

Abstract
Aldosterone plays an important role in the pathogenesis of chronic kidney disease (CKD) by directly damaging renal tubular cells. However, the treatment against aldosterone-induced renal injury is still limited. The present study was performed to examine the protection role of MnTBAP in modulating aldosterone-induced renal tubular injury both in vitro and vivo. MtD is induced by aldosterone in HK-2 cells, as evidenced by decreased expression of mtDNA and reduced mitochondrial membrane potential (MMP), which was markedly ameliorated by treatment with MnTBAP. HK-2 cells treated with MnTBAP demonstrated a reduction in cell apoptosis and improvements in cell phenotypic alterations following aldosterone challenge. Treatment with MnTBAP also inhibited the activation of the NLRP3 inflammasome and subsequent release of pro-inflammatory cytokines, IL-1β and IL-18. In addition, MnTBAP treatment of aldosterone-infused mice significantly improved mitochondrial morphology and function, suppressed the activation of NLRP3 inflammasome, reduced renal tubular cell apoptosis, decreased phenotypic alterations, and ameliorated renal apoptosis. We conclude that MnTBAP treatment ameliorates aldosterone-induced renal injury through regulating MtD and NLRP3 inflammsome signalling axis.
Keywords: Aldosterone, NLRP3 inflammasome, mitochondrial dysfunction, MnTBAP
Introduction
Aldosterone (aldo) is a vital regulator of blood pressure and electrolytic balance through interaction with its receptor, mineralocorticoid receptors (MR), which are expressed ubiquitously in renal tubular cells [1]. Aldosterone increases as the glomerular filtration rate falls, and high aldosterone levels were shown to be adversely associated with sudden cardiac death and all-cause mortality in end-stage renal disease patients [2]. A growing number of studies demonstrated that Aldo plays an important role in the progression of chronic kidney disease (CKD). Exogenous infusion of Aldo elicits glomerular injury and tubulointerstitial fibrosis in the remnant kidney animal model; this was prevented by MR antagonists [3,4]. Aside from its classical action through MR, aldosterone is also associated with inflammation, apoptosis, oxidative stress, and thrombosis [5]. Therefore, it plays an important role in the development of renal injury. It is regarded important and urgent to explore the exact mechanism and novel strategies to prevent the injury.
Mitochondria play an essential role in cellular homeostasis, metabolism, and energy production. Impairment of mitochondrial function, also called mitochondrial dysfunction (MtD), is characterized with reduced ATP production, increased reactive oxygen species (ROS) generation, and release of proapoptotic products, such as mitochondrial DNA (mtDNA) and cytochrome c [6]. Some studies implicate MtD as an important factor in the development of renal fibrosis [7,8]. Furthermore, aldo induces apoptosis and phenotypic alteration of renal tubular cells, which may be associated with mitochondria [9,10]. Mitochondria may be a potential target in protection against Aldo-induced renal tubular injury.
Inflammation is an essential response to tissue damage and to restore homeostasis following a pathogenic stimulus, but uncontrolled or persistent inflammation is often deleterious and causes extensive tissue damage. The Nod-like receptor pyrin domain containing 3 (NLRP3) inflammasome, one of the most well-studied inflammasomes, plays a key role in the activation of sterile inflammation [11]. Following its activation, NLRP3 associates with the adaptor protein apoptosis speck-like protein containing a caspase recruit domain (ASC), which then recruits the effector pro-caspase-1, resulting in the activation of caspase-1 and subsequent cleavage of pro-IL-1β and pro-IL-18 into their mature and active forms [12]. Recently a number of studies have reported that NLRP3 inflammasome participates in the progression of CKD [13,14]. Our previous work has also demonstrated the close relationship between mitochondrial ROS and NLRP3 inflammasome in aldo-induced renal tubular injury [15,16]. Therefore, the role of NLRP3 inflammasome in renal fibrosis warrants further investigation.
In this study, by employing a pharmacological strategy, we investigated the exact role and mechanism of MnTBAP, a synthetic SOD mimic, in modulating aldosterone-induced renal injury both in vitro and vivo. Different from recombinant SOD, which cannot cross biological membranes, MnTBAP is non-immunogenic and crosses the plasma membrane to neutralize superoxide in both extracellular and intracellular compartments. It has a wider clinical application due to its relatively long half-life of scavenging activity.
Materials and methods
Reagents and antibodies
Aldosterone and MnTBAP were purchased from Sigma (St. Louis, MO, USA). Antibodies of anti-E-cadherin, anti-vimentin, and anti-α-SMA were purchased from Abcam (Cambridge, MA, USA). Anti-Nlrp3 antibody and anti-ASC antibody were purchased from adipoGen (San Diego, CA, USA). Anti-IL-18 was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA); Anti- IL-1β antibody was purchased from R&D systems (Minneapolis, MN, USA).
Cell culture and treatments
HK-2 cells were maintained in DMEM/F12 medium, supplemented with 10% fetal bovine serum at 37°C and 5% CO2 in a humidified incubator. HK-2 cells were pretreated with or without MnTBAP (100 µM) for 30 min, followed by incubation with aldosterone (100 nM) for 24 h according to different experiments.
Measurement of mitochondrial membrane potential (Δψm)
The mitochondrial membrane potential in HK-2 cells and isolated mitochondria was measured using the lipophilic cationic probe 5,5’,6,6’-tetrachloro-1,1’,3,3’-tetraethyl-benzimidazol carbocyanine iodide (JC-1, Molecular Probe, Eugene, OR, USA). Following treatment, cells were incubated with JC-1 (300 nM) for 30 min at 37°C and then washed and analysed by flow cytometry according to manufacturer’s instructions.
Real-time reverse transcription polymerase chain reaction (RT-PCR)
Total RNA was isolated from cells or kidney tissue and reverse transcribed to cDNA. RT-PCR analysis was used to quantify mtDNA copy number. RT-PCR was performed according to a previously described protocol [17] using the cDNA as a template and the primer sequences used for PCR amplification are summarized in Table 1.
Table 1.
RT-PCR primer sequences
| Gene | Forward primer sequence (5’-3’) | Reverse primer sequence (5’-3’) |
|---|---|---|
| mtDNA | TTTTATCTGCATCTGAGTTTAATCCTGT | CCACTTCATCTTACCATTTATTATCGC |
| ATP | TCCATCAAAAACATCCAGAAAA | GAGGAGTGAATAGCACCACAAA |
| 18S | TTCGGAACTGAGGCCATGATT | TTTCGCTCTGGTCCGTCTTG |
Western blotting
Western blotting of cells and renal tissue was performed as previously described [18]. Briefly, cells or renal tissue were lysed in protein lysis buffer on ice and protein concentration was quantified. Following blotting, the membrane was incubated overnight with primary antibodies against vimentin, E-cadherin, α-SMA, Nlrp3, ASC, IL-1β, IL-18, or β-actin (all diluted by Tris-buffered saline-Tween 20 [TBST] containing 5% BSA), followed by HRP-labelled secondary antibodies for 2 h. The blots were visualized with an enhanced chemiluminescent system (Amersham, Little Chalfont, Bucks, UK) and band intensity quantified using Quantity One (Bio-Rad, Hercules, CA, USA).
Animal studies
All animal experiments were performed with the approval of the Animal Care Committee at Jiao Tong University. C57BL/6J (wild-type, WT) mice were purchased from Shanghai SLAC Laboratory Animals (Shanghai, China). As described previously [11], all mice underwent left nephrectomy and subsequently given high-salt drinking water following surgery for about 10 days. All mice were then treated randomly with one of the following for four weeks: group 1, vehicle control (0.5% ethanol subcutaneously); group 2, aldosterone (0.75 µg/h subcutaneously); group 3, aldosterone + MnTBAP (0.75 µg/h subcutaneously + 10 mg/kg by intraperitoneal injection, three times per week). All mice were sacrificed at week 4, and kidney samples were collected and immediately frozen in liquid nitrogen for storage at -80°C.
Terminal deoxyribonucleotide transferase (TdT)-mediated dUTP nick-end labelling (TUNEL) assay and annexin V/Propidium iodide staining
Apoptosis in HK-2 cells or kidney tissue was detected using the TUNEL assay (Roche Molecular Biochemicals) according to the manufacturer’s protocol and our previous report [16]. Briefly, slides of cells or deparaffinized kidney tissue section were treated in a permeabilization solution (0.1% Triton X-100 in 0.1% sodium citrate) at 4°C for 2 min, washed twice with PBS, and then incubated with 50 μl TUNEL reaction mixture for 60 min at 37°C in the dark. For quantification, 20 fields were randomly selected in each slide or section, and the number of TUNEL-positive cells was counted in a standard area (1 mm2). Annexin V/Propidium iodide staining method (BD Biosciences, San Diego, CA, USA) was performed as previously described [11].
Electron microscopy assay
To characterize the ultrastructural morphology of mitochondria, kidney tissue was fixed with 2.5% glutaraldehyde at room temperature and cut into 1 mm3 pieces using a scalpel. Following a method described in our previous report [19], we prepared ultrathin sections (60 nm) using a microtome. The sections were then placed on copper grids and stained with uranyl acetate and lead citrate for analysis by electron microscopy.
PAS staining
Harvested kidney tissues from mice were fixed in 4% paraformaldehyde, embedded in paraffin, and cut into 4 µm thick sections. The sections were processed and stained with Periodic acid-Schiff reagent according to our previous report [20]. Following staining, we assessed glomerular injury in experimental mice using light microscopy based on the following semi-quantitative grades: grade 0, normal; grade 1, segmental lesion < 25%; grade 2, 25%-50%; grade, 3, 50%-75%; grade 4, 75%-100%. At least 20 glomeruli were analysed in each group.
Statistical analysis
Data are expressed as the mean ± standard error of the mean (SEM) and were analysed using one-way analysis of variance (ANOVA) followed by Bonferroni’s comparison test. Results with P < 0.05 were considered statistically significant.
Results
MnTBAP ameliorates aldo-induced MtD in HK-2 cells
Treatment with MnTBAP successfully reversed the aldo-induced decrease in MMP according to JC-1 aggregates and JC-1 monomeric ratio (Figure 1A and 1B). Furthermore, MnTBAP also increased expression of mtDNA in HK-2 cells compared with the aldo-treated group (Figure 1C).
Figure 1.
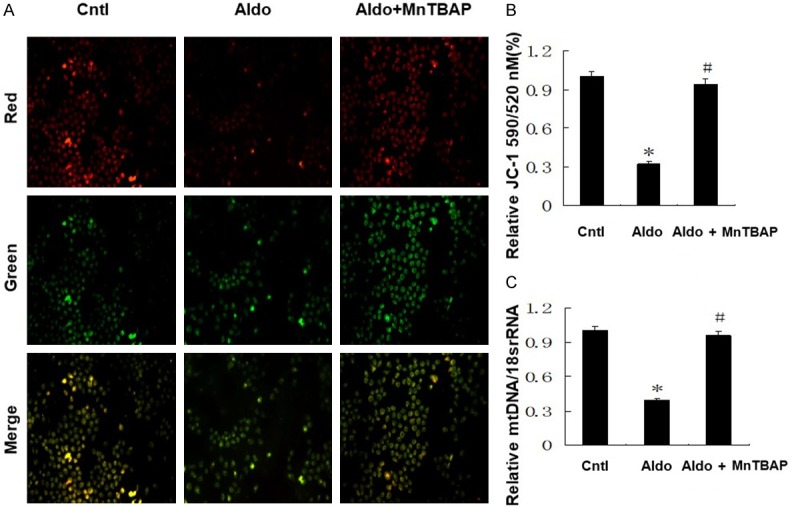
Effects of MnTBAP on Aldo induced MtD. Cells were pretreated with MnTBAP (100 µM) for 30 min, followed by incubation with Aldo (100 nM) for 24 h. A: Representative images of JC-1 staining (×400). B: Quantitation of JC-1 fluorescence by flow cytometry. C: mtDNA copy number following MnTBAP treatment. Data represented as the mean ± SEM (n = 6). *P < 0.01 vs Control; #P < 0.01 versus Aldo-induced group.
MnTBAP attenuates aldo-induced apoptosis and phenotypic alteration in HK-2 cells
To further examine the role of MnTBAP treatment on cell apoptosis, we quantified apoptosis by flow cytometry to measure annexin-V/propidium iodide, and TUNEL staining assays. The apoptosis of HK-2 cells was higher in the aldo-treated group compared with the control group. This was markedly reduced by MnTBAP treatment (Figure 2A and 2B). We also determined the effect of MnTBAP treatment on phenotypic alteration in HK-2 cells induced by aldosterone. We quantified the levels of vimentin and α-SMA by western blots. Vimentin and α-SMA protein expression were significantly increased in the Aldo-treated group compared with the control group. Furthermore, MnTBAP treatment attenuated the induction of vimentin and α-SMA protein expression (Figure 2C and 2D). These results indicated that aldo-treatment induces renal tubular cell injury and that this is reversed by MnTBAP treatment.
Figure 2.
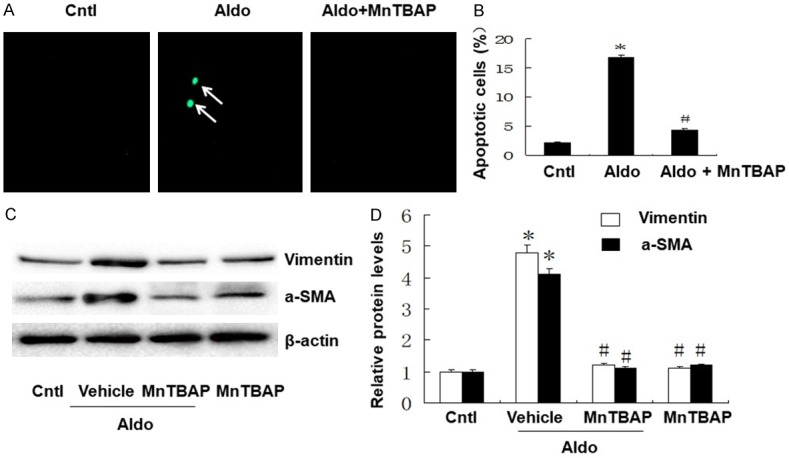
Effects of MnTBAP on Aldo induced HK-2 apoptosis and cell phenotypic alteration. A: TUNEL staining (×400). The arrows indicate TUNEL-positive apoptotic cells. B: HK-2 cell apoptosis following MnTBAP was detected by flow cytometry. Cells were pretreated with MnTBAP (100 µM) for 30 min, followed by incubation with Aldo (100 nM) for 24 h. C: Western blot of vimentin and α-SMA were performed. D: Semi-quantitative analysis of vimentin and α-SMA normalized against β-actin. Data represented as the mean ± SEM (n = 6). *P < 0.01 vs Control; #P < 0.01 versus Aldo-induced group.
MnTBAP attenuates aldo-induced activation of NLRP3 inflammasome in HK-2 cells
Activation of NLRP3 inflammasome induced the cleavage of pro-caspase-1 and the subsequent release of the pro-inflammatory cytokines IL-1β and IL-18. We evaluated the protein expression of NLRP3 and mature IL-1β and IL-18 by western blots. As shown in Figure 3, NLRP3 inflammasome was activated following aldo-treatment, with increased levels of NLRP3, IL-1β and IL-18 proteins. Treatment with MnTBAP significantly decreased aldo-dependent activation of NLRP3 and induction of mature IL-1β and IL-18 protein levels.
Figure 3.
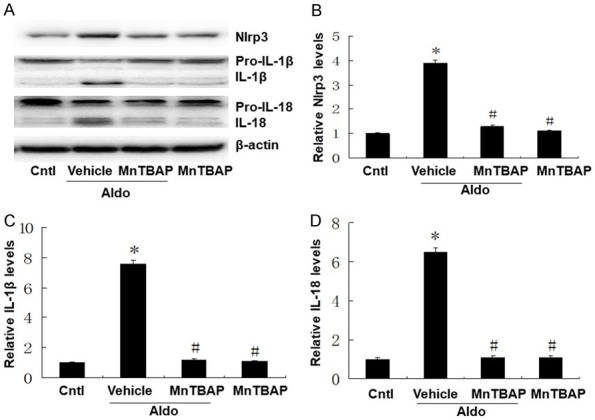
Effects of MnTBAP on Aldo induced NLRP3 inflammasome activation in HK-2 cells. Cells were pretreated with MnTBAP (100 µM) for 30 min, followed by incubation with Aldo (100 nM) for 48 h. (A) Expressions of Nlrp3, pro-IL-1β, IL-1β, pro-IL-18, and IL-18 in HK-2 cells were quantified. Semi-quantitative analysis of Nlrp3 (B), IL-1β (C), and IL-18 (D) normalized against β-actin. Data represented as the mean ± SEM (n = 6). *P < 0.01 vs Control; #P < 0.01 versus Aldo-induced group.
MnTBAP treatment in mice ameliorates aldo-induced renal injury
To characterize the effects of MnTBAP treatment on the pathophysiological phenotypes of aldo-induced injury, we measured apoptosis, glomerular injury, and phenotypic alteration in kidney tissue. As shown in Figure 4, PAS staining and glomerular injury score demonstrated that MnTBAP attenuated glomerular sclerosis induced by aldo-infusion (Figure 4A). Furthermore, TUNEL staining revealed higher apoptosis in the aldo-treated group compared with the control group. This apoptosis was inhibited by MnTBAP treatment (Figure 4B). As shown in Figure 5, we analysed E-cadherin and α-SMA protein expression by western blot to evaluate the phenotypic alterations in kidney tissue. E-cadherin protein expression was significantly decreased and α-SMA protein expression was significantly increased in the aldo-treated group compared to control. However, treatment with MnTBAP attenuated the loss of E-cadherin and induction of α-SMA protein expression (Figure 5B and 5C).
Figure 4.

Effects of MnTBAP on Aldo infused renal injury on day 28. A: Representative photomicrographs of Periodic acid-Schiff-stained kidney sections (magnification, ×400); B: Representative images of TUNEL staining are shown (magnification, ×400). Bar graph indicates glomerular injury score and the mean number of TUNEL-positive tubular cells per field. Data represented as the mean ± SEM (n = 6). *P < 0.01 vs Control; #P < 0.01 versus Aldo-induced group.
Figure 5.
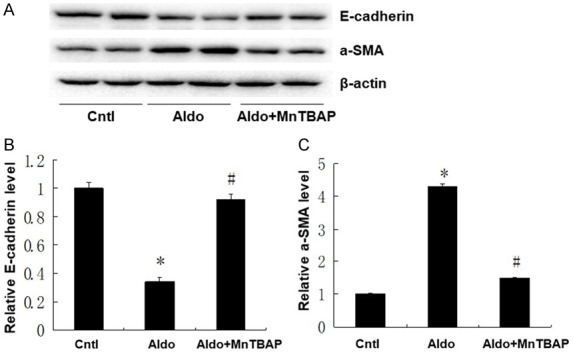
Effects of MnTBAP on Aldo infused cell phenotypic alteration. (A) Expression of α-SMA and E-cadherin protein detected in kidney samples from different groups; Semi-quantitative analysis of E-cadherin (B), and α-SMA (C) normalized against β-actin. Data represented as the mean ± SEM (n = 6). *P < 0.01 vs Control; #P < 0.01 versus Aldo-induced group.
MnTBAP attenuates aldo-induced MtD
We evaluated the effect of MnTBAP treatment on MtD in mice. As shown in Figure 6, observation of the ultrastructural morphology in aldo-treated mice revealed swollen mitochondria with disorganized and fragmented cristae. These effects were accompanied by markedly decreased mtDNA and ATP synthase, which were all significantly reversed by MnTBAP treatment.
Figure 6.
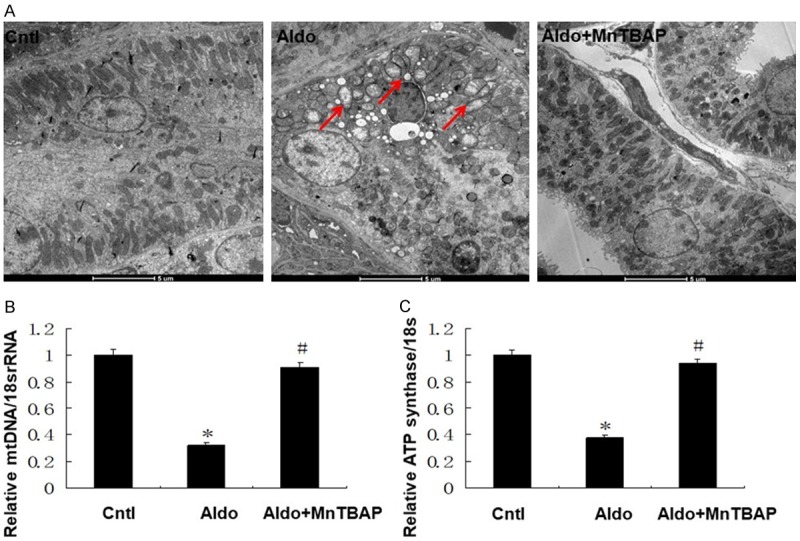
Effects of MnTBAP on Aldo infused mitochondrial dysfunction. Representative electron microscopy photomicrographs of ultrastructural morphology of mitochondria (A), (magnification ×2100). Arrow indicates swollen mitochondria. Semi-quantitative analysis of mtDNA (B) and ATP synthase mRNA (C) expression normalized against 18S performed by real-time PCR. Data represented as the mean ± SEM (n = 6). *P < 0.01 vs Control; #P < 0.01 versus Aldo-induced group.
MnTBAP ameliorates aldo-induced expression of inflammatory cytokines
Following 28 days of aldo-infusion and MnTBAP treatment, we analyzed the expression of NLRP3, ASC, and the inflammasome-regulated inflammatory cytokines. As shown in Figure 7, expression of NLRP3, ASC, IL-1β and IL-18 were significantly increased after consistent aldo-infusion. Treatment with MnTBAP significantly inhibited activation of NLRP3 inflammasome (Figure 7B) and decreased the release of pro-inflammatory cytokines, IL-1β and IL-18 (Figure 7C).
Figure 7.
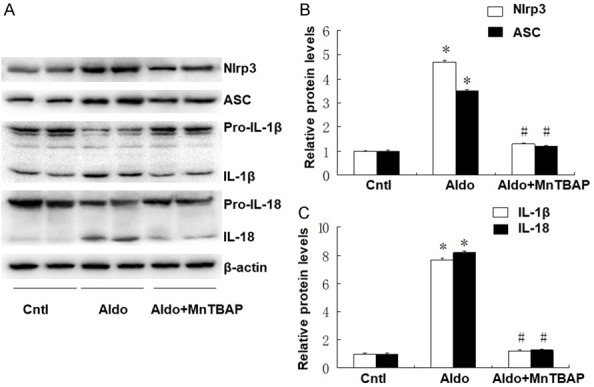
Effects of MnTBAP on Aldo infused NLRP3 inflammasome. (A) Expressions of Nlrp3, ASC, IL-1β, and IL-18 were quantified in kidney samples from different groups; Semi-quantitative analysis of Nlrp3 and ASC (B), IL-1β and IL-18 (C) normalized against β-actin. Data represented as the mean ± SEM (n = 6). *P < 0.01 vs Control; #P < 0.01 versus Aldo-induced group.
Discussion
Aldosterone is essential in the regulation of electrolytes, fluid volume, and the maintenance of blood pressure homeostasis. In recent years, aldosterone has been thought to exert its action through two different mechanisms, one of which is mediated by a genomic process, predominantly through MR, and the other is carried out by non-genomic process, involving both MR-dependent and MR-independent pathways [21]. Although many studies have reported that Aldo is closely related to renal injury, the therapies of Aldo-induced renal injury are limited. In the present study, we demonstrate that MnTBAP reverses Aldo-induced renal tubular injury through attenuation of MtD and NLRP3 inflammasome activation. Our data suggest that MnTBAP is a potential therapy in protection from Aldo-induced renal injury.
In this study, we characterized the role of MnTBAP on Aldo-induced renal tubular injury and its potential mechanism both in vivo and in vitro. We demonstrate that Aldo infusion decreased mtDNA copies and MMP, which was ameliorated by MnTBAP. Aldo also led to the activation of the NLRP3 inflammasome, presented by increased expression of NLRP3, IL-1β, and IL-18 protein as well as cell injury including cell apoptosis and phenotypic alterations. These effects were successfully attenuated by MnTBAP. Since MnTBAP is a type of SOD mimic, which acts as a mitochondrial ROS scavenger, it works towards reversing MtD. Mitochondria are engaged in several metabolic pathways, calcium and iron homeostasis, ROS production, and programmed cell death [22]. Impaired mitochondrial function is closely correlated with cell apoptosis and tissue damage. Mitochondrial dysfunction presents as reduced ATP production, increased accumulation of reactive oxygen species, and release of pro-apoptotic products such as mtDNA and cytochrome c [23]. Recent studies have demonstrated that MtD may be crucial in the pathogenesis of chronic kidney disease [24]. Attenuation of MtD decreased renal tubular cell apoptosis and ameliorated tubulointerstitial fibrosis in 5/6 nephrectomized animals [25,26]. Furthermore, recent studies demonstrated that Aldo exacerbated MtD and promotes epithelial-to-mesenchymal transition of renal tubular cells through several pathways. Aldo treatment decreases the expression of peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α), which maintains normal mitochondrial function [27]. MnTBAP, a synthetic SOD mimic, has been reported to restore SOD activity and improve mitochondrial function [28]. The superoxide dismutase (SOD) family is the main antioxidant system and is very important in oxidative stress modulation. Decreased SOD along with increased oxidative stress in kidney tissue, which usually leads to impaired mitochondrial function, has been considered important in the pathology of renal fibrosis. In this study, we demonstrate attenuation of MtD by MnTBAP significantly ameliorates renal cell apoptosis and phenotypic alteration both in vivo and vitro. A previous study reported that MnTBAP ameliorates MtD and renal fibrosis in mice with 5/6 nephrectomy, which is consistent with our study, although the concentration of MnTBAP differed [26]. Our study suggests that MnTBAP, which restores mitochondrial dysfunction, helps reverse Aldo-induced renal tubular injury.
Inflammation is part of the normal response to tissue damage with the goal of restoring homeostasis. Activation of the inflammasome and subsequent release of pro-inflammation cytokines mediate various disease processes [29]. Aside from its pro-inflammatory effect, NLRP3 also interacts with TGF-β, which has a central role in renal fibrosis [30]. It has been reported that NLRP3 expression and inflammasome-regulated cytokines are elevated in patients with chronic kidney disease as well as in unilateral ureteral obstruction or 5/6 nephrectomy [13]. Our previous report also describes significant elevation of NLRP3 and ASC protein levels and markedly increased mature IL-1β and IL-18 levels following aldosterone administration [19]. Deletion of the NLRP3 inflammasome ameliorates Aldo-induced phenotypic alteration, renal tubular cell apoptosis, and impaired renal function. In many reports, mitochondrial events seem to act upstream of NLRP3 inflammasome activation. Zhou and co-workers demonstrated a correlation between mitochondrial ROS (mtROS) activity and NLRP3 inflammasome activation. Knockdown of voltage-dependent anion channels (VDAC), which are crucial for the generation of mtROS, markedly impair NLRP3 inflammasome activation [31]. Wen and colleagues described that scavenging of overproduced mtROS by mito-TEMPO inhibitd NLRP3 inflammasome activation in angiotensin II-treated renal tubular cell [32]. Our previous study also linked mtROS with the NLRP3 inflammasome activation in Aldo-induced renal tubular injury [11]. In addition, mitochondria work as a platform for inflammasome assembly, which could be facilitated by impaired mitochondrial function [33]. These data suggest a close relationship between MtD and NLRP3 inflammasome activation. In this study, we report the aldosterone decreased the mitochondrial membrane potential, mtDNA expression, and ATP production in renal tubular cells. Application of MnTBAP attenuated MtD and inhibited NLRP3 inflammasome activation as well as the release of mature IL-1β and IL-18. Considering the relationship between MtD and NLRP3 inflammasome, it may suggest that MnTBAP treatment ameliorates aldosterone-induced renal tubular injury and subsequent tubulointerstitial fibrosis possibly through reduced MtD-mediated activation of NLRP3 inflammasome.
In summary, our present study demonstrates that MtD, the NLRP3 inflammasome activation, and subsequent pro-inflammatory cytokine release are involved in aldosterone induced renal tubular injury and tubulointerstitial fibrosis. MnTBAP reverses MtD, inhibits the NLRP3 inflammasome activation and significantly ameliorates renal tubular injury. These data support that MnTBAP treatment ameliorates Aldo-induced renal tubular injury through regulating MtD and NLRP3 signalling axis. Our findings of MnTBAP show its important role in prevention of renal injury.
Acknowledgements
This work was sponsored by Shanghai Pujiang Program (17PJD021), the interdisciplinary program of Shanghai Jiao Tong University (YG2017MS07) and the distinguished Young Scholar of Ninth people’s hospital (jyyq09201701).
Disclosure of conflict of interest
None.
References
- 1.Kiyomoto H, Rafiq K, Mostofa M, Nishiyama A. Possible underlying mechanisms responsible for aldosterone and mineralocorticoid receptor-dependent renal injury. J Pharmacol Sci. 2008;108:399–405. doi: 10.1254/jphs.08r02cr. [DOI] [PubMed] [Google Scholar]
- 2.Volk MJ, Bomback AS, Klemmer PJ. Mineralocorticoid receptor blockade in chronic kidney disease. Curr Hypertens Rep. 2011;13:282–8. doi: 10.1007/s11906-011-0202-2. [DOI] [PubMed] [Google Scholar]
- 3.Sun GP, Kohno M, Guo P, Nagai Y, Miyata K, Fan YY, Kimura S, Kiyomoto H, Ohmori K, Li DT, Abe Y, Nishiyama A. Involvements of rhokinase and TGF-beta pathways in aldosteroneinduced renal injury. J Am Soc Nephrol. 2006;17:2193–201. doi: 10.1681/ASN.2005121375. [DOI] [PubMed] [Google Scholar]
- 4.Nishiyama A, Hitomi H, Rahman A, Kiyomoto H. Drug discovery for overcoming chronic kidney disease (CKD): pharmacological effects of mineralocorticoid-receptor blockers. J Pharmacol Sci. 2009;109:1–6. doi: 10.1254/jphs.08r12fm. [DOI] [PubMed] [Google Scholar]
- 5.Donderski R, Stróżecki P, Sulikowska B, Grajewska M, Miśkowiec I, Stefańska A, Siódmiak J, Odrowąż-Sypniewska G, Manitius J. Aldosterone antagonist therapy andits relationship with inflammation, fibrosis, thrombosis, mineralbone disorder and cardiovascular complications in peritoneal dialysis (PD) patients. Int Urol Nephrol. 2017;49:1867–1873. doi: 10.1007/s11255-017-1655-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N, Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88:529–35. doi: 10.1161/01.res.88.5.529. [DOI] [PubMed] [Google Scholar]
- 7.Tamaki M, Miyashita K, Wakino S, Mitsuishi M, Hayashi K, Itoh H. Chronic kidney disease reduces muscle mitochondria and exercise endurance and its exacerbation by dietary protein through inactivation of pyruvate dehydrogenase. Kidney Int. 2014;85:1330–9. doi: 10.1038/ki.2013.473. [DOI] [PubMed] [Google Scholar]
- 8.Gamboa JL, Billings FT 4th, Bojanowski MT, Gilliam LA, Yu C, Roshanravan B, Roberts LJ 2nd, Himmelfarb J, Ikizler TA, Brown NJ. Mitochondrial dysfunction and oxidative stress in patients with chronic kidney disease. Physiol Rep. 2016;4 doi: 10.14814/phy2.12780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding W, Yang L, Zhang M, Gu Y. Reactive oxygen species-mediated endoplasmic reticulum stress contributes to aldosterone-induced apoptosis in tubular epithelial cells. Biochem Biophys Res Commun. 2012;418:451–6. doi: 10.1016/j.bbrc.2012.01.037. [DOI] [PubMed] [Google Scholar]
- 10.Yuan Y, Chen Y, Zhang P, Huang S, Zhu C, Ding G, Liu B, Yang T, Zhang A. Mitochondrial dysfunction accounts for aldosterone-induced epithelial-to-mesenchymal transition of renal proximal tubular epithelial cells. Free Radic Biol Med. 2012;53:30–43. doi: 10.1016/j.freeradbiomed.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 11.Artlett CM, Thacker JD. Molecular activation of the NLRP3 inflammasome in fibrosis: common threads linking divergent fibrogenic diseases. Antioxid Redox Signal. 2015;22:1162–75. doi: 10.1089/ars.2014.6148. [DOI] [PubMed] [Google Scholar]
- 12.He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41:1012–1021. doi: 10.1016/j.tibs.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vilaysane A, Chun J, Seamone ME, Wang W, Chin R, Hirota S, Li Y, Clark SA, Tschopp J, Trpkov K, Hemmelgarn BR, Beck PL, Muruve DA. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J Am Soc Nephrol. 2010;21:1732–44. doi: 10.1681/ASN.2010020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pulskens WP, Butter LM, Teske GJ, Claessen N, Dessing MC, Flavell RA, Sutterwala FS, Florquin S, Leemans JC. Nlrp3 prevents early renal interstitial edema and vascular permeability in unilateral ureteral obstruction. PLoS One. 2014;9:e85775. doi: 10.1371/journal.pone.0085775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding W, Guo H, Xu C, Wang B, Zhang M, Ding F. Mitochondrial reactive oxygen species-mediated NLRP3 inflammasome activation contributes to aldosterone-induced renal tubular cells injury. Oncotarget. 2016;7:17479–91. doi: 10.18632/oncotarget.8243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo H, Bi X, Zhou P, Zhu S, Ding W. NLRP3 deficiency attenuates renal fibrosis and ameliorates mitochondrial dysfunction in a mouse unilateral ureteral obstruction model of chronic kidney disease. Mediators Inflamm. 2017;2017:8316560. doi: 10.1155/2017/8316560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding W, Wang B, Zhang M, Gu Y. Tempol, a superoxide dismutase-mimetic drug, ameliorates progression of renal disease in CKD mice. Cell Physiol Biochem. 2015;36:2170–82. doi: 10.1159/000430183. [DOI] [PubMed] [Google Scholar]
- 18.Xu C, Ding W, Yang L, Yang M, Zhang M, Gu Y. Contributions of endoplasmic reticulum stress and reactive oxygen species to renal injury in aldosterone/salt induced rats. Nephron Exp Nephrol. 2014;126:25–32. doi: 10.1159/000357777. [DOI] [PubMed] [Google Scholar]
- 19.Ding W, Xu C, Wang B, Zhang M. Rotenone attenuates renal injury in aldosterone-infused rats by inhibiting oxidative stress, mitochondrial dysfunction, and inflammasome activation. Med Sci Monit. 2015;21:3136–43. doi: 10.12659/MSM.895945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding W, Wang B, Zhang M, Gu Y. Involvement of endoplasmic reticulum stress in uremic cardiomyopathy: protective effects of tauroursodeoxycholic acid. Cell Physiol Biochem. 2016;38:141–52. doi: 10.1159/000438616. [DOI] [PubMed] [Google Scholar]
- 21.Kritis AA, Gouta CP, Liaretidou EI, Kallaras KI. Latest aspects of aldosterone action on the heart muscle. J Physiol Pharmacol. 2016;67:21–30. [PubMed] [Google Scholar]
- 22.Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112:481–90. doi: 10.1016/s0092-8674(03)00116-8. [DOI] [PubMed] [Google Scholar]
- 23.Chistiakov DA, Sobenin IA, Revin VV, Orekhov AN, Bobryshev YV. Mitochondrial aging and age-related dysfunction of mitochondria. Biomed Res Int. 2014;2014:238463. doi: 10.1155/2014/238463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Che R, Yuan Y, Huang S, Zhang A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am J Physiol Renal Physiol. 2014;306:367–78. doi: 10.1152/ajprenal.00571.2013. [DOI] [PubMed] [Google Scholar]
- 25.Chen JF, Liu H, Ni HF, Lv LL, Zhang MH, Zhang AH, Tang RN, Chen PS, Liu BC. Improved mitochondrial function underlies the protective effect of pirfenidone against tubulointerstitial fibrosis in 5/6 nephrectomized rats. PLoS One. 2013;8:e83593. doi: 10.1371/journal.pone.0083593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu J, Mao S, Zhang Y, Gong W, Jia Z, Huang S, Zhang A. MnTBAP therapy attenuates renal fibrosis in mice with 5/6 nephrectomy. Oxid Med Cell Longev. 2016;2016:7496930. doi: 10.1155/2016/7496930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao M, Yuan Y, Bai M, Ding G, Jia Z, Huang S, Zhang A. PGC-1alpha overexpression protects against aldosterone-induced podocyte depletion: role of mitochondria. Oncotarget. 2016;7:12150–62. doi: 10.18632/oncotarget.7859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Archer SL. Acquired mitochondrial abnormalities, including epigenetic inhibition of superoxide dismutase 2, in pulmonary hypertension and cancer: therapeutic implications. Adv Exp Med Biol. 2016;903:29–53. doi: 10.1007/978-1-4899-7678-9_3. [DOI] [PubMed] [Google Scholar]
- 29.Anders HJ, Muruve DA. The inflammasomes in kidney disease. J Am Soc Nephrol. 2011;22:1007–18. doi: 10.1681/ASN.2010080798. [DOI] [PubMed] [Google Scholar]
- 30.Chang A, Ko K, Clark MR. The emerging role of the inflammasome in kidney diseases. Curr Opin Nephrol Hypertens. 2014;23:204–10. doi: 10.1097/01.mnh.0000444814.49755.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–5. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 32.Wen Y, Liu Y, Tang T, Lv L, Liu H, Ma K, Liu B. NLRP3 inflammasome activation is involved in Ang II-induced kidney damage via mitochondrial dysfunction. Oncotarget. 2016;7:54290–54302. doi: 10.18632/oncotarget.11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu JW, Lee MS. Mitochondria and the NLRP3 inflammasome: physiological and pathological relevance. Arch Pharm Res. 2016;39:1503–1518. doi: 10.1007/s12272-016-0827-4. [DOI] [PubMed] [Google Scholar]