

Abstract
Ankyrin repeat and KH domain containing 1 (ANKHD1) is a protein with multiple ankyrin repeat domains and a single KH domain, and it is encoded by the ANKHD1 gene in humans. ANKHD1 has been reported to be highly expressed in various cancer tissues, and it is involved in cancer progression, including proliferation and invasion. However, its functional roles in colorectal cancer (CRC) remain unclear. In our study, we first found that high expression of ANKHD1 in CRC tumor tissue was associated with tumor infiltration depth (P=0.03). ANKHD1 was highly expressed in HCT116 and SW480 cells. Downregulation of ANKHD1 inhibited CRC cell proliferation, migration and invasion both in vitro and in vivo. ANKHD1 silencing inhibited the expression of MMP2, MMP9, the mesenchymal marker vimentin, and the epithelial-to-mesenchymal transition (EMT) transcription factors Snail and ZEB1, while increasing the expression of the epithelial marker E-cadherin. As a cofactor of YAP1 in the Hippo signaling pathway, ANKHD1 silencing reduced the expression and increased the phosphorylation of YAP1. Moreover, the phosphorylation of AKT was inhibited when ANKHD1 was knocked down. The mechanism study revealed that the effect of ANKHD1 might be associated with the expression of YAP1 and that AKT signaling and EMT played crucial roles in this process. Overexpression of YAP1 reversed the effect of ANKHD1 silencing on CRC cell proliferation, migration and invasion. In conclusion, these findings suggest that ANKHD1 might act as a novel regulator that promotes CRC cell proliferation, migration and invasion by activating EMT via YAP1.
Keywords: ANKHD1, YAP1, EMT, proliferation, migration, invasion, CRC
Introduction
Colorectal cancer (CRC) is one of the most common malignant tumors in the gastrointestinal tract, the third most commonly diagnosed cancer and the third leading cause of cancer-related death in both males and females in the United States [1]. Surgery and adjuvant therapy, including chemotherapy and radiotherapy, are currently the main treatments for CRC [2]. Despite the advances in treatments, the prognosis of CRC patients is not optimistic, with a 65% five-year relative survival rate for patients diagnosed in 2013 [1]. Metastasis of CRC is one of the main reasons for its high mortality and poor prognosis. Approximately 90% of CRC-related deaths occur as a result of metastatic disease [3]. Epithelial-to-mesenchymal transition (EMT) has been shown to play a vital role in cell metastasis and invasion [4]. Aberrant activation of EMT-inducing transcription factors is an important part of metastasis [5]. Once this conversion occurs, immobile epithelial cells lose their epithelial characteristics and acquire mesenchymal properties, including the ability to migrate and invade [6]. The occurrence of EMT also promotes the progression of malignant tumors [7]. Despite these insights, the molecular mechanisms of EMT are varied, and mechanisms by which it will be possible to influence the metastasis of CRC still need to be explored.
Ankyrin repeat and KH domain containing 1 (ANKHD1) is a protein with multiple ankyrin repeat domains and a single KH domain that is encoded by the ANKHD1 gene. ANKHD1 is a human orthologous protein of MASK and was first identified in Drosophila melanogaster [8]. The presence of multiple ankyrin repeats suggests that it mediates protein-protein interactions and participates in biological processes, including the cell cycle, transcription, differentiation, apoptosis, cell survival, and even the development of cancer [8,9]. ANKHD1 was found to be overexpressed in both malignant cells from patients diagnosed with acute leukemia and multiple myeloma (MM) and several cancer cell lines, including K562 (leukemia), MM1R, MM1S, RPMI 8226 and U266 (MM) and LNCaP (prostate) [10-12]. In leukemia cells, ANKHD1 silencing significantly inhibited cell proliferation and migration [13]. Studies have found that ANKHD1 is a novel component of the Hippo signaling pathway that interacts with YAP1 and promotes cancer progression [12]. However, the expression of ANKHD1 in CRC and its effect on the progression of CRC has not been reported thus far.
Hence, our study aimed to study the effect of ANKHD1 on CRC cell proliferation and metastasis both in vitro and in vivo. Inhibition of ANKHD1 reduced YAP1 expression and inhibited the occurrence of EMT and the phosphorylation of AKT. Then, we investigated the role played by YAP1 in the effect of ANKHD1 on CRC progression.
Materials and methods
Cell culture
The human colorectal carcinoma cell lines HCT8, HCT116, SW480, SW620, LOVO and RKO were purchased from the Shanghai Chinese Academy of Sciences (Shanghai, China) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) (HyClone, Logan, USA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37°C in a humidified atmosphere with 5% CO2. The experiments were performed when the cells reached 70-80% confluence.
Transduction and transient transfection
HCT116 and SW480 cells were transduced with lentivirus-mediated control shRNA or lentivirus-mediated shRNA targeting ANKHD1, designated shNC and shANKHD1, respectively. The lentivirus-mediated shRNA was obtained from Hanbio Biotechnology (Shanghai, China); specific fragments were inserted into shNC (top strand: GATCCGTTCTCCGAACGTGTCACGTAATTCAAGAGATTACGTGACACGTTCGGAGAATTTTTTC; bottom strand: AATTCAAAAAATTCTCCGAACGTGTCACGTAATCTCTTGAATTACGTGACACGTTCGGAGAACG) and shANKHD1 (top strand: GATCCGCACTACTCTTAGCACAAGGATTCAAGAGATCCTTGTGCTAAGAGTAGTGCTTTTTTC; bottom strand: AATTGAAAAAAGCACTACTCTTAGCACAAGGATCTCTTGAATCCTTGTGCTAAGAGTAGTGCG). The cells were subsequently harvested at 72 h postinfection and selected in medium containing 8 μg/ml puromycin (Sigma-Aldrich, St. Louis, Missouri, USA) until all uninfected cells were killed by the puromycin. The stably transfected cell lines were validated by Western blotting and real-time PCR and then used for the subsequent experiments. The YAP1 lentiviral vector (YAP1 cDNA) was purchased from Applied Biological Materials (Richmond, BC, Canada), and transfection was performed using the Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA). The subsequent experiments were performed 48 h after transfection. HCT116-shANKHD1 cells were transfected with the YAP1 lentiviral vector and designated HCT116shANKHD1+YAP1.
Cell proliferation assay
Cell proliferation was measured by the Cell Counting Kit 8 (Beyotime Biotechnology, China) according to the manufacturer’s instructions. The stably transfected cell lines HCT116-shANKHD1 and SW480-shANKHD1 were seeded in 96-well plates. Then, 100 μl of DMEM containing 10% CCK8 reagent was added to each well at 24 h, 48 h, 72 h and 96 h post-cell seeding and cultured for 2 h. The absorbance was measured at a wavelength of 450 nm by a microplate reader (Bio-Rad, Hercules, CA, USA). Three independent experiments were perforfmed.
Cell invasion and migration assay
Cell invasion and migration assays were performed using 8-μm pore size transwell chambers (Corning, New York, USA). For the invasion assay, the upper chamber of the transwell was precoated with Matrigel and incubated at 37°C for 2 h to form the Matrigel layer in the chamber. The stably transfected cells were starved in serum-free DMEM; then, 4 × 104 cells with 0.5% BSA in DMEM were seeded in the upper chamber of the transwell, while complete DMEM with 10% FBS was added to the lower chamber. At 24 h after inoculation, the cells remaining in the upper chamber were disposed of, and the invaded cells were stained by crystal violet. For the cell migration assay, 4 × 104 starved cells with 0.5% BSA in DMEM were seeded in the upper chamber of the transwell, and the lower chamber was filled with complete DMEM with 10% FBS. At 24 h after inoculation, the cells in the upper transwell chamber were disposed of, and the migrated cells were stained by crystal violet. Each experiment was repeated three times independently.
RNA extraction and quantitative real-time PCR
Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), and 1 μg of total RNA from the stably transfected cells was reverse transcribed to cDNA using the Prime Script PRT Reagent Kit (Takara, Kyoto, Japan) according to the manufacturer’s instructions. Quantitative real-time PCR was performed using SYBR-Green Mix and detected by the ABI PRISM 7500 Sequence Detection System (Applied Biosystems, Foster City, CA, USA). The mRNA level of ANKHD1, YAP1, E-cadherin and Vimentin was detected by quantitative real-time PCR, and the amplified transcript level was normalized to that of GAPDH. The primer pairs were as follows: ANKHD1 (forward primer: 5’-CCTGCTTGGAACCCTATGATAAA-3’; reverse primer: 5’-CGTGCCAGGCCAAATCTG-3’), GAPDH (forward primer: 5’-CATGAGAAGTATGACAACAGCCT-3’; reverse primer: 5’-AGTCCTTCCACGATACCAAAGT-3’), YAP1 (forward primer: 5’-CGCTCTTCAACGCCGTCA-3’; reverse primer: 5’-AGTACTGGCCTGTCGGGAGT-3’), E-cadherin (forward primer: 5’-ATTCTGATTCTGCTGCTCTTG-3’; reverse primer: 5’-AGTAGTCATAGTCCTGGTCTT-3’) and Vimentin (forward primer: 5’-GGACCAGCTAACCAACGACA-3’; reverse primer: 5’-AAGGTCAAGACGTGCCAGAG-3’).
Western blot assay
The stably transfected cells and transiently transfected cells were collected at specific times and then lysed by cell lysis buffer for Western blotting. The total protein concentration was detected using a BCA Protein Assay Kit (Beyotime Biotechnology, China). After that, 30 µg of proteins were separated by 8% or 10% SDS-PAGE and transferred to PVDF membranes. The following primary antibodies were used for the Western blots: ANKHD1, YAP1, p-YAP1, ZEB1, Snail, E-cadherin, vimentin, MMP2, MMP9, AKT, p-AKT, Bcl-2 and Bax (1:1000, Abcam, Cambridge, MA, USA), Tubulin and GAPDH (Beyotime Biotechnology, China) were used as internal controls.
In vivo experiments
Male BALB/c nude mice (5 weeks old) were purchased from the SLAC Laboratory Animal Company (Shanghai, China). Then, 4 × 106 HCT116-shANKHD1 cells or HCT116-shNC cells in 0.1 mL of PBS were injected subcutaneously into the right flanks of the nude mice (five mice per group). Tumors were measured every two days by caliper, and the longest diameter (A) and the shortest diameter (B) of every tumor were recorded separately to calculate the tumor volumes according to the following formula: π/6 × A × B2 [14]. For the metastasis assay, 1 × 106 HCT116-shANKHD1 cells or HCT116-shNC cells in 0.1 ml PBS were injected into the nude mice via the tail vein. After 40 days, the lungs and livers were dissected and photographed and then stained with hematoxylin and eosin (H&E). All nude mouse experiments were approved by the ethics committee of Soochow University.
Immunohistochemistry and HE analysis
Immunohistochemistry (IHC) was performed to investigate protein expression levels in cancer tissue. All surgically resected specimens were obtained from patients diagnosed with CRC at the Second Affiliated Hospital of Soochow University from 2009 to 2014. The sections of tissue for IHC were incubated with antibodies against ANKHD1. The intensity of staining of the cancer tissues was scored as follows: 0 (no staining), 1 (weak staining, light yellow), 2 (moderate staining, yellowish brown), and 3 (strong staining, brown). An intensity score ≥ 2 was considered overexpressfion, whereas intensity scores < 2 were cfonsidered indicators of low expression. All slides were evaluated independently by two investigators blinded to the patient identities and clinical outcomes. H&E staining was performed to verify the presence of metastatic cancer nodules.
Statistical analysis
The X2 test was performed to detect the correlation between ANKHD1 expression and clinical parameters [15]. Student’s t-test was performed to determine differences in the data from the in vitro experiments, and the results are expressed as the mean ± standard deviation (SD) of three independent experiments. All statistical analyses were performed using GraphPad Prism 6.0 software (GraphPad Software Inc., San Diego, USA). P < 0.05 was considered statistically significant.
Results
ANKHD1 expression in colorectal cancer tissue sample
To determine the expression of ANKHD1 in CRC, tumor tissues of 136 colorectal cancer patients were used for immunohistochemical staining. Our results showed that ANKHD1 was expressed in 91.2% of the samples (Table 1) and widely expressed in both normal colorectal tissues and colorectal cancer tissues (Figure 1A-C). The expression of ANKHD1 in colorectal cancer was highly correlated with the tumor infiltration depth (P=0.03; Table 1), while its expression was not significantly associated with age, sex, degree of differentiation and lymph node metastasis.
Table 1.
Correlation between tumor ANKHD1 expression and the pathologic features of CRC patients
| Variables | All cases | ANKHD1 expression | P | |
|---|---|---|---|---|
|
| ||||
| Negative (%) | Positive (%) | |||
| Colorectal cancer | 136 | 12 (8.8) | 124 (91.2) | |
| Age | ||||
| < 60 | 35 | 4 (2.9) | 31 (22.8) | 0.372 |
| ≥ 60 | 101 | 8 (5.9) | 93 (68.4) | |
| Gender | ||||
| Male | 82 | 7 (5.1) | 75 (55.1) | 0.558 |
| Female | 54 | 5 (3.7) | 49 (36.1) | |
| Differentiation | ||||
| High/intermediate | 101 | 8 (5.9) | 93 (68.4) | 0.372 |
| Low/null | 35 | 4 (2.9) | 31 (22.8) | |
| Invasion | ||||
| Infiltration of shallow muscularis | 6 | 2 (1.5) | 4 (2.9) | 0.030 |
| Infiltration of deep muscularis | 21 | 3 (2.2) | 15 (11.1) | |
| Infiltration of seroso | 109 | 7 (5.1) | 105 (77.2) | |
| Lymph node metastasis | ||||
| Yes | 56 | 3 (2.2) | 53 (39.0) | 0.189 |
| No | 80 | 9 (6.6) | 71 (52.2) | |
| Nerve invasion | ||||
| Yes | 49 | 4 (2.9) | 45 (33.1) | 0.554 |
| No | 87 | 8 (5.9) | 79 (58.1) | |
| Vascular invasion | ||||
| Yes | 20 | 1 (0.7) | 19 (14.0) | 0.446 |
| No | 116 | 11 (8.1) | 105 (77.2) | |
| Duck’s | ||||
| A | 20 | 4 (2.9) | 16 (11.8) | 0.059 |
| B | 60 | 5 (3.7) | 55 (40.4) | |
| C | 56 | 3 (2.2) | 53 (39.0) | |
Figure 1.
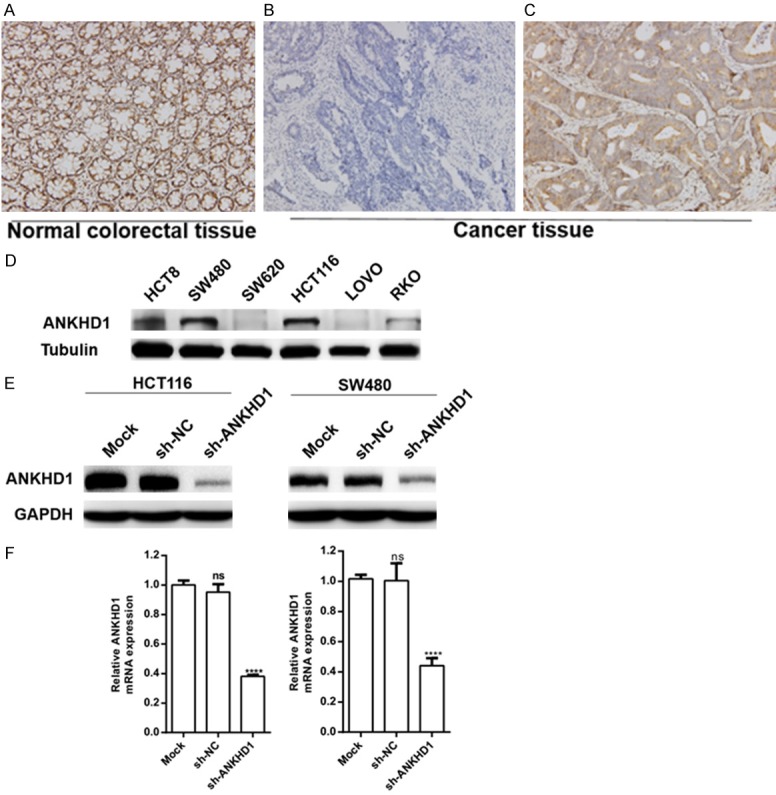
IHC staining of CRC patient tissue samples and the verification of infection efficiency. ANKHD1 was expressed in normal colorectal tissue (A); examples of ANKHD1 negative (B) and positive (C) expression (× 200 magnification) are shown in colorectal cancer. Baseline protein (D) expression of ANKHD1 in 6 CRC cell lines. Protein (E) and mRNA (F) levels of ANKHD1 in HCT116 and SW480 cells transduced with shRNA target ANKHD1 (***P < 0.001).
Construction of cell lines
The baseline expression level of ANKHD1 was detected by Western blot assay in 6 CRC cell lines, including HCT8, SW480, SW620, HCT116, LOVO and RKO. ANKHD1 was highly expressed in SW480 and HCT116 cell lines (Figure 1D). HCT116 and SW480 cells were selected to construct stably transfected cell lines with lentivirus-mediated shRNA targeting ANKHD1 to knockdown ANKHD1 expression. The infection efficiency was verified by Western blotting. Silencing efficiency was 85.26% and 82.64% in HCT116 and SW480 cells, respectively. Analysis was conducted with ImageJ. The mRNA levels exhibited similar silencing efficiency and were reduced by 61.92% and 56.00%, respectively (Figure 1E, 1F).
ANKHD1 silencing inhibits colorectal cancer cell proliferation in vitro
To investigate whether ANKHD1 affects the proliferation of colorectal cancer cells, a CCK8 assay was performed after HCT116 and SW480 cells were stably infected with shANKHD1 for 24, 48, 72 and 96 hours. Our results showed that the silencing of ANKHD1 by shRNA suppressed the proliferation of colorectal cancer cells (Figure 2A, 2B). The relative proliferation rate was significantly reduced by 22.5%, 31.9% and 36.7% in HCT116-shANKHD1 cells at 48 h, 72 h and 96 h compared with HCT116-shNC and HCT116-mock cells (Figure 2A, P < 0.001). This marked inhibition of proliferation was also observed in SW480-shANKHD1 cells, and the relative proliferation rate was significantly reduced by 19.1%, 28.9% and 32.4% at 48 h, 72 h and 96 h compared with SW480-shNC and SW480-mock cells (Figure 2B, P < 0.01), respectively.
Figure 2.

ANKHD1 promotes colorectal cancer cell proliferation in vitro and in vivo. CCK8 assays showed the relative proliferative capacity of specific HCT116 and SW480 cells at 24, 48, 72 and 96 h after seeding in plates (A, B, **P < 0.01; ***P < 0.001). Apoptosis was measured using Annexin V/7-AAD double staining assay and Western blot assay in colorectal cancer cells, ANKHD1 silencing did not significantly affect cells apoptosis (C, D). The nude mouse xenograft model showed that tumors formed by HCT116-shANKHD1 cells had slower growth rates and smaller volumes than tumors formed by HCT116-shNC cells (E, F, *P < 0.05). The data are presented as the means ± SD of three independent experiments.
To determine whether ANKHD1 silencing affects apoptosis of colorectal cancer cells, a flow cytometry-based apoptosis assay was performed. Our results showed that ANKHD1 silencing did not significantly affect apoptosis of colorectal cancer cells compared with control group (Figure 2D). Consistently, ANKHD1 silencing did not significantly affect the expression of Bcl-2 and Bax in colorectal cancer cells (Figure 2C).
ANKHD1 silencing inhibits the growth of xenograft tumors
To further confirm the inhibition of proliferation observed in vitro, we injected HCT116-shANKHD1 and HCT116-shNC cells into the right flanks of nude mice. Consistent with our observation in vitro, the volumes of tumors formed by HCT116-shANKHD1 cells were significantly smaller than those formed by HCT116-shNC cells (Figure 2E, 2F P < 0.05). The results demonstrated that ANKHD1 promoted tumorigenesis in vivo.
ANKHD1 silencing reduced CRC cell migration and invasion
Our migration assay revealed that the downregulation of ANKHD1 markedly suppressed HCT116 and SW480 cell migration (Figure 3A-D shows cell migration reduced by 39.6% and 29.7%, respectively, both P < 0.001). Similarly, an invasion assay also indicated that ANKHD1 silencing significantly reduced cell invasion by 45.4% and 57.5% compared to control HCT116 and SW480 cells, respectively (Figure 3A-D, both P < 0.001). These results suggested that ANKHD1 silencing clearly inhibited CRC cell migration and invasion.
Figure 3.

ANKHD1 promotes colorectal cancer cell migration and invasion in vitro and in vivo. Representative images of the migration and invasion of HCT116 cells (A) and SW480 cells (B) infected with shANKHD1 or shNC or none. ANKHD1 silencing significantly repressed cell migration and invasion (C, D, ***P < 0.001). The cell counts are for the invasion assay with at least five random microscope fields (invasion: × 200 magnification). Cell migration is expressed as the percentage of migrating cells in at least five random microscope fields of view (Analysis conducted with ImageJ software, migration: × 200 magnification). In total, 1 × 106 HCT116-shNC and HCT116-shANKHD1 cells were injected into nude mice through the tail vein, and the mice were reared for 40 days before being sacrificed to evaluate the lung metastases. Representative figures for lung metastatic tumors (E) show that mice injected with HCT116-shANKHD1 cells had fewer metastatic nodules than those injected with HCT116-shNC cells (*P < 0.05). (F) H&E staining of mouse lung metastases and normal lung tissue (× 200 magnification).
ANKHD1 silencing inhibits HCT116 cell lung metastasis in nude mice
To further validate the effect of ANKHD1 silencing on CRC cell migration and invasion, a pulmonary metastasis model was generated by tail vein injections of HCT116-shANKHD1 and HCT116-shNC cells in nude mice. The nude mice were sacrificed after 40 days, and the vital organs were removed and observed. The formation of pulmonary nodules was used to assess the capacity for metastasis. Compared with the HCT116-shANKHD1 group, more and larger migrated nodules were observed in the lungs in the HCT116-shNC group (Figure 3E, P < 0.001). Then, the lung tissue was analyzed by H&E staining to further evaluate the metastases (Figure 3F). These observations further confirmed that ANKHD1 plays an important role in the metastasis and invasion of colorectal cancer.
ANKHD1 silencing inhibited EMT and AKT phosphorylation
Matrix metalloproteinases (MMPs) play a role in ECM degradation and promotes tumor progression, leading to metastasis. Furthermore, MMPs activate TGF-β and promote EMT [16]. Consistently, we found that ANKHD1 silencing clearly reduced MMP2 and MMP9 expression by Western blotting (Figure 4A). As indicated by the Western blot assay, the expression of the epithelial marker protein E-cadherin was increased, while the expression of the mesenchymal marker protein vimentin was decreased in the shANKHD1 group compared with the shNC group and the mock group in both HCT116 and SW480 cells (Figure 4B), these changes also occurred at mRNA level (Figure 4E). The present study showed alterations in EMT-associated transcription factors, including ZEB1 and Snail; Figure 4B shows that ZEB1 and Snail were markedly decreased in the shANKHD1 groups. These results confirmed that ANKHD1 played an important role in the progression of EMT.
Figure 4.

ANKHD1 silencing inhibits EMT and AKT signaling. Western blot analyses of the relative expression levels of MMP2, MMP9 (A), EMT-associated markers E-cadherin and vimentin and EMT transcription factors Snail and ZEB1 (B), AKT (C), and YAP1 (D). GAPDH and Tubulin were detected as internal controls. Real-time PCR detected the expression of ANKHD1, YAP1, E-cadherin and Vimentin at mRNA level (E, *P < 0.05, **P < 0.01).
Our results revealed that AKT phosphorylation was significantly inhibited in the shANKHD1 group compared with the shNC group and the mock group but the expression of AKT remained unchanged (Figure 4C). These results suggest that ANKHD1 silencing inhibits AKT activation.
YAP1 is required for ANKHD1 silencing-induced inhibition of proliferation and migration via AKT signaling and EMT
According to reports, ANKHD1 is a novel member of the Hippo signaling pathway and is essential for the full activity of YAP1 [12,17,18]. Is YAP1 involved in the effects of ANKHD1 on CRC cell proliferation, migration and invasion? Western blot analysis showed that YAP1 was downregulated when ANKHD1 was knocked down (Figure 4D), real-time PCR assay revealed that this inhibition was occurred in transcriptional level (Figure 4E). Due to phosphorylated YAP1 was able to be degraded by ubiquitination, we tested p-YAP1 expression under ANKHD1 was knocked down, and found that p-YAP1 was significantly increased (Figure 4D). The increase of p-YAP1 may enhanced the degradation of YAP1. YAP1 is an important downstream EMT-inducing molecule [14]. Then, HCT116-shANKHD1 cells were transfected with the YAP1 overexpression vector to explore the role of YAP1 in ANKHD1 silencing-mediated inhibition of CRC cell proliferation, migration and invasion. Transfection efficiency was verified by Western blotting, with a 1.64-fold overexpression efficiency in HCT116-shANKHD1 cells (Figure 5A). A CCK8 assay revealed that the upregulation of YAP1 counteracted ANKHD1 silencing-induced inhibition of cell proliferation. The relative proliferation rates were increased by 22.7%, 14.7% and 17.8% in the shANKHD1+YAP1 group compared with the shANKHD1 group at 48 h, 72 h and 96 h, respectively, in HCT116 cells (Figure 5B, P < 0.01). The migration assay indicated that HCT116-shANKHD1 cell migration was increased by 26.7% when the expression of YAP1 was upregulated (Figure 5C, P < 0.001). Similarly, the invasion assay also showed a significant 46.0% increase in invasion (Figure 5C, P < 0.001). These results revealed that ANKHD1 promotes CRC cell proliferation, migration and invasion via YAP1.
Figure 5.

Overexpression of YAP1 reversed the ANKHD1 silencing-induced inhibition of CRC cell proliferation, migration and invasion. (A) Transfection efficiency was verified by Western blotting. (B) CCK8 assays were performed at 24, 48, 72 and 96 h after HCT116-shANKHD1 cells were transfected with the YAP1 vector. The relative proliferation rate was significantly increased in the shANKHD1+YAP1 group compared with the shANKHD1 group (**P < 0.01, ***P < 0.001). (C) Representative images of the migration and invasion of HCT116-shANKHD1 cells infected with the YAP1 vector or not. Cell counts are for the invasion assay with at least five random microscope fields (invasion: × 200 magnification). Cell migration is expressed as the percentage of migrating cells in at least five random microscope fields of view (migration: × 200 magnification). Overexpression of YAP1 increased HCT116-shANKHD1 cell migration and invasion (C, ***P < 0.001). Western blot analyses of the relative expression levels of the EMT-associated markers E-cadherin and vimentin (D), the EMT transcription factors Snail and ZEB1 (E) and AKT (F). The data are presented as the means ± SD of three independent experiments.
Western blot analysis showed that upregulation of YAP1 increased the protein expression of vimentin and the EMT transcription factors Snail and ZEB1, while the expression of E-cadherin was decreased (Figure 5D, 5E). Furthermore, we found that the phosphorylation of AKT in HCT116-shANKHD1+YAP1 cells was activated compared with HCT116-shANKHD1 cells (Figure 5F). These results indicate that ANKHD1 activates EMT and AKT signaling via YAP1. These results suggested that YAP1 is essential for ANKHD1 silencing-induced inhibition of cell proliferation, migration and invasion through the inhibition of AKT signaling and EMT.
Discussion
ANKHD1 is a multiple ankyrin repeat-containing protein that functions as a scaffolding protein. Published studies have revealed that ANKHD1 is highly expressed in cancer cells, including acute leukemia, MM and prostate cancer cells, and that it plays an important role in cancer progression, including in proliferation, migration and invasion [10,11,19]. Nevertheless, the mechanisms by which ANKHD1 affects CRC development remain unclear. The present study retrospectively analyzed ANKHD1 expression in 136 CRC cases. Our research showed that ANKHD1 was expressed in both cancerous and paracancerous nontumor tissues, and the expression of ANKHD1 in CRC tissues was associated with tumor infiltration depth. ANKHD1 acts as a scaffolding protein that is ubiquitously expressed in normal human tissues, plays an important role in proliferation, and mediates protein-protein interactions. Meanwhile, high expression levels of ANKHD1 in cancer promote cancer progression and may become a target of cancer treatment.
Then, we investigated the impacts of shRNA-mediated ANKHD1 silencing in the CRC cell lines HCT116 and SW480. ANKHD1 silencing inhibited the proliferation of CRC cells both in vitro and in vivo in our study, while ANKHD1 silencing did not obviously affect CRC cells apoptosis. Our transwell assay showed that ANKHD1 silencing inhibited CRC cell migration and invasion. Furthermore, a pulmonary metastasis model showed that injection with HCT116-shANKHD1 cells induced fewer lung metastases than HCT116-shNC cells. These results elucidated that ANKHD1 silencing inhibits CRC cell migration and invasion both in vitro and in vivo.
Because degradation of the basement membrane is an essential step for the migration and invasion of most cancers [20], MMPs played an important role in this progress [21,22]. For example, Qin et al. reported that PAD1 promotes EMT and metastasis by regulating MEK1-ERK1/2-MMP2 signaling in triple-negative breast cancer cells [23]. In our study, we found that ANKHD1 silencing reduced the expression levels of MMP2 and MMP9, further confirming that ANKHD1 silencing inhibits CRC metastasis. EMT plays a crucial role in the first step of metastasis [4]. Once EMT occurs, cancer cells acquire a phenotype that is more prone to invasion [24]. The occurrence of EMT is thought to alter the expression of genes the products of which play crucial roles in maintaining the epithelial state, such as E-cadherin [25], and this repression occurs at the transcriptional level via the action of EMT transcription factors, including Snail and ZEB1 [26,27]. Li et al. revealed that the overexpression of MIST1 reversed EMT and reduced migration and invasion in pancreatic cancer cells via modulation of the expression of Snail and E-cadherin [28]. It has been shown that ZEB1 plays an important role in the metastasis of pancreatic cancer [29]. In our study, we found that ANKHD1 silencing increased the expression of the epithelial marker E-cadherin while decreasing the expression of the mesenchymal marker vimentin. ANKHD1 silencing inhibited the occurrence of EMT. Similarly, the expression of the EMT transcription factors Snail and ZEB1 decreased when ANKHD1 silencing inhibited EMT. These results suggest that ANKHD1 silencing reduced CRC cell migration and invasion via inhibition of EMT.
Recent studies have revealed that ANKHD1 is a novel component of the Hippo pathway and plays a role as a cofactor of YAP1 [12,17]. YAP1 is a key molecule in this pathway, when the pathway is activated, YAP1 is phosphorylated and then sequestered in the cytoplasm by 14-3-3 proteins, finally p-YAP1 is ubiquitinated and degraded; while when the pathway is not activated, YAP1 enter the nucleus and regulate gene expression [30-32]. Our study revealed that ANKHD1 silencing reduced the expression of YAP1 at the transcriptional level, and the increase in phosphorylated YAP1 may enhance the degradation of YAP1 by ubiquitination. YAP1 is considered to be an EMT-inducing molecule [14]. Activation of YAP1 stimulates epithelial cells to undergo EMT, while suppressing YAP1 activation enables mesenchymal cells to acquire epithelial characteristics [33-35]. Because YAP1 has established roles in EMT, we predict that ANKHD1 silencing suppresses EMT by inhibiting YAP1 activation. To explore the role of YAP1 in the ANKHD1-YAP1-EMT axis, reversal assays were performed. Our results showed that ANKHD1 silencing induced inhibition of proliferation, migration and invasion were reversed when YAP1 was overexpressed and that the inhibition of EMT caused by ANKHD1 silencing also disappeared. We are convinced that ANKHD1 exerts its proliferative and EMT effects on CRC cells by regulating the downstream molecule YAP1.
The molecular mechanisms by which YAP1 modulates cell proliferation and EMT are varied. In the present study, we found that ANKHD1 silencing markedly inhibited the phosphorylation of AKT. AKT signaling plays a significant role in cell proliferation, invasion and migration. The downregulation of p-AKT inhibits proliferation and invasion in colon [36], pancreas [37] and lung [38] cancers. Moreover, mounting evidence has revealed that AKT activation promotes EMT via upregulated Snail expression and downregulated expression of the epithelial marker E-cadherin [39]. In our study, the inhibition of AKT signaling induced by ANKHD1 silencing was reversed when YAP1 was overexpressed. Taken together, we hypothesized that ANKHD1 silencing inhibits YAP1 action and then suppresses proliferation and EMT via AKT signaling. This molecular mechanism still needs more in-depth research.
In conclusion, we have proposed the concept that the ANKHD1-YAP1-EMT axis might regulate CRC cell proliferation, migration and invasion. The axis conferred decreased proliferative and metastatic capacities on CRC cells when ANKHD1 was knocked down. The knockdown of ANKHD1 attenuated the viability of YAP1, thereby reducing the activity of downstream AKT signaling and inhibiting the occurrence of EMT. Our study elucidates the effect of ANKHD1 on the development of CRC cells and might provide a novel target for anti-colorectal cancer therapy.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No 81672970 and 81673100), the Suzhou Key Medical Center (No LCZX201505), the project of Suzhou Science and Technology Development Plan (No SZS201618 and SS201753), the Health and Family Planning Commission Fund Project of Jiangsu Province (No CXTDA2017016), the Second Affiliated Hospital of Soochow University Preponderant Clinic Discipline Group Project Funding, Graduate Student Scientific Research Innovation Projects of Jiangsu Province (No KYCX18_2534), and Key Scientific Development Program of China (No 2016YFC0904702).
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Fedewa SA, Ahnen DJ, Meester RGS, Barzi A, Jemal A. Colorectal cancer statistics, 2017. CA Cancer J Clin. 2017;67:177–193. doi: 10.3322/caac.21395. [DOI] [PubMed] [Google Scholar]
- 2.Wang Z, Chen Q, Li B, Xie JM, Yang XD, Zhao K, Wu Y, Ye ZY, Chen ZR, Qin ZH, Xing CG. Escin-induced DNA damage promotes escin-induced apoptosis in human colorectal cancer cells via p62 regulation of the ATM/gammaH2AX pathway. Acta Pharmacol Sin. 2018;39:1645–1660. doi: 10.1038/aps.2017.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paauwe M, Schoonderwoerd MJ, Helderman R, Harryvan TJ, Groenewoud A, van Pelt GW, Bor R, Hemmer DM, Versteeg HH, Snaar-Jagalska E, Theuer CP, Hardwick JC, Sier CF, Ten Dijke P, Hawinkels LJ. Endoglin expression on cancer-associated fibroblasts regulates invasion and stimulates colorectal cancer metastasis. Clin Cancer Res. 2018 doi: 10.1158/1078-0432.CCR-18-0329. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 4.Stankic M, Pavlovic S, Chin Y, Brogi E, Padua D, Norton L, Massague J, Benezra R. TGF-beta-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-toepithelial transition. Cell Rep. 2013;5:1228–1242. doi: 10.1016/j.celrep.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jolly MK, Ware KE, Gilja S, Somarelli JA, Levine H. EMT and MET: necessary or permissive for metastasis? Mol Oncol. 2017;11:755–769. doi: 10.1002/1878-0261.12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ye X, Weinberg RA. Epithelial-mesenchymal plasticity: a central regulator of cancer progression. Trends Cell Biol. 2015;25:675–686. doi: 10.1016/j.tcb.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelialmesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22:725–736. doi: 10.1016/j.ccr.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith RK, Carroll PM, Allard JD, Simon MA. MASK, a large ankyrin repeat and KH domaincontaining protein involved in drosophila receptor tyrosine kinase signaling. Development. 2002;129:71–82. doi: 10.1242/dev.129.1.71. [DOI] [PubMed] [Google Scholar]
- 9.Hollenbeck JJ, Danner DJ, Landgren RM, Rainbolt TK, Roberts DS. Designed ankyrin repeat proteins as scaffolds for multivalent recognition. Biomacromolecules. 2012;13:1996–2002. doi: 10.1021/bm300455f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Traina F, Favaro PM, Medina Sde S, Duarte Ada S, Winnischofer SM, Costa FF, Saad ST. ANKHD1, ankyrin repeat and KH domain containing 1, is overexpressed in acute leukemias and is associated with SHP2 in K562 cells. Biochim Biophys Acta. 2006;1762:828–834. doi: 10.1016/j.bbadis.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 11.Dhyani A, Duarte AS, Machado-Neto JA, Favaro P, Ortega MM, Olalla Saad ST. ANKHD1 regulates cell cycle progression and proliferation in multiple myeloma cells. FEBS Lett. 2012;586:4311–4318. doi: 10.1016/j.febslet.2012.10.037. [DOI] [PubMed] [Google Scholar]
- 12.Machado-Neto JA, Lazarini M, Favaro P, Franchi GC Jr, Nowill AE, Saad ST, Traina F. ANKHD1, a novel component of the Hippo signaling pathway, promotes YAP1 activation and cell cycle progression in prostate cancer cells. Exp Cell Res. 2014;324:137–145. doi: 10.1016/j.yexcr.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 13.Machado-Neto JA, Lazarini M, Favaro P, de Melo Campos P, Scopim-Ribeiro R, Franchi Junior GC, Nowill AE, Lima PR, Costa FF, Benichou S, Olalla Saad ST, Traina F. ANKHD1 silencing inhibits stathmin 1 activity, cell proliferation and migration of leukemia cells. Biochim Biophys Acta. 2015;1853:583–593. doi: 10.1016/j.bbamcr.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 14.Shi J, Li F, Yao X, Mou T, Xu Z, Han Z, Chen S, Li W, Yu J, Qi X. The HER4-YAP1 axis promotes trastuzumab resistance in HER2-positive gastric cancer by inducing epithelial and mesenchymal transition. Oncogene. 2018;37:3022–3038. doi: 10.1038/s41388-018-0204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Yang X, Wu Y, Zhao K, Ye Z, Zhu J, Xu X, Zhao X, Xing C. B7-H3 promotes gastric cancer cell migration and invasion. Oncotarget. 2017;8:71725–71735. doi: 10.18632/oncotarget.17847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011;278:16–27. doi: 10.1111/j.1742-4658.2010.07919.x. [DOI] [PubMed] [Google Scholar]
- 17.Sidor CM, Brain R, Thompson BJ. Mask proteins are cofactors of Yorkie/YAP in the Hippo pathway. Curr Biol. 2013;23:223–228. doi: 10.1016/j.cub.2012.11.061. [DOI] [PubMed] [Google Scholar]
- 18.Sansores-Garcia L, Atkins M, Moya IM, Shahmoradgoli M, Tao C, Mills GB, Halder G. Mask is required for the activity of the Hippo pathway effector Yki/YAP. Curr Biol. 2013;23:229–235. doi: 10.1016/j.cub.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poulin F, Brueschke A, Sonenberg N. Gene fusion and overlapping reading frames in the mammalian genes for 4E-BP3 and MASK. J Biol Chem. 2003;278:52290–52297. doi: 10.1074/jbc.M310761200. [DOI] [PubMed] [Google Scholar]
- 20.Mook OR, Frederiks WM, Van Noorden CJ. The role of gelatinases in colorectal cancer progression and metastasis. Biochim Biophys Acta. 2004;1705:69–89. doi: 10.1016/j.bbcan.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 21.Chabottaux V, Noel A. Breast cancer progression: insights into multifaceted matrix metalloproteinases. Clin Exp Metastasis. 2007;24:647–656. doi: 10.1007/s10585-007-9113-7. [DOI] [PubMed] [Google Scholar]
- 22.Hidalgo M, Eckhardt SG. Development of matrix metalloproteinase inhibitors in cancer therapy. J Natl Cancer Inst. 2001;93:178–193. doi: 10.1093/jnci/93.3.178. [DOI] [PubMed] [Google Scholar]
- 23.Qin H, Liu X, Li F, Miao L, Li T, Xu B, An X, Muth A, Thompson PR, Coonrod SA, Zhang X. PAD1 promotes epithelial-mesenchymal transition and metastasis in triple-negative breast cancer cells by regulating MEK1-ERK1/2-MMP2 signaling. Cancer Lett. 2017;409:30–41. doi: 10.1016/j.canlet.2017.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 25.Aiello NM, Maddipati R, Norgard RJ, Balli D, Li J, Yuan S, Yamazoe T, Black T, Sahmoud A, Furth EE, Bar-Sagi D, Stanger BZ. EMT subtype influences epithelial plasticity and mode of cell migration. Dev Cell. 2018;45:681–695. e4. doi: 10.1016/j.devcel.2018.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166:21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Zou X, Qian W, Weng X, Zhang L, Zhang L, Wang S, Cao X, Ma L, Wei G, Wu Y, Hou Z. Enhanced PAPSS2/VCAN sulfation axis is essential for snail-mediated breast cancer cell migration and metastasis. Cell Death Differ. 2018 doi: 10.1038/s41418-018-0147-y. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li X, Chen H, Liu Z, Ye Z, Gou S, Wang C. Overexpression of MIST1 reverses the epithelial-mesenchymal transition and reduces the tumorigenicity of pancreatic cancer cells via the Snail/E-cadherin pathway. Cancer Lett. 2018;431:96–104. doi: 10.1016/j.canlet.2018.05.043. [DOI] [PubMed] [Google Scholar]
- 29.Deng SJ, Chen HY, Ye Z, Deng SC, Zhu S, Zeng Z, He C, Liu ML, Huang K, Zhong JX, Xu FY, Li Q, Liu Y, Wang CY, Zhao G. Hypoxia-induced LncRNA-BX111 promotes metastasis and progression of pancreatic cancer through regulating ZEB1 transcription. Oncogene. 2018;37:5811–5828. doi: 10.1038/s41388-018-0382-1. [DOI] [PubMed] [Google Scholar]
- 30.Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee DH, Park JO, Kim TS, Kim SK, Kim T, Kim M, Park GS, Kim JH, Kuninaka S, Olson EN, Saya H, Kim SY, Lee H, Lim DS. LATS-YAP/TAZ controls lineage specification by regulating TGFβ signaling and Hnf4α expression during liver development. Nat Commun. 2016;7:11961. doi: 10.1038/ncomms11961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oh H, Irvine KD. Yorkie: the final destination of hippo signaling. Trends Cell Biol. 2010;20:410–417. doi: 10.1016/j.tcb.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oh SH, Swiderska-Syn M, Jewell ML, Premont RT, Diehl AM. Liver regeneration requires Yap1-TGFbeta-dependent epithelial-mesenchymal transition in hepatocytes. J Hepatol. 2018;69:359–367. doi: 10.1016/j.jhep.2018.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Serrao A, Jenkins LM, Chumanevich AA, Horst B, Liang J, Gatza ML, Lee NY, Roninson IB, Broude EV, Mythreye K. Mediator kinase CDK8/CDK19 drives YAP1-dependent BMP4-induced EMT in cancer. Oncogene. 2018;37:4792–4808. doi: 10.1038/s41388-018-0316-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dubois F, Keller M, Calvayrac O, Soncin F, Hoa L, Hergovich A, Parrini MC, Mazieres J, Vaisse-Lesteven M, Camonis J, Levallet G, Zalcman G. RASSF1A suppresses the invasion and metastatic potential of human non-small cell lung cancer cells by inhibiting yap activation through the GEF-H1/RhoB pathway. Cancer Res. 2016;76:1627–1640. doi: 10.1158/0008-5472.CAN-15-1008. [DOI] [PubMed] [Google Scholar]
- 36.Gao Y, Xiao X, Zhang C, Yu W, Guo W, Zhang Z, Li Z, Feng X, Hao J, Zhang K, Xiao B, Chen M, Huang W, Xiong S, Wu X, Deng W. Melatonin synergizes the chemotherapeutic effect of 5-fluorouracil in colon cancer by suppressing PI3K/AKT and NF-kappaB/iNOS signaling pathways. J Pineal Res. 2017;62 doi: 10.1111/jpi.12380. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y, Kuramitsu Y, Baron B, Kitagawa T, Tokuda K, Akada J, Maehara SI, Maehara Y, Nakamura K. PI3K inhibitor LY294002, as opposed to wortmannin, enhances AKT phosphorylation in gemcitabine-resistant pancreatic cancer cells. Int J Oncol. 2017;50:606–612. doi: 10.3892/ijo.2016.3804. [DOI] [PubMed] [Google Scholar]
- 38.Pan H, Jiang T, Cheng N, Wang Q, Ren S, Li X, Zhao C, Zhang L, Cai W, Zhou C. Long noncoding RNA BC087858 induces non-T790M mutation acquired resistance to EGFR-TKIs by activating PI3K/AKT and MEK/ERK pathways and EMT in non-small-cell lung cancer. Oncotarget. 2016;7:49948–49960. doi: 10.18632/oncotarget.10521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma YS, Wu ZJ, Bai RZ, Dong H, Xie BX, Wu XH, Hang XS, Liu AN, Jiang XH, Wang GR, Jiang JJ, Xu WH, Chen XP, Tan GH, Fu D, Liu JB, Liu Q. DRR1 promotes glioblastoma cell invasion and epithelial-mesenchymal transition via regulating AKT activation. Cancer Lett. 2018;423:86–94. doi: 10.1016/j.canlet.2018.03.015. [DOI] [PubMed] [Google Scholar]