

Abstract
Ribonucleotide reductase small subunit M2B (RRM2B) plays an essential role in maintaining mitochondrial homeostasis. Mitochondrial permeability transition pore (MPTP) is a key regulator of mitochondrial homeostasis. MPTP contributes to cell death and is crucial in cancer progression. RRM2B’s relation to MPTP is not well known, and the role of RRM2B in cancer progression is controversial. Here, our aim was to study the role of RRM2B in regulating MPTP and the association between RRM2B and clinicopathological manifestations in breast cancer. Analysis of Rrm2b-/- mice cells found changes consistent with MPTP opening, including mitochondrial swelling and upregulation of cyclophilin D (CypD), a protein that activates MPTP opening. Silencing of RRM2B gene expression in MCF7 and KB cell lines led to MPTP opening. Accordingly, dysfunctional oxidative phosphorylation and elevated superoxide levels were also detected in RRM2B-silenced MCF7 and KB cell lines, which was consistent with the findings by gene set enrichment analysis of 159 breast cancer cases that genes involving respiratory electron transport were enriched in high-RRM2B breast cancer, and genes involving biologic oxidation were enriched in low-RRM2B breast cancers. A metabolomic study revealed that spermine levels in RRM2B-silenced MCF7 and KB cells were only 5% and 8% of control levels, respectively. Addition of exogenous spermine to RRM2B-silenced MCF7 and KB cells was able to reverse the MPTP opening induced by RRM2B deficiency. These results suggest that RRM2B may induce MPTP opening through reducing spermine levels. Immunohistochemical analysis of 148 breast cancer cases showed that RRM2B and CypD protein levels were inversely correlated in breast cancer specimens (P<0.05), so were their associated clinicopathologic parameters that high-level RRM2B expression was associated with better clinicopathological features. We conclude that RRM2B deficiency leads to MPTP opening mediated by spermine. Coupling of low RRM2B and high CypD expression is associated with aggressive manifestations of breast cancer.
Keywords: Ribonucleotide reductase small subunit M2B, mitochondrial permeability transition pore, cyclophilin D, spermine, aggressive manifestations of breast cancer
Introduction
Ribonucleotide reductase (RR) is the rate limiting enzyme in the synthesis and repair of DNA. Human RR consists of large subunits (R1) and small subunits (R2). There are two known smaller subunits, RRM2 and RRM2B, that bind large subunits to form an active heterodimeric tetramer. Studies have shown that increased RR activity is highly correlated with cancer growth rate [1]. Therefore, RR is considered a therapeutic target in cancer therapy. However, recent studies demonstrated that these three subunits regulate cancer progression in different ways. RRM1 has been described as a suppressor of tumor initiation, and high-level RRM1 expression correlates with longer lifespan and later disease recurrence for patients diagnosed with early stage non-small cell lung cancer [2,3]. In contrast to the tumor-suppressing role of RRM1, RRM2 shows oncogenic activity [4,5]. However, the role of RRM2B in mutagenesis and tumorigenesis is controversial [6-8]. These discrepancies led us to seek further mechanistic insight into the association between RRM2B and cancer.
Mutant RRM2B has been reported to cause severe mitochondrial DNA depletion [9]. Our recent studies showed that RRM2B can interact with pyrroline-5-carboxylate reductase (a mitochondrial protein) and is involved in regulation of anti-oxidative stress [10]. RRM2B is also involved in other mitochondrial functions, such as ATP synthesis and membrane potential (Δm) maintenance [11]. However, RRM2B’s relation to mitochondrial permeability transition pore (MPTP) is not known. MPTP is a nonselective voltage-dependent mitochondrial channel [12]. Studies showed the MPTP can initiate cell death; thus, it was thought to be a promising tool for blocking cancer progression [13]. Recently, a model for the pore structure has been developed. This model involves adenine nucleotide translocase in the inner mitochondrial membrane and a voltage-dependent anion channel in the outer mitochondrial membrane, together forming a continuous channel across the inner membrane space under the control of cyclophilin D (CypD) [14]. Numerous studies have emphasized the importance of CypD in the formation and regulation of MPTP, and subsequent cell death [15]. However, CypD is upregulated in cancer cells derived from estrogen-responsive tissues such as breast, uterus, and ovary [16]. This paradox makes MPTP and CypD valuable research focuses to help understanding MPTP regulation in cancer.
Our aim was to study the role of RRM2B in regulating MPTP and the association between RRM2B and clinicopathological manifestations in breast cancer.
Materials and methods
Cell culture and transfection
MCF-7 (breast cancer cells) and KB (a subline of the cervical carcinoma cell line HeLa) were obtained from ATCC (American Type Culture Collection, Manassas, VA). Frozen aliquots were stored in liquid nitrogen vapor phase, and cells were cultured for no longer than 6 months after thawing. Cell lines were authenticated by ATCC before delivery and not re-authenticated in our laboratory. Cells were cultured on tissue culture plates in Dulbecco’s Modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin in a 5% CO2 atmosphere at 37°C. MCF7 and KB cells were transduced with pTRIPZ lentiviruses expressing vector, shNS, or shRRM2B and cultured as described previously [17]. The MCF7/KB-shRR-M2B cells were validated for loss of RRM2B expression using Western blot analysis.
Mouse husbandry
Generation of Rrm2b-/- mice by the gene trap method has been described [18]. Rrm2b+/- mice in mixed genetic background (129/SvEv-Brd and C57BL/6J) were obtained from the Texas A&M Institute for Genomic Medicine (TI-GM). Murine studies were carried out in accordance with protocols approved by the Institutional Animal Care and Use Committee (IAC-UC) of City of Hope. Generation of Rrm2b+/+, Rrm2b-/+, and Rrm2b-/- mice, and husbandry was performed with approved protocols as described previously [19].
Electron microscopy
Mice were anesthetized by intraperitoneal injection of ketamine/xylazine and gaseous isoflurane. Mice kidneys were fixed in situ by cardiac perfusion with phosphate-buffered saline (PBS) containing heparin, followed by 2.5% glutaraldehyde/2.5% paraformaldehyde in PBS. Dissected kidneys were cut into 1- to 2-mm3 pieces and visualized with a Twin Tecnai 120 KV transmission electron microscope (TEM; FEI, Hillsboro, OR, USA).
Mitochondrial permeability transition pore assay and live-cell imaging
MCF7- and KB-derived cell lines stably expressing shNS or shRRM2B were incubated with or without 1 mM spermine (supplemented with 1 mM aminoguanidine to prevent spermine degradation by serum amine oxidases [20] for 24 hours before applying the image-iTTM live mitochondrial transition pore assay kit (I35103, Invitrogen-Life Technologies, Grand Island, NY), which was performed according to the manufacturer’s instructions, to visualize the mitochondrial transition pores in the cells. In brief, cells were stained with acetoxymethyl ester of calcein dye (calcein AM, green) and MitoTracker Red CMXRos (red) to visualize the mitochondria. Subsequently, the cells were incubated with CoCl2 solution, which quenches the calcein fluorescence within the cytoplasm but not within the mitochondria if the MPTP is tightly closed. When the MPTP is open, the Co2+ ions can enter the mitochondria and quench the mitochondrial calcein fluorescence. Therefore, healthy mitochondria appear an orange/yellow color. Mitochondrial pore opening results in a loss of the green fluorescence and then the red fluorescence appears.
Oxygen consumption rate measurement
Oxygen consumption rate (OCRs) were measured using the Seahorse Biosciences extracellular flux analyzer (XF24) (North Billerica, MA). Cells (2 × 104) were seeded in 24-well plates in XF24 and incubated for 20 to 24 h prior to performing the XF assay, as described previously [21]. The presented data were normalized to the final cell number.
Isolation of mitochondria
Intact mitochondria from cells were isolated using a Mitochondria Isolation Kit (Ab110170, MitoSciences, Eugene, Oregon) according to the manufacturer’s instructions. About 0.2 g of cell pellet was collected and homogenized in 1 mL Buffer 1 solution (0.25 M sucrose, 1 mM EGTA, 10 mM HEPES, pH 7.4) at 4°C. The mitochondrial pellet was obtained by low-speed centrifugation (750 g) of the homogenate, followed by high-speed centrifugation (11,000 g) of the supernatant. The final mitochondrial pellet was resuspended in 200 μL Buffer 2 solution (70 mM sucrose, 220 mM mannitol, 1 mM EDTA, 2 mM HEPES, pH 7.4).
Western blot analysis and antibodies
Western blots were performed as described previously [17]. Briefly, the protein concentrations of mitochondrial extracts were determined using a Bio-Rad Protein Assay Kit (5000001, Hercules, California). About 20 to 40 μg of protein were mixed with an equal volume of 2 × SDS loading buffer, boiled for 5 min, and then separated by tris-glycine SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. The membranes were blocked with 5% nonfat milk in PBST (PBS containing 0.05% Tween 20) and incubated with primary antibodies at 4°C overnight. The membranes were then washed three times with PBST for 10 min and then incubated with HRP-labeled secondary antibodies for 2 hours at room temperature. Immunoblots were visualized using VersaDoc 5000 Imaging System (Bio-Rad). Three independent experiments were performed, and representative blots were presented.
A rabbit polyclonal antibody against RRM2B (600-401-B67S, Rockland Immunochemical, Gilbertsville, PA) was used for Western blot analysis and immunohistochemical (IHC) staining. An antibody against CypD (LS-C412621, Abcam, Cambridge, MA) was used for immunohistochemical staining. Total OXPHOS human Western blot antibody cocktail (ab110413, MitoSciences Inc., Eugene, OR) was used to detect the protein levels for the five OXPHOS core components. The antibodies for Ki-67 (ab156956, Abcam, Cambridge, MA) and HER2 (ab16901, Abcam, Cambridge, MA) were used for IHC staining.
Mitochondrial complex I and II activity measurements
Complex I activity was determined using the Complex I Enzyme Activity Microplate Assay Kit (MS141, MitoSciences, Eugene, Oregon), based on the manufacturer’s recommended protocol and settings. Complex II activity was quantified based on established protocols described in previous studies [22].
Measurement of mitochondrial ATP
Cells were harvested and washed using PBS. ATP levels were quantified using the ATP bioluminescent somatic cell assay kit (FLASC-1KT, Sigma-Aldrich, St. Louis, MO), according to the manufacturer’s instructions.
Mitochondrial superoxide measurements
MitoSOXTM Red mitochondrial superoxide indicator for live-cell imaging (M36008, Molecular Probes, Carlsbad, CA) was used to detect mitochondrial superoxide. The cells were cultured in 10-cm dishes in regular growth media until 70% to 80% confluent. The cells were incubated for 30 min with 10 µM MitoSOX Red, then washed twice with PBS and analyzed for fluorescence by flow cytometry. At least 50,000 events were collected on a FACScalibur flow cytometer (Beckton Dickinson, Rockville, MD) and analyzed using Cellquest (Beckton Dickinson). High fluorescence was calculated by setting the gate on the control cells such that the peak reached a minimum, and all experimental samples were compared to this control gate.
Measurement of metabolite levels
Metabolomic analysis of cell samples was performed using high performance liquid chromatography time-of-flight mass spectrometry (HPLC-TOFMS). MCF7- and KBderived cell lines stably expressing shNS or shRRM2B were processed using a two-step metabolite extraction procedure [23] and analyzed by HPLC-TOFMS [24]. The acquired data files from HPLC-TOFMS analysis were processed using an Agilent Mass-Hunter Qualitative Analysis Program (vB.03.00, Agilent) and XCMS package [25], respectively. The resulting data sheet was used for further analysis.
Human breast tissue collection and immunohistochemical analysis
The protocol for human subjects’ leftover tissue collection and testing was reviewed and approved by the Institutional Review Board of the Second Affiliated Hospital of Zhejiang University (Hangzhou, Zhejiang, China). The research was carried out according to The Code of Ethics of the World Medical Association (Declaration of Helsinki). Informed consent was obtained from the patients whose tissues were used. Some eligible patients were excluded due to (i) lack of informed consent; (ii) multiple cancers; (iii) lack of histologic diagnosis; (iv) lack of follow-up information; or (v) missing of tissue samples. Details for clinicopathologic information collecting and follow-up were described in our previous publication [26]. The demographic distribution of patients is listed in Table 1 in detail.
Table 1.
Demographic distribution of RRM2B and CypD in breast cancer patients
| RRM2B | CypD | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| High (%*) | Low | p value† | High (%*) | Low | p value† | |
| Age | ||||||
| <50 | 38 (54.3) | 32 | 32 (50.5) | 32 | ||
| ≥50 | 59 (64.1) | 33 | 0.205 | 58 (65.5) | 26 | 0.019 |
| pT stage‡ | ||||||
| T0-T1 | 35 (72.9) | 13 | 24 (55.9) | 19 | ||
| T2-T4 | 57 (54.8) | 47 | 0.034 | 62 (63.9) | 35 | 0.364 |
| pN stage‡ | ||||||
| 0 | 47 (55.3) | 38 | 39 (50.0) | 39 | ||
| 1-2 | 40 (65.6) | 21 | 42 (77.8) | 12 | ||
| 3 | 10 (62.5) | 6 | 0.446 | 9 (56.3) | 7 | 0.004 |
| Grade | ||||||
| 1 = Well | 12 (60.0) | 8 | 10 (55.6) | 8 | ||
| 2 = Mod | 42 (62.7) | 25 | 37 (58.7) | 26 | ||
| 3 = Poor | 38 (57.6) | 28 | 0.834 | 38 (64.4) | 21 | 0.725 |
| ER | ||||||
| Negative | 28 (51.9) | 26 | 35 (68.6) | 16 | ||
| Positive | 64 (68.1) | 30 | 0.050 | 46 (54.1) | 39 | 0.095 |
| PR | ||||||
| Negative | 39 (57.4) | 29 | 42 (66.7) | 21 | ||
| Positive | 53 (63.1) | 31 | 0.471 | 43 (56.6) | 33 | 0.225 |
| HER2 | ||||||
| Negative | 63 (58.3) | 45 | 58 (58.0) | 42 | ||
| Positive | 14 (63.6) | 8 | 0.645 | 14 (63.6) | 8 | 0.627 |
| Ki67 | ||||||
| Negative | 31 (48.4) | 33 | 36 (60.0) | 24 | ||
| Positive | 61 (67.8) | 29 | 0.016 | 51 (62.2) | 31 | 0.791 |
| Molecular subtype | ||||||
| Luminal A | 27 (52.9) | 24 | 27 (56.3) | 21 | ||
| Luminal B/HER2- | 24 (72.7) | 9 | 16 (53.3) | 14 | ||
| Luminal B/HER2+ | 5 (55.6) | 4 | 4 (44.4) | 5 | ||
| Basal-like TNBC | 15 (55.6) | 12 | 18 (75.0) | 6 | ||
| HER2-positive | 7 (77.8) | 2 | 0.091 | 8 (90.0) | 1 | 0.166 |
| CD44/CD24 status | ||||||
| CD44+/CD24- | 48 (57.8) | 35 | 47 (62.7) | 28 | ||
| Others | 40 (63.5) | 23 | 0.750 | 35 (60.3) | 23 | 0.792 |
Note: There are 1, 9, 15, 14, 35, 12 and 1 missing cases in Grade, pT stage, ER, PR, HER2, Ki67 and CD44/CD24 status, respectively. There are 132 cases could be classified on molecular subtype.
%represents rate of RRM2-High, which is equal to NHigh/(NHigh+NLow) × 100%.
pT and pN stage are pathological stage.
In this study, no patient has > or = 4 lymph nodes involvement.
p values were based on Pearson Chi square test.
The primary breast cancer tissue specimens were assembled and built into a multiple-tissue array as described previously [8,27]. To exclude bias, the IHC conditions for CypD, RRM2B, Ki-67 and HER2 were determined and optimized using test slides including breast cancer and normal breast tissues and then applied to the multiple-tissue array. All proteins were detected on 4-m sections of tissue from the multiple-tissue array using a previously described deparaffinization and staining protocol [8,27].
Immunoreactivity in the cytoplasm was scored according to the percentage and intensity of the staining. The staining intensity (0: negative, 1: weak, 2: moderate, and 3: strong) was defined as low (negative or weak) or high (moderate or strong). To exclude observer bias, all the slides resulting from the immunohistochemical reactions were evaluated individually and independently by two observers in a blinded manner. Discrepancies were re-estimated after joint review of the two readers, and missing samples were left blank. A total of 162 breast cancer patients met the inclusion criteria, and 148 samples were stained for both RRM2B and CypD.
Gene set enrichment analysis (GSEA)
The detailed GSEA protocol can be obtained from the Broad Institute Gene Set Enrichment Analysis website (www.broad.mit.edu/gsea) and related reference [28]. The GSEA software v2.0.13 was run on a JAVA 7.0 platform. A published breast cancer microarray dataset (Pawitan, GSE1459) was used for analysis [29]. The dataset (gct) and phenotype label (cls) files were created and loaded into the GSEA software, and gene sets were downloaded from the website of the Broad Institute. The number of permutations was set to 1000, and phenotype label was RRM2B expression levels. The ranked list metric was generated by calculating the signal-to-noise ratio (High vs Low) and Pearson model (continues levels), which is based on the difference of means scaled according to the standard deviation.
Statistical analysis
Data analysis was conducted using JMP 8.0 software (SAS, Cary, NC). Categorical data were compared by χ2 analysis, Fisher’s exact test, or the binomial test of proportions. Statistical significance was set as P<0.05. All tests were two-tailed.
Results
Rrm2b knockout mice exhibit mitochondrial swelling and high CypD expression
To clarify the role of RRM2B in regulating MPTP, we first used electron microscopy to observe mitochondrial morphology changes in Rrm2b knockout mice. Our results showed that, in comparison with kidney tissues from Rrm2b+/+ mice and Rrm2b+/- mice, tissues from Rrm2b-/- mice showed dramatic structural changes in the mitochondria, including an enlarged and rounded morphology with a less electron-dense and more cristae-free matrix. The mitochondria were abnormally shaped and disorganized, and their number was significantly reduced due to the rupture of swollen mitochondria (Figure 1, left panel). These phenotypes are indicators of mitochondrial swelling, which is a marker of MPTP opening. IHC on the same kidney tissues showed that CypD, a protein which could promote MPTP opening, was barely expressed in Rrm2b+/+ or Rrm2b+/- mice but was strongly expressed in Rrm2b-/- mice (Figure 1, right panel, CypD). Hematoxylin and eosin (H&E) staining revealed a normal cortex with normal glomeruli and tubules in Rrm2b+/+ and Rrm2b+/- mice, but obvious tubular atrophy in Rrm2b-/- mice (Figure 1, right panel, H&E).
Figure 1.
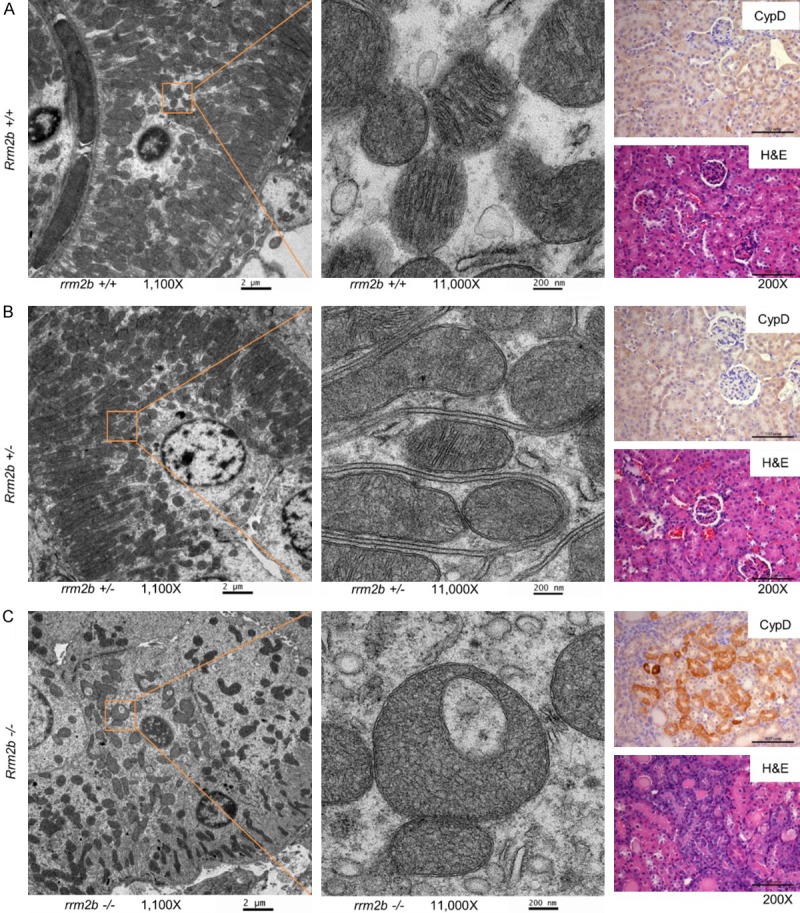
Rrm2b-knockout mice exhibit mitochondrial swelling and high Cyclophilin D (CypD) expression. Left panel, scanning electron microscope images at 1100-fold magnification (scale bar: 2 µm). A 10 × higher magnification of the area marked by the orange square is located next to the 1100-fold magnification image. Right panel, immunohistochemistry (IHC) stained with anti-CypD (CypD) and hematoxylin and eosin (H&E)-stained images of kidney tissue from Rrm2b+/+ (A), Rrm2b+/- (B), or Rrm2b-/- (C) mice. Scale bar corresponds to 100 µm.
This data indicates that Rrm2b deficiency causes mitochondrial changes consistent with MPTP opening, including mitochondrial swelling and upregulation of CypD.
RRM2B gene silencing promotes mitochondrial permeability transition pore opening
To make certain that RRM2 deficiency could lead to MPTP opening, we used a live-cell mitochondrial assay to determine the MPTP status in MCF7 breast cancer cells with or without RRM2B silencing by shRNA. This live-cell mitochondrial assay uses a green fluorescent dye (calcein AM) that is maintained in mitochondria when the MPTP is closed. Mitochondria were also stained with MitoTracker Red. Our study showed that in control cells, which were transfected with either vector only or a non-silencing shRNA (shNS), most cells were stained both green and red, indicating a closed MPTP status (Figure 2, upper and middle rows). In cells that were transfected by shRNA targeting RRM2B (shRRM2B), there was a greater population of predominately red cells than predominately green cells, indicating greater MPTP opening in comparison with control cells (Figure 2, lower row). This result indicates that RRM2B gene silencing leads to MPTP pore opening.
Figure 2.
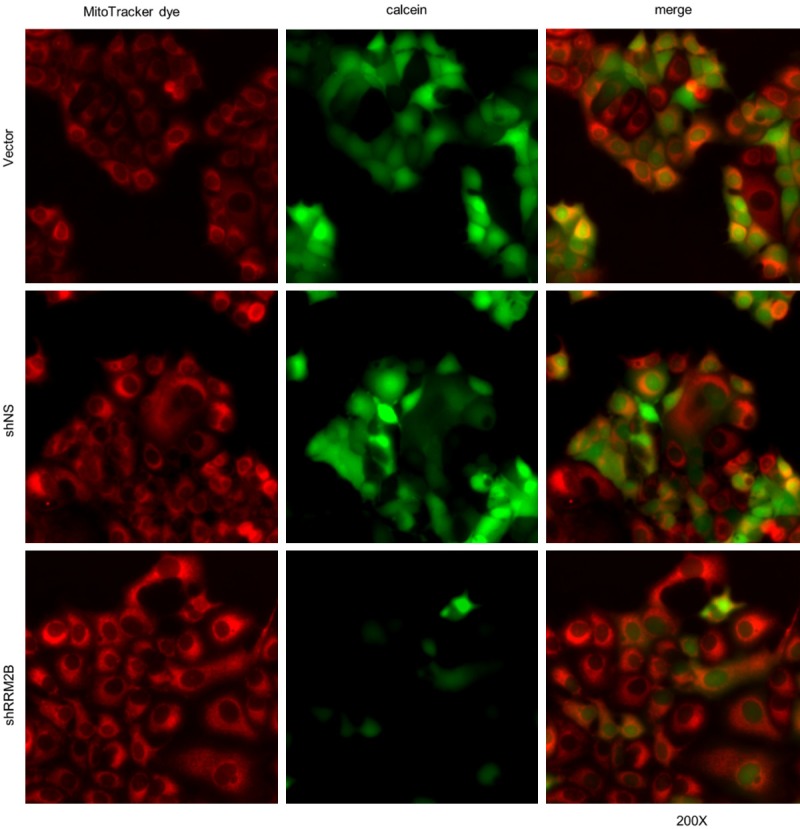
RRM2B gene silencing promotes mitochondrial permeability transition pore (MPTP) opening. Live-cell fluorescence microscopy analysis of MPTP activity in MCF7 cells expressing vector, shNS or shRRM2B. All mitochondria are stained red with MitoTracker Red CMXRos (left panel). Healthy cells are stained green with calcein AM (middle panel), which is concentrated in the mitochondria. Merged images are presented on the right panel.
RRM2B silencing results in dysfunctional oxidative phosphorylation and enhanced ROS production
MPTP opening in vitro leads to a collapse of the proton motive force, ATP hydrolysis, disruption of ionic homeostasis, and mitochondrial swelling. Therefore, we next examined whether RRM2B silencing altered mitochondrial physiological function in cell lines. Function was initially assessed by real-time monitoring of mitochondrial respiration. Oxidative phosphorylation (OX-PHOS) was measured by assessing the oxygen consumption rate (OCR) in the presence of sh-RRM2B in MCF7 and KB cells. The OCR was determined by sequential addition of oligomycin (ATP synthase inhibitor), FCCP (uncoupler of oxidative phosphorylation), and rotenone (complex I inhibitor) to cells, which allowed us to determine the ATP-coupled respiration, leaked respiration (proton leak), and maximal respiration capacity. RRM2B silencing in MCF7 cells decreased the basal mitochondrial OCR (Figure 3A, left panel, MCF7). In addition, the maximal mitochondrial respiration capacity (i.e., FCCP-stimulated OCR) was significantly lower in RRM2B-silenced cells than in control cells (Figure 3A, left panel, MCF7). The same phenomenon was observed in KB cells (Figure 3A, right panel, KB). To determine whether RRM2B silencing influenced the expression of the electron transport complex, we measured the expression of mitochondrial electron transport complex I, II, III, IV, and V core proteins by Western blot analysis. The complex III and complex II core protein levels of cells expressing sh-RRM2B were clearly reduced compared to cells expressing shNS, indicating that the protein expression of complex III and II core genes was inhibited by RRM2B silencing (Figure 3B, complex III, left panel; complex II in left panel for KB cells, complex II in right panel for MCF7). To further clarify whether RRM2B causes functional changes in the level of oxidative phosphorylation, the activities of the electron transport complexes I and II and the ATP levels were determined in cells expressing shNS and shRRM2B. These experiments revealed a statistically significant decrease in complex II activity (Figure 3C, right panel, MCF7 and KB cells) and ATP levels (Figure 3D, MCF7 cells) in cells expressing shRRM2B, indicating that RRM2B silencing reduced mitochondrial respiration and total ATP levels. RRM2B silencing, however, did not affect complex I activity (Figure 3C, left panel), consistent with unchanged complex I protein expression after RRM2B-silencing in MCF7 and KB cells (Figure 3B). MPTP opening might directly or indirectly increase ROS production in vivo [30]. Therefore, steady-state levels of mitochondrial superoxides were measured in cells expressing empty vector, shNS, or shRRM2B by use of MitoSOXTM Red in live cells. The percentage of MitoSOXTM-stained cells was significantly increased in RRM2B-silenced MCF7 cells compared to the vector and shNS controls (Figure 3E). These findings suggested that RRM2B silencing disrupts oxidative phosphorylation and enhances ROS production. Gene set enrichment analysis (GSEA) of 159 cases of human breast cancer also showed RRM2B expression is positively associated with gene signatures linked with mitochondrial upregulation of respiratory electron transport (Figure 3F, and negatively associated with gene signatures linked with biologic oxidation (Figure 3G).
Figure 3.
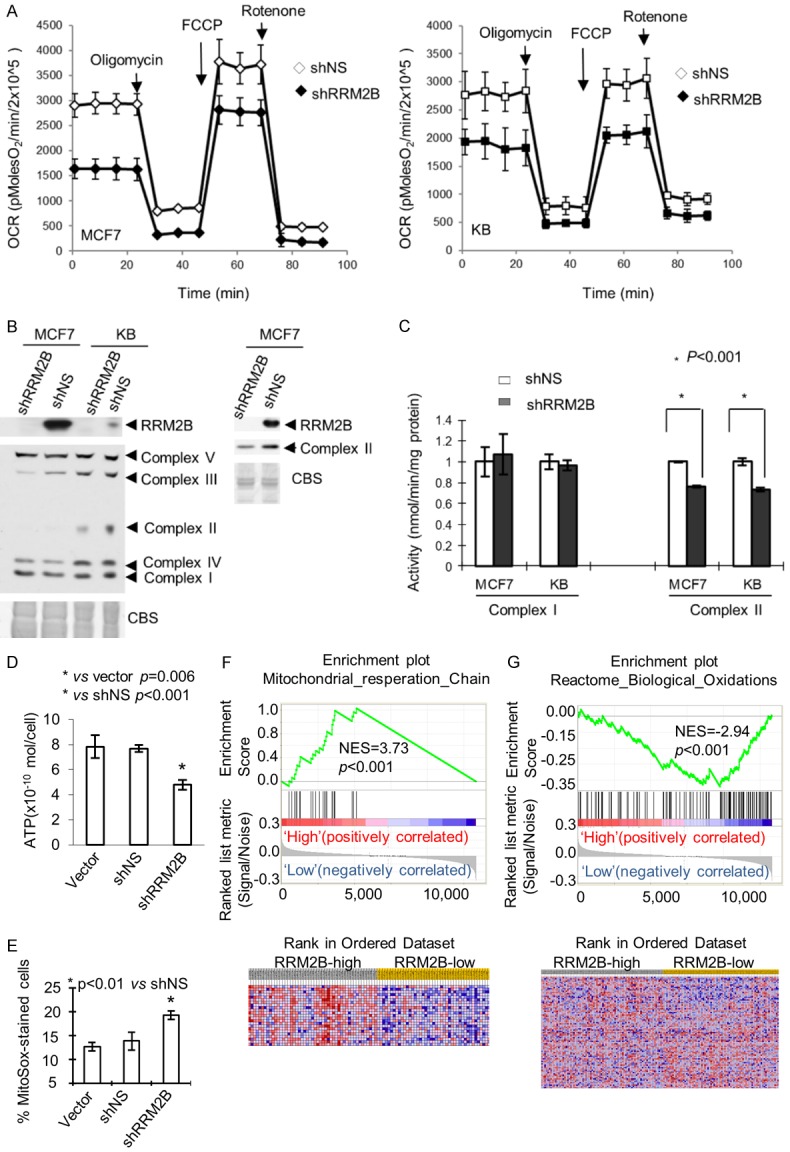
RRM2B silencing results in dysfunctional oxidative phosphorylation and enhanced ROS production. A: Effect of RRM2B-silencing on oxygen consumption rate (OCR) was measured in MCF7 and KB cells expressing shNS and shRRM2B with a Seahorse Extracellular Flux (XF-24) analyzer. Arrows indicate the time of addition of oligomycin (5 μg/ml), FCC P (1 μM), or rotenone (1 μM). B: Western blot analysis shows reduced expression of the core genes of the oxidative phosphorylation (OXPHOS) complex II and III in RRM2B-silenced cells. C: OXPHOS complex I and II activity in RRM2B-silenced MCF7 and KB cells. OXPHOS activity was measured spectrophotometrically in mitochondrial suspensions. D: Total cellular ATP levels were decreased in RRM2B-silenced MCF7 cells. MCF7 cells expressing empty vector, shNS, and shRRM2B were lysed and ATP levels were measured using chemiluminescence. E: RRM2B-silenced cells exhibit increased superoxide levels as indicated by MitoSox Red. Mitochondrial superoxide levels were determined in shRRM2B and control (vector or shNS) cells (MCF7) by MitoSox staining and analyzed by flow cytometry. P value for t test is reported. F: Respiratory electron transport gene signature was associated with high RRM2B expression in breast cancer patients. G: Genes of biologic oxidation were enriched in low-RRM2B breast cancers. Columns represent individual samples, and rows represent each gene. Each cell in the matrix represents the expression level of a gene in an individual sample. Detailed methodological information for gene set enrichment analysis (GSEA) is available from the Broad Institute (www.broad.mit.edu/gsea). The phenotype labels were based on RRM2B expression levels (continues value). Red indicates a high level of expression and blue indicates a low level of expression. An NES (normalized enrichment score) greater than 1 indicates enrichment of the gene set in high-RRM2B breast cancers. The ranked list metric was generated by Pearson model.
RRM2B gene silencing-induced MPTP opening can be rescued by exogenous spermine
Spermine is one of the most powerful physiological MPTP inhibitors. To explore the possible mechanisms underlying the involvement of RRM2B in MPTP status, we performed a metabolomic study in MCF7 and KB cells with or without RRM2B-silencing. These experiments revealed a significantly reduced number of metabolites associated with polyamine synthesis reactions, including ketoglutaramic acid, N-acetyl-L-glutamate 5-semialdehyde, spermidine, and spermine in both RRM2B-silenced cell lines (Figure 4A). Among hundreds of metabolites, the change in the spermine level was the most dramatic in the RRM2B-silenced MCF7 and KB cells in comparison with their controls, respectively. The spermine level in the RRM2B-silenced cells was 5% and 8% that of control MCF7 and KB cells, respectively (Figure 4A, spermine), and the precursor spermidine level was also reduced to 47% and 37% that of control MCF7 and KB cells, respectively (Figure 4A, spermidine). These findings suggested that RRM2B silencing led to lower spermine levels. Therefore, we used the same live-cell mitochondrial assay as we used in Figure 2 to determine the MPTP status in cell lines with or without RRM2B silencing in response to spermine treatment. Our study again showed that in control cells transfected with shNS, most cells were stained both green and red (shown as yellow), indicating a closed MPTP status (Figure 4B, left panels). In cells transfected with shRRM2B, more red cells showed up, indicating more MPTP opening, in comparison with control cells (Figure 4B, middle panels). Interestingly, addition of spermine in the medium prevented MPTP opening in the RRM2B-silenced cells (Figure 4B, right panels). These results suggest that RRM2B may induce MPTP opening through down-regulation of spermine levels.
Figure 4.
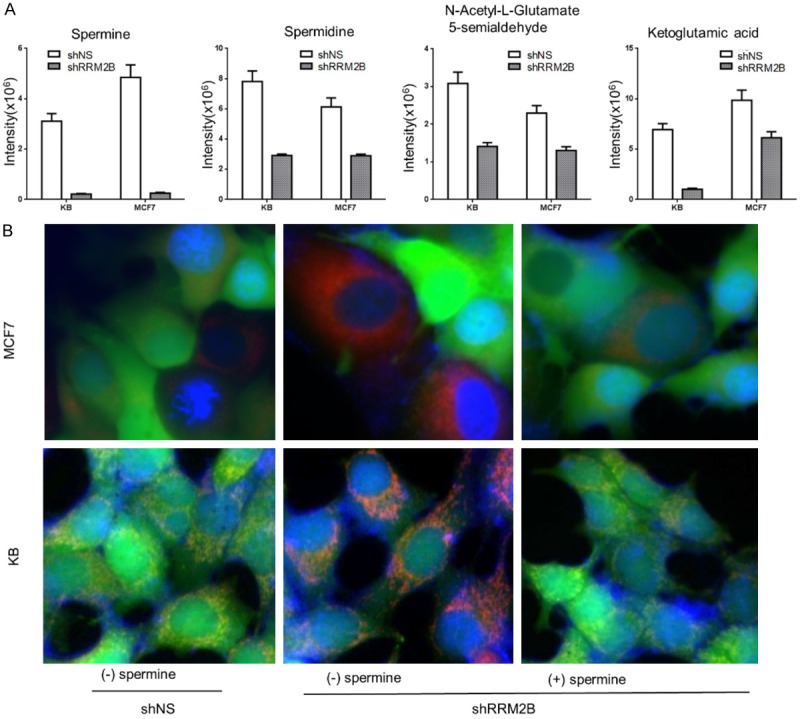
Spermine reduction is involved in MPTP opening induced by RRM2B silencing. A: Levels of spermine, spermidine, lN-acetyl-L-glutamate 5-semialdehyde, and ketoglutamic acid were reduced by RRM2B silencing in MCF7 and KB cells. B: RRM2B maintains MPTP integrity via spermine. Live-cell fluorescence microscopy analysis of mitochondrial transition pore activity in MCF7 and KB cells expressing shNS and shRRM2B (with or without spermine). Nuclei are stained blue with Hoechst 33342. All mitochondria are stained red with MitoTracker Red, and healthy cells are stained green with calcein AM, which is concentrated in the mitochondria. Merged images are presented.
RRM2B and CypD are inversely correlated in protein expression levels as well as in their associated clinical pathologic parameters where RRM2B is associated with better clinical pathologic parameters
To further confirm the negative correlation between RRM2B expression and MPTP opening, the protein expression levels of RRM2B and CypD were determined in 148 human breast cancer samples by IHC. Quality control of IHC was maintained by following protocols as in our previous studies [8]. A representative IHC image was shown in Figure 5A, which showed that RRM2B and CypD expression were inversely correlated in breast cancer patient samples. Patients who expressed higher levels of RRM2B in breast cancer tissue would express lower levels of CypD. Similarly, the patients who expressed lower levels of RRM2B in breast cancer tissue had higher expression levels of CypD. In other words, RRM2B inversely correlated with CypD expression in breast cancer patients (Trends P = 0.02; Figure 5B). We also found that higher expression of RRM2B was significantly associated with a lower tumor staging and decreased staining of the proliferation marker Ki67 (Figure 5C and 5D). In contrast, increased CypD expression was significantly associated with lymph node metastasis and tended to occur in the more malignant subtype triple-negative breast cancer (TNBC) (Figure 5E and 5F). These findings suggest that RRM2B and CypD protein levels are inversely correlated, so are their associated clinical pathologic parameters.
Figure 5.
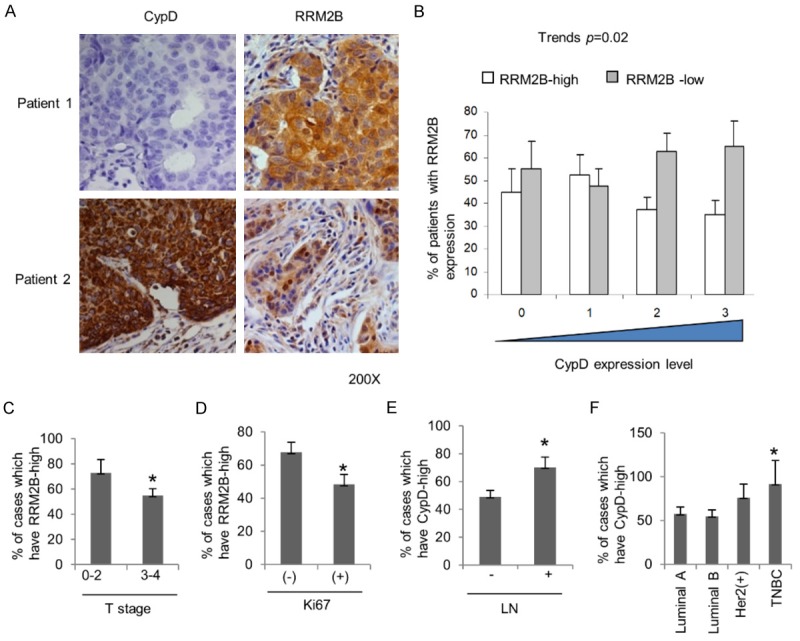
RRM2B and CypD are inversely correlated in protein expression levels as well as in their associated clinical pathologic parameters. A: Representative immunohistochemistry (IHC) image from two breast cancer patients (Patient 1, Patient 2). B: Negative correlation between RRM2B and CypD protein expression levels in 148 breast cancer patients. C: High RRM2B protein expression was associated with lower level T stage. D: High RRM2B protein expression was associated with lower expression of Ki67. E: High CypD protein expression was associated with lymph node metastasis (LN). F: High CypD protein expression tended to happen in triple negative breast cancer (TNBC). *P<0.05 comparing to control (the other group).
Discussion
To our knowledge, this study represents the first investigation of RRM2B’s relation to MPTP and the associated clinical significance in breast cancer. Here, we found that RRM2B deficiency could reduce spermine levels, which resulted in MPTP opening in cancer cell lines. Consistent with this, the increased expression of MPTP promoter CypD protein and the classical consequences of MPTP opening, including mitochondrial matrix swelling, reduction of ATPase activity, uncoupling of OXPHOS, enhanced oxidation and loss of respiratory control [31] were clearly observed in Rrm2b knock-out mice or RRM2B silenced cancer cell lines. The importance of RRM2B involving MPTP regulation is further illustrated by the inverse correlation of its expression as well as clinicopathologic manifestation with CypD in breast cancer patient. Both low expression of RRM2B and high expression of CypD were associated with aggressive tumor phenotype characterized by higher tumor staging, increased proliferative rate, lymph node metastasis and tendency to occur in more malignant subtype triple-negative breast cancer (TNBC). In line with our results, RRM2B has previously been related to favorable outcome for patients with nasopharyngeal carcinoma, colorectal cancer and lung cancer [8,32,33] and cyclophilins play a role in the development of tumors [16,34,35].
Beside CypD, studies in cancer cell lines and tumor models have shown that different MPTP activating proteins are overexpressed [36,37]. For example, the expression levels of VDAC isoforms are significantly higher in malignant tumor cells [38]. These studies suggested MPTP opening might promote tumor cells proliferation rather than death, which is contrast to the previous concept that MPTP activating proteins make MPTP open, thus lead to cell death and better clinical outcome. One explanation for this paradox is that reactive oxygen species (ROS) can promote several hallmarks of cancer, such as proliferation, invasion, and metastasis [39], which is well supported by our findings.
In our study, Rrm2b-knockout mice/cell lines exhibited swollen mitochondria and membrane rupture, deficient OXPHOS and increased ROS. Deficient OXPHOS may lead to more electrons prematurely exiting the respiratory chain to augment mitochondrial superoxide generation. Electron leakage can occur at complex I, complex II, or complex III [40]. Our findings indicate that the ROS induced by RRM2B silencing might be produced from complexes II and III because the core protein expression of these two complexes is significantly reduced by RRM2B inhibition. Disruption of complex III by RRM2B silencing is consistent with a report showing that complex III activity was deficient in the muscle mitochondria of a patient with neonatal trunk hypotonia, who had barely detectable levels of muscle RRM2B [9].
Mitochondria are not only the major sites of ROS production, they are also the major ROS targets. Enhanced generation of mitochondrial ROS can induce MPTP opening, and this opening can be prevented by antioxidant treatment [41]. Therefore, the OXPHOS deficiency and increased levels of superoxide in RRM2B-silenced cells are likely to trigger MPTP opening. MPTP opening results in mitochondrial dysfunction with uncoupled OXPHOS and ATP hydrolysis [42], in turn leading to more oxidant release in a process termed the “vicious cycle”. Our study suggests that this “vicious cycle”, under condition of RRM2B deficiency, may result from a lack of spermine, because spermine, an antioxidant, decreased dramatically when RRM2B was inhibited. It has been reported that naturally occurring polyamines, including spermine, exert protective effects against MPT in isolated mitochondria in the presence of various inducing agents [43]. Several mechanisms have been proposed to explain this protective action. The free radical scavenging action of spermine is considered an important mechanism [44-46]. Spermine also prevents oxidation of several mitochondrial components--such as thiols, glutathione, and pyridine nucleotides, which are involved in MPTP opening [44]. Consistent with these reports, our findings indicate that RRM2B regulates MPTP status through regulating spermine levels. Taken together, we found that RRM2B and CypD protein levels are inversely correlated, so are their associated clinicopathologic parameters. The underlying mechanism was through MPTP. These findings highlight the value to routinely detect both RRM2B and CypD by immunohistochemistry in breast cancer patients for predicting disease progression.
Acknowledgements
This work was financially supported by the “TMU Research Center of Cancer Translational Medicine” from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan.
Disclosure of conflict of interest
None.
References
- 1.Elford HL, Freese M, Passamani E, Morris HP. Ribonucleotide reductase and cell proliferation. I. Variations of ribonucleotide reductase activity with tumor growth rate in a series of rat hepatomas. J Biol Chem. 1970;245:5228–5233. [PubMed] [Google Scholar]
- 2.Fan H, Huang A, Villegas C, Wright JA. The R1 component of mammalian ribonucleotide reductase has malignancy-suppressing activity as demonstrated by gene transfer experiments. Proc Natl Acad Sci U S A. 1997;94:13181–13186. doi: 10.1073/pnas.94.24.13181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bepler G, Sharma S, Cantor A, Gautam A, Haura E, Simon G, Sharma A, Sommers E, Robinson L. RRM1 and PTEN as prognostic parameters for overall and disease-free survival in patients with non-small-cell lung cancer. J Clin Oncol. 2004;22:1878–1885. doi: 10.1200/JCO.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. RNA interference targeting the M2 subunit of ribonucleotide reductase enhances pancreatic adenocarcinoma chemosensitivity to gemcitabine. Oncogene. 2004;23:1539–1548. doi: 10.1038/sj.onc.1207272. [DOI] [PubMed] [Google Scholar]
- 5.Zhang K, Hu S, Wu J, Chen L, Lu J, Wang X, Liu X, Zhou B, Yen Y. Overexpression of RRM2 decreases thrombspondin-1 and increases VEGF production in human cancer cells in vitro and in vivo: implication of RRM2 in angiogenesis. Mol Cancer. 2009;8:11. doi: 10.1186/1476-4598-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tian H, Ge C, Li H, Zhao F, Hou H, Chen T, Jiang G, Xie H, Cui Y, Yao M, Li J. Ribonucleotide reductase M2B inhibits cell migration and spreading by early growth response protein 1-mediated phosphatase and tensin homolog/Akt1 pathway in hepatocellular carcinoma. Hepatology. 2014;59:1459–1470. doi: 10.1002/hep.26929. [DOI] [PubMed] [Google Scholar]
- 7.Link PA, Baer MR, James SR, Jones DA, Karpf AR. p53-inducible ribonucleotide reductase (p53R2/RRM2B) is a DNA hypomethylation-independent decitabine gene target that correlates with clinical response in myelodysplastic syndrome/acute myelogenous leukemia. Cancer Res. 2008;68:9358–9366. doi: 10.1158/0008-5472.CAN-08-1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsu NY, Wu JY, Liu X, Yen Y, Chen CY, Chou MC, Lin CH, Lee H, Cheng YW. Expression status of ribonucleotide reductase small subunits hRRM2/p53R2 as prognostic biomarkers in stage I and II non-small cell lung cancer. Anticancer Res. 2011;31:3475–3481. [PubMed] [Google Scholar]
- 9.Bourdon A, Minai L, Serre V, Jais JP, Sarzi E, Aubert S, Chretien D, de Lonlay P, Paquis-Flucklinger V, Arakawa H, Nakamura Y, Munnich A, Rotig A. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat Genet. 2007;39:776–780. doi: 10.1038/ng2040. [DOI] [PubMed] [Google Scholar]
- 10.Kuo ML, Lee MB, Tang M, den Besten W, Hu S, Sweredoski MJ, Hess S, Chou CM, Changou CA, Su M, Jia W, Su L, Yen Y. PYCR1 and PYCR2 interact and collaborate with RRM2B to protect cells from overt oxidative stress. Sci Rep. 2016;6:18846. doi: 10.1038/srep18846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X, Liu X, Xue L, Zhang K, Kuo ML, Hu S, Zhou B, Ann D, Zhang S, Yen Y. Ribonucleotide reductase subunit p53R2 regulates mitochondria homeostasis and function in KB and PC-3 cancer cells. Biochem Biophys Res Commun. 2011;410:102–107. doi: 10.1016/j.bbrc.2011.05.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brenner C, Moulin M. Physiological roles of the permeability transition pore. Circ Res. 2012;111:1237–1247. doi: 10.1161/CIRCRESAHA.112.265942. [DOI] [PubMed] [Google Scholar]
- 13.Suh DH, Kim MK, Kim HS, Chung HH, Song YS. Mitochondrial permeability transition pore as a selective target for anti-cancer therapy. Front Oncol. 2013;3:41. doi: 10.3389/fonc.2013.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elrod JW, Molkentin JD. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ J. 2013;77:1111–1122. doi: 10.1253/circj.cj-13-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fayaz SM, Raj YV, Krishnamurthy RG. CypD: the key to the death door. CNS Neurol Disord Drug Targets. 2015;14:654–663. doi: 10.2174/1871527314666150429113239. [DOI] [PubMed] [Google Scholar]
- 16.Schubert A, Grimm S. Cyclophilin D, a component of the permeability transition-pore, is an apoptosis repressor. Cancer Res. 2004;64:85–93. doi: 10.1158/0008-5472.can-03-0476. [DOI] [PubMed] [Google Scholar]
- 17.Kuo ML, Sy AJ, Xue L, Chi M, Lee MT, Yen T, Chiang MI, Chang L, Chu P, Yen Y. RRM2B suppresses activation of the oxidative stress pathway and is up-regulated by p53 during senescence. Sci Rep. 2012;2:822. doi: 10.1038/srep00822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Powell DR, Desai U, Sparks MJ, Hansen G, Gay J, Schrick J, Shi ZZ, Hicks J, Vogel P. Rapid development of glomerular injury and renal failure in mice lacking p53R2. Pediatr Nephrol. 2005;20:432–440. doi: 10.1007/s00467-004-1696-5. [DOI] [PubMed] [Google Scholar]
- 19.Chang L, Guo R, Huang Q, Yen Y. Chromosomal instability triggered by Rrm2b loss leads to IL-6 secretion and plasmacytic neoplasms. Cell Rep. 2013;3:1389–1397. doi: 10.1016/j.celrep.2013.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hyvonen MT, Uimari A, Keinanen TA, Heikkinen S, Pellinen R, Wahlfors T, Korhonen A, Narvanen A, Wahlfors J, Alhonen L, Janne J. Polyamine-regulated unproductive splicing and translation of spermidine/spermine N1-acetyltransferase. RNA. 2006;12:1569–1582. doi: 10.1261/rna.39806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin TC, Chen YR, Kensicki E, Li AY, Kong M, Li Y, Mohney RP, Shen HM, Stiles B, Mizushima N, Lin LI, Ann DK. Autophagy: resetting glutamine-dependent metabolism and oxygen consumption. Autophagy. 2012;8:1477–1493. doi: 10.4161/auto.21228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lesnefsky EJ, Tandler B, Ye J, Slabe TJ, Turkaly J, Hoppel CL. Myocardial ischemia decreases oxidative phosphorylation through cytochrome oxidase in subsarcolemmal mitochondria. Am J Physiol. 1997;273:H1544–1554. doi: 10.1152/ajpheart.1997.273.3.H1544. [DOI] [PubMed] [Google Scholar]
- 23.Dettmer K, Nurnberger N, Kaspar H, Gruber MA, Almstetter MF, Oefner PJ. Metabolite extraction from adherently growing mammalian cells for metabolomics studies: optimization of harvesting and extraction protocols. Anal Bioanal Chem. 2011;399:1127–1139. doi: 10.1007/s00216-010-4425-x. [DOI] [PubMed] [Google Scholar]
- 24.Goodson SG, Qiu Y, Sutton KA, Xie G, Jia W, O’Brien DA. Metabolic substrates exhibit differential effects on functional parameters of mouse sperm capacitation. Biol Reprod. 2012;87:75. doi: 10.1095/biolreprod.112.102673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith CA, Want EJ, O’Maille G, Abagyan R, Siuzdak G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem. 2006;78:779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 26.Qiu F, Chen YR, Liu X, Chu CY, Shen LJ, Xu J, Gaur S, Forman HJ, Zhang H, Zheng S, Yen Y, Huang J, Kung HJ, Ann DK. Arginine starvation impairs mitochondrial respiratory function in ASS1-deficient breast cancer cells. Sci Signal. 2014;7:ra31. doi: 10.1126/scisignal.2004761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang Q, Li F, Liu X, Li W, Shi W, Liu FF, O’Sullivan B, He Z, Peng Y, Tan AC, Zhou L, Shen J, Han G, Wang XJ, Thorburn J, Thorburn A, Jimeno A, Raben D, Bedford JS, Li CY. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat Med. 2011;17:860–866. doi: 10.1038/nm.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smeds J, Miller LD, Bjohle J, Hall P, Klaar S, Liu ET, Pawitan Y, Ploner A, Bergh J. Gene profile and response to treatment. Ann Oncol. 2005;16(Suppl 2):ii195–202. doi: 10.1093/annonc/mdi737. [DOI] [PubMed] [Google Scholar]
- 30.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bernardi P, Di Lisa F. The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J Mol Cell Cardiol. 2015;78:100–106. doi: 10.1016/j.yjmcc.2014.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu X, Lai L, Wang X, Xue L, Leora S, Wu J, Hu S, Zhang K, Kuo ML, Zhou L, Zhang H, Wang Y, Wang Y, Zhou B, Nelson RA, Zheng S, Zhang S, Chu P, Yen Y. Ribonucleotide reductase small subunit M2B prognoses better survival in colorectal cancer. Cancer Res. 2011;71:3202–3213. doi: 10.1158/0008-5472.CAN-11-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen J, Li S, Xiao Y, Zou X, Zhang X, Zhu M, Cai M, Xie D. p53R2 as a novel prognostic biomarker in nasopharyngeal carcinoma. BMC Cancer. 2017;17:846. doi: 10.1186/s12885-017-3858-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar P, Mark PJ, Ward BK, Minchin RF, Ratajczak T. Estradiol-regulated expression of the immunophilins cyclophilin 40 and FKBP52 in MCF-7 breast cancer cells. Biochem Biophys Res Commun. 2001;284:219–225. doi: 10.1006/bbrc.2001.4952. [DOI] [PubMed] [Google Scholar]
- 35.Ryo A, Liou YC, Wulf G, Nakamura M, Lee SW, Lu KP. PIN1 is an E2F target gene essential for Neu/Ras-induced transformation of mammary epithelial cells. Mol Cell Biol. 2002;22:5281–5295. doi: 10.1128/MCB.22.15.5281-5295.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brenner C, Grimm S. The permeability transition pore complex in cancer cell death. Oncogene. 2006;25:4744–4756. doi: 10.1038/sj.onc.1209609. [DOI] [PubMed] [Google Scholar]
- 37.Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. 2010;9:447–464. doi: 10.1038/nrd3137. [DOI] [PubMed] [Google Scholar]
- 38.Shinohara Y, Ishida T, Hino M, Yamazaki N, Baba Y, Terada H. Characterization of porin isoforms expressed in tumor cells. Eur J Biochem. 2000;267:6067–6073. doi: 10.1046/j.1432-1327.2000.01687.x. [DOI] [PubMed] [Google Scholar]
- 39.Bonora M, Pinton P. The mitochondrial permeability transition pore and cancer: molecular mechanisms involved in cell death. Front Oncol. 2014;4:302. doi: 10.3389/fonc.2014.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. 2011;11:389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Assaly R, de Tassigny A, Paradis S, Jacquin S, Berdeaux A, Morin D. Oxidative stress, mitochondrial permeability transition pore opening and cell death during hypoxia-reoxygenation in adult cardiomyocytes. Eur J Pharmacol. 2012;675:6–14. doi: 10.1016/j.ejphar.2011.11.036. [DOI] [PubMed] [Google Scholar]
- 42.Javadov S, Karmazyn M, Escobales N. Mitochondrial permeability transition pore opening as a promising therapeutic target in cardiac diseases. J Pharmacol Exp Ther. 2009;330:670–678. doi: 10.1124/jpet.109.153213. [DOI] [PubMed] [Google Scholar]
- 43.Cao XH, Zhao SS, Liu DY, Wang Z, Niu LL, Hou LH, Wang CL. ROS-Ca(2+) is associated with mitochondria permeability transition pore involved in surfactin-induced MCF-7 cells apoptosis. Chem Biol Interact. 2011;190:16–27. doi: 10.1016/j.cbi.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 44.Sava IG, Battaglia V, Rossi CA, Salvi M, Toninello A. Free radical scavenging action of the natural polyamine spermine in rat liver mitochondria. Free Radic Biol Med. 2006;41:1272–1281. doi: 10.1016/j.freeradbiomed.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 45.Casero RA Jr, Marton LJ. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat Rev Drug Discov. 2007;6:373–390. doi: 10.1038/nrd2243. [DOI] [PubMed] [Google Scholar]
- 46.Ha HC, Sirisoma NS, Kuppusamy P, Zweier JL, Woster PM, Casero RA Jr. The natural polyamine spermine functions directly as a free radical scavenger. Proc Natl Acad Sci U S A. 1998;95:11140–11145. doi: 10.1073/pnas.95.19.11140. [DOI] [PMC free article] [PubMed] [Google Scholar]