y—H insertion chemoselectivity obtained with the purified protein were lower than those achieved in the whole cell reactions, clearly indicating a favorable effect of the intracellular environment on the catalytic performance of this biocatalyst. This effect can be in part attributed to the efficient reduction of Mb(H64V,V68A,H93S)[Co(ppIX)] within the reducing intracellular milieu, as suggested by UV-vis spectroscopy analysis of cell lysates (Co(II):Co(III) > 9:1). Through thermal denaturation experiments using circular dichroism (CD), we further determined that the Mb(H64V,V68A,H93S)[Co(ppIX)] variant has an apparent melting temperature (Tm) of 60.0°C ± 0.2 (Figure 2b). This Tm value is comparable to that of Mb(H64V,V68A) (66.0°C ± 1.0),18 indicating that remodeling of the cofactor environment in this variant did not significantly affect the stability of the metalloprotein.
To gain further insights into the chemoselectivity properties of Mb(H64V,V68A,H93S)[Co(ppIX)], this and other selected variants were tested in an intramolecular competition experiment using 4-(dimethylsilyl)aniline 5a, in which N—H insertion competes for Si—H insertion (Table 5). These studies showed that Mb(H64V,V68A,H93S)[Co(ppIX)] exhibits a strong preference toward the N—H insertion reaction over the Si—H insertion reaction (89% selectiv.), a feature shared by the Fe-containing or histidine-ligated counterpart (86–92% selectivity; Table 5, Entries 2–3 vs. 4). In comparison, the parent variant Mb(H64V,V68A) is more active (72% vs. 23% yield for Mb(H64V,V68A,H93S)[Co(ppIX)]) but less selective toward N—H insertion over the Si—H insertion and double insertion reactions (Table 4, Entries 1 vs. 4). From these and the previous experiments, it can be derived that the general order of reactivity for the Mb(H64V,V68A,H93S)[Co(ppIX)] variant is: C=C > N—H > Si—H. This chemoselectivity is inverted compared to that of the parent Mb variant Mb(H64V,V68A) (N—H > Si—H >> C=C) or other hemoproteins (Table 2) and organometallic catalysts (N—H >> C=C).10a-c
Table 5.
Catalytic activity and selectivity of selected Mb(H64V,V68A) variants in the reaction with 4-(dimethylsilyl)aniline (5a) and EDA.a
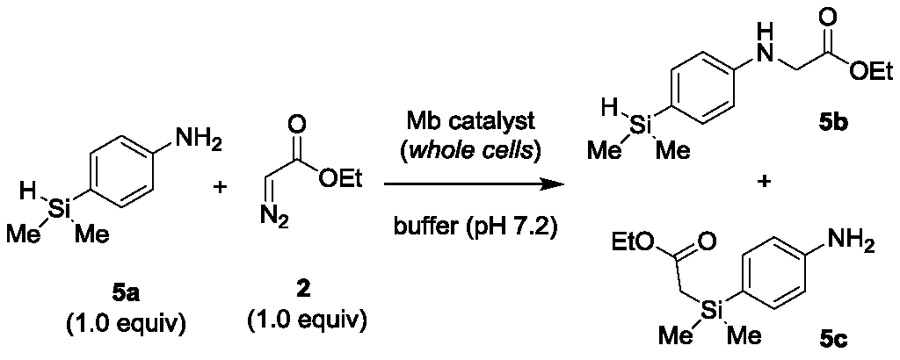 | |||||
|---|---|---|---|---|---|
| Entry | Protein | Cofactor | % yieldb |
% 5bc |
% 5cc |
| 1 | Mb(H64V,V68A) | Fe(ppIX) | 72 | 76 | 1 |
| 2 | Mb(H64V,V68A,H93S) | Fe(ppIX) | 25 | 86 | 3 |
| 3 | Mb(H64V,V68A) | Co(ppIX) | 33 | 92 | 5 |
| 4 | Mb(H64V,V68A,H93S) | Co(ppIX) | 23 | 89 | 5 |
Reaction conditions: 10 mM 6a, 10 mM EDA, E. coli whole cells expressing Mb variants (OD600 = 40) in 50 mM phosphate buffer (pH 7.2).
Based on GC conversion using calibration curves with isolated 5b and 5c.
The remainder to 100% corresponds to double insertion product(s).
To evaluate the effect of the altered cofactor environment on the catalyst reactivity toward other types of Y—H insertions, further tests were carried out using 4-amino-thiophenol (6a), in which both N—H and S—H insertion reactions are possible (Table 6). Interestingly, these experiments showed that whereas Mb(H64V,V68A) favors the N—H insertion reaction (~4:1 ratio for 6b:6c; Table 6, Entries 1–2), the Mb(H64V,V68A,H93S)[Co(ppIX)] variant features inverted chemoselectivity, favoring the S—H insertion process (6b:6c ratio from 1:2 to 1:4; Table 6, Entries 3–4). In addition, Mb(H64V,V68A,H93S)[Co(ppIX)] shows higher selectivity against formation of the double insertion byproduct (0% vs. 9–15%).
Table 6.
Catalytic activity and selectivity of Mb(H64V,V68A) and Mb(H64V,V68A,H93S)[Co(ppIX)] in the reaction with 4-amino-thiophenol (6a) and EDA.a
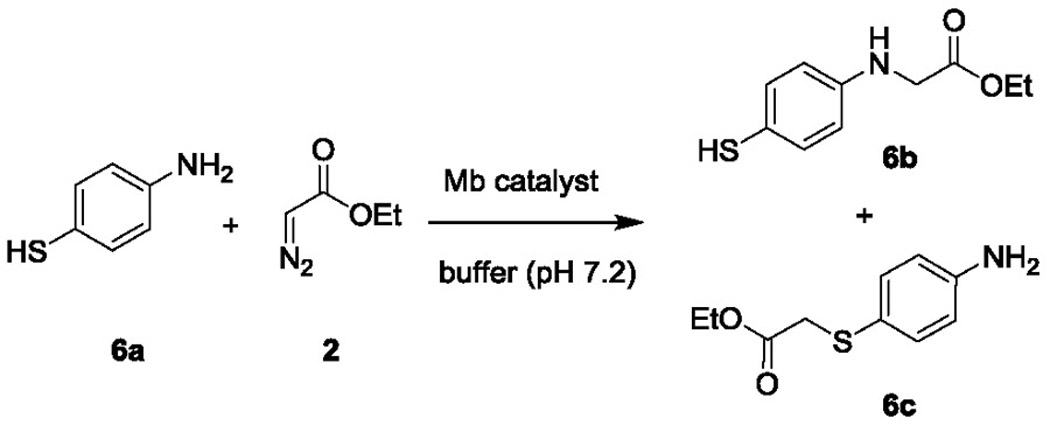 | ||||||
|---|---|---|---|---|---|---|
| Entry | Protein | Equiv, EDA |
% yieldb |
% 6b |
% 6c |
% otherc |
| 1 | Mb(H64V,V68A) | 1 | 43 | 73 | 18 | 9 |
| 2 | Mb(H64V,V68A) | 2 | 76 | 70 | 15 | 15 |
| 3 | Mb(H64V,V68A,H93S) [Co(ppIX)] |
1 | 27 | 20 | 80 | 0 |
| 4 | Mb(H64V,V68A,H93S) [Co(ppIX)] |
2 | 55 | 35 | 65 | 0 |
Reaction conditions: 3 mM 5a, 3 or 6 mM EDA, 10 mM Na2S2O4, 10 mM TCEP, 20 μM purified Mb variant in 50 mM phosphate buffer (pH 7.2).
Based on HPLC conversion using calibration curves with isolated 6b and 6c.
Double insertion product(s).
Neither Mb(H64V,V68A) nor Mb(H64V,V68A,H93S)[Co(ppIX)] were found to catalyze O—H insertion reactions, as determined in experiments with phenol and EDA (no formation of ethyl 2-phenoxyacetate) and the lack of reactivity with water in the presence of EDA alone (no formation of ethyl 2-hydroxyacetate). This is unlike Rh- and other transition metal-based catalysts, which readily react with alcohols or water to give carbene O—H insertion products.1a,10e,19
Having identified a biocatalyst with high chemoselectivity for cyclopropanation over N—H/Si—H insertion, we were interested in examining the impact of the modified cofactor environment on the substrate scope, catalytic activity, and selectivity of this biocatalyst in olefin cyclopropanation, as compared to the parent catalyst Mb(H64V,V68A). For this purpose, we tested a panel of representative vinylarene substrates (7a-15a) previously characterized in the context of Mb(H64V,V68A)-catalyzed cyclopropanation with EDA.2a,2b As summarized in Scheme 1, all of these compounds could be converted by the Mb(H64V,V68A,H93S)[Co(ppIX)] variant into the corresponding cyclopropanation products 7b-15b in moderate to good efficiency (30–88% product conversion). The results with 7b-9b indicate that substitutions at the ortho-, meta-, and para-position of the benzene ring are well tolerated by the Mb variant. Both electronrich (11a) and electrondeficient (12a) styrene derivatives were efficiently converted (70–88% yield). More sterically hindered α,α-disubstituted and heterocycle-containing olefins also successfully participated in the reaction to afford the corresponding cyclopropanes 13b-15b in good yields (38–59%). Thus, despite a generally reduced catalytic activity compared to Mb(H64V,V68A),2a,2b the Mb(H64V,V68A,H93S)[Co(ppIX)] variant features a comparably broad substrate scope across structurally different vinylarenes.
Scheme 1. Substrate scope for Mb(H64V,V68A,H93S)[(Co(ppIX)]-catalyzed cyclopropanation of vinylarenes with EDA.a.

a Reaction conditions: 10 mM olefin, 20 mM EDA, E. coli whole cells expressing Mb variants (OD600 = 80) in 50 mM phosphate buffer (pH 7.2). b Based on GC conversion. c For trans-(1S,2S) product. d Using 5 mM olefin and 10 mM EDA. Enantiomeric excess not determined.
Across all the tested substrates, Mb(H64V,V68A,H93S)[Co(ppIX)] was found to favor the formation of the trans-cyclopropanation product, but with overall reduced diastereoselectivity (i.e., from 6:4 to 2:1 ratio for trans:cis) compared to Mb(H64V,V68A) (d.r. >98:2).2a,2b As an exception, 7b was obtained with high diastereoselectivity (98:2 trans:cis). Similarly, Mb(H64V,V68A,H93S)[Co(ppIX)] maintains preference for formation of the (1S,2S)-configured product but with significantly reduced enantioselectivity (e.g., 11–57% ee for 7b-11b) compared to Mb(H64V,V68A) (95–99% ee)2a,2b, especially for the α,α-disubstituted olefins (3–4% ee for 13b-15b). Interestingly, parallel experiments with Mb(H64V,V68A)[Co(ppIX)] showed that this metallo-substituted variant displays higher trans-(1S,2S)-selectivity in these reactions compared to Mb(H64V,V68A,H93S)[Co(ppIX)] (Scheme S1). Thus, the general reduction in stereoselectivity observed with Mb(H64V,V68A,H93S)[Co(ppIX)] vs. Mb(H64V,V68A) can be attributed to both the metal substitution and the mutation of the proximal ligand (His→Ser). This result is not entirely surprising, given that (a) the active site mutations in Mb(H64V,V68A) were optimized for high trans-(1S,2S)-selectivity in the presence of the native histidine-ligated heme cofactor, and (b) both the metal substitution and the structural change induced by the His93→Ser substitution are likely to alter the orientation of the metalloporphyrin within the core of the protein, thereby affecting the asymmetric induction effect imposed by the protein scaffold during this reaction. Indeed, a significant shift in diastereoselectivity for styrene cyclopropanation with EDA was also noted upon substitution of the proximal cysteine with serine in a cytochrome P450 enzyme (22:78 vs. 76:24 cis:trans, respectively).3b Altogether, the results above showed that Mb(H64V,V68A,H93S)[Co(ppIX)] could be successfully applied to the cyclopropanation of a variety of aryl-substituted olefins, albeit re-optimization of its active site would be required for enhancing its stereoselectivity,
Conclusion
In summary, we have developed a novel biocatalytic strategy, based on an engineered myoglobin-based carbene transferase, for the chemoselective cyclopropanation of olefin substrates bearing unprotected Y—H groups (Y= N, Si). This transformation, unattainable using previously available carbene transfer (bio)catalysts, opens the way to the selective carbene-mediated functionalization of olefins in the presence of inherently more reactive Y—H groups. As emerged from the analysis of a systematic library of cofactor modified Mb variants, the unique and atypical reactivity of Mb(H64V,V68A,H93S)[Co(ppIX)] compared to Mb and other hemoproteins is specifically induced by the combination of a non-native metal (Co) and non-native residue (Ser) at the proximal axial site of the metalloporphyrin cofactor. While featuring inverted chemoselectivity in carbene transfer reactions compared to the histidine-ligated Fe-containing counterpart (i.e., C=C > N—H > Si—H vs. N—H > Si—H >> C=C and S—H > N—H vs. N—H > S—H), Mb(H64V,V68A,H93S)[Co(ppIX)] features comparable stability, ease of expression in E. coli, and broad substrate scope in the cyclopropanation of vinylarenes with EDA. Further engineering of this scaffold (e.g., through active site mutagenesis)2a,2b is expected to improve its stereoselectivity, which is currently modest. The present studies highlight the functional plasticity of myoglobin as a carbene transferase and illustrate how modulation of the cofactor environment within this metalloprotein scaffold can provide a promising strategy for tuning its reactivity in the context of abiotic carbene transfer reactions. Further studies are warranted to elucidate the factors underlying the atypical reactivity of the Mb(H64V,V68A,H93S)[Co(ppIX)] catalyst.
Experimental Section
General Information.
All chemicals and reagents were purchased from commercial suppliers (Sigma-Aldrich, Alfa Aesar, TCI, Oakwood Chemicals, Combi Blocks) and used without any further purification, unless otherwise stated. EDA was purchased from Sigma-Aldrich as 87% m/v solution in dichloromethane. Dimethyl(4-vinylphenyl)silane 3a and 4-(dimethylsilyl)aniline 4a were synthesized according to a previously reported procedure.6 Authentic racemic standards for the cyclopropanation products 7b-15b and 20b were synthesized and characterized as described previously.2a,2b All dry organic reactions were carried out under argon in oven-dried glassware with magnetic stirring using standard gas-tight syringes, cannulae and septa. 1H and 13C NMR spectra were measured on Bruker DPX-400 (operating at 400 MHz for 1H and 100 MHz for 13C) or Bruker DPX-500 (operating at 500 MHz for 1H and 125 MHz for 13C). Tetramethylsilane (TMS) served as the internal standard (0 ppm) for 1H NMR and the residual proton signal of the solvents was used as the internal standard for 13C NMR. HRMS analyses were performed using a Q Exactive Plus Mass Spectrometer at the Proteomics Facility of the University of Rochester. Silica gel chromatography purifications were carried out using AMD Silica Gel 60 230–400 mesh. Thin Layer Chromatography (TLC) were carried out using Merck Millipore TLC silica gel 60 F254 glass plates.
Growth Media.
Enriched M9 media was prepared as follows. For 1 L, 770 mL deionized H2O was added with 200 mL M9 salts (5x) solution, 20 mL glucose (20% m/v), 10 mL casamino acids (20% m/v), 1 mL MgSO4 (2 M), and 100 μL CaCl2 (1 M). The M9 salts (5x) solution was prepared by dissolving 34 g Na2HPO4, 15 g K2HPO4, 2.5 g NaCl, 5 g NH4Cl in 1 L deionized H2O and sterilized by autoclaving. The casamino acids and MgSO4 solutions were autoclaved separately. The CaCl2 and glucose stock solutions were sterilized by filtration. Enriched M9 agar plates were prepared by adding 17 g agar to 1 L of enriched M9 media containing all of the aforementioned components at the specified concentrations with the exception of glucose and CaCl2, which were added immediately prior to plating. To media and plates, ampicillin was added to a final concentration of 100 mg/L and chloramphenicol was added to a final concentration of 34 mg/L.
Recombinant expression of myoglobins containing artificial metalloporphyrins.
All myoglobin variants were expressed from pET22-based vectors containing the target gene under a T7 promoter. The recombinant protein was engineered with a C-terminal polyhistidine tag. E. coli C41(DE3) cells expressing the heme transporter ChuA (T7 promoter) and GroES/EL chaperon proteins (araBAD promoter) were prepared via transformation with the plasmid pGroES/EL-ChuA.16 E. coli C41(DE3) cells containing the pGroES/EL-ChuA plasmid were transformed with the pET22 vector encoding the desired Mb variant and selected on M9 agar plates containing ampicillin (100 mg/L) and chloramphenicol (34 mg/L). Single colonies were used to inoculate 5 mL of enriched M9 media supplemented with ampicillin and chloramphenicol followed by incubation at 37°C with shaking (180 rpm) for 10 to 15 hours. At an OD600 of 0.8 to 1.0, the desired cofactor was added directly to the expression culture to a final concentration of about 30 mg/L followed by induction of protein expression with IPTG and arabinose at final concentrations of 1 mM. Cells were incubated at 20°C with shaking (180 rpm) for 20 to 24 hours. After harvesting, the cell pellets were resuspended in 20 mL of Ni NTA Lysis Buffer (50 mM KPi, 250 mM NaCl, 10 mM histidine, pH = 8.0) lysed by sonication, and clarified by centrifugation (14,000 rpm, 4°C, 30 min). When using E. coli in whole cell reactions, harvested cells were resuspended in 50 mM KPi pH 7.2 and stored at 4°C for no longer than 12 hours.
Protein Purification.
Clarified lysate was transferred to a Ni-NTA column equilibrated with Ni-NTA Lysis Buffer. The resin was washed with 50 mL of Ni-NTA Lysis Buffer followed by 50 mL of Ni-NTA Wash Buffer (50 mM KPi, 250 mM NaCl, 20 mM Histidine, pH = 8.0). Proteins were eluted with Ni-NTA Elution Buffer (50 mM KPi, 250 mM NaCl, 250 mM Histidine, pH = 7.0). After elution from the Ni-NTA column, the protein was buffer exchanged against 50 mM KPi buffer (pH 7.0) using 10 kDa Centricon filters. The concentrations of the Mb variants were calculated using the following extinction coefficients: ε410 = 157 mM−1 cm−1 for Fe(ppIX)-containing variants, ε470 = 60 mM−1 cm−1 for Mn(ppIX)-containing variants, ε424 = 152.5 mM−1 cm−1 for Co(ppIX)-containing variants, and ε405 = 122 mM−1 cm−1 for Rh(mpIX)-containing variants.
General Procedure for Biocatalytic Reactions with Whole Cells and Purified Protein.
Under standard reaction conditions, 500 μL-scale reactions were carried out using 10–20 μM Mb variant, 10 mM substrate, 10 mM EDA and 10 mM sodium dithionite. TCEP (10 mM) was also added to the reactions with 6a to prevent disulfide formation. In a typical procedure, a solution containing the desired myoglobin variant in potassium phosphate buffer (50 mM, pH 7.0) with sodium dithionite was prepared in an anaerobic chamber. Reactions were initiated by addition of 10 μL of olefin followed by the addition of 10 µL of EDA from 0.5 M stock solutions, and the reaction mixtures were stirred in the chamber for 12 hours at room temperature. Whole-cell reactions were performed using E. coli C41(DE3) cells prepared using the method described above. Cell suspensions were transferred to an anaerobic chamber and diluted to the indicated cell density (OD600: 40–80) with 50 mM KPi pH 7.2. Reactions were initiated by addition of 10 μL of olefin followed by the addition of 10 µL of EDA from 0.5 M stock solutions, and the mixtures were stirred in the chamber for 12 hours at room temperature.
Product Analysis.
The reactions with 1a, 3a, 4a, and 5a were analyzed by adding 20 µL of internal standard (benzodioxole, 50 mM in ethanol) to the reaction mixture, followed by extraction with 400 µL of dichloromethane and analysis by gas chromatography (GC). GC analyses were carried out using a Shimadzu GC-2010 gas chromatograph equipped with a FID detector and a Chiral Cyclosil-B column (30 m x 0.25 mm x 0.25 μm film). Separation method: 1 μL injection, injector temp.: 200 °C, detector temp: 300 °C. Gradient: column temperature set at 120 °C for 3 min, then to 150 °C at 0.8 °C/min, then to 245°C at 25 °C/min. Total run time was 28.60 min. For the reactions with 6a, the DCM extraction solution was evaporated and the residue was redissolved in 300 uL methanol containing 10 mM TCEP, followed by HPLC analysis using a Shimadzu LC-2010A instrument equipped with a UV-vis detector. Analytical conditions: Thermo Fisher Scientific Hypersil GOLD C18 column (250 × 4.6 mm); UV detection: 254 nm; flow rate: 1.0 mL/min: solvent A: 1% trifluoroacetic acid in H2O; solvent B: 1% trifluoroacetic acid in acetonitrile; gradient: 0–3 min: 30% B; 3–21 mins: 30% B to 90% B; 21–24 mins: 90% B; 24–24.5 mins: 90% B to 30% B; 24.5–30 mins: 30% B. Retention times: 3.82 min for 4-aminothiophenol; 4.89 min for S-H insertion product; 13.57 min for N-H insertion product; 14.91 min for double insertion product. Calibration curves for quantification of each single insertion product were constructed using authentic standards prepared synthetically or enzymatically as described above. All measurements were performed at least in duplicate.
CD analyses and Tm Determination.
Circular dichroism (CD) spectroscopy and thermal denaturation analysis were performed using a JASCO J-1100 CD spectrophotometer equipped with variable temperature/wavelength denaturation software. Far UV CD spectra (250–190 nm) were obtained using 3 μM solutions of purified Mb variant in 50 mM potassium phosphate buffer (pH 7.0) and recorded at 20°C at a scan rate of 50 nm/min with a bandwidth of 1 nm and an averaging time of 10 seconds per measurement. Thermal denaturation curves were measured by monitoring the change in molar ellipticity at 222 nm (θ222) over a temperature range from 20°C to 100°C. The temperature increase was set to 0.5°C per minute with an equilibration time of 10 seconds. Data integration time for the melt curve was set to 4 seconds with a bandwidth of 1 nm. Linear baselines for the folded (θf) and unfolded state (θu) were generated. Melting temperature (Tm) was determined using the low temperature (θf = mf T + bf ) and high temperature (θu = mu T + bu) equations fitted to the experimental data before and after global unfolding, respectively. These data were normalized and converted to fraction of folded protein (Ff) vs. temperature plots and the resulting curve was fitted to a sigmoidal equation (θfit) via nonlinear regression analysis in SigmaPlot, from which apparent melting temperatures were derived.
Ethyl 2-(dimethyl(phenyl)silyl)acetate (1b).
Ethyl 2-(dimethyl(phenyl)silyl)acetate 1b was synthesized according to a modification of a reported procedure.6 Dimethyl(phenyl)silane 1a (136 mg, 2.00 mmol, 1 equiv.) and Rh2(OAc)4 (4.4 mg, 10 μmol, 1 mol%) were added to a flame dried 10 mL round bottom flask under argon atmosphere, dissolved in anhydrous DCM (4 mL), and subsequently cooled to −78 °C. A solution of EDA (87%, 131 mg, 1.00 mmol, 1 equiv.) in anhydrous DCM (1 mL) was added dropwise with a syringe pump over a period of 30 min. The reaction mixture was allowed to reach room temperature and stirred for 16 h. The solvent was removed under reduced pressure and the crude product purified via column chromatography on silica gel (0–7.5% EtOAc/pentanes) to yield ethyl 2-(dimethyl(phenyl)silyl)acetate 1b as a clear oil (99.4 mg, 0.45 mmol, 45% yield). Rf = 0.34 (7.5% EtOAc/pentanes). 1H NMR (CDCl3, 500 MHz): δ 7.51 (m, 2H), 7.34 (m, 3H), 3.98 (q, J = 7.2, 2H), 2.06 (s, 2H), 1.12 (t, J = 7.0 Hz, 3H), 0.36 (s, 6H). 13C NMR (CDCl3, 125 MHz): δ 172.7, 137.7, 134.1, 130.0, 128.4, 60.4, 26.7, 14.7, −2.6. GC-MS m/z (% relative intensity): 207 (46.5), 177 (29.9), 165 (84.3), 145 (71.6), 135 (100.0)
Ethyl 2-(4-(dimethylsilyl)phenyl)cyclopropanecarboxylate (3b).
E. coli C41(DE3) cells expressing the variant Mb(H64V,V68A,H93S)[Co(ppIX)] were prepared and harvested according to the protocol described above. In an anaerobic chamber (0 ppm O2, 2.5% H2 atmosphere), the whole cell suspension was diluted to an OD600 of 80 with argon purged in phosphate buffer (50 mM , pH 7.2). A solution of dimethyl(4-vinylphenyl)silane 3a (50.0 mg, 0.31 mmol, 1 equiv.) in methanol (0.75 mL) was added dropwise and while stirring to 28.5 mL of the whole cell suspension in an Erlenmeyer flask. The reaction was initiated by the dropwise addition of a solution of EDA (87%, 29.5 mg, 0.23 mmol, 0.75 equiv.) in methanol (0.75 mL). The reaction mixture was stirred at room temperature for 16 hours and extracted with DCM (3×30 mL). The combined organic layers were dried over sodium sulfate and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (0–7.5% Et2O/pentanes) and the fractions analyzed by gas chromatography with chiral column to separate the cis- (4.5 mg, 0.017 mmol, 1%) 3b-cis and trans-isomers (3.9 mg, 0.015 mmol, 1%) 3b-trans and as clear oils. The configuration of 3b was assigned by comparison to the splitting pattern in the 1H NMR in alignment with the previously determined cis and trans isomers of 10b (see supporting information). 3b-cis: Rf = 0.44 (7.5% Et2O/pentanes). 1H NMR (CD2Cl2, 500 MHz): δ 7.43 (d, J = 8.0 Hz, 2H), 7.08 (d, J = 7.5 Hz, 2H), 4.36 (m, 1H), 4.11 (q, J = 7.2 Hz, 2H), 4.45 (m, 1H), 1.87 (m. 1H), 1.54 (m, 1H), 1.29 (m, 1H), 1.23 (t, J = 7.0 Hz, 3H), 0.30 (d, J = 4.0 Hz, 6H). 13C NMR (CD2Cl2, 125 MHz): δ 171.2, 138.6, 135.8, 134.1, 129.4, 60.6, 25.9, 22.2, 14.4, 11.5, −3.6. GC-MS m/z (% relative intensity): 248 (79.0), 233 (33.0), 201 (44.6), 144 (55.6), 129 (59.8), 115 (100.0). 3b-trans: Rf = 0.61 (7.5% Et2O/pentanes). 1H NMR (CD2Cl2, 500 MHz): δ 7.43 (d, J = 8.0 Hz, 2H), 7.08 (d, J = 7.5 Hz, 2H), 4.36 (m, 1H), 4.11 (q, J = 7.2 Hz, 2H), 4.45 (m, 1H), 1.87 (m. 1H), 1.54 (m, 1H), 1.29 (m, 1H), 1.23 (t, J = 7.0 Hz, 3H), 0.30 (d, J = 4.0 Hz, 6H). 13C NMR (CD2Cl2, 125 MHz): δ 173.5, 142.0, 135.8, 134.7, 126.2, 61.2, 26.5, 24.8, 17.4, 14.6, −3.5. GC-MS m/z (% relative intensity): 248 (83.3), 233 (33.5), 201 (43.7), 114 (56.6), 129 (62.7), 115 (100.0). HRMS (ESI) m/z: [M + H]+ Calcd. For C14H21O2Si 249.1311; Found 249.1303.
Ethyl 2-(dimethyl(4-vinylphenyl)silyl)acetate (3c).
Ethyl 2-(dimethyl(4-vinylphenyl)silyl)acetate 3c was synthesized according to a modification of a previously reported procedure.6 Dimethyl(4-vinylphenyl)silane 3a (150 mg, 0.92 mmol, 1 equiv.) and Rh2(OAc)4 (4.0 mg, 9 μmol, 1 mol%) were added to a flame dried 10 mL round bottom flask under argon atmosphere, dissolved in anhydrous DCM (4 mL), and subsequently cooled to −78 °C. A solution of EDA (106 mg, 0.9 mmol, 1 equiv.) in anhydrous DCM (1 mL) was added dropwise with a syringe pump over a period of 30 min. The reaction mixture was allowed to reach room temperature and stirred for 16 h. The solvent was removed under reduced pressure and the crude product purified via column chromatography on silica gel (0–4% EtOAc/hexanes) to yield ethyl 2-(dimethyl(4-vinylphenyl)silyl)acetate 3c as a clear oil (36.7 mg, 0.15 mmol, 15% yield). Rf = 0.48 (4% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz): δ 7.50 (d, J = 8.0, 2H), 7.41 (d, J = 8.0, 2H), 6.72 (dd, J = 11.0, 17.5, 1H), 5.79 (d, J = 17.5, 1H), 5.28 (d, J = 11.0, 1H), 4.05 (q, J = 7.2, 2H), 2.11 (s, 2H), 2.27 (t, J = 7.3 Hz, 3H), 0.40 (s, 6H). 13C NMR (CDCl3, 125 MHz): δ 172.7, 138.8, 136.8, 133.9, 129.0, 125.8, 114.7, 60.1, 26.4, 14.5, −2.6. GC-MS m/z (% relative intensity): 233 (76.6), 191 (87.2), 163 (44.2), 161 (100.0), 147 (39.7), 145 (42.7). HRMS (ESI) m/z: [M + H]+ Caldc for C14H21O2Si 249.1311; Found 249.1306.
Ethyl 2-(4-aminophenyl)cyclopropanecarboxylate (4b).
Compound 4b was prepared according to the procedure described in Scheme S2. Di-tert-butyl dicarbonate/Boc2O (1.174 g, 5.38 mmol. 3.2 equiv.) and N,N-diisopropylethyl-amine/DIPEA (651 mg, 5.04 mmol, 3 equiv.) were added to a solution of dimethyl(4-vinylphenyl)silane 3a (200 mg, 1.68 mmol, 1 equiv.) in DCM (30 mL). The reaction mixture was stirred over night at room temperature and, after that, refluxed at 40 °C until completion, which was monitored by TLC. To remove excess Boc2O, the solvent was evaporated and the crude residue taken up in 10 mL of a 1:1 mixture of THF:water before addition of glycine (246 mg, 4.30 mmol) and Na2CO3 (910 mg, 8.60 mmol). After stirring overnight, THF was removed in vacuo and the aqueous layer extracted with DCM (3×10 mL). The combined organic layers were washed with brine (10 mL), dried over sodium sulfate, filtered and concentrated under reduced pressure. Flash column chromatography on silica gel (25% Et2O/pentanes) furnished tert-butyl (4-vinylphenyl)carbamate 16 as white flaky solid (284 mg, 1.29 mmol, 77 % yield). Rf = 0.63 (25% Et2O/pentanes). 1H NMR (CDCl3, 500 MHz): δ 7.30–7.19 (m, 4H), 6.59 (dd, J = 11.0, 17.5 Hz, 1H) 6.43 (br, 1H), 5.58 (d, J = 17.5 Hz, 1H), 5.09 (d, J = 11.0 Hz, 1H), 1.45 (s, 9H). GC-MS m/z (% relative intensity): 219 (19.4), 163 (100.0), 119 (76.9).
To a flame dried 10 mL round bottom flask, equipped with a stir bar, were added tert-butyl (4-vinylphenyl)carbamate 16 (100 mg, 0.46 mmol, 1 equiv.), Rh2(OAc)4 (4.0 mg, 9 μmol, 2 mol%) and anhydrous DCM (2.5 mL) under argon. After that, a solution of EDA (87%, 119 mg, 0.9 mmol, 2 equiv.) in anhydrous DCM (2.5 mL) was slowly added dropwise with a syringe pump over a period of 4 hours. The resulting reaction mixture was stirred at room temperature for 12 hours, after which the solvent was removed under reduced pressure and the crude product purified via column chromatography on silica gel (0–20% Et2O/pentanes) to afford ethyl 2-(4-((tert-butoxycarbonyl)amino)phenyl)cyclopropanecarboxylate 17 as a colorless oil (23 mg, 0.075 mmol, 16% yield). Rf = 0.36 (25% Et2O/pentanes). 1H NMR (CDCl3, 500 MHz): δ 7.26 (d, J = 10.5 Hz, 2H), 7.02 (d, J = 10.5 Hz, 2H), 6.48 (br, 1H), 4.16 (q, J = 9.0 Hz, 2H), 2.47 (m, 1H), 1.83 (m, 1H), 1.54 (m, 1H), 1.51 (s, 9H), 1.28 (t, J = 9.0 Hz, 3H), 1.26 (m, 1H). 13C NMR (CDCl3, 125 MHz): δ 173.6, 152.9, 137.0, 134.8, 126.9, 118.8, 80.6, 60.8, 28.5, 25.9, 24.1, 17.0, 14.4. GC-MS m/z (% relative intensity): 305 (16.8), 249 (70.2), 205 (22.1), 176 (51.2), 158 (36.6), 132 (100.0).
A solution of 2-(4-((tert-butoxycarbonyl)amino)phenyl)cyclopropanecarboxylate 17 (20 mg, 0.065 mmol, 1 equiv.) in DCM (0.5 mL) was cooled to 0 °C. Trifluoroacetic acid (0.5 mL) was added dropwise and the reaction mixture stirred for 2 hours before removing the solvent under reduced pressure. The crude product was taken up in 1.5 mL DCM, transferred to an Eppendorf tube and washed with NaHCO3 (1 mL). The aqueous layer was extracted with DCM (2×1 mL) and the combined organic layers were dried over sodium sulfate, centrifuged, the supernatant decanted and the solvent removed in vacuo to yield ethyl 2-(4-aminophenyl)cyclopropanecarboxylate 4b as yellow solid (11.3 mg, 0.055 mmol, 85 % yield). Rf = 0.43 (40% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz): δ 6.91 (d, J = 8.0 Hz, 2H), 6.61 (d, J = 8.5 Hz, 2H), 4.16 (q, J = 7.0, 2H), 3.60 (br, 2H), 2.43 (m, 1H), 1.79 (m, 1H), 1.52 (m, 1H), 1.27 (t, J = 7.0 Hz, 3H), 1.23 (m, 1H). 13C NMR (CDCl3, 125 MHz): δ 173.9, 145.1, 130.0, 127.4, 115.3, 60.7, 26.0, 23.9, 16.7, 14.4. GC-MS m/z (% relative intensity): 205 (44.0), 176 (23.6), 160 (21.1), 132 (100.0).
Ethyl 2-((4-vinylphenyl)amino)acetate (4c).
E. coli C41(DE3) cells expressing the variant Mb(H64V,V68A,H93C) were prepared and harvested according to the protocol described above. In an anaerobic chamber (0 ppm O2, 2.5% H2 atmosphere), the whole cell suspension was diluted to an OD600 of 80 with argon-purged 50 mM phosphate buffer (pH 7.2). A solution of 4-vinylaniline 4a (48.0 mg, 0.40 mmol, 1 equiv.) in methanol (1.0 mL) was added dropwise and while stirring to 38 mL of the whole cell suspension in an Erlenmeyer flask. The reaction was initiated by the dropwise addition of a solution of EDA (87%, 52.4 mg, 0.40 mmol, 1 equiv.) in methanol (1.0 mL). The reaction mixture was stirred at room temperature for 16 hours and extracted with DCM (3×40 mL). The combined organic layers were dried over sodium sulfate and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (0–20% Et2O/pentanes) to yield ethyl 2-((4-vinylphenyl)amino)acetate 4c (20.0 mg, 0.10 mmol, 25%) as white solid. Rf = 0.31 (20% Et2O/pentanes). 1H NMR (CDCl3, 500 MHz): δ 7.23 (d, J = 8.0 Hz, 2H), 6.53 (d, J = 8.0 Hz, 2H), 5.51 (d, J = 17.5 Hz, 1H), 5.01 (d, J = 10.5 Hz, 1H), 4.32 (br, 1H), 4.21 (q, J = 6.7 Hz, 2H), 3.87 (d, J = 4.5 Hz, 2H), 1.27 (t, J = 7.0 Hz, 3H). 13C NMR (CDCl3, 125 MHz): δ 171.1, 146.9, 136.7, 128.1, 127.5, 113.0, 111.0, 61.5, 45.9, 14.3. GC-MS m/z (% relative intensity): 205 (25.8), 132 (100.0).
Ethyl 2-((4-(dimethylsilyl)phenyl)amino)acetate (5b).
E. coli C41(DE3) cells expressing the variant Mb(H64V,V68A) were prepared and harvested according to the protocol described above. Under argon atmosphere, the cells were resuspended in argon purged 50 mM phosphate buffer (pH 7.2) and diluted to an OD600 of 40. A solution of 4-(dimethylsilyl)aniline 5a (60.5 mg, 0.40 mmol, 1 equiv.) in ethanol (1.0 mL) was added dropwise and while stirring to 38 mL of the whole cell suspension in an Erlenmeyer flask. A solution of EDA (87%, 52.6 mg, 0.40 mmol, 1 equiv.) in ethanol (1.0 mL) was added dropwise over 4 hours. The reaction mixture was stirred at room temperature for 16 hours and extracted with DCM (3×40 mL). The combined organic layers were dried over sodium sulfate and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (0–20% EtOAc/hexanes) to yield ethyl 2-(4-(dimethylsilyl)-phenyl)cyclopropanecarboxylate 5b (40.0 mg, 0.17 mmol, 42%) as clear oil. Rf = 0.48 (20% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz): δ 7.37 (d, J = 8.5 Hz, 2H), 6.62 (d, J = 8.5 Hz, 2H), 4.39 (m, 1H), 4.26 (q, J = 7.0 Hz, 2H), 3.91 (s, 2H), 1.31 (t, J = 7.3 Hz, 3H), 0.30 (d, J = 3.5 Hz, 6H). 13C NMR (CDCl3, 125 MHz): δ 171.1, 148.0, 135.4, 124.8, 122.8, 61.5, 45.7, 14.3, −3.3. GC-MS m/z (% relative intensity): 237 (32.5), 164 (100.0). HRMS (ESI) m/z: [M + H]+ Calcd for C12H20NO2Si 238.1263; Found 238.1260.
Ethyl 2-((4-aminophenyl)dimethylsilyl)acetate (5c).
Compound 5c was prepared according to the procedure described in Scheme S3, which was adapted from a previously reported procedure.6 A solution of 4-(dimethylsilyl)aniline 5a (50 mg, 0.33 mmol, 1 equiv.) and pyridine (52 mg, 0.66 mmol, 2 equiv.) in anhydrous DCM (1 mL) was stirred for 10 min, followed by the addition of benzyl chloroformate (68 mg, 0.40 mmol, 1.2 equiv.). The reaction mixture was stirred overnight, after which the solvent was removed under reduced pressure. Benzyl (4-(dimethylsilyl)phenyl)carbamate 18 was obtained as a white flaky solid (62 mg, 0.22 mmol, 66% yield) after purification by column chromatography on silica gel (5% EtOAc/hexanes). Rf = 0.38 (5% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz): δ 7.49–7.35 (m, 9H), 6.71 (br, 1H), 5.21 (s, 2H), 4.41 (quin, J = 3.5 Hz, 1H), 0.33 (d, J = 3.5 Hz, 6H). 13C NMR (CDCl3, 125 MHz): δ 153.3, 138.9, 136.1, 135.1, 132.0, 128.8, 128.5, 128.5, 118.2, 67.2, −3.5. GC-MS m/z (% relative intensity): 285 (82.5), 241 (25.9), 226 (34.0), 162 (51.7), 150 (100.0).
Benzyl (4-(dimethylsilyl)phenyl)carbamate 18 (50 mg, 0.18 mmol, 1 equiv.) and Rh2(OAc)4 (1 mg, 2 μmol, 1 mol%) were added to a flame dried 5 mL round bottom flask under argon atmosphere, dissolved in anhydrous DCM (1 mL), and subsequently cooled to −78 °C. A solution of EDA (23.6 mg, 0.18 mmol, 1 equiv.) in anhydrous DCM (1 mL) was added dropwise with a syringe pump over a period of 30 min. The reaction mixture was allowed to reach room temperature and stirred for 16 h. The solvent was removed under reduced pressure and the crude product purified via column chromatography on silica gel (0–20% EtOAc/hexanes) to afford ethyl 2-((4-(((benzyloxy)carbonyl)amino)phenyl)dimethylsilyl)acetate 19 as a clear oil (6.0 mg, 0.018 mmol, 9% yield). Rf = 0.49 (20% EtOAc/hexanes). 1H NMR (CDCl3, 400 MHz): δ 7.48–7.34 (m, 9H), 6.70 (br, 1H), 5.20 (s, 2H), 4.03 (q, J = 7.1 Hz, 2H). 2.08 (s, 2H), 1.16 (t, J = 7.1 Hz, 3H), 0.38 (s, 6H). 13C NMR (CDCl3, 125 MHz): δ 172.8, 153.2, 130.1, 136.1, 134.7, 131.5, 128.8, 128.6, 128.5, 118.1, 67.3, 60.1, 26.5, 14.5, −2.6. APCI-MS (TLC) m/z: 372, 284.
A flame dried 5 mL round bottom flask was charged with ethyl 2-((4-(((benzyloxy)carbonyl)amino)phenyl)dimethylsilyl)acetate 19 (10 mg, 0.027 mmol, 1 equiv.), 10% Pd/C (4 mg) and anhydrous EtOH (1 mL) under argon atmosphere. The reaction mixture was purged with H2 for 30 min and stirred overnight under H2 atmosphere. After exchanging the atmosphere with argon, the residue was diluted with EtOAc, filtered over celite, concentrated under reduced pressure and purified by column chromatography on silica gel (10–50% EtOAc/hexanes) to give ethyl 2-((4-aminophenyl)dimethylsilyl)acetate 5c as yellow oil (4.1 mg, 0.017 mmol, 64 %). Rf = 0.55 (50% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz): δ 6.96 (d, J = 5.0, 2H), 6.59 (d, J = 5.0 Hz, 2H), 4.24 (q, J = 5.0 Hz, 2H), 2.67 (s, 2H), 1.95 (t, J = 5.0 Hz, 3H), 1.31 (s, 6H). 13C NMR spectrum could not be collected due to the instability of the product 5c in the NMR solvent. GC-MS m/z (% relative intensity): 237 (20.6), 222 (27.4), 180 (69.1), 150 (100.0), 136 (31.8). HRMS (ESI) m/z: [M + H]+ Calcd for C12H20NO2Si 238.1263; Found 238.1256.
Ethyl 2-((4-mercaptophenyl)amino)acetate (6b).
This compound was synthesized according to a modification of a previously reported procedure (Scheme S4).20 4-Aminothiophenol 6a (200 mg, 1.60 mmol, 1 equiv.) was dissolved in a minimal amount of MeOH (2 mL) and DMSO (60 μL) and treated with I2 (162 mg, 0.64 mmol, 0.4 equiv.). After consumption of the starting material (3 h), the solvent was removed under reduced pressure and the crude product purified by column chromatography on silica gel (20–80% EtOAc/hexanes) to yield 4,4’-disulfanediyldianiline (20) as yellow solid (104 mg, 0.42 mmol, 52% yield). Rf = 0.26 (50% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz): δ 7.22 (d, J = 7.0 Hz, 2H), 6.55 (d, J = 8.5 Hz, 2H), 3.75 (br, 4H). 13C NMR (CDCl3, 125 MHz): δ 147.7, 134.4, 124.7, 115.5. GC-MS m/z (% relative intensity): 248 (38.5), 124 (78.9), 125 (100.0).
Ethyl 2-((4-mercaptophenyl)amino)acetate. To a flame dried 10 mL round bottom flask, equipped with a stir bar, were added 4,4’-disulfanediyldianiline 20 (48 mg, 0.19 mmol, 1 equiv.), Rh2(OAc)4 (2 mg, 4 μmol, 2 mol%), and anhydrous DCM (1 mL) under argon. After that, a solution of EDA (43 mg, 0.38 mmol, 2 equiv.) in anhydrous DCM (1 mL) was slowly added dropwise over a period of 30 min. The resulting reaction mixture was stirred at room temperature for 16 hours. The solvent was removed under reduced pressure and the crude product purified via column chromatography on silica gel (20–80% EtOAc/hexanes) to afford ethyl 2-((4-mercaptophenyl)amino)acetate (6b) as a yellow solid (20 mg, 0.089 mmol, 47% yield). Rf = 0.31 (50% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz): δ 7.25 (d, J = 7.5 Hz, 2H), 6.47 (d, J = 7.0 Hz, 2H), 4.41 (br, 1H), 4.22 (q, J = 6.5 Hz, 2H), 4.09 (br, 1H), 3.85 (s, 2H), 1.27 (t, J = 6.3 Hz, 3H). 13C NMR (CDCl3, 125 MHz): δ 170.8, 147.5, 134.2, 125.4, 113.3, 61.6, 45.2, 14.3. GC-MS m/z (% relative intensity): 211 (36.0), 128 (100.0), 136 (23.8), 109 (11.3).
Ethyl 2-((4-aminophenyl)thio)acetate (6c).
This compound was synthesized according to a modification of a previously reported procedure (Scheme S5).21 A flame dried 25 mL 2-neck round bottom flask was charged with 4-aminothiophenol 6a (200 mg, 1.60 mmol, 1 equiv.) and anhydrous THF (10 mL) under argon. The solution was cooled to −30 °C and sodium hydride (66 mg, 1.66 mmol, 1.04 equiv.) was added in portions. After the reaction mixture was allowed to reach 0 °C, it was cooled again to −30 °C for the addition of ethyl bromoacetate (264 mg, 1.58 mmol, 0.99 equiv.), and stirred overnight at room temperature. The solvent was removed under reduced pressure and the crude product taken up in EtOAc (20 mL) and water (20 mL). The aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with aq. saturated NaHCO3 (10 mL), brine (10 mL) and water (10 mL), dried over Na2SO4 and concentrated under reduced pressure. Purification by column chromatography on silica gel (0–40% EtOAc/hexanes) afforded ethyl 2-((4-aminophenyl)thio)acetate (6c) as a yellow oil (268 mg, 1.26 mmol, 79% yield). Rf = 0.54 (20% EtOAc/hexanes). 1H NMR (CDCl3, 500 MHz): δ 7.29 (d, J = 6.5 Hz, 2H), 6.60 (d, J = 7.0 Hz, 2H), 4.13 (t, J = 6.7 Hz, 2H), 3.76 (br, 2H), 3.45 (s, 2H), 1.21 (t, J = 6.5 Hz, 3H). 13C NMR (CDCl3, 125 MHz): δ 170.3, 147.0, 135.0, 121.8, 115.6, 61.4, 39.3, 14.2. GC-MS m/z (% relative intensity): 211 (84.2), 212.1 (10.9), 138 (25.8), 124 (100.0).
Supplementary Material
ACKNOWLEDGMENT
This work was supported in part by the U.S. National Science Foundation (NSF) grant CHE-1609550 and in part by the U.S. National Institute of Health (NIH) grant GM098628. E.J.M. acknowledges support from the NIH Graduate Training Grant T32GM118283. MS instrumentation was supported by the U.S. NSF grant CHE-0946653. The authors are grateful to Dr. Jermaine Jenkins and the Structure Biology & Biophysics Facility at the University of Rochester for providing access to the CD instrumentation.
Footnotes
Supporting Information. Additional experimental data, synthetic schemes, and NMR spectra for all new compounds.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Miller DJ; Moody CJ Synthetic Applications of the O-H Insertion Reactions of Carbenes and Carbenoids Derived from Diazocarbonyl and Related Diazo-Compounds. Tetrahedron 1995, 51, 10811–10843; [Google Scholar]; (b) Doyle MP; Forbes DC Recent Advances in Asymmetric Catalytic Metal Carbene Transformations. Chem. Rev. 1998, 98, 911–936; [DOI] [PubMed] [Google Scholar]; (c) Lebel H; Marcoux JF; Molinaro C; Charette AB Stereoselective Cyclopropanation Reactions. Chem. Rev. 2003, 103, 977–1050; [DOI] [PubMed] [Google Scholar]; (d) Zhang ZH; Wang JB Recent Studies on the Reactions of Alpha-Diazocarbonyl Compounds. Tetrahedron 2008, 64, 6577–6605; [Google Scholar]; (e) Davies HML; Morton D Guiding Principles for Site Selective and Stereoselective Intermolecular C-H Functionalization by Donor/Acceptor Rhodium Carbenes. Chem. Soc. Rev. 2011, 40, 1857–1869; [DOI] [PubMed] [Google Scholar]; (f) Zhu SF; Zhou QL Transition-Metal-Catalyzed Enantioselective Heteroatom-Hydrogen Bond Insertion Reactions. Acc. Chem. Res. 2012, 45, 1365–1377. [DOI] [PubMed] [Google Scholar]
- (2).(a) Bordeaux M; Tyagi V; Fasan R Highly Diastereoselective and Enantioselective Olefin Cyclopropanation Using Engineered Myoglobin-Based Catalysts. Angew. Chem. Int. Ed 2015, 54, 1744–1748; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bajaj P; Sreenilayam G; Tyagi V; Fasan R Gram-Scale Synthesis of Chiral Cyclopropane-Containing Drugs and Drug Precursors with Engineered Myoglobin Catalysts Featuring Complementary Stereoselectivity. Angew. Chem. Int. Ed. 2016, 55, 16110–16114; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sreenilayam G; Moore EJ; Steck V; Fasan R Metal Substitution Modulates the Reactivity and Extends the Reaction Scope of Myoglobin Carbene Transfer Catalysts. Adv. Synth. Cat. 2017, 359, 2076–2089; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Tinoco A; Steck V; Tyagi V; Fasan R Highly Diastereo- and Enantioselective Synthesis of Trifluoromethyl-Substituted Cyclopropanes Via Myoglobin-Catalyzed Transfer of Trifluoromethylcarbene. J. Am. Chem. Soc. 2017, 139, 5293–5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Coelho PS; Brustad EM; Kannan A; Arnold FH Olefin Cyclopropanation Via Carbene Transfer Catalyzed by Engineered Cytochrome P450 Enzymes. Science 2013, 339, 307–310; [DOI] [PubMed] [Google Scholar]; (b) Coelho PS; Wang ZJ; Ener ME; Baril SA; Kannan A; Arnold FH; Brustad EM A Serine-Substituted P450 Catalyzes Highly Efficient Carbene Transfer to Olefins in Vivo. Nat. Chem. Biol 2013, 9, 485–487; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gober JG; Rydeen AE; Gibson-O’Grady EJ; Leuthaeuser JB; Fetrow JS; Brustad EM Mutating a Highly Conserved Residue in Diverse Cytochrome P450s Facilitates Diastereoselective Olefin Cyclopropanation. Chembiochem 2016, 17, 394–397; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Gober JG; Ghodge SV; Bogart JW; Wever WJ; Watkins RR; Brustad EM; Bowers AA P450-Mediated Non-Natural Cyclopropanation of Dehydroalanine-Containing Thiopeptides. ACS Chem. Biol 2017, 2, 1726–1731. [DOI] [PubMed] [Google Scholar]
- (4).(a) Wang ZJ; Peck NE; Renata H; Arnold FH Cytochrome P450-Catalyzed Insertion of Carbenoids into N-H Bonds. Chem. Sci 2014, 5, 598–601; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sreenilayam G; Fasan R Myoglobin-Catalyzed Intermolecular Carbene N-H Insertion with Arylamine Substrates. Chem. Commun 2015, 51, 1532–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Tyagi V; Bonn RB; Fasan R Intermolecular Carbene S-H Insertion Catalysed by Engineered Myoglobin-Based Catalysts. Chem. Sci 2015, 6, 2488–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kan SBJ; Lewis RD; Chen K; Arnold FH Directed Evolution of Cytochrome C for Carbon-Silicon Bond Formation: Bringing Silicon to Life. Science 2016, 354, 1048–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).(a) Sreenilayam G; Moore EJ; Steck V; Fasan R Stereoselective Olefin Cyclopropanation under Aerobic Conditions with an Artificial Enzyme Incorporating an Iron-Chlorin E6 Cofactor. ACS Catal 2017, 7, 7629–7633; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Key HM; Dydio P; Clark DS; Hartwig JF Abiological Catalysis by Artificial Haem Proteins Containing Noble Metals in Place of Iron. Nature 2016, 534, 534–537; [DOI] [PubMed] [Google Scholar]; (c) Reynolds EW; McHenry MW; Cannac F; Gober JG; Snow CD; Brustad EM An Evolved Orthogonal Enzyme/Cofactor Pair. J. Am. Chem. Soc 2016, 138, 12451–12458; [DOI] [PubMed] [Google Scholar]; (d) Reynolds EW; Schwochert TD; McHenry MW; Watters JW; Brustad EM Orthogonal Expression of an Artificial Metalloenzyme for Abiotic Catalysis. Chembiochem 2017, 18, 2380–2384; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wolf MW; Vargas DA; Lehnert N Engineering of Rumb: Toward a Green Catalyst for Carbene Insertion Reactions. Inorg. Chem 2017, 56, 5623–5635; [DOI] [PubMed] [Google Scholar]; (f) Ohora K; Meichin H; Zhao LM; Wolf MW; Nakayama A; Hasegawa J; Lehnert N; Hayashi T Catalytic Cyclopropanation by Myoglobin Reconstituted with Iron Porphycene: Acceleration of Catalysis Due to Rapid Formation of the Carbene Species. J. Am. Chem. Soc 2017, 139, 17265–17268. [DOI] [PubMed] [Google Scholar]
- (8).(a) Srivastava P; Yang H; Ellis-Guardiola K; Lewis JC Engineering a Dirhodium Artificial Metalloenzyme for Selective Olefin Cyclopropanation. Nat. Commun 2015, 6, 7789; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rioz-Martinez A; Oelerich J; Segaud N; Roelfes G DNA-Accelerated Catalysis of Carbene-Transfer Reactions by a DNA/Cationic Iron Porphyrin Hybrid. Angew. Chem. Int. Ed 2016, 55, 14136–14140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Talele TT The “Cyclopropyl Fragment” Is a Versatile Player That Frequently Appears in Preclinical/Clinical Drug Molecules. J. Med. Chem 2016, 59, 8712–8756. [DOI] [PubMed] [Google Scholar]
- (10).(a) Galardon E; Le Maux P; Simonneaux G Cyclopropanation of Alkenes, N-H and S-H Insertion of Ethyl Diazoacetate Catalysed by Ruthenium Porphyrin Complexes. Tetrahedron 2000, 56, 615–621; [Google Scholar]; (b) Aviv I; Gross Z Iron Corroles and Porphyrins as Very Efficient and Highly Selective Catalysts for the Reactions of Alpha-Diazo Esters with Amines. Synlett 2006, 951–953; [Google Scholar]; (c) Baumann LK; Mbuvi HM; Du G; Woo LK Iron Porphyrin Catalyzed N-H Insertion Reactions with Ethyl Diazoacetate. Organometallics 2007, 26, 3995–4002; [Google Scholar]; (d) Noels AF; Demonceau A; Petiniot N; Hubert AJ; Teyssie P Transition-Metal-Catalyzed Reactions of Diazo-Compounds, Efficient Synthesis of Functionalized Ethers by Carbene Insertion into the Hydroxylic Bond of Alcohols. Tetrahedron 1982, 38, 2733–2739; [Google Scholar]; (e) Zhu SF; Cai Y; Mao HX; Xie JH; Zhou QL Enantioselective Iron-Catalysed O-H Bond Insertions. Nat. Chem 2010, 2, 546–551. [DOI] [PubMed] [Google Scholar]
- (11).(a) Bagheri V; Doyle MP; Taunton J; Claxton EE A New and General-Synthesis of Alpha-Silyl Carbonyl-Compounds by Si-H Insertion from Transition-Metal Catalyzed-Reactions of Diazo Esters and Diazo Ketones. J. Org. Chem 1988, 53, 6158–6160; [Google Scholar]; (b) Andrey O; Landais Y; Planchenault D A One-Pot Synthesis of Alpha-(Alkoxysilyl)Acetic Esters. Tetrahedron Lett 1993, 34, 2927–2930; [Google Scholar]; (c) Wang JC; Xu ZJ; Guo Z; Deng QH; Zhou CY; Wan XL; Che CM Highly Enantioselective Intermolecular Carbene Insertion to C-H and Si-H Bonds Catalyzed by a Chiral Iridium(Iii) Complex of a D-4-Symmetric Halterman Porphyrin Ligand. Chem. Commun 2012, 48, 4299–4301; [DOI] [PubMed] [Google Scholar]; (d) Keipour H; Ollevier T Iron-Catalyzed Carbene Insertion Reactions of Alpha-Diazoesters into Si-H Bonds. Organic Letters 2017, 19, 5736–5739. [DOI] [PubMed] [Google Scholar]
- (12).(a) Sambasivan R; Ball ZT Metallopeptides for Asymmetric Dirhodium Catalysis. J. Am. Chem. Soc 2010, 132, 9289–9291; [DOI] [PubMed] [Google Scholar]; (b) Yang H; Srivastava P; Zhang C; Lewis JC A General Method for Artificial Metalloenzyme Formation through Strain-Promoted Azide-Alkyne Cycloaddition. Chembiochem 2014, 15, 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zhang KD; El Damaty S; Fasan R P450 Fingerprinting Method for Rapid Discovery of Terpene Hydroxylating P450 Catalysts with Diversified Regioselectivity. J. Am. Chem. Soc 2011, 133, 3242–3245. [DOI] [PubMed] [Google Scholar]
- (14).Wei Y; Tinoco A; Steck V; Fasan R; Zhang Y Cyclopropanations Via Heme Carbenes: Basic Mechanism and Effects of Carbene Substituent, Protein Axial Ligand, and Porphyrin Substitution. J. Am. Chem. Soc 2018, 140, 1649–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Vojtechovsky J; Chu K; Berendzen J; Sweet RM; Schlichting I Crystal Structures of Myoglobin-Ligand Complexes at near-Atomic Resolution. Biophys. J 1999, 77, 2153–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Bordeaux M; Singh R; Fasan R Intramolecular C(Sp(3))H Amination of Arylsulfonyl Azides with Engineered and Artificial Myoglobin-Based Catalysts. Bioorg. Med. Chem 2014, 22, 5697–5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kunze C; Bommer M; Hagen WR; Uksa M; Dobbek H; Schubert T; Diekert G Cobamide-Mediated Enzymatic Reductive Dehalogenation Via Long-Range Electron Transfer. Nat. Commun. 2017, 8, ARTN 15858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Moore EJ; Zorine D; Hansen WA; Khare SD; Fasan R Enzyme Stabilization Via Computationally Guided Protein Stapling. Proc. Natl. Acad. Sci. USA 2017, 114, 12472–12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Aller E; Brown DS; Cox GG; Miller DJ; Moody CJ Diastereoselectivity in the O-H Insertion Reactions of Rhodium Carbenoids Derived from Phenyldiazoacetates of Chiral Alcohols - Preparation of Alpha-Hydroxy and Alpha-Alkoxy Esters. J. Org. Chem 1995, 60, 4449–4460. [Google Scholar]
- (20).Bettanin L; Saba S; Galetto FZ; Mike GA; Rafique J; Braga AL Solvent- and Metal-Free Selective Oxidation of Thiols to Disulfides Using I-2/Dmso Catalytic System. Tetrahedron Lett 2017, 58, 4713–4716. [Google Scholar]
- (21).Chen BB; Chen HY; Gesicki GJ In Pat. Appl. EP0891325-B1; GD Searle LLC: 1996. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.