

Abstract
Activating mutations in GTPase protein KRAS occurs in approximately 90% of pancreatic cancers. Mutated KRAS lead to constitutive activation of RAF/MEK/ERK and PI3K/Akt pathways in pancreatic cancer. There is currently no effective KRAS-targeted therapeutics available in the clinic for treating this subset of cancer. In this study we demonstrate that combination of a plant-isolated triterpenoid compound AMR-MeOAc with a low concentration of an antiapoptotic protein inhibitor, FL118 exhibited synergistic cytotoxic activity against pancreatic cancer cells with either mutant KRAS (HPAF-II, KRASG12D) or wild type KRAS (BxPC-3, KRASWT). In pancreatic cancer cells with mutant KRASG12D, AMR-MeOAc and FL118 acting together to inhibit the constitutive KRASG12D mutant activity, increase the reactive oxygen species (ROS) formation, apoptosis induction, and decrease of the expression of survivin and XIAP, while strongly inducing Bax. These effects were also associated with the decrease of B-RAF, ERK and p-ERK. Additionally, AMR-MeOAc and FL118 alone or in combination inhibited the constitutive activation of NF-κB in BxPC-3 cells, which suggests that inhibition of NF-κB in BxPC-3 cells by AMR-MeOAc and FL118 may also be a part of the mechanism of action, when pancreatic cancer cells possess wild type KRAS. Together, the novel combination treatment might provide an effective strategy to overcome the KRASG12D mutant-mediated and NF-κB activation-mediated resistance in pancreatic cancer with either KRASG12D mutation or NF-κB activation/wild type KRAS.
Keywords: FL118, AMR-MeOAc, pancreatic cancer, KRASG12D, survivin, XIAP, ERK, NF-κB, reactive oxygen species (ROS)
Introduction
Pancreatic cancer is one of the most aggressive and treatment-resistant tumors, and is currently the fourth leading cause of cancer-related death in the United States [1]. Activating mutations of the KRAS oncogene represent one of the most prevalent genetic alterations in cancer, which is mutated in over 90% of pancreatic cancer [2]. KRAS is a membrane-bound GTPase that cycles between an active GTP-bound form and an inactive GDP-bound form, resultant from the hydrolysis of the bound GTP [3,4]. Switching between these two states is controlled by two classes of proteins: guanosine nucleotide exchange factors known as GEFs and GTPase-activating proteins known as GAPs. As their name suggests, GEFs assist with the exchange of bound GDP with GTP, whereas GAPs stimulate the hydrolytic ability of RAS to convert bound GTP to GDP [5]. Mutations in KRAS have been shown to lock active KRAS in the GTP bound conformation and activate the RAS-RAF-MEK-ERK signaling pathway [6]. Although significant progress has been made in the understanding of KRAS mutation and KRAS mutation-mediated therapeutic resistance, there is still no effective KRAS-targeted therapy available in the clinic. Several strategies have been designed to target mutant KRAS, including direct targeting by inhibiting its GTPase activity [6]. The most promising strategy was thought to be the indirect targeting by inhibition of Ras farnesylation, which would block the prenylation of KRAS, which is required for proper reaching of the Ras site of action at the inner surface of the plasma membrane for inhibiting its activity or downstream effectors [7]. Several natural and synthetic compounds were shown to suppress cell growth and inhibited the enzyme farnesyl transferase that is responsible for addition of the farnesyl group to Ras; such compounds have shown potent antitumor activity on transgenic mouse models [8]. Unfortunately, these compounds did not exhibit clinical efficacy as single agents [9,10]. Lack of clinical efficacy was found to be due to the prenylation of KRAS through alternative mechanisms, involving geranylgeranyltransferase I (GGTase I) to correct localization to the membrane [9,11]. Dual inhibitors of GGTase and FTase, such as L-778, 123 were therefore developed. However, their toxicities in clinical trials prohibited their further clinical development [11].
Another strategy to inhibit mutant KRAS is to inhibit the interaction between KRAS and cyclic GMP phosphodiesterase δ (PDEδ), which mediates correct localization and signaling by farnesylated KRAS [12]. The other approach utilized was to inhibit components of its key effector signaling molecules through combination therapies. Among numerous downstream effectors of KRAS, the well characterized effector signaling pathways are RAF/MEK/ERK and PI3K/AKT cascades, as well as the GEFs for the RAS-like (Ral) small GTPases (RalGEFs); these signaling pathways ultimately control cancer cell growth and/or survival [13-15]. Activated KRAS can inhibit the apoptotic signaling cascade through its effector PI3K, which in turn activates AKT [16,17]. AKT is known to be a potent pro-survival kinase that inhibits apoptosis via several mechanisms, including the phosphorylation and subsequent inactivation of the pro-apoptotic Bcl-2 family protein BAD, the induction of the anti-apoptotic protein Bcl-XL overexpression and the inhibitory phosphorylation of the initiator caspase-9 [18,19]. Akt induces the activation of NF-κB and inhibition of NF-κB (IκB) kinase α (IKKα) [20]. Recently, several synthetic lethal interactions have been identified in large-scale screening based on shRNA constructs. The study shows that a noncanonical IκB kinase TBK1 is involved in NF-κB signaling for survival [21]. Other synthetic lethal interactions in pancreatic cancer cells with mutant KRAS include the dual inhibition of Bcl-XL and MEK [22]. Despite these promising developments, targeting mutant KRAS-driven cancers remains one of the most difficult challenges in anticancer therapy due to several obstacles, including limited understanding of RAS-mediated signaling transduction feedback loops, pathway redundancy, tumor heterogeneity, and unclear mechanisms of how RAS proteins activate their downstream targets, as well as unresolved structures of protein complexes formed on the RAS oncoprotein [23]. Gemcitabine (2’-deoxy-2’-difluorodeoxycytidine), a purine analog, is the standard chemotherapeutic drug for pancreatic cancer therapy. The response rate however, is only about 12% with a median patient’s survival of about six months with this drug [24]. FOLFIRINOX chemotherapy (5-flurouracil, leucovorin, irinotecan and oxaliplatin combination) was recently used for the treatment of pancreatic cancer, which increased survival of patients by four months with severe toxicity in comparison with gemcitabine [25]. Therefore, new therapeutic strategies are urgently needed to improve the outcomes. Additionally, it is also essential to fill in these knowledge gaps in order to develop more effective agents for targeting KRAS-mutant cancers.
Previous studies demonstrated that AMR-MeOAc, a triterpene amooranin monoacetate isolated from Amoora rohituka stem bark, inhibits mutation-activated KRASG12D through ERK, Akt and survivin, and caused pancreatic cancer HPAF-II cell death [26]. FL118 is a novel camptothecin derivative with different mechanism of action, and shows a wide range of anticancer activities. Studies show that FL118 effectively inhibits the expression of multiple cancer survival proteins including survivin, Mcl-1, XIAP, and cIAP2 in a p53 status-independent manner in colorectal, head and neck, ovarian, prostate and lung cancer cells [27]. FL118 exhibits superior antitumor activity in human tumor xenograft models in comparison with irinotecan, topotecan, doxorubicin, 5-FU, gemcitabine, docetaxel, oxaliplatin, cytoxan and cisplatin tested [27]. Notably, in the cancer cells with wild type p53, FL118 activates p53-dependent senescence and induced MdmX protein degradation irrespective of ATM, p53 and p21 status in colon cancer cells [28]. In addition, our studies demonstrate that FL118 shows superior activity and overcomes irinotecan and topotecan resistance in human tumor xenograft models [29]. Recent studies indicate that FL118 alone or in combination with gemcitabine can effectively inhibit pancreatic cancer tumor growth in both pancreatic cancer cell line-established tumor and pancreatic cancer patient-derived xenografts in animal models [30]; the present study was conducted to determine if a low concentration of FL118 can enhance the effect of AMR-MeOAc and overcome KRASG12D-mediated resistance in pancreatic cancer cells as well as the mechanism of action, and thus provide the experimental basis for potential clinical application of this combination.
Materials and methods
Cells, vectors and cell culture
Human pancreatic adenocarcinoma HPAF-II cells with mutated KRASG12D and BxPC-3 cells with wild type KRAS were purchased from American Type Culture Collection (ATCC, Manassas, VA). HPAF-II cells were stably transfected with lentiviral vector encoding KRAS-specific shRNA or control shRNA, respectively. Cells were maintained in RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 100 U/mL penicillin, and 0.1 μg/mL streptomycin.
Cell viability
Cell viability was assessed using MTT assay as previously reported [31]. Briefly, human pancreatic cancer cell lines HPAF-II and BxPC-3 cells were cultured in RPMI-1640 at 37°C and 5% CO2. Cells were seeded in 96-well microplates at a density of 4 × 104 cells/well and incubated overnight. The cells were then treated with AMR-MeOAc (Figure 1A) and FL118 (Figure 1B) at various concentrations for 48 h. After drug treatment, 20 μl MTT solution (5 mg/ml in PBS) was added to each well and incubated for 4 h at 37°C. The formed formazan crystals were dissolved in 100 μl DMSO and mixed thoroughly for 20 min at room temperature. Cell viability was determined by measuring absorbance at 570 nm in a microplate reader (VersaMax, Molecular Devices). The IC50 value was generated from the log dose-response curves for cells using the Graphpad Prism version 5 for Windows (Graphpad Software, La Jolla, CA, USA).
Figure 1.
Chemical structure of AMR-MeOAc (A) and FL118 (B).
Cell treatment and combination index (CI) calculation
Cells were treated with 0.001-100 µM AMR-MeOAc and 0.001-100 nM FL118 alone and in combination, which is the so-called “fixed ratio one another”. Cell viability assay data obtained from cells treated as above were used to analyze the combined drug effects using the CalcuSyn software (Biosoft, Ferguson, MO, USA) to determine whether the combination was synergistic. This approach is based upon the Chou-Talalay equation [32], which calculates a combination index (CI). The formula for the classic isobologram is given by: CI = CA,X/ICX,A + CB,X/ICX,B, where, CA,X and CB,X indicate the concentrations of drug A and drug B, used in combination to achieve x% drug effect. ICX,A and ICX,B were the concentrations of individual agents necessary to achieve the same effect. CI of 1 indicated an additive effect between the two agents, whereas CI < 1 indicates, synergism and CI > 1 indicates antagonism, respectively [33].
RAS pull down assay
HPAF-II cells were treated with AMR-MeOAc and FL118 alone or in combination for 0, 6, 12, 24, or 48 h. RAS GTP levels were determined using a RAS activation assay kit (Millipore) according to the manufacturer’s instructions. Cell lysates (1 mg) were incubated with 10 μg Raf-1-RBD for 45 min at 4°C on a rotating wheel and centrifuged for 15 sec at 14,000 × g to pellet agarose beads. After discarding the supernatant, agarose beads were washed three times with 500 μl of lysis buffer and the pellets were resuspended in 2X Laemmli sample buffer (1 M Tris-HCl, 10% SDS, 1 M DTT and 1% bromophenol blue), boiled for 5 min, and centrifuged at 14,000 × g. The pull-down RAS-GTP was subjected to 12% SDS-PAGE and the pulled-down active KRAS was revealed by immunoblot analysis with KRASG12D and total RAS antibodies.
Flow cytometric analysis of cell cycle and apoptosis
Measurement of cell cycle was conducted by propidium iodide (PI) staining and flow cytometry as previously described [34]. Briefly, cells were treated with 3 µM AMR-MeOAc and 8 nM FL118 alone or in combination for 48 h, and single cell suspensions were fixed in ice-cold 70% ethanol overnight, washed twice in PBS and then cells were incubated with 100 μg/ml DNase-free RNase A (Sigma-Aldrich) in PBS and PI staining solution (Sigma-Aldrich) (50 μg/ml) for at least 30 min in dark at room temperature, and monitored with the FL3 channel in a FACS CaliburTM flow cytometer. The cell cycle data was analyzed with CellQuest software (Tree Star, Oregon, US). Cells apoptosis were measured by Annexin V-FITC (fluorescein isothiocyanate)/PI staining as described previously [27].
Detection of reactive oxygen species (ROS) generation
Intracellular ROS generation was detected using flow cytometry with Dichloro-dihydro-fluorescein diacetate (H2DCF-DA)) (Invitrogen). Cells were treated with AMR-MeOAc, FL118 alone and in combination for 0, 12 h. After treatment, cells were harvested with trypsin, washed once with PBS, resuspended in 10 µM H2DCF-DA and incubated at 37°C for 30 min in the dark. In the antioxidant combination experiments, cells were pretreated with 5 mM N-acetyl-L-cysteine (NAC) for 1 h before adding the drugs. The samples were then immediately assayed with the FL1 channel by flow cytometry. The experiments were repeated at least two times.
Lentiviral shRNA infection
HPAF-II cells (5 × 105/well) were plated in 6-well plates. After 24 h, cell culture medium was removed and cells were infected with lentiviral vector encoding KRAS-specific shRNA (Thermo Fisher Scientific),) with the sequence of TGC TGT TGA CAG TGA GCG AGC ATA CTA GTA CAA GTG GTA ATA GTG AAG CCA CAG ATG TAT TAC CAC TTG TAC TAG TAT GCC TGC CTA CTG CCT CGG A and one control non-silencing shRNA using a 1:10 dilution of virus in medium for 16 h. In order to select stably transfected clones, medium containing viral particles were replaced with culture medium containing 5 µg/ml puromycin to select for 72 h; then stable clones were expanded. Cell lysates were prepared following KRAS shRNA expression to assess gene suppression. To determine differential viability effects in HPAF-II versus HPAF-II KRAS shRNA and HPAF-II control shRNA cells, the mean cell viability was calculated 48 h after seeding the cells. Results were grouped together for HPAF-II versus HPAF-II KRAS shRNA and HPAF-II control shRNA cells.
Real-time quantitative RT-PCR (real-time RT-qPCR)
The real-time RT-qPCR was performed similar to previously described [35]. Briefly, total RNA was extracted from drug-treated and control cells using TRI REAGENT RT (Molecular Research Center, Inc.). RNA concentration was measured using an ND-1000 spectrophotometer (Thermo Fisher Scientific Inc., USA). RNA (2 μg) was converted to cDNA using anchored oligo (dT) primers (RevertAid First Strand cDNA Synthesis Kit, Thermo Scientific) following the manufacturer’s instructions. Real time PCR was conducted to measure survivin mRNA expression. Newly synthesized cDNA (2 μl) was used as a template for the reaction in a total volume of 20 μl reactants using the iTaq SYBR Green Supermix with ROX (Bio-Rad, Hercules, CA) and analyzed on an Applied Biosystems 7300 Real Time PCR System and normalized to 18S. The sequences of oligonucleotides (primers) used in real-time qPCR reactions for survivin were: 5’-GGC AGC CCT TTC TCA AGG ACC ACC-3’ (forward) and 5’-GAT GGC ACG GCG CAC TTT CTT CGC-3’ (reverse) for amplifying survivin product. 18S was used as an internal control. The real-time qPCR condition is 95°C for 3 min as a pre-denature step, followed by 40 PCR cycles at 95°C for 15 s and 60°C for 45 s. A dissociation curve was created at the completion of the PCR in order to ensure that the reaction produced the correct products as anticipated. The results of the relative mRNA expression (survivin to 18S) in the HPAF-II cells were analyzed using the ΔΔC t method [36].
Nuclear and cytoplasmic fractionation
Cells were washed in ice-cold PBS twice and incubated on ice for 10 min after treatment with hypotonic cytoplasmic lysis buffer (20 mM Hepes pH 7.6, 10 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM DTT, 0.1% NP-40, 20% glycerol) plus proteinase inhibitors. Nuclei were pelleted at 400 × g at 4°C for 4 min. Supernatants were collected, and the nuclear pellet was washed twice with hypotonic cytoplasmic lysis buffer. Nuclear lysis buffer (hypotonic cytoplasmic lysis buffer plus 500 mM NaCl) was added to the pellet, and samples were incubated for 30 min on ice. Following centrifugation at 16,000 × g at 4°C for 15 min, the supernatant was collected as the nuclear fraction extracts.
Western blot analysis
Western blots were performed as previously described [34]. Briefly, cell lysates were isolated from cells with RIPA lysis buffer. The protein concentration was determined by BCA method. Equal amounts (20 to 50 μg) of cell extract were subjected to electrophoresis in 7-12% SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA). The membranes were blocked with 5% milk and then incubated with antibodies for survivin, Bax, Bcl-2, Akt, p-AktSer473, ERK, p-ERK, B-RAF, NF-κB p65, IκBα, p-IκBα, KRASG12D, RAS and GAPDH overnight at 4°C. Subsequently, the membranes were incubated with HRP-conjugated anti-mouse or -rabbit secondary antibodies at room temperature for 1 h. The protein bands were visualized using an enhanced chemiluminescence reagent (ECL) kit (PerkinElmer, Inc), according to the manufacturer’s instructions.
Electrophoretic mobility shift assay (EMSA)
EMSA for NF-κB was performed using LightshiftTM Chemiluminiscent EMSA kit (Thermo Fisher Scientific) following manufacturer’s protocol. Briefly, DNA was biotin-labeled using the Biotin 3’ end labeling kit (Thermo Fisher Scientific) in a 50 μl reaction buffer, 5 pmol of double stranded NF-κB oligonucleotide (5’-AGTTGAGGGGACTTTCCCAGGC-3’ and 3’-TCAACTCCCCTGAAAGGGTCCG-5’) (Promega) incubated in a microfuge tube with 10 μl of 5x TdT (terminal deoxynucleotidyl transferase) buffer, 5 μl of 5 μM biotin-N4-CTP, 10 U of diluted TdT, 25 μl of ultrapure water at 37°C for 30 min. The reaction was stopped with 2.5 μl of 0.2 M EDTA. To extract labeled DNA, 50 μl of chloroform: isoamyl alcohol (24:1) was added to each tube and centrifuged at 13,000 × g. The top aqueous phase containing the labeled DNA was used for binding reactions. Each binding reaction contained 1X-binding buffer (100 mM Tris, 500 mM KCl, 10 mM Dithiothretol, pH 7.5), 2.5% glycerol, 5 mM MgCl2, 50 ng/μl poly (dI-dC), 0.05% NP-40, 2.5 μg of nuclear extract and 20-50 fm of biotin-end-labeled target DNA. The contents were incubated at room temperature for 20 min. Then, 5 μl of 5X loading buffer was added to this reaction mixture. The resultant samples were subjected to gel electrophoresis on a non-denatured native polyacrylamide gel, followed by a transferring to a nylon membrane. After transfer was completed, DNA was cross-linked to the membrane at 120 mJ/cm2 using a UV cross-linker equipped with 254-nm bulb. The biotin end-labeled DNA was detected using streptavidin-horseradish peroxidase conjugate and a chemiluminiscent substrate. The result was visualized by exposing the membrane to X-ray film (MIDSCI), followed by X-ray film development using a film processor.
Statistical analysis
Data were evaluated using the Student’s t-test and GraphPad Prism version 5. P < 0.05 was considered as statistically significant. Data are presented as means ± SD as indicated. For all graphs: * represents P < 0.05; ** represents P < 0.01; *** represents P < 0.001.
Results
Synergistic antineoplastic effects induced by AMR-MeOAc in combination with a low nM FL118 in pancreatic cancer cells
To determine the effects of AMR-MeOAc and FL118 on the growth inhibition of human pancreatic cancer cells with different KRAS status (HPAF-II with mutant KRASG12D and BxPC-3 with wild type KRAS) were exposed to increasing concentrations of AMR-MeOAc and FL118 for 48 h as single agent to reflect the cell growth inhibition. The concentration that needs to inhibit 50% of HPAF-II and BxPC-3 cell growth was found to be 14.69 µM, 12.45 µM for AMR-MeOAc (Figure 2A, 2C) and 10.21 nM, 4.17 nM for FL118 (Figure 2B, 2D), respectively. The Figure 2 results indicate that HPAF-II and BxPC-3 cells showed similar sensitivity to AMR-MeOAc, while BxPC-3 cells were relatively more sensitive to FL118.
Figure 2.
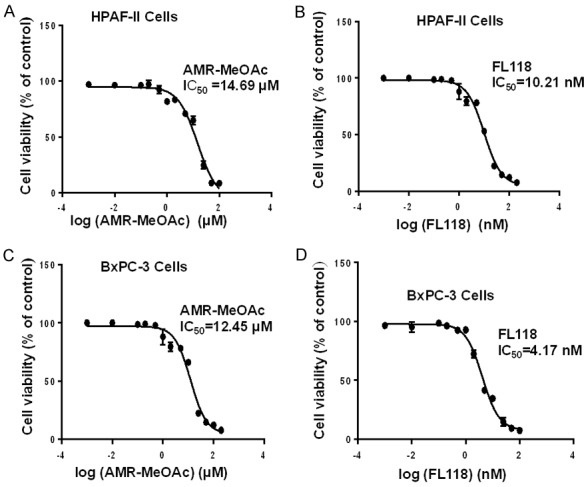
Antiproliferative activity of AMR-MeOAc and FL118 in pancreatic cancer cells. A and B are HPAF-II cell viability curves upon treatment with AMR-MeOAc and FL118, respecitvely. C and D are BxPC-3 cell viability curves upon treatment with AMR-MeOAc and FL118, respectively. Their corresponding IC50 are shown.
To evaluate the synergistic effect of their combination, cells were treated with fixed ratio one another over a range of drug concentrations in experiments (see the method section for details). The drugs combination index (CI) was calculated at 50% cell growth inhibition using Calcusyn software, which is based on Chou and Talalay’s method [32]. As shown in Table 1, a synergistic effect was observed in a broad range of concentrations of AMR-MeOAc with FL118 in both cell types. Of note, CI < 1, = 1, and > 1 indicated synergistic, additive and antagonistic effect, respectively.
Table 1.
Effect of AMR-MeOAc and FL118 in combination in pancreatic cancer cells
| Cell lines | AMR-MeOAc/FL118 | AMR-MeOAc (µM) | FL118 (nM) | MTT Assay |
|---|---|---|---|---|
| µM/µM | IC50 | IC50 | CI at IC50 | |
| HPAF-II | 3:0.0072 | 14.69 | 10.21 | 0.92 |
| 6:0.0035 | 0.75 | |||
| 7:0.0025 | 0.72 | |||
| BxPC-3 | 4:0.0018 | 12.45 | 4.17 | 0.77 |
| 6:0.001 | 0.73 |
The combination index was calculated using the formula: CI = CA,X/ICX,A + CB,X/ICX,B. The combination index (CI) < 1, < 0.5, > 1 represents synergism, strong synergism and antagonistic effects. Synergistic drug combination index of AMR-MeOAc: FL118 (8+20 nM) in HPAF-II cells and AMR-MeOAc: FL118 (8+20 nM) in BxPC-3 cells showing about 40% cell death were used in rest of the assays.
AMR-MeOAc inhibits mutant KRASG12D activity as well as downstream MAPK and AKT signaling
Next, we examined whether the growth inhibitory activity of AMR-MeOAc and FL118 was associated with downregulation of RAS signaling. Using the RAS-GTP pull-down assay and HPAF-II cells, we showed that AMR-MeOAc significantly inhibits active RAS-GTP in a time-dependent manner; this decrease was more pronounced in combination with FL118 than those observed with AMR-MeOAc alone at the 48 h time point (Figure 3A). It is known that activated RAS binds to RAF and promotes the later activation, then phosphorylates and activates the MEK1 and MEK2 dual specificity protein kinases, which in turn phosphorylate the ERK1/2 mitogen-activated protein kinases (MAPKs) [25]. Therefore, we evaluated RAF/ERK signaling changes induced by AMR-MeOAc and/or FL118 treatment at 12, 24 and 48 h. The inhibition of KRAS by AMR-MeOAc or FL118 alone and in combination resulted in a significant time-dependent inhibition of the B-RAF, ERK and p-ERK, two downstream effectors of KRAS (Figure 3B). The AKT pathway is an important cancer cell survival pathway. AKT can be activated through constitutively active RAS and Src [37]. Our studies showed that AMR-MeOAc decreased the expression of p-AKT and overrode the slight increase of p-AKT by FL118 at the 12 h time point in pancreatic cancer cells (Figure 3B), suggesting that AMR-MeOAc counteracts AKT signaling.
Figure 3.

Effects of AMR-MeOAc and FL118 alone and in combination on mutant KRASG12D activity and downstream signaling. A. HPAF-II cells were treated with 8 µM AMR-MeOAc or 20 nM FL118 alone and in combination for 6, 12, 24, and 48 h. RAS Pull-Down assays and Western blot analysis using KRAS (G12D) and total Ras antibodies were performed to measure the activated KRAS and total RAS protein expressions. B. HPAF-II cells were treated for 12, 24 and 48 h with 8 µM AMR-MeOAc and 20 nM FL118 either alone or in combination, and analyzed for the expressions of B-RAF, ERK, p-ERK and p-AKT by Western blots. C. Cell cycle distribution after AMR-MeOAc and FL118 treatment alone or in combination. After HPAF-II cells treated with 3 µM AMR-MeOAc and 10 nM FL118 alone or in combination for 48 h, cell cycle distribution was analyzed by flow cytometry. D. The percentage of cells at each phase of cell cycle (G0/G1, S and G2/M) was quantified and represented in histogram. Each bar is the mean ± SD derived from three independent experiments.
AMR-MeOAc in combination with FL118 causes cell cycle arrest into S and G2/M phases, and induces apoptosis and reactive oxygen species (ROS) generation
To further determine whether AMR-MeOAc-inhibited cell proliferation alone or in combination with FL118 was involved in cell cycle arrest, HPAF-II cells were exposed to AMR-MeOAc and FL118, alone or in combination for 48 h and cell-cycle distribution was determined by flow cytometric analysis. This study revealed that AMR-MeOAc in combination with FL118 caused S and G2/M phase cell cycle arrest (Figure 3C, 3D). To determine if the cytotoxic effects of AMR-MeOAc alone or in combination with FL118 were due to induction of apoptosis, both HPAF-II and BxPC-3 cells were treated with the compounds alone and in combination for 48 h. Cell apoptosis was then determined by Annexin V-FITC and PI staining, followed by flow cytometry analysis. We found that AMR-MeOAc (µM level) and FL118 (nM level) in turn induced 28.9%, 10.3% apoptosis in HPAF-II cells and 27.1%, 17.3% apoptosis in BxPC-3 cells, while combination treatment resulted in significant increased percentage of apoptosis with 38.1% and 32.6% in HPAF-II and BxPC-3 cells, respectively (Figure 4A, 4B). Furthermore, AMR-MeOAc and FL118 treatment alone or in combination induced cytochrome c release from mitochondria to cytosol (Figure 4C), cleavage of procaspase 3, and PARP (Figure 4D), suggesting cell apoptosis following treatment. Additionally, high levels of reactive oxygen species (ROS) are known to induce apoptosis through different pathways. It is known that mitochondrial metabolism and ROS production are essential for oncogenic KRAS-mediated cell proliferation and tumorigenesis [38]. We determined ROS formation in HPAF-II cells after exposure to AMR-MeOAc alone or in combination with FL118 in the presence or absence of the antioxidant N-acetylcysteine (NAC). Interestingly, while no clear ROS generation was induced in cells exposed to AMR-MeOAc or FL118 alone (Figure 5A), combination treatment significantly increased ROS generation at 12 h (Figure 5A), indicating a possibility that further increased level of ROS beyond a threshold, triggering apoptosis in KRAS mutant cells. To investigate a possible role of ROS production in the apoptotic effect of AMR-MeOAc and FL118, we determined whether antioxidants could at least partially block ROS production and apoptosis. For this purpose, cells were pretreated with 5 mM NAC, an antioxidant, for 1 h before treatment with AMR-MeOAc and FL118 combination. The amount of ROS production was measured 12 h after treatment. Combination treatments induced ROS production was substantially reduced by pretreatment with NAC (Figure 5B). Flow cytometric analyses showed that NAC partially decreases AMR-MeOAc and FL118-induced apoptosis (Figure 5C). Our findings suggest that AMR-MeOAc and FL118 combination induces ROS production, which is involved in apoptosis induction.
Figure 4.

AMR-MeOAc and FL118 induce apoptosis. A. HPAF-II cells were treated with AMR-MeOAc (8 µM) and FL118 (20 nM) alone or in combination for 48 h. Cell apoptosis was measured using Annexin V-FITC/PI staining and flow cytometry. B. BxPC-3 cells were treated with AMR-MeOAc (8 µM) and FL118 (20 nM) alone or in combination for 48 h. Cell apoptosis was measured using Annexin V-FITC+PI flow cytometry. C. AMR-MeOAc and FL118 induce cytochrome c release into the cytosolic fraction in HPAF-II cells. D. Caspase 3 activation and PARP cleavage in HPAF-II cells after AMR-MeOAc and FL118 treatment.
Figure 5.

ROS production in HPAF-II cells after treatment with AMR-MeOAc and FL118. A. HPAF-II cells were treated with AMR-MeOAc and FL118 individually or in combination for 12 h. Cells were then stained with Dichloro-dihydro-fluorescein diacetate (H2DCF-DA), followed by flow cytometry analysis. Representative image histograms of ROS generation are shown. B. Cells were treated with AMR-MeOAc and FL118 in combination for 12 h in the presence or absence of 1 h NAC pretreatment and the intracellular ROS levels were measured by flow cytometry. C. Pretreatment with 5 mM NAC for 1 h blocked combination treatment-mediated apoptosis. Each bar is the mean ± SD derived from three independent experiments. D. Western blot analysis of apoptosis regulation-associated proteins in cell lysates prepared at the indicated time points after AMR-MeOAc and FL118 treatment of HPAF-II cells alone or in combinaiton. E. Survivin mRNA expression was analyzed by real time RT-qPCR after drug treatment as shown. Each bar represents the mean ± SD of triplicate samples, **represents P < 0.01; *** represents P < 0.001.
AMR-MeOAc and FL118 modulates the expression of apoptosis regulatory proteins
It has been reported that FL118 downregulates the expression of several antiapoptotic proteins, including survivin, Mcl-1, cIAP2 and XIAP [27]. We determined whether the decreased cell proliferation and increased caspase 3 and PARP cleavages were associated with the modulation of these apoptotic regulatory proteins. We found that AMR-MeOAc and FL118 decreased the expressions of KRASG12D, survivin, Bcl-xL and XIAP, but elevated the expression of Bax (Figure 5D). Consistent with these observations, previous research showed that RAS/RAF/MEK/ERK and PI3K/AKT positively regulates the transcription of survivin [39]. In our studies we found a strong synergistic reduction in survivin protein expression on AMR-MeOAc plus FL118 at 48 h. Consistently, real-time RT-qPCR analysis also revealed a downregulation of survivin transcript levels alone or in combination treatment at 48 h (Figure 5E).
Effect of AMR-MeOAc and FL118 on cell growth and apoptosis of HPAF-II cells after KRAS depletion
Next, we studied the effect of AMR-MeOAc and FL118 on cell growth and apoptosis after knockdown of KRAS. We found that knockdown of KRASG12D itself in HPAF-II cells decreases cell proliferation (Figure 6A, 6B). We therefore examined whether the expression of cell proliferation and survival-relevant antiapoptotic proteins were also altered in the cells after KRASG12D silencing. Our studies indicated that the expression of antiapoptotic regulators (survivin, XIAP, Bcl-xL) and cell cycle regulator c-Myc were decreased (Figure 6C). To further study the effect of KRASG12D silencing on AMR-MeOAc and FL118-mediated cell growth inhibition, we determined cell growth curves after treatments of KRASG12D-silenced HPAF-II cells with AMR-MeOAc and FL118 alone and in combination. We found that silencing of KRASG12D results in a modest decrease of AMR-MeOAc IC50 (13.12 µM Figure 6D left panel versus 14.69 µM shown in Figure 2A) and FL118 IC50 (8.42 nM Figure 6D right panel versus 10.21 nM shown in Figure 2B). Next, we determined the effect of KRASG12D silencing on AMR-MeOAc and FL118-induced apoptosis alone and in combination. We found that silencing of KRASG12D significantly sensitizes FL118 induction of cell apoptosis (12.4%+7.1% in Figure 6E versus 6.2%+4.1% in Figure 4A), while AMR-MeOAc-induced apoptosis is unaffected by KRASG12D silencing (18.6%+8.8% in Figure 6E versus 18.3%+10.6% in Figure 4A). Overall, the apoptosis induced by AMR-MeOAc and FL118 combinational treatment in KRASG12D-silenced HPAF-II cells versus parental HPAF-II cells are similar: 35.6% (23.7%+11.9% in Figure 6E) versus 38.1% (23.4%+14.7% in Figure 4A). These results suggest that combination treatment exhibited similar effectiveness in KRASG12D mutant pancreatic cancer cells and KRAS wild-type pancreatic cancer cells.
Figure 6.

Apoptotic cell death in KRASG12D-silenced HPAF-II cells versus in the parental HPAF-II cells. A. Western blot analysis of KRAS shRNA-mediated silencing of oncogenic KRASG12D in HPAF-II cells. B. Growth inhibition of HPAF-II, KRAS-silenced HPAF-II pooled cells and control shRNA cells. KRAS shRNA-silencing cells showed reduced cell growth compared to parental and vector control cells. Each bar is the mean ± SD derived from three independent experiments, * represents P < 0.05. C. KRASG12D silencing in HPAF-II cells was associated with decreased expression of downstream signaling effector molecules. D. The IC50 of AMR-MeOAc and FL118 after KRAS-silencing in HPAF-II cells. E. AMR-MeOAc and FL118 induce apoptosis in KRAS-silencing HPAF-II cells.
AMR-MeOAc and FL118 inhibit the nuclear translocation and DNA binding of NF-κB p65 in BxPC-3 cells
NF-κB is constitutively activated in approximately 67% of pancreatic adenocarcinomas and in other solid tumors, as well as in pancreatic cancer cell lines as a treatment resistant factor [40,41]. We found that constitutive NF-κB p65 activation with the DNA binding activity in BxPC-3 cells but not in HPAF-II cells (Figure 7A). To examine the effect of AMR-MeOAc and FL118 on NF-κB constitutive activation, BxPC-3 cells were treated with AMR-MeOAc (8 µM) and FL118 (20 nM) alone or in combination for 24 and 48 h. We found that treatment of cells with AMR-MeOAc or FL118 alone and in combination resulted in nuclear depletion and cytoplasmic accumulation of NF-κB p65 (comparison of Figure 7B versus 7C). This effect correlated with time-dependent increase in the levels of total IκBα and a significant decrease of serine 32-phosphorylated IκBα (p-IκBα, Figure 7D). Next, we studied whether the translocation of NF-κB p65 from nucleus to cytoplasm induced by AMR-MeOAc and FL118 treatment alone and in combination leads to a decrease of specific DNA binding by NF-κB p65 as a marker of decrease treatment resistance. Electrophoretic mobility shift assay (EMSA) analysis showed that AMR-MeOAc and FL118 decreased NF-κB p65 DNA-binding activity in time-dependent manner using the nuclear extracts prepared from BxPC-3 cells (Figure 7E). Combination of AMR-MeOAc and FL118 treatment inhibited the NF-κB activation/DNA binding stronger than each drug alone (Figure 7E). Since there is no constitutive activation of NF-κB p65 in pancreatic cancer HPAF-II cells (Figure 7A), HPAF-II cells were first treated with 10 ng/ml TNFα for 1 h to activate NF-κB p65, followed by AMR-MeOAc (8 µM) and FL118 (20 nM) treatment for 16 h alone and in combination. In this experimental condition, we obtained the result from HPAF-II cells similar to the result shown in the Figure 7E for the constitutive NF-κB p65 activation in BxPC-3 cells (Figure 7F). These results indicate that AMR-MeOAc and FL118 could abrogate both the constitutive and cytokine-induced NF-κB p65 activation/DNA binding in pancreatic cancer cells.
Figure 7.

Inhibitory effects of AMR-MeOAc and FL118 on NF-κB activation. A. Nuclear extracts from HPAF-II and BxPC-3 cells were analyzed for NF-κB DNA binding activity by EMSA using biotin-labeled NF-κB oligonucleotide. B-D. BxPC-3 cells were treated with AMR-MeOAc (8 µM) and FL118 (20 nM) alone and in combination as shown. Cytosolic and nuclear extracts were fractionated and analyzed for NF-κB p65, IκBα and p-IκBα by Western blotting. E. Nuclear extracts of BxPC-3 cells were analyzed for NF-κB p65 DNA binding activity by EMSA using biotin labeled NF-κB oligonucleotide after treatment with AMR-MeOAc (8 µM) and FL118 (20 nM) alone and in combination. F. Left panel (control reactions): 1, biotin-labeled consensus NF-κB positive control DNA probe only; 2, biotin-labeled consensus NF-κB positive control DNA probe plus nuclear extract; and 3, biotin-labeled consensus NF-κB positive control DNA probe plus nuclear extract together with 200-fold molar excess of biotin-unlabeled consensus NF-κB positive control DNA probe. Right panel: HPAF-II cells were grown to 50% confluence and were further grown in the absence of FBS for 12 h; cells were then treated with 10 ng/ml TNFα for 1 h, followed by AMR-MeOAc (8 µM) and FL118 (20 nM) treatment alone and in combination for 16 h. Nuclear extracts were then prepared for NF-κB p65 DNA binding activity by EMSA using biotin-labeled NF-κB oligonucleotide.
Discussion
KRAS activating mutations are reported to occur in over 90% of pancreatic carcinomas [42]. There are no effective KRAS-targeted therapies that have been developed to date for this genetically defined subset of cancers. Therefore, finding novel and potential drugs and molecular targets to inhibit oncogenic KRAS would fill an unmet need in pancreatic cancer therapy. A number of agents that have been developed as molecular cancer therapeutics are under clinical investigation. Unfortunately, they did not exhibit clinical efficacy as single agents due to their inability to inhibit the KRAS downstream signaling pathways or due to the development of resistance to standard therapy [43]. Therefore, combination of molecular targeting agents that are mechanistically complementary to overcome monotherapy may represent an effective strategy and has become a common therapeutic approach. AMR-MeOAc was shown to be an active agent in pancreatic cancer cells with mutant KRASG12D [26]. Accumulating experimental data indicates that FL118 exhibits antitumor activity and completely remove small and large human tumor xenografts in animal models by inhibiting cancer survival and proliferation-associated antiapoptotic proteins [27]. In the present study, we have investigated whether AMR-MeOAc in combination with a low concentration of FL118 sensitizes AMR-MeOAc effects on pancreatic cancer cell growth inhibition and apoptosis induction as well as the underlying mechanisms. We found that AMR-MeOAc alone inhibits mutant KRASG12D activity; AMR-MeOAc-induced KRASG12D inhibition was enhanced by FL118, which appeared to be due to the suppression of downstream signaling in the context of mutant KRAS activity. AMR-MeOAc and FL118 in combination significantly enhanced apoptosis in HPAF-II cells. We further found that AMR-MeOAc and FL118 alone or in combination treatment induced very similar apoptosis in pancreatic cancer cell with mutant KRASG12D (HPAF-II) or with wild type KRAS (BxPC-3), but the latter may be involved in the abrogation of NF-κB constitutive activation by AMR-MeOAc and FL118. To further verify the role of mutant KRAS activity in the efficacy of AMR-MeOAc or FL118 alone and in combination treatment, we silenced KRASG12D expression in HPAF-II cells with lentiviral infection particle containing KRAS-specific shRNA; we then analyzed its effect on apoptosis induced by AMR-MeOAc and FL118 alone and in combination. We found that in comparison with the data obtained with the parental HPAF-II (Figure 4A), silencing of KRASG12D in HPAF-II cells slightly increase FL118 sensitivity to induce apoptosis, while it showed no effect on AMR-MeOAc induced apoptosis (Figure 6E). However, the total early and later apoptosis induced by AMR-MeOAc and FL118 combination treatment maintained almost the same in both KRASG12D-silencing (Figure 6E) versus un-silencing (Figure 4A) HPAF-II cells. We also found that silencing of KRASG12D itself in HPAF-II cells induces the downregulation of c-Myc, Bcl-xL, XIAP and survivin (Figure 6C). This suggests the active signaling pathway cross-talk and the KRASG12D relevance, because AMR-MeOAc and FL118 treatment of the parental HPAF-II cells alone or in combination decreased the expressions of KRASG12D, survivin, XIAP and Bcl-xL, but increased the expression of proapoptotic protein Bax (Figure 5D).
Accumulation of ROS was observed in AMR-MeOAc and FL118 combination treatment (Figure 5A); ROS scavenger NAC partially attenuated the drug treatment-induced apoptosis, suggesting the involvement of drug-induced ROS in apoptosis in HPAF-II cells. Several studies have demonstrated that targeting both the RAS/RAF/ERK and PI3K/AKT pathways is highly beneficial in different tumor types [44]. In ovarian cancer, co-targeting of the PI3K/mTOR and RAS/MEK pathways demonstrated a synergistic inhibition of proliferation and induction of cell death [45]. Activating mutations of KRAS results in increased KRAS oncogenic activity and in turn activates the RAF/MEK/ERK and PI3K/PTEN/AKT/mTOR cascades, which play critical roles in cell growth, inhibiting apoptosis, promoting metastasis and chemotherapeutic drug resistance, suggesting the nature of their important therapeutic targets [13,46,47]. It is possible that activation of the RAS/RAF/MEK/ERK and RAS/PI3K/AKT survival pathways by KRAS mutations may complicate chemotherapy because tumor cells may not be responsive to treatment with single inhibitor. In this respect, development of novel combination approaches that target the KRASG12D activity and activated downstream signaling molecules would be a potentially effective approach to resolving this challenge. Accordingly, AMR-MeOAc and FL118 alone or in combination resulted in marked inhibition of KRASG12D-GTP, B-RAF, ERK, p-ERK1/2, and p-AKT, suggesting the advantage of these two anticancer agent combination synergistic inhibition of KRASG12D activity.
NF-κB activation in RAS-transformed cells is mediated through both ERK and AKT pathways [48]. Activation of NF-κB in cancer results in the induction of cancer cell proliferation, survival, angiogenesis, metastasis, epithelial to mesenchymal transition, and inflammation; this could also increase the expression of multiple antiapoptotic proteins (survivin, XIAP and Bcl-x) that contribute to these responses [49-52]. Interestingly, in this study we found that AMR-MeOAc and FL118 treatment alone or in combination inhibits both the constitutive and cytokine-induced activation of NF-κB. Given that the pancreatic cancer cell line BxPC-3 but not HPAF-II has shown constitutive activation of NF-κB, it is possible that the inhibition of NF-κB in BxPC-3 by AMR-MeOAc and FL118 treatment involved in drug mechanism of action in BxPC-3 cells but not HPAF-II cells since the later has an active NF-κB only when certain stimuli such as the cytokine TNFα treatment. Additionally, several studies have demonstrated that ROS are involved in modulating the nuclear translocation and transcriptional activity of NF-κB, and inhibition of ROS can block NF-κB activity [53,54]. Our results showed that AMR-MeOAc and FL118 alone did not cause clear accumulation of ROS, whereas their combination treatment induced generation of ROS (Figure 5A), suggesting that downregulation of NF-κB and apoptotic cell death may take place in pancreatic cells through both ROS-dependent and ROS-independent pathways.
In summary, it appears that multiple mechanisms are involved in the combination of AMR-MeOAc with a low concentration of FL118 for pancreatic cancer cell growth inhibition and apoptosis induction. However, it remains to be known whether such a drug combination could increase the efficacy to inhibit pancreatic tumor growth in animal models. Unfortunately, due to the lack of AMR-MeOAc for in vivo studies, we are unable to test this in vivo. Our recent studies in pancreatic cancer treated with FL118 alone or in combination with other cytotoxic agents (cisplatin, gemcitabine) obtained exciting results indicating their ability to eliminate pancreatic cancer patient-derived xenograft (PDX) tumors that are relatively resistant to both FL118 and gemcitabine [30], indicating the powerful drug combination treatment consequences.
In conclusion, following our recent studies in pancreatic cancer with FL118 alone or in combination with other cytotoxic agents (cisplatin, gemcitabine) [30], our current studies contribute new insights into the synergistic interaction of AMR-MeOAc and FL118 combination treatment, which appears to be involved in multiple mechanisms of action. FL118 in combination with other therapeutic or chemopreventive agents appears to be a promising therapeutic strategy for pancreatic cancer with or without KRAS mutations.
Acknowledgements
This study was supported by the NIH/NCI grant R03CA182552. We thank Roswell Park Comprehensive Cancer Center Flow Cytometry Core for their assistance with data analysis, which was supported by NCI grant (P30CA016056). We thank Ms. Amanda Hess for editorial proofreading and grammar check of this manuscript.
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014;39:91–100. doi: 10.1016/j.tibs.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giehl K. Oncogenic Ras in tumour progression and metastasis. Biol Chem. 2005;386:193–205. doi: 10.1515/BC.2005.025. [DOI] [PubMed] [Google Scholar]
- 4.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B, Starling N. Colorectal cancer. Lancet. 2010;375:1030–1047. doi: 10.1016/S0140-6736(10)60353-4. [DOI] [PubMed] [Google Scholar]
- 6.Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010;10:842–857. doi: 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ellis CA, Clark G. The importance of being K-Ras. Cell Signal. 2000;12:425–434. doi: 10.1016/s0898-6568(00)00084-x. [DOI] [PubMed] [Google Scholar]
- 8.Kohl NE, Omer CA, Conner MW, Anthony NJ, Davide JP, deSolms SJ, Giuliani EA, Gomez RP, Graham SL, Hamilton K, et al. Inhibition of farnesyltransferase induces regression of mammary and salivary carcinomas in ras transgenic mice. Nat Med. 1995;1:792–797. doi: 10.1038/nm0895-792. [DOI] [PubMed] [Google Scholar]
- 9.Gysin S, Salt M, Young A, McCormick F. Therapeutic strategies for targeting ras proteins. Genes Cancer. 2011;2:359–372. doi: 10.1177/1947601911412376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berndt N, Hamilton AD, Sebti SM. Targeting protein prenylation for cancer therapy. Nat Rev Cancer. 2011;11:775–791. doi: 10.1038/nrc3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lobell RB, Liu D, Buser CA, Davide JP, DePuy E, Hamilton K, Koblan KS, Lee Y, Mosser S, Motzel SL, Abbruzzese JL, Fuchs CS, Rowinsky EK, Rubin EH, Sharma S, Deutsch PJ, Mazina KE, Morrison BW, Wildonger L, Yao SL, Kohl NE. Preclinical and clinical pharmacodynamic assessment of L-778,123, a dual inhibitor of farnesyl: protein transferase and geranylgeranyl: protein transferase type-I. Mol Cancer Ther. 2002;1:747–758. [PubMed] [Google Scholar]
- 12.Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, Hahn SA, Triola G, Wittinghofer A, Bastiaens PI, Waldmann H. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature. 2013;497:638–642. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- 13.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 14.Shields JM, Pruitt K, McFall A, Shaub A, Der CJ. Understanding Ras: ‘it ain’t over ‘til it’s over’. Trends Cell Biol. 2000;10:147–154. doi: 10.1016/s0962-8924(00)01740-2. [DOI] [PubMed] [Google Scholar]
- 15.Pruitt K, Der CJ. Ras and Rho regulation of the cell cycle and oncogenesis. Cancer Lett. 2001;171:1–10. doi: 10.1016/s0304-3835(01)00528-6. [DOI] [PubMed] [Google Scholar]
- 16.Cox AD, Der CJ. The dark side of Ras: regulation of apoptosis. Oncogene. 2003;22:8999–9006. doi: 10.1038/sj.onc.1207111. [DOI] [PubMed] [Google Scholar]
- 17.Downward J. Ras signalling and apoptosis. Curr Opin Genet Dev. 1998;8:49–54. doi: 10.1016/s0959-437x(98)80061-0. [DOI] [PubMed] [Google Scholar]
- 18.Kasper S, Breitenbuecher F, Reis H, Brandau S, Worm K, Kohler J, Paul A, Trarbach T, Schmid KW, Schuler M. Oncogenic RAS simultaneously protects against anti-EGFR antibody-dependent cellular cytotoxicity and EGFR signaling blockade. Oncogene. 2013;32:2873–2881. doi: 10.1038/onc.2012.302. [DOI] [PubMed] [Google Scholar]
- 19.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–465. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 20.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 21.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, Frohling S, Chan EM, Sos ML, Michel K, Mermel C, Silver SJ, Weir BA, Reiling JH, Sheng Q, Gupta PB, Wadlow RC, Le H, Hoersch S, Wittner BS, Ramaswamy S, Livingston DM, Sabatini DM, Meyerson M, Thomas RK, Lander ES, Mesirov JP, Root DE, Gilliland DG, Jacks T, Hahn WC. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, Greninger P, Brown RD, Godfrey JT, Cohoon TJ, Song Y, Lifshits E, Hung KE, Shioda T, Dias-Santagata D, Singh A, Settleman J, Benes CH, Mino-Kenudson M, Wong KK, Engelman JA. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013;23:121–128. doi: 10.1016/j.ccr.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell. 2014;25:272–281. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 24.Kindler HL, Niedzwiecki D, Hollis D, Sutherland S, Schrag D, Hurwitz H, Innocenti F, Mulcahy MF, O’Reilly E, Wozniak TF, Picus J, Bhargava P, Mayer RJ, Schilsky RL, Goldberg RM. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the Cancer and Leukemia Group B (CALGB 80303) J. Clin. Oncol. 2010;28:3617–3622. doi: 10.1200/JCO.2010.28.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardiere C, Bennouna J, Bachet JB, Khemissa-Akouz F, Pere-Verge D, Delbaldo C, Assenat E, Chauffert B, Michel P, Montoto-Grillot C, Ducreux M Groupe Tumeurs Digestives of Unicancer; PRODIGE Intergroup. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 26.Rabi T, Venkatanarashiman M. Novel synthetic oleanane triterpenoid AMR-MeOAc inhibits K-Ras through ERK, Akt and survivin in pancreatic cancer cells. Phytomedicine. 2014;21:491–496. doi: 10.1016/j.phymed.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 27.Ling X, Cao S, Cheng Q, Keefe JT, Rustum YM, Li F. A novel small molecule FL118 that selectively inhibits survivin, Mcl-1, XIAP and cIAP2 in a p53-independent manner, shows superior antitumor activity. PLoS One. 2012;7:e45571. doi: 10.1371/journal.pone.0045571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ling X, Xu C, Fan C, Zhong K, Li F, Wang X. FL118 induces p53-dependent senescence in colorectal cancer cells by promoting degradation of MdmX. Cancer Res. 2014;74:7487–7497. doi: 10.1158/0008-5472.CAN-14-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ling X, Liu XJ, Zhong K, Smith N, Prey J, Li F. FL118, a novel camptothecin analogue, overcomes irinotecan and topotecan resistance in human tumor xenograft models. Am J Transl Res. 2015;7:1765–1781. [PMC free article] [PubMed] [Google Scholar]
- 30.Ling X, Wu W, Fan C, Xu C, Liao J, Rich LJ, Huang RY, Repasky EA, Wang X, Li F. An ABCG2 non-substrate anticancer agent FL118 targets drug-resistant cancer stem-like cells and overcomes treatment resistance of human pancreatic cancer. J Exp Clin Cancer Res. 2018;37:240. doi: 10.1186/s13046-018-0899-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ling X, Calinski D, Chanan-Khan AA, Zhou M, Li F. Cancer cell sensitivity to bortezomib is associated with survivin expression and p53 status but not cancer cell types. J Exp Clin Cancer Res. 2010;29:8. doi: 10.1186/1756-9966-29-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 33.Nie P, Hu W, Zhang T, Yang Y, Hou B, Zou Z. Synergistic induction of erlotinib-mediated apoptosis by resveratrol in human non-smallcell lung cancer cells by down-regulating survivin and up-regulating PUMA. Cell Physiol Biochem. 2015;35:2255–2271. doi: 10.1159/000374030. [DOI] [PubMed] [Google Scholar]
- 34.Ling X, Bernacki RJ, Brattain MG, Li F. Induction of survivin expression by taxol (paclitaxel) is an early event which is independent on taxol-mediated G2/M arrest. J Biol Chem. 2004;279:15196–15203. doi: 10.1074/jbc.M310947200. [DOI] [PubMed] [Google Scholar]
- 35.Wu J, Apontes P, Song L, Liang P, Yang L, Li F. Molecular mechanism of upregulation of survivin transcription by the AT-rich DNA-binding ligand, Hoechst33342: evidence for survivin involvement in drug resistance. Nucleic Acids Res. 2007;35:2390–2402. doi: 10.1093/nar/gkm149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 37.Liu AX, Testa JR, Hamilton TC, Jove R, Nicosia SV, Cheng JQ. AKT2, a member of the protein kinase B family, is activated by growth factors, v-Ha-ras, and v-src through phosphatidylinositol 3-kinase in human ovarian epithelial cancer cells. Cancer Res. 1998;58:2973–2977. [PubMed] [Google Scholar]
- 38.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boidot R, Vegran F, Lizard-Nacol S. Transcriptional regulation of the survivin gene. Mol Biol Rep. 2014;41:233–240. doi: 10.1007/s11033-013-2856-0. [DOI] [PubMed] [Google Scholar]
- 40.Arlt A, Vorndamm J, Breitenbroich M, Folsch UR, Kalthoff H, Schmidt WE, Schafer H. Inhibition of NF-kappaB sensitizes human pancreatic carcinoma cells to apoptosis induced by etoposide (VP16) or doxorubicin. Oncogene. 2001;20:859–868. doi: 10.1038/sj.onc.1204168. [DOI] [PubMed] [Google Scholar]
- 41.Arlt A, Schafer H. NFkappaB-dependent chemoresistance in solid tumors. Int J Clin Pharmacol Ther. 2002;40:336–347. doi: 10.5414/cpp40336. [DOI] [PubMed] [Google Scholar]
- 42.Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 43.Hausenloy DJ, Mocanu MM, Yellon DM. Cross-talk between the survival kinases during early reperfusion: its contribution to ischemic preconditioning. Cardiovasc Res. 2004;63:305–312. doi: 10.1016/j.cardiores.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 44.Migliardi G, Sassi F, Torti D, Galimi F, Zanella ER, Buscarino M, Ribero D, Muratore A, Massucco P, Pisacane A, Risio M, Capussotti L, Marsoni S, Di Nicolantonio F, Bardelli A, Comoglio PM, Trusolino L, Bertotti A. Inhibition of MEK and PI3K/mTOR suppresses tumor growth but does not cause tumor regression in patient-derived xenografts of RAS-mutant colorectal carcinomas. Clin Cancer Res. 2012;18:2515–2525. doi: 10.1158/1078-0432.CCR-11-2683. [DOI] [PubMed] [Google Scholar]
- 45.Sheppard KE, Cullinane C, Hannan KM, Wall M, Chan J, Barber F, Foo J, Cameron D, Neilsen A, Ng P, Ellul J, Kleinschmidt M, Kinross KM, Bowtell DD, Christensen JG, Hicks RJ, Johnstone RW, McArthur GA, Hannan RD, Phillips WA, Pearson RB. Synergistic inhibition of ovarian cancer cell growth by combining selective PI3K/mTOR and RAS/ERK pathway inhibitors. Eur J Cancer. 2013;49:3936–3944. doi: 10.1016/j.ejca.2013.08.007. [DOI] [PubMed] [Google Scholar]
- 46.Shankar S, Chen Q, Srivastava RK. Inhibition of PI3K/AKT and MEK/ERK pathways act synergistically to enhance antiangiogenic effects of EGCG through activation of FOXO transcription factor. J Mol Signal. 2008;3:7. doi: 10.1186/1750-2187-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin S, Shen JN, Wang J, Huang G, Zhou JG. Oridonin induced apoptosis through Akt and MAPKs signaling pathways in human osteosarcoma cells. Cancer Biol Ther. 2007;6:261–268. doi: 10.4161/cbt.6.2.3621. [DOI] [PubMed] [Google Scholar]
- 48.Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol. 2010;2:a000109. doi: 10.1101/cshperspect.a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stehlik C, de Martin R, Kumabashiri I, Schmid JA, Binder BR, Lipp J. Nuclear factor (NF)-kappaB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor alpha-induced apoptosis. J Exp Med. 1998;188:211–216. doi: 10.1084/jem.188.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Erl W, Hansson GK, de Martin R, Draude G, Weber KS, Weber C. Nuclear factor-kappa B regulates induction of apoptosis and inhibitor of apoptosis protein-1 expression in vascular smooth muscle cells. Circ Res. 1999;84:668–677. doi: 10.1161/01.res.84.6.668. [DOI] [PubMed] [Google Scholar]
- 51.Kawakami H, Tomita M, Matsuda T, Ohta T, Tanaka Y, Fujii M, Hatano M, Tokuhisa T, Mori N. Transcriptional activation of survivin through the NF-kappaB pathway by human Tcell leukemia virus type I tax. Int J Cancer. 2005;115:967–974. doi: 10.1002/ijc.20954. [DOI] [PubMed] [Google Scholar]
- 52.Lin MT, Chang CC, Chen ST, Chang HL, Su JL, Chau YP, Kuo ML. Cyr61 expression confers resistance to apoptosis in breast cancer MCF-7 cells by a mechanism of NF-kappaBdependent XIAP up-regulation. J Biol Chem. 2004;279:24015–24023. doi: 10.1074/jbc.M402305200. [DOI] [PubMed] [Google Scholar]
- 53.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 54.Flohe L, Brigelius-Flohe R, Saliou C, Traber MG, Packer L. Redox regulation of NF-kappa B activation. Free Radic Biol Med. 1997;22:1115–1126. doi: 10.1016/s0891-5849(96)00501-1. [DOI] [PubMed] [Google Scholar]