

Abstract
Derangements in tendon matrisome are pathognomonic for musculoskeletal disorders including rotator cuff tendinopathies (RCT). Collagen type-1 accounts for more than 85% of the dry weight of tendon extracellular matrix (ECM). The understanding of basic tendon physiology, organization of ECM, structure and function of component biomolecules of matrisome and the underlying regulatory mechanisms reveal the pathological events associated with RCT. Histomorphological evidence from RCT patients and animal models illustrate that ECM disorganization is the major hallmark in tendinopathy where a significant decrease in type-1 collagen is prevalent. However, the molecular events and regulatory signals associated with the regulation of collagen organization and its composition switch in response to pathological stimuli are largely unknown. The elucidation of various regulatory signalling pathways associated with collagen type-1 gene expression could benefit to develop novel promising therapeutic approaches to restore the tendon ECM. The major focus of the article is to critically evaluate tendon architecture regarding type-1 collagen, the molecular events associated with gene expression, secretion and maturation, the possible mechanisms of type-1 collagen regulation and its translational significance in RCT management.
Keywords: Rotator cuff tendinopathies, shoulder injury, type-1 collagen, tendon matrisome, tendon biology
Introduction
Tendinopathy accounts for about 50% of both work-related and sports-related injuries. The most vexing clinical issue associated with tendinopathies is its asymptomatic onset leading to tendon rupture [1]. Rotator cuff tendinopathy (RCT) is the most common tendon disorder of the shoulder which causes pain and dysfunction. Even though the multifactorial etiology of RCT is being studied, the biochemical and molecular events behind the pathogenesis remain unclear [2]. Rotator cuff tendinopathies can result from impingement, partial/full thickness tears, biceps tendinopathies, and frozen shoulders [3]. The understanding of basic biology, histology, molecular composition, and the associated signalling pathways are necessary in order to understand the pathological events associated with RCT. The current knowledge on the pathogenesis of RCT is almost entirely based on the findings in animal models and surgically removed tendons from patients and cadaver specimen [1].
Recently, we reported that the disorganization of tendon extracellular matrix (ECM) due to alteration of collagen composition and existence of inflammation without the active involvement of inflammatory cells are the major hallmarks of RCT [4-6]. The delay in restoring the altered Collagen I to III ratio was found to be another reason underlying the persistence of RCT [7]. In addition, increased cellular density, alterations in cellular morphology from spindle to round, apoptosis and necrosis have also been reported [4,8]. Rotator cuff (RC) tendons being hypovascular rely mostly on the metabolome secreted from the local environment and their autocrine/paracrine effects to maintain the ECM homeostasis [10]. Moreover, the mechanical load exhibited by the tenocytes acts as a trigger to upregulate the expression of various mediators of remodeling and adaptation. However, in RCT the tenocytes fail to withstand the stress and the resulting impaired mechanobiologic-response affects the tendon homeostasis [9,10,13]. Remarkably, the altered tendon matrisome is the root cause for RCT [7] and the exploration of matrisome integrity and homeostasis in relation to RCT is the focus of this article.
Tendon ECM structure-the hierarchical organization
In its natural state, tendon tissue is composed of 70% water while more than 85% of the dry weight is constituted by collagen I [14]. The cells are limited and more than 90% of the available cells are fibroblasts [2,15]. Since collagen I is the major component of tendon ECM, the structural and mechanical properties of tendon tissue depend largely on the quantity and quality of type I collagen. The hierarchical organization of collagen molecules is peculiar to tendon tissue. The collagen molecules associate to form fibrils, fibrils to fiber bundles, fiber bundles to fascicles and several fascicles constitute to form tendon units. The tendon units align in parallel to the long axis of tendon which is evident from basic histological staining. The collagen fibril is the basic structural unit of tendon and is formed by end-to-end organization of collagen type I (1 nm diameter) molecules. However, the fibril diameter varies from 10-500 nm depending on age, anatomical location, extent of physical activities and genetics. The usual trend is the increase in fibril diameter with age. Collagen fibrils constitute to form fibers (1-20 μm) which are enclosed by endotendons. Endotendons are thin connective tissues supplied with blood vessels, lymph vessels and nerves. The fibers organize to form fascicles (20-200 μm) where fascicle bundles are covered by epitendon. Similar to endotendon, epitendon surrounds a functional tendon unit (~500 μm) and is also supplied with blood, lymph and nerves [12]. Along with epitendon, paratendon also covers tendon tissue at certain sites. However, epitendon and paratendon make up to form peritendion which is responsible for minimizing the friction between the adjacent tissues [16]. The hierarchical organization of collagen molecules in the RC tendon tissues are displayed in Figure 1.
Figure 1.
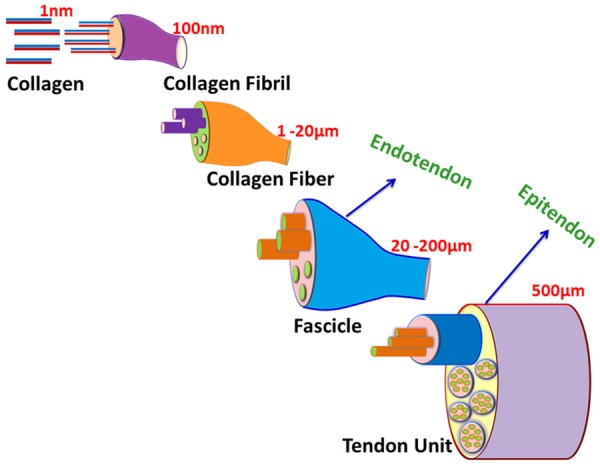
The hierarchical organization of collagen molecules forming tendon tissue. The basic unit for tendon architecture is collagen (especially type I) which aligns in end-to-end fashion to form fibrils which associated to form fibers which in turn give rise to fascicles. Several fascicles associate to from a functional tendon unit.
Biosynthesis and secretion of type I collagen-a key to RC tendon function
Type I collagen constitutes more than 95% of the total collagen content of RC tendon tissue, where the remaining 5% are mostly collagen type III and type V [14,17]. Type III collagen is mostly concentrated to the regions of endotendon and epitendon and also in injured tendon [18,19]. On the other hand, type V collagen intercalates with type I at the core and functions to regulate the fibril growth [20]. Besides, collagen type II, type VI, type IX and type XI also exist in minute levels but their exact function in RC tendon is largely unknown [21]. Type I collagen is the major collagen in RC tendon ECM and the major focus of tenocyte metabolism is to maintain matrisome integrity by regulating type I collagen.
Tenoblasts are the fibroblasts of tendon tissue which actively synthesize ECM components, especially collagen I [22]. Tenoblasts are the mechanoresponsive cells in post-natal tendon tissue which maintain tendon homeostasis by regulating ECM integrity [10]. The actual mechanism behind the synthesis and the regulatory events during conditions like embryonic development, regeneration, repair and inflammation are poorly known. However, the collagen molecules secreted by these cells attain a thermodynamically stable conformation by self-assembly [23]. The tenoblast cells secrete collagen precursor, the procollagen, as large aggregates in the lumen of cis-Golgi complex. The large dimension of procollagen-aggregate creates a hurdle in loading them into small transport vehicles. It was elucidated that the cis-Golgi procollagen moves to medial-Golgi cisternae and then to trans-Golgi in a wavy fashion for secretion to ECM [24]. To date, there are more than 27 different types of collagen have been characterized where type I and III are significant for RC tendons [23].
All fibrillar collagen subtypes are made of repeating units of Gly-X-Y, where X and Y can be any amino acid residues, but usually Proline (Pro) and Hydroxyproline (HyPro), respectively (Figure 2A). The repeating triplet motif forms a left-handed helix which in turn organizes a right-handed triple helix by interacting with two other left-handed helices. The collagen molecules can be homotrimeric or heterotrimeric depending on the collagen subtype. For instance, collagen type I is a heterotrimer comprising two α-1 chains and one α-2 chain while type III collagen is made up of three α-2 chains (Figure 2B) [25]. The triple helical domain is 300 nm in length connecting N and C globular domains which do not necessarily follow Gly-X-Y repeating sequence. The proteolytic cleavage of N and C globular domains release collage triple helical molecule. The collagen units have short telopeptides on their both ends which assemble in an end-to-end fashion to form the fibril. The fibrils can be made up of type I and/or type III collagens in RC tendon tissue [26].
Figure 2.
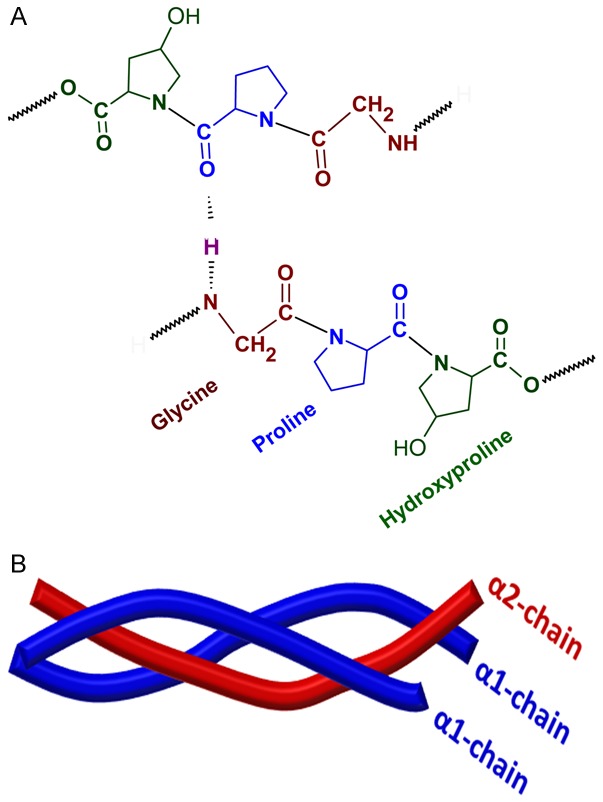
The molecular organization of collagen type I. A. The building units of collagen comprising repeating units of Gly-X-Y which is stabilized by hydrogen bonds. X and Y can be any amino acids, but usually Proline and Hydroxyproline respectively. B. The triple helical structure of collagen type I collagen comprising of two α-chains and one β-chain.
After synthesis, the procollagen type I translocates to ER (endoplasmic reticulum) lumen where the trimerization takes place, which is mediated by several molecular chaperones like HSP47 [26]. The procollagen contains propeptides at their C (carboxy-terminal propeptide of procollagen type I, PICP) and N (amino-terminal propeptide of procollagen type I, PINP) terminal ends which are proteolytically cleaved to integrate with the fibril. The cleaved fragments are released in the blood stream where the circulatory levels of the cleaved fragments represent the index of collagen synthesis. The 1:1 stoichiometry for PICP and PINP in the blood stream exist, but for PIIICP and PIIINP it may vary. This is because the proteolysis is incomplete during fibril formation, but completes during collagen type III degradation [29]. PICP is responsible for folding and chain selection for the nucleation/growth of fibrils. PICP facilitates the folding and collagen fibril formation by associating procollagen chains by disulphide bond formation within individual C propeptides before their cleavage. Protein disulphide isomerase (PDI) is the enzyme responsible for association of the procollagen chains with inter-chain disulphide bonds (Figure 3A). However, the directional folding from C to N terminal side also occurs independent of PDI reaction [23].
Figure 3.
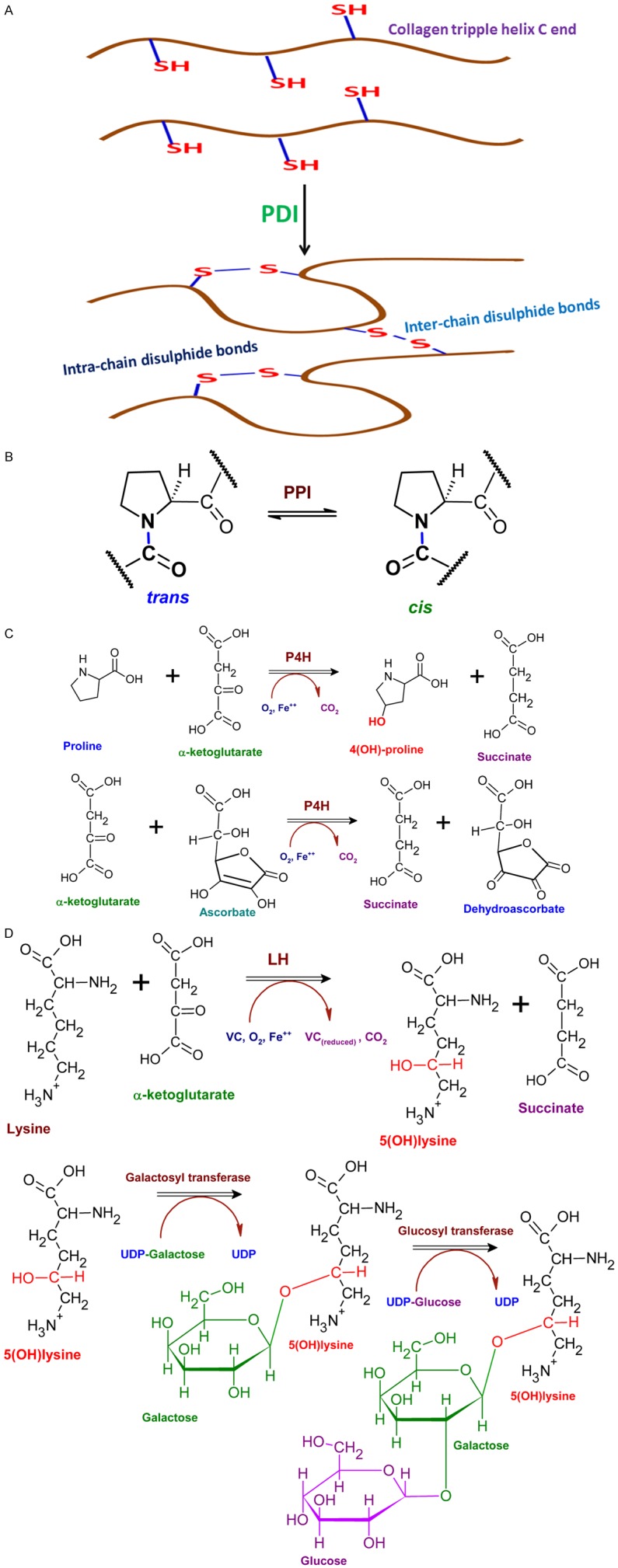
Enzyme activities associated with collagen type I formation. A. PDI catalyzes the formation of inter-chain and intra-chain disulphide bonds especially at the C terminal end of procollagen which is inevitable for the proper organization of triple helix. B. PPI converts cis-Pro residues to trans isomers which is essential for proper folding and fitting into triple helix. C. P4H is an iron-containing enzyme responsible for hydroxylation of Pro residues. Oxygen, proline residue, and α-ketoglutarate interact with the enzyme and the proline residue is hydroxylated while the α-ketoglutarate undergoes oxidative decarboxylation to form succinate and carbon dioxide. The role of vitamin C is shown as another reaction which prolyl 4-hydroxylase catalyzes the oxidative decarboxylation of α-ketoglutarate. But, in this reaction the iron is oxidized which inactivates enzyme. Vitamin C (ascorbate) reduces the iron at the vicinity of the enzyme to restore its active state. D. LH catalyzes the hydroxylation of Lys residues at the 5th C atom to from 5(OH)lysine and LH acts like P4H enzyme. The 5(OH)lysine acts as a template for the addition of galactose moiety followed by glucose moiety to the procollagen by the hydroxylysl galactose transferase and galactosyl hydroxylysl glucose transferase respectively. UDP-Galactose/Glucose acts as sugar donors and this reaction is crucial for trafficking of procollagen from Golgi to ECM.
Additional modifications are required for maintaining the thermodynamic stability of the type I collagen triple helix. Peptidyl proline cis-trans isomerase (PPI) converts cis-Pro residues to trans isomers which is essential for proper folding and fitting into triple helix (Figure 3B) [30]. Moreover, the hydroxylation of Pro residues is required for the stability of triple helix as evident by the increased temperature for denaturation of collagen I with hydroxylated Pro residues. Prolyl-4-hydroxylase (P4H), a tetrameric enzyme with two-α and two-β subunits where the β subunit possess catalytic domain, catalyzes the Pro hydroxylation reaction. The β subunit possesses PDI activity also. The enzyme requires vitamin C (VC) for its activity which depends on high cell density to induce collagen pathway (Figure 3C). The cell density stabilizes the effect of triple helix in regard to the changes in cellular redox potential [31]. Hydroxylation holds an extensive network of water molecules within the triple helix by coordinately bridging them within and between the chains [23]. Similar sort of hydroxylation occurs in Lys residues as well. The added -OH groups act as cues for attaching galactose and glucose moieties in the ER by specific transferases (Figure 3D) [23].
The glycosylation of procollagens occurs in ER and Golgi complex. Galactose and glucose residues added to the hydroxylysine and the oligosaccharide units added to Asn residues at the C terminal end of procollagen is required for proper alignment for its transport to ECM. Usually the hydroxylation of Pro and Lys residues in the middle portion are hydroxylated by membrane bound hydroxylases. Once these events are completed, the type I procollagen is secreted to extracellular space by exocytosis. In the extracellular space, the N and C terminal propeptides are removed by procollagen peptidases resulting in tropocollagen, which consists of mostly triple helix. The N and C terminal trimmed procollagen polymerize to form fibrils in the extracellular space. This assembly is accelerated by lysyl oxidase, an extracellular enzyme which creates aldehyde groups in Lys and contributes to crosslinking and fibril stability. The maturation of collagen molecules requires additional post-translational modifications [24]. The molecular and cellular events associated with collagen fibril formation by tenoblast/tenocytes are depicted in Figure 4.
Figure 4.
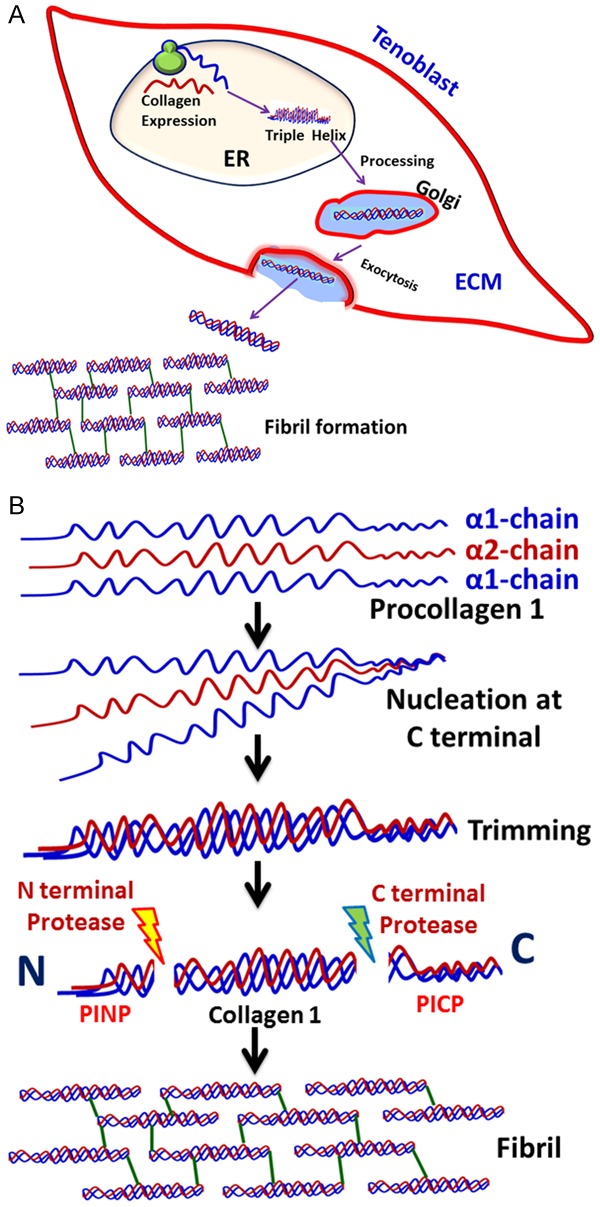
Secretion of fibrils to ECM in tenoblasts. A. The collagen gene expression and assembly of triple helix occurs in the ER lumen of tenoblasts. The procollagen is then processed in Golgi apparatus and is transported to plasma membrane for exocytosis of mature collagen to ECM where the fibril formation takes place. B. The molecular events in the assembly of fibril. The procollagen type I subunits were synthesized in the ER lumen which align to form triple helix by nucleating at the C terminal end by disulphide formation. The collagen triple helix matures by the trimming of PICP and PINP by corresponding proteases. This triple helix aligns at end-to-end fashion in the ECM to form fibrils.
The procollagen type I protein needs to attain its natively folded conformation to reside in the ER. The loading of cargo (here procollagen type 1) is under the control of two proteins COPI and COPII (coat protein complex I and II). COPI and COPII coat the outer surface of transport vesicles [32]. The fibrils being ~300 nm diameter and the 60-90 nm vesicles suggest the possibilities of alternate carriers for lager cargos like collagen type I [23]. Evidence from freeze fracture technique, light/video electron microscopy and GFP-tagging experiments revealed the transport of procollagen as saccular structure budded from ER membrane and the ECM-targeted proteins are segregated from other targets in early stage [29-31]. The COPII vesicles are necessary for the export to procollagen type I loaded cargo at the ER exit site.
COPII coat assembly requires six polypeptides. Initially, GDP-bound Sar1 (Secretion-associated RAS-related protein 1) polypeptide interacts with Sec12 and exchange its GDP with GTP [36]. This activates Sar1-GTP to insert its hydrophobic domain to ER outer-membrane where it recruits and binds with Sec23/Sec24 protein coated complex. The binding is between Sec24 and GTP-bound Sar1. This assembly forms the first layer of coat protein in the cytoplasmic face of ER. Binding of Sec24 facilitates the cargo to COPII in the transmembrane through interactions with hydrophobic, aromatic and acidic motifs [37]. COPII pre-budding complex then recruits Sec13/Sec31 dimer and Sec31 directly interacts with Sec23 via a proline-rich domain. Sec23 also serves as a GTPase activation protein (GAP) for Sar1 and the GAP activity is regulated by the interaction of Sec31 through the proline-rich domain. The two events, Sar1 GTPase activity and outer coat layer recruitment, complete the biogenesis of COPII and trigger the budding. These coat proteins are highly conserved in all eukaryotes [38].
Transport ANd Golgi Organization 1 (TANGO1) protein (1907 amino acids) is also reported to be involved in the trafficking of larger cargoes like collagen fibrils to exit ER. This is evident from the silencing experimental data on HeLa cells which confirmed that the depletion of TANGO1 significantly affected collagen type VII without affecting general protein secretion [35,36]. Interestingly, type 1 collagen was also affected in TANGO1 knockout mice [41]. However, TANGO1 plays a role in synchronizing the sorting of type I collagen from other types [42]. TANGO1 resides at the exit site of ER and is comprised of SR3-like domain and a coiled-coil domain at N-terminus, a cytoplasmic domain and a C-terminus Pro-rich domain. TANGO1 bears two closely located hydrophobic domains; a transmembrane domain and a partially embedded domain in the outer or inner leaflet [40]. Collagen VII binding to SH3 domain of TANGO1 in ER lumen facilitates the binding of Sec23/Sec24 via Pro-rich domain. This binding blocks the Pro-rich domain-mediated binding of Sec31 to Sec23 and delays Sar1-GTP hydrolysis. This temporary block provides enough time for COPII to accommodate larger cargoes like collagen fibrils. After loading to COPII, the collagen molecule dissociates from TANGO1 which couples the dissociation of TANGO1 from Sec23/Sec24. Now, Sec23/Sec24 is exposed for receiving Sec13/Sec31 to trigger budding. In short, TANGO1 provides enough time to load larger cargos to COPII carrier. Another protein, cTAGE5, binds to TANGO1 Sec23/Sec24 to facilitate effective packing of fibrils [34,36].
Defects in the ER transport of collagen type I fibrils can affect the integrity of RC tendon ECM and lead to poor quality and low quantity collagens. But, the studies relating ER defects and RC tendon disorders are rare in the literature. However, the active role of ER proteins in the collagen metabolism is clearly understood [43]. Moreover, the age-related alterations in ER transport of collagen fibrils has been demonstrated in horse tendon [44]. Since RCT are characterized by alterations in the collagen ratio, the possibilities of ER defects should not be overlooked. Extensive research is needed in these aspects on RC tenoblasts/tenocytes which would have translational significance in RCT management.
The transport of collagen cargo in the anterograde direction across the Golgi apparatus of mammalian cells is largely unknown and is still under debate. The cisternae of Golgi apparatus can mediate sequential post-translational modifications of procollagen type I molecules [38]. The glycosylation enzymes are in the cisternae where the COP coated vesicles are associated. Interestingly, the procollagen can move across the Golgi without being detached from cisternae. The maturation of cisternae results in the retrograde transport of local enzymes especially mannosidase I and sialyl-transferase [23]. Once budded from ER exit site, they shed the coat and undergo homotypic (between same compartments) or heterotypic fusion (between different compartments). The SNARE [soluble NSF (N-ethylmaleimide-sensitive factor) attachment protein receptors] proteins especially, v-SNAREs (Vesicle-SNSAREs) and t-SNAREs (target-localized SNAREs) aid in the fusion leading to the formation of continuous compartment in ER [24].
Evidence from electron microscopy of fibroblast revealed the release of procollagen fibrils from Golgi apparatus as secretory vacuoles [23]. The procollagen cargo from trans-Golgi-network (TGN) is transported to endosomes and lysosomes in a process mediated by clathrin-coated vesicles [45]. Clathrin-independent endosomal transport system and Ca2+-based sorting of secretory cargo have also been reported [46]. But, the underlying mechanism and transport to the plasma membrane of procollagen is unknown. However, the involvement of protein kinase C-δ (PKCδ) in collagen type I export has been identified in smooth muscle cells [47]. Still, the mechanism of PKCδ activation and the effect of PKCδ system in tendon cells are yet to be discovered.
Interestingly, the cross section of tendon tissues revealed the existence of collagen fibrils in the cytoplasm surrounded by cell membrane. This finding revealed the presence of cellular recesses for the secretion of collagen [48]. The single fibrils released through the recesses assemble to form higher order structures thereby contributing to tendon growth. The long collagen fibrils co-exist with several short ones within the cell and are referred as fibripositor (fibril-depositor). Fibripositor extends from the side of tenoblasts; align through the long axis of tendon and projects out to ECM through recesses. The carriers push the fibril out of the cell and subsequently fuse with the plasma membrane to form a new fibripositor. It can also fuse with the existing fibripositor. The procollagen molecules are added to the base of fibripositor facilitating the end-to-end fusion to form fibrils [49].
ECM processing of procollagen I-the final make-up
The proteolytic removal of globular C and N terminal peptides from procollagen type I limits the effective concentration required for fibril formation which in turn triggers self-assembly. The cleavage occurs during or following secretion to ECM. The enzymatic cleavage is performed by type-specific MMPs (metalloproteinases), like ADAMTS (A disintegrin and metalloproteinase with thrombospondin motif) especially ADAMTS-2, ADAMTS-3 and ADAMTS13, bone morphogenetic protein 1 (BMP1) and tolloid-like families like furin-like proprotein convertases (FLPC) [50]. The presence of C-propeptide increases the solubility of procollagen in ECM and prevents immature fibril formation. On the other hand, the N-propeptide directs the diameter and shape and contributes to increase in surface-to-volume ratio [50]. N-propeptides are completely removed from type I, II and III procollagens where the cleavage is rapid in types I and II but this process is relatively slow in type III. The delay in the removal of N-terminal propeptide accumulates partially-processed procollagen called pN-collagen (without C-propeptides with N-propeptides). The retention of N-propeptides in pN-collagen prevents its access to the center of fibril. Whereas, N-propeptides cause the incorporation of type III collagen to the surface and thus limits the fibril diameter. That is why, collagen III is usually associated with the surface of type I [51].
Electron microscopy data revealed the collagen self-assembly with a quarter stagger axial D periodicity of approximately 67 nm which appear as characteristic striations in the ECM images [50]. Micro-fibrils are formed by the sequence of events including nucleation, organization and elongation towards the long axis of tendon. It is believed that the N-terminal propeptides of type V are required for the occurrence of nucleation of type I and II collagens as these projects to the gap between adjacent fibrils and prevents the lateral growth by enhancing steric hindrance and charge interactions [52]. Fibril-associated collagens with interrupted triple helices (FACIT) like type XII and XIV (non-fibrillar collagen) were also reported to have a regulatory role in fibril formation. These non-fibrillar collagens aid in orchestrating matrix components with type I collagens by interacting with proteoglycans, fibromodulin, decorin and others [49,50]. Rentz and colleagues reported the association of adhesive protein SPARC (secreted protein acidic and rich in cysteine) in the processing and incorporation of procollagen type I in tendons [51,52]. Indeed, there is evidence for the involvement of several proteins and other biomolecules in the maturation of collagen type I in RC tendon. The functions of some of such molecules are given in Table 1. In a nut shell, the collagen fibril assembly owes to unique chemical and physical interactions resulting in a multi-hierarchical architecture which maintains the integrity of ECM in the RC tendon.
Table 1.
Non-collagenous biomolecules associated with RC tendon matrisome
| Biomolecules | Function | References |
|---|---|---|
| Small Leucine-Rich Proteoglycans (SLRPs) | Regulation of fibrillogenesis | [105] |
| Decorin | Mediator of fibrillogenesis by inhibiting fibril formation by lateral fusion, contribute to alignment, stability, strength and elasticity to collagen fibrils via surface interactions | [106] |
| Biglycan | Regulation of fibrillogenesis during development | [107] |
| Fibromodulin | Regulation of fibril fusion by inhibiting lateral fusion | [108] |
| Lumican | Regulation of fibril fusion | [109] |
| Cartilage oligomeric matrix protein (COMP) | Enhancer of collagen fibrillogenesis, mediates interaction between different components of ECM | [110] |
| Elastin | Stretch and recoil | [12] |
| Fibronectin | Cell adhesion | [111] |
| Laminins | Cell differentiation and migration | [112] |
| Tenascins | Load bearing and regeneration | [113] |
| Growth factors | Growth, repair and regeneration | [114] |
| MMPs | Repair and regeneration | [115] |
Matrisome composed of thousands of proteins and their co-ordinated interactions that are needed to sustain the ECM homeostasis. According to human matrisome database (HMD), 1139 proteins were associated with ECM, of which 128 proteins attribute solely to human ECM where 76 proteins associate with core matrisome and 52 proteins with ECM-associated components. Twelve proteoglycans, 18 collagens, and 46 glycoproteins comprise to form the core. ECM-associated components are constituted by 16 ECM-affiliated components, 24 regulators and 12 secreted factors [57]. However, the proteomics of RC tendon matrisome has not been reported yet. Recently, we have reported some of the key protein mediators of RC tendon matrisome in regard to the miRNA mediated regulation of glenohumeral arthritis [7]. The identification of regulatory proteins of RC tendon ECM and the knowledge of their alterations associated with RCT could have translational impact to pave ways for the development of novel therapeutic strategies.
Collagen type I gene expression-a felicitous point for tendon ECM regulation
Human pro-COL1A1 (18 kb) and pro-COL1A2 (38 kb) genes are located in 17q21.31-22.05 and in 7q21.3-22.1, respectively [58]. Even though both sub-units (COL1A1 and COL1A2) are found on different chromosomal locations, the transcripts of these two genes are maintained as 2:1 ratio at the cellular levels. Both the genes contain several repressor and enhancer elements in their promoter regions, and binding sites for basal, cell-specific and cytokine-mediated transcription factors. Moreover, the cytokine-responsive elements (CREs) have also been identified and located in several tissues. Epigenetic regulation by DNA methylation and subsequent downregulation of type I collagen genes were also described [59]. The gene expression of collagen type I in RC tendons is also under strict regulation. However, it should be noted that the regulatory signals alter significantly depending on the type and severity of RCT. Since collagen I is the major component of RC tendon, the regulatory mediators mainly focus on the restoration of collagen I after RCT. Several regulatory molecules are associated with collagen type I gene expression and the functions of the common ones are shown in Table 2.
Table 2.
Regulators of collagen type I gene expression
| Proteins | Function | References |
|---|---|---|
| CCAAT binding factor (CBF) | Interacts with CCAAT box located at -100 to -96 bp and enhance collagen I expression. | [116] |
| Nuclear factor-1 | Binds between -350 and -300 bp of COL1A2 and act as a positive TF. | [117] |
| Inhibitor protein 1 & 2 | Competes with CBF for binding to promoter and inhibits collagen I expression. | [59] |
| CCAAT/enhancer binding proteins | Basal and cytokine modulated Type I collagen gene expression. | [59] |
| Specificity factor 1 (SP1) | A zinc-finger family transcription factor that binds to GC-rich regions located between -303 and -271 bp in COL1A2 gene promoter and modulate collagen I expression. | [71] |
| Activator protein 1 (AP1) | AP1 binds at +292 to +670-bp region of first intron of human COL1A1 and functions in transcriptional regulation. | [118] |
| NF-κβ | Inhibits COL1A1 and COL1A2 expression. | [76] |
| Smad7 | Smad binding element, CAGACA, is located at -263 to -258 bp, and Smad7 mediates COL1A2 gene transcription. | [59] |
| p300/CBP | p300/CBP are adaptor with intrinsic HAT activity that modulates Type I collagen gene transcription. | [119] |
| Myb | Modulation of Type I collagen gene expression in cell type dependent manner. | [120] |
| c-Myc | Suppress Type I collagen transcription. | [121] |
| c-Krox | Regulator of COL1A1. | [59] |
| Basic transcription element binding protein | Enhance COL1A1 expression. | [122] |
| Protease nexin 1 (PN1) | PN-1 increases the level of collagen transcription and decreases its degradation. | [123] |
| Protein phosphatase 2A | Enhance COL1A1 expression. | [124] |
| Extracellular signal-regulated kinase 1/2 | Represses Type I collagen synthesis. | [59] |
| CBFA1 | Transcription factor for osteoblast-specific gene expression of Type I collagen. | [125] |
| Connective tissue growth factor | Fibroblast proliferation and wound healing by stimulation of Type I collagen gene expression. | [126] |
Since hypoxia is often associated with RCT, the hypoxic signaling elicited by the activation of hypoxia inducible factor (HIF) plays a crucial role in collagen homeostasis. The effect of HIF in collagen cross linking can be attributed to its action on lysyl oxidase (LOX) gene. HIF-1 and HIF-2 upregulate LOX where the effect of HIF-2 is superior due to its effective binding with the hypoxia response elements (HRE) of LOX promoter [60]. However, HIF-1α has been reported to upregulate collagen genes by activating NOTCH1, NOTCH1 ligand (JAGGED1) and hairy and enhancer of split-1 (HES1) [61]. There are four NOTCH (1-4) receptors and their corresponding ligands Delta-like (1-4) and JAGGED (JAG1 and 2) have been characterized, so far, in mammalian system. On binding with ligand the presenilin proteases (PSEN1/2) (part of γ-secretase complex, GSC) cleave intracellular domain of NOTCH (Notch intracellular domain, NICD) which translocates to nucleus and interacts with immunoglobulin κ J region (RBP-Jκ) and Mastermind-like (Maml) proteins which in turn trigger the transcription of HES and HEY (mammalian counterparts of drosophila HES1) genes [58,59]. RBP-Jκ, also known as CSL (CBF-1, Su(H), Lag-1), is responsible for the binding of the complex to DNA. Interestingly, the NICD hyperactivity inhibits differentiation of osteoblasts by controlling collagen type 1 [64]. However, the effect of NOTCH signaling and its activation by hypoxia in RCT have not been studied yet; which opens up tremendous research opportunities especially in tendon matrix biology.
Growth factors (GFs) are another class of mediators that have active role in tendon matrix homeostasis, thereby play significant role in healing after RCT. The involvement of several GFs and their specific functions have been characterized in tendon tissues (Table 3). Among them, TGF-β signaling pathway has direct impact on tendon ECM regeneration which proceeds through a class of intracellular signaling proteins called Smads. Upon binding to its receptor TGF-β activates downstream Smad2 and Smad3 and facilitates to form heteromers by binding to the co-Smad, Smad4. This Smad complex translocates to nucleus and regulates the expression of ECM-associated genes, especially Col1A1. The hyperactivity of Smad complex is controlled by inhibitory Smads (I-Smads), which are Smad6 and Smad7. I-Smads interfere Smad2 and Smad3 phosphorylation thereby block the interaction of TGF-β receptors (negative-feedback) [65]. Moreover, TGF-β activator protein (TAP) binds to TGF-β cis-element and the TGF-β-responsive sequences (TBRS) (between -174 and -84 bp) of downstream transcription start site of Col1A1 gene. In addition, TGF-β1 induces type 1 collagen gene expression by the epigenetic inhibition of DNMT1, DNMT3a and global DNMT activity which result in DNA demethylation of the COL1A1 promoter [66]. There are reports projecting the operation of TGF-β signaling in tendon tissue [67,12]. However, the translational aspects of TGF-β with respect to COL1 gene expression in RCT are limited in the literature.
Table 3.
Role of growth factors in RCT
| Growth factor | Function | References |
|---|---|---|
| PDGF | Secretion of other GFs | [127] |
| Cell proliferation | ||
| Tendon healing | ||
| DNA synthesis | ||
| TGF-β | Tendon healing | [12] |
| Tendon matrix deposition | ||
| Scarring and fibrosis | ||
| VEGF | Endothelial cell proliferation | [128] |
| Angiogenesis | ||
| bFGF | Tendon cell migration and proliferation | [12] |
| Angiogenesis | ||
| Collagen type 1 gene expression | ||
| BMPs | Tenogenesis (BMP-12) | [129] |
| Tendon callus formation (BMP-13 & 14) | ||
| Mechanical strength | ||
| Tenoblast proliferation | ||
| Collagen type 1 and type 3 gene expression | ||
| IGF1 | Mechanical strength | [130] |
Most inflammatory signals converge to the transcription factor NF-κB which upregulate a battery of pro-inflammatory genes [68]. However, NF-κB downregulates COL1A1 by interfering the activities of zinc fingers, Sp1 (specific protein-1) and Sp3 which act as trans-activators of COL1A1 at an upstream 51 bp region (-112/-61). The p65 (RelA) subunit of NF-κB is responsible for the trans-inhibiting effect [69]. TGF-β-responsive activator sequences located 174 and 84 bp upstream from the initiation site of COL1A1 have binding sites for Sp1 and NF-1 (nuclear factor-1) [70]. Similar effect of TGF-β trans-activation of COL1A2 has also been established which confirms the role of TGF-β in ECM remodeling by upregulating COL1 synthesis [65,67]. Apart from Sp, zinc finger proteins like c-Krox were also reported to upregulate COL1A1 by -112/-61 upstream sequence [72].
NF-κB associated transcription factors share a conserved domain of ~300 N-terminal amino acids called Rel homology domain (RHD) which bears NLS (nuclear localization sequence). RHD facilitates DNA binding, dimerization and association with inhibitory IκB proteins. However, p65 subunit possesses C-terminal trans-activation domain for eliciting the transcriptional activities [69]. NF-κB is associated with IκB family of inhibitors in unchallenged cells thereby sequestering in the cytoplasm. It has been estimated that more than 200 signals can elicit the activation of NF-κB by inducing IκB dissociation which exposes the NLS permitting their nuclear entry and promoter binding. These signals include IL-1, IL-6, TNF-α, TREM-1 and others [65,69,70]. However, the release of NF-κB by IκB degradation is not sufficient for eliciting the transcriptional control but, requires other factors like Sp1. This explains NF-κB-mediated downregulation of COL1A1 due to its interaction with Sp1 [75]. The p65 subunit also has an inhibitory effect on collagen gene expression [69].
TNF-α elicits the regulatory effects on COL1A1 and COL1A2 in fibroblasts and other cell types which is mediated by NF-κB family of transcription factors [72,73]. TNF-α-inhibitory responsive element (TαRE) has been mapped in the upstream promoter region between 378 and 345 of COL1A1 which host the binding site for a school of DNA binding complexes like p20C/EBPβ, p35C/EBPβ, and C/EBPδ. Interestingly, this sequence co-localizes with TGF-β-responsive element suggesting that the inhibitory effects of TNF-α proceeds by interfering TGF-β-induced activation, nuclear transport and DNA binding effects of Smad complexes [78]. Also, the inhibitory Smad7 has been reported to be activated by p65-mediated activation of TNF-α in rat hepatic stellate cells [79]. Thus, TNF-α signaling converges to NF-κB activation and shuts down COL1 synthesis. The operation of TNF-α and NF-κB signaling for collagen gene regulation has not been well established in tendon tissues, however, the elevation of these signals with a concomitant downregulation of collagen type 1 has been found to be associated with mechanically stressed tendon [80].
The prevalence of RCT in elder population is higher than in the younger subjects which often coincide with inflammatory (such as glenohumeral arthritis) or metabolic disorders (such as diabetes) [77,78]. The etiology of RCT and collagen regulation associated with aging has not been elucidated yet. However, greater significance has been gained to the concomitant increase in advanced glycation end products (AGEs) and its implication to age-associated structural alterations in the ECM of several tissues. AGE signaling with its receptor RAGE also converges to NF-κB and subsequent inflammation. However, based on the type of signals NF-κB elicits different transcriptional outputs. This can be explained by ‘NF-κB barcode hypothesis’ which states that depending on dynamics of cellular network signal-specific post-translational modifications, “the barcodes” to NF-κB are induced which act as a signature for the specific gene expression pattern [83].
Apart from AGEs, RAGE shares a multitude of ligand subset which includes high mobility group box-1 (HMGB1), S100 family, and lipopolysaccharide (LPS) [80-82]. The expression of RAGE was also found to be enhanced with aging which has a strong correlation with diabetes and hyperglycemia [87]. In the present context, the collagen I and III expression have been found to be decreased with concomitant increase of RAGE in the arterial wall of RAGE null mice suggesting its effect on collagen production [88]. However, contrasting results were obtained in fibroblasts from different tissue sources [65,85]. Recently, we reported that in the RC tendon tissues the decrease in the ratio of collagen 1 to 3 was a major cause for ECM disorganization [7]. Interestingly, we found an upregulation of RAGE/HMGB1 axis with a proportionate increase in TREM1 expression [4]. On correlating the increased TREM1 with decreased COL1 expression, we speculate that TREM1-mediated NF-κB activation proceeds through a different barcode other than RelA which warrants further investigation.
Metalloproteinase activity influences the expression of collagen types in RC tendon tissues following injury. Like other tissues, tendons also have a basal level of MMPs under normal conditions where a rapid upregulation occurs immediately after the injury. Most cell types are capable of MMP secretion in response to the signals like cytokines, hormones, and cell-cell/cell-ECM contact. These signals result in the activation of either NF-κB, Smad, focal adhesion kinase (FAK) or Wnt. Moreover, epigenetic stabilization of chromatin and mRNAs significantly contribute to MMP expression [90]. Control of MMP expression at the transcriptional level is prevalent in most tissues. Promoters of MMPs have cis-elements which favor binding for diverse trans-activators like AP-1, polyomavirus enhancer A binding protein-3 (PEA3), Sp-1, β-catenin/Tcf-4, NF-κB and others [91]. Depending on the composition of cis-elements there are 3 major categories of MMP promoters which are summarized in Table 4 [91,92].
Table 4.
Major categories of MMP promoters, their features and function
| Groups | Features | Function |
|---|---|---|
| Group I | TATA box ~-30 bp | Regulates most MMPs |
| AP-1 binding site ~-70 bp | ||
| PEA3 binding site adjacent to that of AP1 | ||
| Group II | TATA box present | Regulates MMP-8, MMP11 and MMP-21 |
| AP-1 binding site absent | ||
| Group III | No TATA box | Regulates MMP-2, MMP14 and MMP-28 |
| Multiple start site for transcription | ||
| Requires SP-1 family of TF | ||
| Expression mediates through GC box |
RCT is associated with the upregulation of MMPs especially MMP-1, MMP-8, MMP-13 and gelatinases (MMP-2 and MMP-9) [89,90]. Our findings from rotator cuff tendon of RCT human and RCT rat models revealed an upregulation of MMP-9 where other MMPs were expressed in basal level [7]. This suggests the potential role of MMP-9 in RCT pathology which proceeds through the alteration of COL1 to COL3 ratio. Among the signals that triggers MMP-9 expression, TGF-β is considered to play a potential role in the pathology of several diseases especially cancer [95]. MMP-9 has been associated with several biological responses like wound healing, inflammation, metastasis, vasculogenesis and development [96]. MMP-9 is secreted by immune cells like neutrophils and macrophages and non-immune cells like fibroblasts which are released upon injury/stress. Fibroblasts from most tissue types express MMP-9 when stimulated with IL-1β and TNF-α via ERK1/2 and NF-κB signaling [97].
Apart from NF-κB, PEA3, AP-1, Sp-1 and serum amyloid A-activating factor -1 (SAF-1) also regulate MMP-9 transcription [98]. Reactive oxygen species activates MMP-9 through NF-κB while thrombospondins through AP-1 [99]. TNF-α triggers the expression of MMP-9 by the activation of protein kinase C (PKC) pathway [97]. MMP-9 expression has been associated with overuse tendinopathies of Achilles tendon mostly due to the increase in peripheral circulation of leukocytes [100]. Actual mechanism behind the expression of MMP-9 in shoulder tendon is largely unknown. However, the decrease in COL1 in RC tendon ECM after injury is the major histological hallmark suggesting the COL1 degradation to be the basis for RCT pathology [101].
Tenocyte differentiation is mediated by the transcription factor Scleraxis (Scx) which is induced by TGFβ. On binding with type I and type II transmembrane serine/threonine kinases TGFβ initiates activation and phosphorylation of intracellular effectors (Smad2 and 3, Erk1/2 and p38 MAP kinases). Activation of Smad3 induces Scx upregulation and Mohawk (Mkx). Mkx is required for tendon development and maturation [102]. Apart from these, Smad3 upregulates COL1 and activates healing responses [103]. Moreover, Scx upregulates Tenomodulin (Tnmd) and COL1 which are inevitable for tenocyte proliferation [104]. The mechanism behind the activation of Scx and the execution of downstream signaling warrants more research, especially in regard to RCT.
The integration of various signals associated with the regulation of type-1 collagen is displayed in Figure 5.
Figure 5.
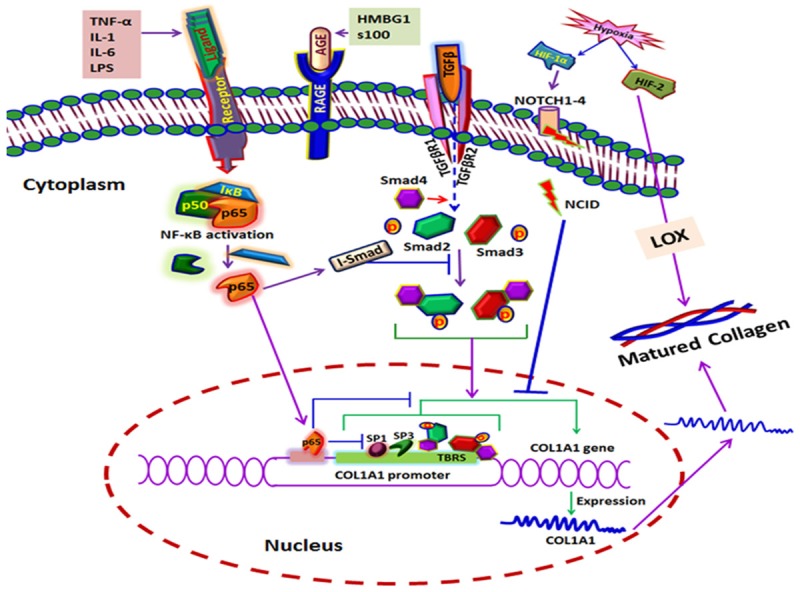
The integration of various signals associated with the regulation of type-1 collagen. Most of the inhibitory signals converge to NF-κB while the stimulatory signals proceed through Smads.
Summary and future directions
RC tendon is composed of mainly type 1 collagen in which the tendon cells (tenocytes and tenoblasts) are dispersed. The cell density in normal RC tendon is low and a drastic increase in cellularity has been observed histologically after RCT. This shift in cellularity is thought to be responsible for the pathology. Most of the immune and inflammatory cells were characterized in RCT tendon; however, the possibility of existence of other cellular phenotypes should not be neglected. Several theories exist regarding the pathological basis of tendon ECM; the mechanistic models for explaining such theories are still limited. Understanding the pathological basis of collagen gene alteration and COL1 to COL3 ratio warrants extensive research in regard to tendon molecular biology and genetics. The signals involved and the underlying genomics and proteomics in the regulation of collagen expression in tendons (normal and pathological) are still unknown. The establishment of a tendon matrisome database, tendon genomic database and tendon epigenomic database are needed at this time for complete understanding of RCT pathology. Novel approaches like tendon matrisome remodeling and tendon matrisome engineering should be practiced from in vitro to in vivo animal models and to be translated to clinical arena. The immunomodulatory approaches to activate the self-healing responses of RC tendons after injury could have promising clinical outcomes. The restoration of tendon ECM and homeostasis of collagen type 1 could help the sufferers to gain the tendon function after injury. Stem cell-based approaches are also appreciated. Limited information on the basic science of tendon biology, lack of proper animal models and control specimens offer hurdles to RC tendon research. In conclusion, the basic research on RC tendon pathology is still in its infancy which warrants multidisciplinary approach involving expertise in surgery, basic sciences, biomaterials sciences and genetics which would unravel the mystery and pave ways to the development of effective translational strategies for RCT management.
Acknowledgements
This work was supported by Creighton University LB692 grant to MFD and LB506 grant to DKA from the State of Nebraska. The research work of DKA is also supported by grants from the National Institutes of Health. The contents of this original research article are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health or the State of Nebraska.
Disclosure of conflict of interest
None.
References
- 1.Joseph M, Maresh CM, McCarthy MB, Kraemer WJ, Ledgard F, Arciero CL, Anderson JM, Nindl BC, Mazzocca AD. Histological and molecular analysis of the biceps tendon long head posttenotomy. J Orthop Res. 2009;27:1379–1385. doi: 10.1002/jor.20868. [DOI] [PubMed] [Google Scholar]
- 2.DE Giorgi S, Saracino M, Castagna A. Degenerative disease in rotator cuff tears: what are the biochemical and histological changes? Joints. 2014;2:26–8. [PMC free article] [PubMed] [Google Scholar]
- 3.Reuther KE, Thomas SJ, Tucker JJ, Yannascoli SM, Caro AC, Vafa RP, Liu SS, Gordon JA, Bhatt PR, Kuntz AF, Soslowsky LJ. Scapular dyskinesis is detrimental to shoulder tendon properties and joint mechanics in a rat model. J Orthop Res. 2014;32:1436–1443. doi: 10.1002/jor.22693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thankam FG, Dilisio MF, Dietz NE, Agrawal DK. TREM-1, HMGB1 and RAGE in the shoulder tendon: dual mechanisms for inflammation based on the coincidence of glenohumeral arthritis. PLoS One. 2016;11:e0165492. doi: 10.1371/journal.pone.0165492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Millar NL, Akbar M, Campbell AL, Reilly JH, Kerr SC, McLean M, Frleta-Gilchrist M, Fazzi UG, Leach WJ, Rooney BP, Crowe LA, Murrell GA, McInnes IB. IL-17A mediates inflammatory and tissue remodelling events in early human tendinopathy. Sci Rep. 2016;6:27149. doi: 10.1038/srep27149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thankam FG, Roesch ZK, Dilisio MF, Radwan MM, Kovilam A, Gross RM, Agrawal DK. Association of inflammatory responses and ECM disorganization with HMGB1 upregulation and NLRP3 inflammasome activation in the injured rotator cuff tendon. Sci Rep. 2018;8:8918. doi: 10.1038/s41598-018-27250-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thankam FG, Boosani CS, Dilisio MF, Dietz NE, Agrawal DK. MicroRNAs associated with shoulder tendon matrisome disorganization in glenohumeral arthritis. PLoS One. 2016;11:e0168077. doi: 10.1371/journal.pone.0168077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Järvinen M, Józsa L, Kannus P, Järvinen TL, Kvist M, Leadbetter W. Histopathological findings in chronic tendon disorders. Scand J Med Sci Sports. 1997;7:86–95. doi: 10.1111/j.1600-0838.1997.tb00124.x. [DOI] [PubMed] [Google Scholar]
- 9.Lian Ø, Scott A, Engebretsen L, Bahr R, Duronio V, Khan K. Excessive apoptosis in patellar tendinopathy in athletes. Am J Sports Med. 2007;35:605–611. doi: 10.1177/0363546506295702. [DOI] [PubMed] [Google Scholar]
- 10.Kjaer M, Magnusson P, Krogsgaard M, Boysen Møller J, Olesen J, Heinemeier K, Hansen M, Haraldsson B, Koskinen S, Esmarck B, Langberg H. Extracellular matrix adaptation of tendon and skeletal muscle to exercise. J Anat. 2006;208:445–450. doi: 10.1111/j.1469-7580.2006.00549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma P, Maffulli N. Tendon injury and tendinopathy: healing and repair. J Bone Joint Surg Am. 2005;87:187–202. doi: 10.2106/JBJS.D.01850. [DOI] [PubMed] [Google Scholar]
- 12.Wang JH. Mechanobiology of tendon. J Biomech. 2006;39:1563–1582. doi: 10.1016/j.jbiomech.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 13.Freedman BR, Rodriguez AB, Leiphart RJ, Newton JB, Ban E, Sarver JJ, Mauck RL, Shenoy VB, Soslowsky LJ. Dynamic loading and tendon healing affect multiscale tendon properties and ECM stress transmission. Sci Rep. 2018;8:8918. doi: 10.1038/s41598-018-29060-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thankam FG, Dilisio MF, Agrawal DK. Immunobiological factors aggravating the fatty infiltration on tendons and muscles in rotator cuff lesions. Mol Cell Biochem. 2016;417:17–33. doi: 10.1007/s11010-016-2710-5. [DOI] [PubMed] [Google Scholar]
- 15.Thankam FG, Chandra IS, Kovilam AN, Diaz CG, Volberding BT, Dilisio MF, Radwan MM, Gross RM, Agrawal DK. Amplification of mitochondrial activity in the healing response following rotator cuff tendon injury. Sci Rep. 2018;8:17027. doi: 10.1038/s41598-018-35391-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schatzker J, Brånemark PI. Intravital observations on the microvascular anatomy and microcirculation of the tendon. Acta Orthop Scand Suppl. 1969;126:1–23. doi: 10.3109/ort.1969.40.suppl-126.01. [DOI] [PubMed] [Google Scholar]
- 17.Thankam FG, Evan DK, Agrawal DK, Dilisio MF. Collagen type III content of the long head of the biceps tendon as an indicator of glenohumeral arthritis. Mol Cell Biochem. 2018 doi: 10.1007/s11010-018-3449-y. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 18.Duance VC, Restall DJ, Beard H, Bourne FJ, Bailey AJ. The location of three collagen types in skeletal muscle. FEBS Lett. 1977;79:248–252. doi: 10.1016/0014-5793(77)80797-7. [DOI] [PubMed] [Google Scholar]
- 19.Fan L, Sarkar K, Franks DJ, Uhthoff HK. Estimation of total collagen and types I and III collagen in canine rotator cuff tendons. Calcif Tissue Int. 1997;61:223–9. doi: 10.1007/s002239900327. [DOI] [PubMed] [Google Scholar]
- 20.Birk DE, Fitch JM, Babiarz JP, Doane KJ, Linsenmayer TF. Collagen fibrillogenesis in vitro: interaction of types I and V collagen regulates fibril diameter. J Cell Sci. 1990;95:649–657. doi: 10.1242/jcs.95.4.649. [DOI] [PubMed] [Google Scholar]
- 21.Benjamin M, Ralphs JR. Functional and developmental anatomy of tendons and ligaments. Research Gate. 1995;1995:185–203. [Google Scholar]
- 22.Quigley AS, Bancelin S, Deska-Gauthier D, Légaré F, Kreplak L, Veres SP. In tendons, differing physiological requirements lead to functionally distinct nanostructures. Sci Rep. 2018;8:4409. doi: 10.1038/s41598-018-22741-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Canty EG. Procollagen trafficking, processing and fibrillogenesis. J Cell Sci. 2005;118:1341–1353. doi: 10.1242/jcs.01731. [DOI] [PubMed] [Google Scholar]
- 24.Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J. Overview of the secretory pathway. 2000 [Google Scholar]
- 25.Chang SW, Shefelbine SJ, Buehler MJ. Structural and mechanical differences between collagen homo- and heterotrimers: relevance for the molecular origin of brittle bone disease. Biophys J. 2012;102:640–648. doi: 10.1016/j.bpj.2011.11.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fleischmajer R, Perlish JS, Timpl R, Olsen BR. Procollagen intermediates during tendon fibrillogenesis. J Histochem Cytochem. 1988;36:1425–32. doi: 10.1177/36.11.3049791. [DOI] [PubMed] [Google Scholar]
- 27.Nagata K. Hsp47: a collagen-specific molecular chaperone. Trends Biochem Sci. 1996;21:23–26. doi: 10.1016/0968-0004(96)80881-4. [DOI] [PubMed] [Google Scholar]
- 28.Hendershot LM, Bulleid NJ. Protein-specific chaperones: the role of hsp47 begins to gel. Curr Biol. 2000;10:R912–R915. doi: 10.1016/s0960-9822(00)00850-2. [DOI] [PubMed] [Google Scholar]
- 29.Lopez B, Gonzalez A, Diez J. Circulating biomarkers of collagen metabolism in cardiac diseases. Circulation. 2010;121:1645–1654. doi: 10.1161/CIRCULATIONAHA.109.912774. [DOI] [PubMed] [Google Scholar]
- 30.Galat A. Peptidylproline cis-trans-isomerases: immunophilins. Eur J Biochem. 1993;216:689–707. doi: 10.1111/j.1432-1033.1993.tb18189.x. [DOI] [PubMed] [Google Scholar]
- 31.Schwarz RI. Collagen I and the fibroblast: high protein expression requires a new paradigm of post-transcriptional, feedback regulation. Biochem Biophys Rep. 2015;3:38–44. doi: 10.1016/j.bbrep.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fokin AI, Brodsky IB, Burakov AV, Nadezhdina ES. Interaction of early secretory pathway and Golgi membranes with microtubules and microtubule motors. Biochem Mosc. 2014;79:879–893. doi: 10.1134/S0006297914090053. [DOI] [PubMed] [Google Scholar]
- 33.Bannykh SI, Rowe T, Balch WE. The organization of endoplasmic reticulum export complexes. J Cell Biol. 1996;135:19–35. doi: 10.1083/jcb.135.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mironov AA, Mironov AA Jr, Beznoussenko GV, Trucco A, Lupetti P, Smith JD, Geerts WJ, Koster AJ, Burger KN, Martone ME, Deerinck TJ, Ellisman MH, Luini A. ER-to-Golgi carriers arise through direct en bloc protrusion and multistage maturation of specialized ER exit domains. Dev Cell. 2003;5:583–594. doi: 10.1016/s1534-5807(03)00294-6. [DOI] [PubMed] [Google Scholar]
- 35.Stephens DJ, Pepperkok R. Imaging of procollagen transport reveals COPI-dependent cargo sorting during ER-to-Golgi transport in mammalian cells. J Cell Sci. 2002;115:1149–1160. doi: 10.1242/jcs.115.6.1149. [DOI] [PubMed] [Google Scholar]
- 36.Bi X, Corpina RA, Goldberg J. Structure of the Sec23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature. 2002;419:271–277. doi: 10.1038/nature01040. [DOI] [PubMed] [Google Scholar]
- 37.Wendeler MW, Paccaud JP, Hauri HP. Role of Sec24 isoforms in selective export of membrane proteins from the endoplasmic reticulum. EMBO Rep. 2007;8:258–264. doi: 10.1038/sj.embor.7400893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malhotra V, Erlmann P. Protein export at the ER: loading big collagens into COPII carriers: protein export at the ER. EMBO J. 2011;30:3475–3480. doi: 10.1038/emboj.2011.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bard F, Casano L, Mallabiabarrena A, Wallace E, Saito K, Kitayama H, Guizzunti G, Hu Y, Wendler F, DasGupta R, Perrimon N, Malhotra V. Functional genomics reveals genes involved in protein secretion and Golgi organization. Nature. 2006;439:604–607. doi: 10.1038/nature04377. [DOI] [PubMed] [Google Scholar]
- 40.Saito K, Chen M, Bard F, Chen S, Zhou H, Woodley D, Polischuk R, Schekman R, Malhotra V. TANGO1 facilitates cargo loading at endoplasmic reticulum exit sites. Cell. 2009;136:891–902. doi: 10.1016/j.cell.2008.12.025. [DOI] [PubMed] [Google Scholar]
- 41.Saito K, Katada T. Mechanisms for exporting large-sized cargoes from the endoplasmic reticulum. Cell Mol Life Sci. 2015;72:3709–3720. doi: 10.1007/s00018-015-1952-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ishikawa Y, Ito S, Nagata K, Sakai LY, Bächinger HP. Intracellular mechanisms of molecular recognition and sorting for transport of large extracellular matrix molecules. Proc Natl Acad Sci U S A. 2016;113:E6036–E6044. doi: 10.1073/pnas.1609571113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tiwari P, Kumar A, Das RN, Malhotra V, VijayRaghavan K. A tendon cell specific RNAi screen reveals novel candidates essential for muscle tendon interaction. PLoS One. 2015;10:e0140976. doi: 10.1371/journal.pone.0140976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peffers MJ, Thorpe CT, Collins JA, Eong R, Wei TK, Screen HR, Clegg PD. Proteomic analysis reveals age-related changes in tendon matrix composition, with age- and injury-specific matrix fragmentation. J Biol Chem. 2014;289:25867–25878. doi: 10.1074/jbc.M114.566554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pfeffer SR. Cargo carriers from the Golgi to the cell surface: cargo carriers from the Golgi to the cell surface. EMBO J. 2012;31:3954–3955. doi: 10.1038/emboj.2012.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.von Blume J, Alleaume AM, Cantero-Recasens G, Curwin A, Carreras-Sureda A, Zimmermann T, van Galen J, Wakana Y, Valverde MA, Malhotra V. ADF/cofilin regulates secretory cargo sorting at the TGN via the Ca2+ ATPase SPCA1. Dev Cell. 2011;20:652–662. doi: 10.1016/j.devcel.2011.03.014. [DOI] [PubMed] [Google Scholar]
- 47.Lengfeld J, Wang Q, Zohlman A, Salvarezza S, Morgan S, Ren J, Kato K, Rodriguez-Boulan E, Liu B. Protein kinase C regulates the release of collagen type I from vascular smooth muscle cells via regulation of Cdc42. Mol Biol Cell. 2012;23:1955–1963. doi: 10.1091/mbc.E11-06-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Birk DE, Trelstad RL. Extracellular compartments in tendon morphogenesis: collagen fibril, bundle, and macroaggregate formation. J Cell Biol. 1986;103:231–240. doi: 10.1083/jcb.103.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kadler KE, Holmes DF, Graham H, Starborg T. Tip-mediated fusion involving unipolar collagen fibrils accounts for rapid fibril elongation, the occurrence of fibrillar branched networks in skin and the paucity of collagen fibril ends in vertebrates. Matrix Biol. 2000;19:359–65. doi: 10.1016/s0945-053x(00)00082-2. [DOI] [PubMed] [Google Scholar]
- 50.Mouw JK, Ou G, Weaver VM. Extracellular matrix assembly: a multiscale deconstruction. Nat Rev Mol Cell Biol. 2014;15:771–785. doi: 10.1038/nrm3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hulmes DJ. Building collagen molecules, fibrils, and suprafibrillar structures. J Struct Biol. 2002;137:2–10. doi: 10.1006/jsbi.2002.4450. [DOI] [PubMed] [Google Scholar]
- 52.Wenstrup RJ, Florer JB, Brunskill EW, Bell SM, Chervoneva I, Birk DE. Type V collagen controls the initiation of collagen fibril assembly. J Biol Chem. 2004;279:53331–53337. doi: 10.1074/jbc.M409622200. [DOI] [PubMed] [Google Scholar]
- 53.Ansorge HL, Meng X, Zhang G, Veit G, Sun M, Klement JF, Beason DP, Soslowsky LJ, Koch M, Birk DE. Type XIV collagen regulates fibrillogenesis: premature collagen fibril growth and tissue dysfunction in null mice. J Biol Chem. 2009;284:8427–8438. doi: 10.1074/jbc.M805582200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Connizzo BK, Yannascoli SM, Soslowsky LJ. Structure-function relationships of postnatal tendon development: a parallel to healing. Matrix Biol. 2013;32:106–116. doi: 10.1016/j.matbio.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rentz TJ, Poobalarahi F, Bornstein P, Sage EH, Bradshaw AD. SPARC regulates processing of procollagen I and collagen fibrillogenesis in dermal fibroblasts. J Biol Chem. 2007;282:22062–22071. doi: 10.1074/jbc.M700167200. [DOI] [PubMed] [Google Scholar]
- 56.Gehwolf R, Wagner A, Lehner C, Bradshaw AD, Scharler C, Niestrawska JA, Holzapfel GA, Bauer HC, Tempfer H, Traweger A. Pleiotropic roles of the matricellular protein Sparc in tendon maturation and ageing. Sci Rep. 2016;6:32635. doi: 10.1038/srep32635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nebuloni M, Albarello L, Andolfo A, Magagnotti C, Genovese L, Locatelli I, Tonon G, Longhi E, Zerbi P, Allevi R, Podestà A, Puricelli L, Milani P, Soldarini A, Salonia A, Alfano M. Insight on colorectal carcinoma infiltration by studying perilesional extracellular matrix. Sci Rep. 2016;6:22522. doi: 10.1038/srep22522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Retief E, Parker MI, Retief AE. Regional chromosome mapping of human collagen genes alpha 2(I) and alpha 1(I) (COLIA2 and COLIA1) Hum Genet. 1985;69:304–308. doi: 10.1007/BF00291646. [DOI] [PubMed] [Google Scholar]
- 59.Ghosh AK. Factors involved in the regulation of type I collagen gene expression: implication in fibrosis. Exp Biol Med (Maywood) 2002;227:301–14. doi: 10.1177/153537020222700502. [DOI] [PubMed] [Google Scholar]
- 60.Wang V, Davis DA, Yarchoan R. Identification of functional hypoxia inducible factor response elements in the human lysyl oxidase gene promoter. Biochem Biophys Res Commun. 2017;490:480–485. doi: 10.1016/j.bbrc.2017.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu Z, Li C, Meng X, Bai Y, Qi J, Wang J, Zhou Q, Zhang W, Zhang X. Hypoxia-inducible factor-l? Mediates aggrecan and collagen? Expression via NOTCH1 signaling in nucleus pulposus cells during intervertebral disc degeneration. Biochem Biophys Res Commun. 2017;488:554–561. doi: 10.1016/j.bbrc.2017.05.086. [DOI] [PubMed] [Google Scholar]
- 62.Zanotti S, Canalis E. Notch regulation of bone development and remodeling and related skeletal disorders. Calcif Tissue Int. 2012;90:69–75. doi: 10.1007/s00223-011-9541-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang H, Tian Y, Wang J, Phillips KL, Binch AL, Dunn S, Cross A, Chiverton N, Zheng Z, Shapiro IM, Le Maitre CL, Risbud MV. Inflammatory cytokines induce NOTCH signaling in nucleus pulposus cells: implications in intervertebral disc degeneration. J Biol Chem. 2013;288:16761–16774. doi: 10.1074/jbc.M112.446633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zanotti S, Smerdel-Ramoya A, Stadmeyer L, Durant D, Radtke F, Canalis E. Notch inhibits osteoblast differentiation and causes osteopenia. Endocrinology. 2008;149:3890–3899. doi: 10.1210/en.2008-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li X. Physical therapy and rehabilitation after rotator cuff repai: a review of current concepts. Int J Phys Med Rehabil. 2013:1. [Google Scholar]
- 66.Pan X, Chen Z, Huang R, Yao Y, Ma G. Transforming growth factor β1 induces the expression of collagen type i by DNA methylation in cardiac fibroblasts. PLoS One. 2013;8:e60335. doi: 10.1371/journal.pone.0060335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huisman E, Lu A, McCormack RG, Scott A. Enhanced collagen type I synthesis by human tenocytes subjected to periodic in vitro mechanical stimulation. BMC Musculoskelet Disord. 2014;15:386. doi: 10.1186/1471-2474-15-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 69.Beauchef G, Bigot N, Kypriotou M, Renard E, Poree B, Widom R, Dompmartin-Blanchere A, Oddos T, Maquart FX, Demoor M, Boumediene K, Galera P. The p65 subunit of NF-κB inhibits COL1A1 gene transcription in human dermal and scleroderma fibroblasts through its recruitment on promoter by protein interaction with transcriptional activators (c-Krox, Sp1, and Sp3) J Biol Chem. 2012;287:3462–78. doi: 10.1074/jbc.M111.286443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jimenez SA, Varga J, Olsen A, Li L, Diaz A, Herhal J, Koch J. Functional analysis of human alpha 1(I) procollagen gene promoter. Differential activity in collagen-producing and -nonproducing cells and response to transforming growth factor beta 1. J Biol Chem. 1994;269:12684–12691. [PubMed] [Google Scholar]
- 71.Tamaki T, Ohnishi K, Hartl C, LeRoy EC, Trojanowska M. Characterization of a GC-rich region containing Sp1 binding site(s) as a constitutive responsive element of the 2(I) collagen gene in human fibroblasts. J Biol Chem. 1995;270:4299–4304. doi: 10.1074/jbc.270.9.4299. [DOI] [PubMed] [Google Scholar]
- 72.Kypriotou M, Beauchef G, Chadjichristos C, Widom R, Renard E, Jimenez SA, Korn J, Maquart FX, Oddos T, Von Stetten O, Pujol JP, Galéra P. Human collagen Krox up-regulates type I collagen expression in normal and scleroderma fibroblasts through interaction with Sp1 and Sp3 transcription factors. J Biol Chem. 2007;282:32000–32014. doi: 10.1074/jbc.M705197200. [DOI] [PubMed] [Google Scholar]
- 73.Thankam FG, Dilisio MF, Dougherty KA, Dietz NE, Agrawal DK. Triggering receptor expressed on myeloid cells and 5’ adenosine monophosphate-activated protein kinase in the inflammatory response: a potential therapeutic target. Expert Rev Clin Immunol. 2016;12:1239–1249. doi: 10.1080/1744666X.2016.1196138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 75.Rippe RA, Schrum LW, Stefanovic B, Solís-Herruzo JA, Brenner DA. NF-kappaB inhibits expression of the alpha1(I) collagen gene. DNA Cell Biol. 1999;18:751–761. doi: 10.1089/104454999314890. [DOI] [PubMed] [Google Scholar]
- 76.Kouba DJ, Chung KY, Nishiyama T, Vindevoghel L, Kon A, Klement JF, Uitto J, Mauviel A. Nuclear factor-kappa B mediates TNF-alpha inhibitory effect on alpha 2(I) collagen (COL1A2) gene transcription in human dermal fibroblasts. J Immunol Baltim Md 1950. 1999;162:4226–4234. [PubMed] [Google Scholar]
- 77.Mori K, Hatamochi A, Ueki H, Olsen A, Jimenez SA. The transcription of human alpha 1(I) procollagen gene (COL1A1) is suppressed by tumour necrosis factor-alpha through proximal short promoter elements: evidence for suppression mechanisms mediated by two nuclear-factor binding sites. Biochem J. 1996;319:811–816. doi: 10.1042/bj3190811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Varela-Rey M, Montiel-Duarte C, Osés-Prieto JA, López-Zabalza MJ, Jaffrèzou JP, Rojkind M, Iraburu MJ. p38 MAPK mediates the regulation of alpha1(I) procollagen mRNA levels by TNF-alpha and TGF-beta in a cell line of rat hepatic stellate cells(1) FEBS Lett. 2002;528:133–138. doi: 10.1016/s0014-5793(02)03276-3. [DOI] [PubMed] [Google Scholar]
- 79.Bitzer M, von Gersdorff G, Liang D, Dominguez-Rosales A, Beg AA, Rojkind M, Böttinger EP. A mechanism of suppression of TGF-beta/SMAD signaling by NF-kappa B/RelA. Genes Dev. 2000;14:187–197. [PMC free article] [PubMed] [Google Scholar]
- 80.Chen K, Li P, Zhao H, Yan X, Ma Y. Effects of tumor necrosis factor inhibitor on stress-shielded tendons. Orthopedics. 2017;40:49–55. doi: 10.3928/01477447-20160926-03. [DOI] [PubMed] [Google Scholar]
- 81.Chatzikyrkou C, Haller H, Menne J. Fixed-dose combination of lercanidipine/enalapril in the treatment of hypertension in the elderly. Aging Health. 2009;5:141–149. [Google Scholar]
- 82.Nové-Josserand L, Walch G, Adeleine P, Courpron P. Effect of age on the natural history of the shoulder: a clinical and radiological study in the elderly. Rev Chir Orthop Reparatrice Appar Mot. 2005;91:508–514. doi: 10.1016/s0035-1040(05)84440-x. [DOI] [PubMed] [Google Scholar]
- 83.Peng Y, Kim JM, Park HS, Yang A, Islam C, Lakatta EG, Lin L. AGE-RAGE signal generates a specific NF-κB RelA “barcode” that directs collagen I expression. Sci Rep. 2016;6:18822. doi: 10.1038/srep18822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ibrahim ZA, Armour CL, Phipps S, Sukkar MB. RAGE and TLRs: relatives, friends or neighbours? Mol Immunol. 2013;56:739–744. doi: 10.1016/j.molimm.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 85.Lin L. RAGE on the toll road? Cell Mol Immunol. 2006;3:351–358. [PubMed] [Google Scholar]
- 86.Yamamoto Y, Yamamoto H. RAGE-mediated inflammation, type 2 diabetes, and diabetic vascular complication. Front Endocrinol (Lausanne) 2013;4:105. doi: 10.3389/fendo.2013.00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yamamoto Y, Yamagishi S, Yonekura H, Doi T, Tsuji H, Kato I, Takasawa S, Okamoto H, Abedin J, Tanaka N, Sakurai S, Migita H, Unoki H, Wang H, Zenda T, Wu PS, Segawa Y, Higashide T, Kawasaki K, Yamamoto H. Roles of the AGERAGE system in vascular injury in diabetes. Ann N Y Acad Sci. 2000;902:163–170. doi: 10.1111/j.1749-6632.2000.tb06311.x. discussion 170-172. [DOI] [PubMed] [Google Scholar]
- 88.Grözinger G, Schmehl J, Bantleon R, Kehlbach R, Mehra T, Claussen C, Wiesinger B. Decreased neointimal extracellular matrix formation in RAGE-knockout mice after microvascular denudation. Cardiovasc Intervent Radiol. 2012;35:1439–1447. doi: 10.1007/s00270-011-0319-3. [DOI] [PubMed] [Google Scholar]
- 89.Szoka L, Karna E, Palka JA. UVC inhibits collagen biosynthesis through up-regulation of NF-κB p65 signaling in cultured fibroblasts. J Photochem Photobiol B. 2013;129:143–148. doi: 10.1016/j.jphotobiol.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 90.Caley MP, Martins VL, O’Toole EA. Metalloproteinases and wound healing. Adv Wound Care (New Rochelle) 2015;4:225–234. doi: 10.1089/wound.2014.0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yan C, Boyd DD. Regulation of matrix metalloproteinase gene expression. J Cell Physiol. 2007;211:19–26. doi: 10.1002/jcp.20948. [DOI] [PubMed] [Google Scholar]
- 92.Reunanen N, Kähäri VM. Matrix metalloproteinases in cancer cell invasion. Essays in Biochemistry. 2002;38:21. doi: 10.1042/bse0380021. [DOI] [PubMed] [Google Scholar]
- 93.Del Buono A, Oliva F, Osti L, Maffulli N. Metalloproteases and tendinopathy. Muscles Ligaments Tendons J. 2013;3:51–7. doi: 10.11138/mltj/2013.3.1.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jacob J, Eisemon E, Sheibani-Rad S, Patel A, Jacob T, Houeka J. Matrix metalloproteinase levels as a marker for rotator cuff tears. Orthopedics. 2012;35:e474–e478. doi: 10.3928/01477447-20120327-18. [DOI] [PubMed] [Google Scholar]
- 95.Yao J, Xiong S, Klos K, Nguyen N, Grijalva R, Li P, Yu D. Multiple signaling pathways involved in activation of matrix metalloproteinase-9 (MMP-9) by heregulin-beta1 in human breast cancer cells. Oncogene. 2001;20:8066–74. doi: 10.1038/sj.onc.1204944. [DOI] [PubMed] [Google Scholar]
- 96.Ardi VC, Van den Steen PE, Opdenakker G, Schweighofer B, Deryugina EI, Quigley JP. Neutrophil MMP-9 proenzyme, unencumbered by TIMP-1, undergoes efficient activation in vivo and catalytically induces angiogenesis via a basic fibroblast growth factor (FGF-2)/FGFR-2 pathway. J Biol Chem. 2009;284:25854–25866. doi: 10.1074/jbc.M109.033472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yabluchanskiy A, Ma Y, Iyer RP, Hall ME, Lindsey ML. Matrix metalloproteinase-9: many shades of function in cardiovascular disease. Physiology. 2013;28:391–403. doi: 10.1152/physiol.00029.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Crawford HC, Matrisian LM. Mechanisms controlling the transcription of matrix metalloproteinase genes in normal and neoplastic cells. Enzyme Protein. 1996;49:20–37. doi: 10.1159/000468614. [DOI] [PubMed] [Google Scholar]
- 99.Li YF, Xu XB, Chen XH, Wei G, He B, Wang JD. The nuclear factor-κB pathway is involved in matrix metalloproteinase-9 expression in RU486-induced endometrium breakdown in mice. Hum Reprod. 2012;27:2096–2106. doi: 10.1093/humrep/des110. [DOI] [PubMed] [Google Scholar]
- 100.Magra M. Matrix metalloproteases: a role in overuse tendinopathies. Br J Sports Med. 2005;39:789–791. doi: 10.1136/bjsm.2005.017855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Riley G. Tendinopathy--from basic science to treatment. Nat Clin Pract Rheumatol. 2008;4:82–89. doi: 10.1038/ncprheum0700. [DOI] [PubMed] [Google Scholar]
- 102.Ito Y, Toriuchi N, Yoshitaka T, Ueno-Kudoh H, Sato T, Yokoyama S, Nishida K, Akimoto T, Takahashi M, Miyaki S, Asahara H. The Mohawk homeobox gene is a critical regulator of tendon differentiation. Proc Natl Acad Sci U S A. 2010;107:10538–10542. doi: 10.1073/pnas.1000525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Berthet E, Chen C, Butcher K, Schneider RA, Alliston T, Amirtharajah M. Smad3 binds scleraxis and mohawk and regulates tendon matrix organization. J Orthop Res. 2013;31:1475–1483. doi: 10.1002/jor.22382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yoshimoto Y, Takimoto A, Watanabe H, Hiraki Y, Kondoh G, Shukunami C. Scleraxis is required for maturation of tissue domains for proper integration of the musculoskeletal system. Sci Rep. 2017;7:45010. doi: 10.1038/srep45010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Watanabe T, Hosaka Y, Yamamoto E, Ueda H, Sugawara K, Takahashi H, Takehana K. Control of the collagen fibril diameter in the equine superficial digital flexor tendon in horses by decorin. J Vet Med Sci. 2005;67:855–860. doi: 10.1292/jvms.67.855. [DOI] [PubMed] [Google Scholar]
- 106.Banos CC, Thomas AH, Kuo CK. Collagen fibrillogenesis in tendon development: current models and regulation of fibril assembly. Birth Defects Res Part C Embryo Today Rev. 2008;84:228–244. doi: 10.1002/bdrc.20130. [DOI] [PubMed] [Google Scholar]
- 107.Ameye L. Abnormal collagen fibrils in tendons of biglycan/fibromodulin-deficient mice lead to gait impairment, ectopic ossification, and osteoarthritis. FASEB J. 2002;16:673–680. doi: 10.1096/fj.01-0848com. [DOI] [PubMed] [Google Scholar]
- 108.Goldberg M, Septier D, Oldberg A, Young MF, Ameye LG. Fibromodulin-deficient mice display impaired collagen fibrillogenesis in predentin as well as altered dentin mineralization and enamel formation. J Histochem Cytochem. 2006;54:525–537. doi: 10.1369/jhc.5A6650.2005. [DOI] [PubMed] [Google Scholar]
- 109.Ezura Y, Chakravarti S, Oldberg Å, Chervoneva I, Birk DE. Differential expression of lumican and fibromodulin regulate collagen fibrillogenesis in developing mouse tendons. J Cell Biol. 2000;151:779–788. doi: 10.1083/jcb.151.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hansen U, Platz N, Becker A, Bruckner P, Paulsson M, Zaucke F. A secreted variant of cartilage oligomeric matrix protein carrying a chondrodysplasia-causing mutation (p.H587R) disrupts collagen fibrillogenesis. Arthritis Rheum. 2011;63:159–167. doi: 10.1002/art.30073. [DOI] [PubMed] [Google Scholar]
- 111.Sharma P, Maffulli N. Tendon injury and tendinopathy: healing and repair. J Bone Joint Surg Am. 2005;87:187–202. doi: 10.2106/JBJS.D.01850. [DOI] [PubMed] [Google Scholar]
- 112.Eckes B, Nischt R, Krieg T. Cell-matrix interactions in dermal repair and scarring. Fibrogenesis Tissue Repair. 2010;3:4. doi: 10.1186/1755-1536-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kular JK, Basu S, Sharma RI. The extracellular matrix: structure, composition, age-related differences, tools for analysis and applications for tissue engineering. J Tissue Eng. 2014;5:204173141455711. doi: 10.1177/2041731414557112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kim SH, Turnbull J, Guimond S. Extracellular matrix and cell signalling: the dynamic cooperation of integrin, proteoglycan and growth factor receptor. J Endocrinol. 2011;209:139–151. doi: 10.1530/JOE-10-0377. [DOI] [PubMed] [Google Scholar]
- 115.Bosman FT, Stamenkovic I. Functional structure and composition of the extracellular matrix: Structure and composition of the extracellular matrix. J Pathol. 2003;200:423–428. doi: 10.1002/path.1437. [DOI] [PubMed] [Google Scholar]
- 116.Saitta B, Gaidarova S, Cicchillitti L, Jimenez SA. CCAAT binding transcription factor binds and regulates human COL1A1 promoter activity in human dermal fibroblasts: demonstration of increased binding in systemic sclerosis fibroblasts. Arthritis Rheum. 2000;43:2219–2229. doi: 10.1002/1529-0131(200010)43:10<2219::AID-ANR9>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 117.Rossi P, Karsenty G, Roberts AB, Roche NS, Sporn MB, de Crombrugghe B. A nuclear factor 1 binding site mediates the transcriptional activation of a type I collagen promoter by transforming growth factor-beta. Cell. 1988;52:405–414. doi: 10.1016/s0092-8674(88)80033-3. [DOI] [PubMed] [Google Scholar]
- 118.Liska DJ, Slack JL, Bornstein P. A highly conserved intronic sequence is involved in transcriptional regulation of the alpha 1(I) collagen gene. Cell Regul. 1990;1:487–498. doi: 10.1091/mbc.1.6.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Vo N, Goodman RH. CREB-binding protein and p300 in transcriptional regulation. J Biol Chem. 2001;276:13505–13508. doi: 10.1074/jbc.R000025200. [DOI] [PubMed] [Google Scholar]
- 120.Kypreos KE, Nugent MA, Sonenshein GE. Basic fibroblast growth factor-induced decrease in type I collagen gene transcription is mediated by B-myb. Cell Growth Differ. 1998;9:723–30. [PubMed] [Google Scholar]
- 121.Yang BS, Geddes TJ, Pogulis RJ, de Crombrugghe B, Freytag SO. Transcriptional suppression of cellular gene expression by c-Myc. Mol Cell Biol. 1991;11:2291–2295. doi: 10.1128/mcb.11.4.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kobayashi A, Sogawa K, Imataka H, Fujii-Kuriyama Y. Analysis of functional domains of a GC box-binding protein, BTEB. J Biochem (Tokyo) 1995;117:91–95. doi: 10.1093/oxfordjournals.jbchem.a124727. [DOI] [PubMed] [Google Scholar]
- 123.Bergman BL, Scott RW, Bajpai A, Watts S, Baker JB. Inhibition of tumor-cell-mediated extracellular matrix destruction by a fibroblast proteinase inhibitor, protease nexin I. Proc Natl Acad Sci U S A. 1986;83:996–1000. doi: 10.1073/pnas.83.4.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang Q, Raghow R. Okadaic acid-induced transcriptional downregulation of type I collagen gene expression is mediated by protein phosphatase 2A. Mol Cell Biochem. 1996;158:33–42. doi: 10.1007/BF00225880. [DOI] [PubMed] [Google Scholar]
- 125.Kern B, Shen J, Starbuck M, Karsenty G. Cbfa1 contributes to the osteoblast-specific expression of type I collagen genes. J Biol Chem. 2001;276:7101–7107. doi: 10.1074/jbc.M006215200. [DOI] [PubMed] [Google Scholar]
- 126.Shi-wen X, Pennington D, Holmes A, Leask A, Bradham D, Beauchamp JR, Fonseca C, du Bois RM, Martin GR, Black CM, Abraham DJ. Autocrine overexpression of CTGF maintains fibrosis: RDA analysis of fibrosis genes in systemic sclerosis. Exp Cell Res. 2000;259:213–224. doi: 10.1006/excr.2000.4972. [DOI] [PubMed] [Google Scholar]
- 127.Raghavan SS, Woon CY, Kraus A, Megerle K, Pham H, Chang J. Optimization of human tendon tissue engineering: synergistic effects of growth factors for use in tendon scaffold repopulation. Plast Reconstr Surg. 2012;129:479–489. doi: 10.1097/PRS.0b013e31823aeb94. [DOI] [PubMed] [Google Scholar]
- 128.Halper J. Advances in the use of growth factors for treatment of disorders of soft tissues. In: Halper J, editor. Progress in heritable soft connective tissue diseases. Dordrecht: Springer Netherlands; 2014. pp. 59–76. [Google Scholar]
- 129.Nourissat G, Berenbaum F, Duprez D. Tendon injury: from biology to tendon repair. Nat Rev Rheumatol. 2015;11:223–233. doi: 10.1038/nrrheum.2015.26. [DOI] [PubMed] [Google Scholar]
- 130.Dahlgren LA, Mohammed HO, Nixon AJ. Temporal expression of growth factors and matrix molecules in healing tendon lesions. J Orthop Res. 2005;23:84–92. doi: 10.1016/j.orthres.2004.05.007. [DOI] [PubMed] [Google Scholar]