

Abstract
MicroRNAs (miRNAs) play critical roles in the tumorigenesis and progression of oral squamous cell carcinoma (OSCC). MiR-106a* functions as a tumor suppressor miRNA in several cancers; however, its role in OSCC has not been elucidated. We investigated the role of miR-106a* in human OSCC and explored its relevant mechanisms. The expression of miR-106a* was significantly downregulated in OSCC tissues and cell lines. The overexpression of miR-106a* inhibited OSCC cell proliferation and the cell cycle G1-S transition, and induced apoptosis. In contrast, inhibition of miR-106a* promoted cell proliferation and G1-S transition and suppressed apoptosis. The expression of miR-106a* inversely correlated with methyl-CpG binding protein 2 (MeCP2) expression in OSCC tissues. Using a luciferase reporter assay, MeCP2 was determined to be a direct target of miR-106a*. Overexpression of miR-106a* decreased MeCP2 expression at both the mRNA and protein levels, while inhibition of miR-106a* increased MeCP2 expression. Importantly, overexpression of MeCP2 eliminated the effects of miR-106a* overexpression in OSCC cells and silencing of MeCP2 recapitulated the cellular and molecular effects observed with miR-106a* overexpression. MeCP2 may promote OSCC cell proliferation by activating the Wnt/β-Catenin signaling pathway. Taken together, our study demonstrated that miR-106a* inhibited OSCC cell proliferation by suppression of the Wnt/β-Catenin signaling pathway and induced apoptosis through regulation of Caspase 3/9 expression via targeting MeCP2. These findings suggest that miR-106a* acted as a tumor suppressor in the progression of OSCC and may be a potential new target for OSCC diagnosis and therapy.
Keywords: Oral squamous cell carcinoma (OSCC), methyl-CpG binding protein 2 (MeCP2), miR-106a*, proliferation, apoptosis
Introduction
Oral squamous cell carcinoma (OSCC), which originates from the squamous epithelium of the gingiva, tongue, and floor of mouth, is a common head and neck cancer that has a poor prognosis due to recurrence [1]. More than 90% of all oral cancers are diagnosed as OSCC with it being ranked as the sixth most common cancer worldwide and having high mortality rates [2,3]. Although systemic therapeutic strategies, including surgery, radiotherapy, and chemotherapy, have been developed for treating patients with OSCC, the 5-year survival rate remains less than 50% due to the lack of effective treatments [4]. OSCC progression involves a multistep transformational change involving multiple type of genes, including oncogenes, tumor suppressor genes, and cancer-related genes [5]. Therefore, to improve the efficacy of treatment of OSCC, a better understanding of the molecular mechanisms involved in OSCC carcinogenesis and progression is needed.
MicroRNAs (miRNAs) are highly conserved, endogenous non-coding, single-stranded RNAs of 18-24 nucleotides that can serve as pivotal gene regulators in mammals and other multicellular organisms [6,7]. Regulation of gene expression by miRNAs may occur at the posttranscriptional or translational levels through the binding to complimentary sequences of the 3’-untranslated regions (3’-UTRs) of target mRNAs and can influence various physiological and pathological processes [8-10]. Numerous studies have reported that miRNAs are able to act as oncogenes or tumor suppressors and participate in the development of cancers by regulating tumor cell proliferation, survival, differentiation, apoptosis, metabolism, and other biological processes by suppressing transcription or degrading the mRNAs of oncogenes or tumor suppressor genes [11-13]. Previous studies have shown that the dysregulation of miRNAs plays an important role in OSCC progression. Recently, several studies found that miR-106a* serves as a tumor suppressor gene in esophageal carcinoma and renal carcinoma [14,15]. However, the roles and molecular mechanisms of miR-106a* in the development and progression of OSCC remain to be elucidated.
In the current study, we examined the expression of miR-106a* in clinical human OSCC tissues and their matched adjacent normal tissues and investigated the function of miR-106a* in OSCC cell lines. We found that the expression of miR-106a* was significantly downregulated in OSCC tissues and correlated with clinicopathological characteristics. In addition, our results showed that Methyl-CpG binding protein 2 (MeCP2) was overexpressed in OSCC tissues compared with that of matched adjacent normal tissues. We hypothesized that miR-106a* was able to target MeCP2, which was confirmed using bioinformatics software (RegRNA and TargetScan). MeCP2, a member of methyl-CpG-binding domain (MBD) family, is an abundantly present mammalian protein with two main domains, the MBD and a transcriptional repression domain (TRD) [16]. MeCP2 is reported to be a master regulator of gene expression by binding to methylated DNA or gene promoters [17]. Emerging evidence demonstrates that MeCP2 acts as a key oncogene in several cancers, including liver cancer, colorectal cancer, and gastric cancer [18-20]. We found that miR-106a* potently inhibited human OSCC cell proliferation, induced G1-S cell cycle arrest, and promoted cell apoptosis. More importantly, to our knowledge, we provide evidence for the first time that MeCP2 was a direct and functional target of miR-106a*. Our findings suggest that miR-106a* may be a novel therapeutic target for OSCC therapy.
Materials and methods
Human OSCC samples
Human OSCC samples (n = 68) and adjacent normal tissues were collected at the Department of Stomatology, the First Affiliated Hospital, Xi’an Jiaotong University, China. Informed consent was obtained from each patient prior to surgery. The tissues were stored at -80°C. Clinicopathological data on the patients were obtained by reviewing pathology records. The study was approved by the Ethical Committee of the First Affiliated Hospital, Xi’an Jiaotong University and committee guidelines were followed.
Cell culture
Human OSCC cell lines, Cal-27, Tca-8113, and HEK293, were purchased from the Cell Bank (Shanghai Genechem Co., Ltd., Shanghai, China). The cell lines have been screened and authenticated by the Cell Bank. The cell lines were grown in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS; Gibco BRL, NY, USA) and cultured at 37°C in an incubator under 5% CO2.
RNA extraction and quantitative real-time PCR
Total RNA of the human OSCC tissue specimens and cultured cells was extracted using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocols. Expression of miR-106a* and MeCP2 mRNA was measured using an SYBR Premix Ex Taq II Kit (Takara, China). Real-time quantitative reverse transcription PCR (qRT-PCR) was performed using an iCycler iQ Multicolor qRT-PCR System (Bio-Rad, USA). The data was normalized to RNU6B (U6) or GAPDH gene expression. The primers and their sequences included the miR-106a* reverse-transcription primer (5’-GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGACGTAAGAA-3’), miR-106a* PCR forward primer (5’-ATCCAGTGCGTGTCGTG-3’), miR-106a* PCR reverse primer (5’-TGCTCTGCAATGTAAGCAC-3’), U6 reverse-transcription primer (5’-CGCTTCACGAATTTGCGTGTCAT-3’), U6 PCR forward primer (5’-GCTTCGGCAGCACATATACTAAAAT-3’), U6 PCR reverse primer (5’-CGCTTCACGAATTTGCGTGTCAT-3’), MeCP2 PCR forward primer (5’-GCCGAGAGCTATGGACAGCA-3’), MeCP2 PCR reverse primer, (5’-CCAACCTCAGACAGGTTTCCAG-3’), GAPDH PCR forward primer, (5’-GAAGGTGAAGGTCGGAGTCA-3’), GAPDH PCR reverse primer, (5’-TTGAGGTCAATGAAGGGGTC-3’). The data were statistically analyzed using Student’s t-test and graphed with the Graphpad Prism 5.0 software. Chi-square test was used to analyze the relationships between miR-106a* expression and clinicopathological characteristics. The correlation between miR-106a* and MeCP2 in OSCC tissues was estimated using Pearson’s correlation analysis.
Anti-miR-106a*/MeCP2 siRNA synthesis and transfection
A small interfering RNA (siRNA) oligonucleotide, anti-miR-106a*, was synthesized by Gene Pharma (Shanghai, China) and used as an miR-106a* inhibitor. The anti-miR-106a* sequence was 5’-GUAAGAAGUGCUUACAUUGCAG-3’. Scramble siRNA (anti-miR-Control) with the sequence 5’-CAGUACUUUUGUGUAGUACAA-3’ was used as a negative control. The RNA oligonucleotides were transfected into human OSCC Tca-8113 cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). To silence the MeCP2 gene, specific siRNAs were used. Human MeCP2 siRNAs of sense (5’-GCUUAAGCAAAGGAAAUCUTT-3’) and antisense (5’-AGAUUUCCUUUGCUUAAGCTT-3’); and negative control siRNA (NC-siRNA) of sense (5’-UUCUCCGAACGUGUCACGUTT-3’) and antisense (5’-ACGUGACACGUUCGGAGAATT-3’) were synthesized by GenePharma Corporation. The siRNAs were diluted to 70 nM and transfected into OSCC Tca-8113 cells using Lipofectamine 2000.
Vector construction and transfection
The Hsa-miR-106a* precursor expression vector (miR-106a*) and the empty vector (Control vector) were constructed using synthetic oligonucleotides and then inserted into plasmid pcDNA6.2-GW/Empiric according to the manufacturer’s instructions. Full-length complementary human MeCP2 DNA was cloned into the pCMV2-GV146 vector (Genechem Co. Ltd, Shanghai, China). The plasmids were transfected into the cells using Lipofectamine 2000 according to the manufacturer’s instructions.
Dual-luciferase reporter assay
The binding site for miR-106a* in the 3’-UTR of MeCP2 was constructed by Beijing AuGCT DNA-SYN Biotechnology Co. Ltd (Beijing, China), cloned into the pmirGLO Dual-Luciferase vector, and named MeCP2-WT. The mutated 3’-UTR sequences of MeCP2 were also cloned into the pmirGLO Dual-Luciferase vector and named MeCP2-MT. The pre-miR-106a* expression vector and WT or MT reporter plasmids were cotransfected into HEK293 cells. The cells were harvested 24 h post transfection. The Dual-Luciferase Assay System (Promega, Madison, WI, USA) was used according to the manufacturer’s protocol for detecting reporter activity. The data were analyzed using Student’s t-test.
MTT proliferation assay
Human OSCC Tca-8113 cells (3,500 cells/well) in 100 μl DMEM were plated into 96-well plates and incubated for 24 h. The cells were transfected with Control vector, miR-106a*, anti-miR-Control, anti-miR-106a*, NC-siRNA (70 nM), MeCP2 siRNA (70 nM), or miR-106a*+MeCP2 expression vectors for 24, 48, and 72 h. Cell viability was evaluated using MTT assays and a FLUOstar OPTIMA microplate reader (BMG Labtech, Germany) at a wavelength of 492 nm. The data were statistically analyzed using Student’s t-test or one-way ANOVA.
Cell counting assay
To measure cell proliferation, 2.0 × 105 cells were seed into 60-mm diameter plates and cultured for 24 h. Tca-8113 cells were transfected with Control vector, miR-106a*, anti-miR-Control, anti-miR-106a*, NC-siRNA (70 nM), MeCP2 siRNA (70 nM), or miR-106a*+MeCP2 expression vectors. The number of cells was determined at 24, 48, and 72 h post transfection using a Countess Automated Cell Counter (Life Technologies Corp., Carlsbad, USA). The data were analyzed using Student’s t-test or one-way ANOVA.
Cell cycle analysis
The Tca-8113 cells were seeded into 6-well plates and transfected for 48 h. The cells were harvested, washed in PBS, and fixed in 70% ice-cold ethanol at 4°C. The cells were then washed in PBS and stained with 50 μg/ml propidium iodide containing 50 μg/ml RNase A (DNase free) for 15 min at room temperature. The stained cells were analyzed by fluorescence-activated cell sorting (BD Biosciences, USA). The cell-cycle populations were analyzed by using ModFit software. The data were analyzed using Student’s t-test or one-way ANOVA.
Apoptosis analysis
Tca-8113 cells were plated in triplicate into 6-well plates and transfected for 48 h. Cell apoptosis was evaluated using an Annexin-V FITC Apoptosis Detection Kit (Invitrogen, USA) according to the manufacturer’s instructions. The stained cells were counted using a flow cytometer (BD Biosciences, USA) and analyzed for apoptosis using the ModFit software. The data were analyzed using Student’s t-test or one-way ANOVA.
Western blotting
Human OSCC cells or the clinical tissue samples were lysed using RIPA lysis buffer (Sigma, USA). Total protein was separated using 10% SDS polyacrylamide gels and electroblotted to nitrocellulose membranes. The membranes were incubated with primary antibodies at 4°C. The primary antibodies included rabbit polyclonal anti-MeCP2 (1:1,000; Santa Cruz Biotechnology, CA, USA), rabbit polyclonal anti-Wnt (1:1,000; Santa Cruz Biotechnology), rabbit monoclonal anti-β-Catenin (1:1,000; Cell Signaling Technology, USA), mouse monoclonal anti-Cyclin D1 (1:1,000; Santa Cruz Biotechnology), rabbit monoclonal anti-Caspase-9 (1:1,000; Cell Signaling Technology), rabbit monoclonal anti-Caspase-3 (1:1,000; Cell Signaling Technology), and mouse monoclonal anti-GAPDH (1:5,000; Santa Cruz Biotechnology). The membranes were incubated in the dark with ECL (Pierce, Rockford, IL, USA) for chemiluminescence detection. The luminescent signal was detected by CCD camera, recorded and quantified with Syngene GBox (Syngene, UK).
Statistical analysis
All data were analyzed using SPSS 21.0 statistical software. The experiments were performed at a minimum in triplicate. The data are depicted as mean ± SEM from three independent experiments. All data were statistically analyzed using Student’s t-test or one-way ANOVA. Chi-square test was used to analyze the relationships between miR-106a* expression and clinicopathological characteristics. The correlation between miR-106a* and MeCP2 in OSCC tissues was estimated using Pearson’s correlation analysis. P < 0.05 was considered statistically significant.
Results
MiR-106a* was frequently downregulated in human OSCC and correlated with the clinicopathological features
To explore the role of miR-106a* in the progression of OSCC, we first performed qRT-PCR to examine miR-106a* expression in clinical OSCC specimens (n = 68), adjacent normal tissues (n = 68), and tumor cell lines. The results showed that miR-106a* expression was remarkably downregulated in 76.5% (52/68) of the OSCC specimens compared to that of the adjacent normal tissues (Figure 1A, P < 0.001). Additional evaluation revealed the relationship between miR-106a* expression and the clinicopathological features of OSCC patients. As shown in Table 1, low miR-106a* expression was associated with poor tumor histology [well: 65.6% (21/32); moderate: 78.9% (15/19); poor: 94.1% (16/17)]. However, miR-106a* expression was not associated with age, gender, or pTNM stage. Additionally, miR-106a* expression was significantly downregulated in human OSCC cell lines (Cal-27 and Tca-8113) as compared with that in normal oral mucosa cells (Figure 1B, P < 0.001). Therefore, miR-106a* may act as a potential tumor suppressor in OSCC.
Figure 1.
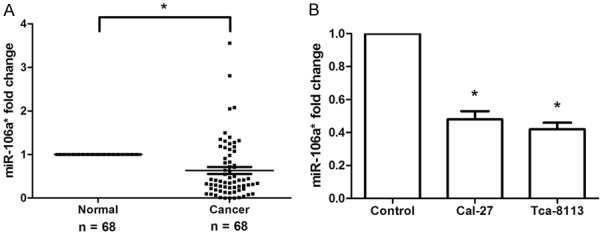
miR-106a* was downregulated in human OSCC tissues and cell lines. A. qRT-PCR analysis revealed that miR-106a* expression was significantly decreased in 68 human OSCC tissues as compared with its adjacent normal tissues. B. Expression of miR-106a* was remarkably reduced in human OSCC cell lines (Cal-27 and Tca-8113) as compared with normal oral mucosa cells (Control) was analyzed using qRT-PCR. *P < 0.001.
Table 1.
The correlation between miR-106a* expression and clinicopathological features of OSCC patients
| Characteristics | Number of cases | miR-106a* expression | P-value | |
|---|---|---|---|---|
|
| ||||
| High (n = 16) | Low (n = 52) | |||
| Age | 0.728 | |||
| ≥ 60 years | 29 | 6 | 23 | |
| < 60 years | 39 | 10 | 29 | |
| Gender | 0.635 | |||
| Male | 43 | 9 | 34 | |
| Female | 25 | 7 | 18 | |
| Histology | 0.017 | |||
| Well | 32 | 11 | 21 | |
| Moderate | 19 | 4 | 15 | |
| Poor | 17 | 1 | 16 | |
| pTNM Stage | 0.536 | |||
| I | 20 | 5 | 15 | |
| II | 16 | 3 | 13 | |
| III | 13 | 4 | 9 | |
| IV | 19 | 4 | 15 | |
MiR-106a* inhibited human OSCC cell proliferation and induced G1-S arrest and apoptosis
We investigated the function of miR-106a* in human OSCC by transfecting Tca-8113 cells with the miR-106a* precursor expression vector, empty vector, miR-106a* antisense oligonucleotides, or negative control oligonucleotides. The expression of miR-106a* was measured after transfection using qRT-PCR. The results showed that the miR-106a* expression was significantly increased in Tca-8133 cells transfected with miR-106a* vector compared with that of cells transfected with empty vector (Figure 2A, P < 0.01). However, there were no significant differences between the anti-miR-106a*-transfected group of cells and the anti-miR-Control-transfected group of cells (Figure 2B). MTT assays revealed that overexpression of miR-106a* remarkably suppressed the cell activity of OSCC Tca-8113 cells at 48 and 72 h post transfection and that anti-miR-106a* treatment promoted Tca-8113 the cell activity at 48 and 72 h post transfection (Figure 2C, 2D). A similar trend was observed using cell counting assays. MiR-106a* overexpression significantly inhibited Tca-8113 cell-proliferation while anti-miR-106a*-treatment promoted cell growth (Figure 2E, 2F). Since the cell cycle is involved in controlling cell proliferation, we evaluated the cell-cycle 48 h post transfection using a flow cytometer. Our results demonstrated that miR-106a* overexpression resulted in a marked accumulation of Tca-8113 cells in the G1/G0 phase and a decrease of S and G2/M phase populations (Figure 2G); however, inhibition of miR-106a* caused a significant decrease in the G1/G0 phase population and increased the S and G2/M phase populations (Figure 2H). Evaluation of cell apoptosis showed that the proportions of early-apoptotic and late-apoptotic cells were remarkably increased when miR-106a* was overexpressed (Figure 2I) and significantly decreased when cells were transfected with anti-miR-106a* (Figure 2J). Our findings demonstrated that miR-106a* inhibited human OSCC cell proliferation and induced G1-S cell cycle arrest and cell apoptosis.
Figure 2.
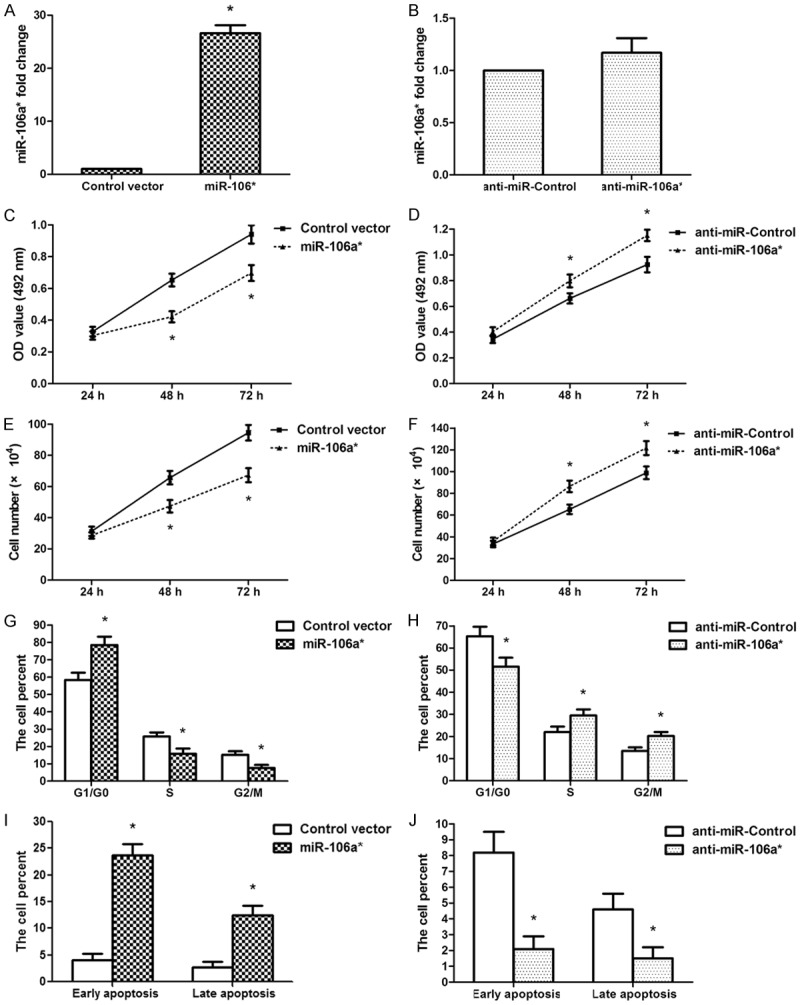
In human OSCC Tca-8113 cells, miR-106a* inhibited cell proliferation and induced G1-S cell cycle arrest. A. Expression of miR-106a* was significantly upregulated in Tca-8113 cells after transfection with miR-106a* expression vector. B. The expression of miR-106a* was no difference in Tca-8113 cells following anti-miR-106a* treatment. C. MTT assays demonstrated that miR-106a* overexpression decreased the cell activity at 48 and 72 h post transfection. D. Anti-miR-106a* increased cell activity at 48 and 72 h post transfection. E. Cell counting assays revealed that miR-106a* overexpression suppressed Tca-8113 cell proliferation. F. Anti-miR-106a*-treatment promoted Tca-8113 cell growth. G. Flow cytometry analysis revealed that the percentages of cells in G1/G0, S, and G2/M phases following the overexpression of miR-106a*. H. The percentage of cells in G1/G0, S, and G2/M phases following the transfection of cells with anti-miR-106a*. I. The data revealed that the percentages of early apoptosis and late apoptosis increased in cells after inducing the overexpression of miR-106a*. J. The data showed that the percentages of early apoptotic and late apoptotic cell decreased following the transfection of cell with anti-miR-106a*. *P < 0.01, n = 3.
MeCP2 was a direct target of miR-106a*
Bioinformatics databases (miRBase and RegRNA) were used to identify several potential target genes of miR-106a*. Among the candidates, MeCP2 was selected for the further analysis. A binding site for miR-106a* was identified in the 3’-UTR of MeCP2 mRNA located at nucleotides 9613-9645 bp (Figure 3A). To verify whether miR-106a* directly targeted MeCP2, a dual-luciferase reporter system containing WT or MT 3’-UTR of MeCP2 was used. HEK293 cells were cotransfected with reporter pmirGLO plasmids and pre-miR-106a* or empty vector (Control vector). It was determined that pre-miR-106a*/WT-MeCP2-UTR-transfected cells demonstrated a significant reduction in luciferase activity, while pre-miR-106a*/MT-MeCP2-UTR-transfected cells failed to show a similar decrease in relative luciferase activity (Figure 3B). This suggested that miR-106a* directly targeted the 3’-UTR of MeCP2. We then measured MeCP2 expression at the mRNA and protein levels. The results demonstrated that the expression of MeCP2 was remarkably upregulated with both mRNA and protein in human OSCC tissues as compared with that in adjacent normal tissues (Figure 3C, 3D, P < 0.001). The association between miR-106a* and MeCP2 was assessed using the data obtained from qRT-PCR assays. A significant inverse correlation was identified between MeCP2 mRNA and miR-106a* expressions (Figure 3E, P < 0.001, r = -0.6881, n = 68).
Figure 3.
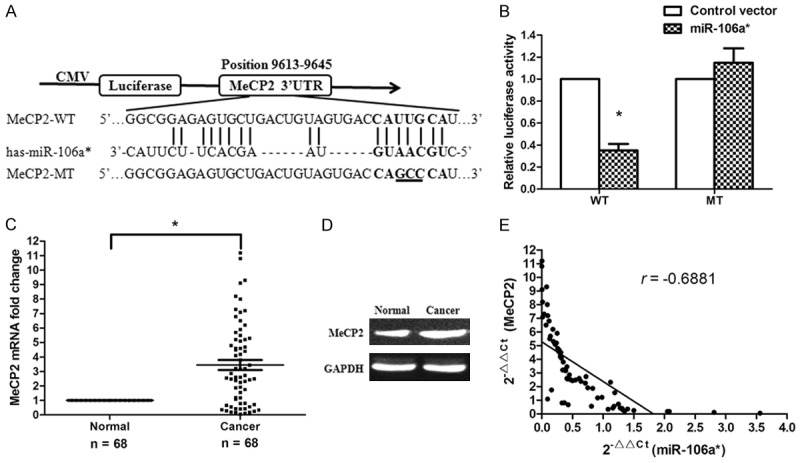
MiR-106a* directly targeted the MeCP2 gene. A. Bioinformatics predicted interactions between miR-106a* and its binding sites in the 3’-UTR of MeCP2. B. The luciferase reporter plasmid containing wild or mutant type MeCP2-3’-UTR was cotransfected into HEK293 cells, in combination with an miR-106a*-expressing plasmid or Control vector. Luciferase activity was evaluated using dual luciferase assays. *P < 0.01. C. MeCP2 mRNA expression was remarkably upregulated in 68 human OSCC tissues. *P < 0.001. D. MeCP2 protein levels were measured by western blotting (n = 18). E. Levels of miR-106a* and MeCP2 were inversely correlated. The 2-ΔΔCt values from qRT-PCR analysis of miR-106a* and MeCP2 were subjected to a Pearson’s correlation analysis (P < 0.001, r = -0.6881, n = 68).
MiR-106a* suppressed OSCC cell growth and induced apoptosis through the Wnt/β-Catenin signaling pathway by targeting MeCP2
Overexpression of miR-106a* significantly downregulated the expression of MeCP2 mRNA in Tca-8113 cells, while anti-miR-106a*-treatment remarkably increased MeCP2 mRNA expression (Figure 4A, P < 0.001). Similar trends were observed with the protein levels (Figure 4B). To further investigate the potential molecular mechanisms of miR-106a* underlying regulation of cell proliferation, cell cycle transition, and apoptosis, western blot analysis was conducted to determine the protein levels of Wnt/β-Catenin signaling pathway-related proteins, G1 regulators, and Caspase 3/9. Our results showed that miR-106a* overexpression decreased the expression levels of Wnt, β-Catenin, and Cyclin D1 proteins in Tca-8113 cells. In contrast, treatment with anti-miR-106a* promoted Wnt, β-Catenin, and Cyclin D1 protein expression. Moreover, we found that miR-106a* also promoted Caspase 3/9 protein expression (Figure 4B). These results indicated that miR-106a* may modulate OSCC cell proliferation, cell cycle, and apoptosis by regulating the MeCP2/Wnt/β-Catenin signaling pathway.
Figure 4.
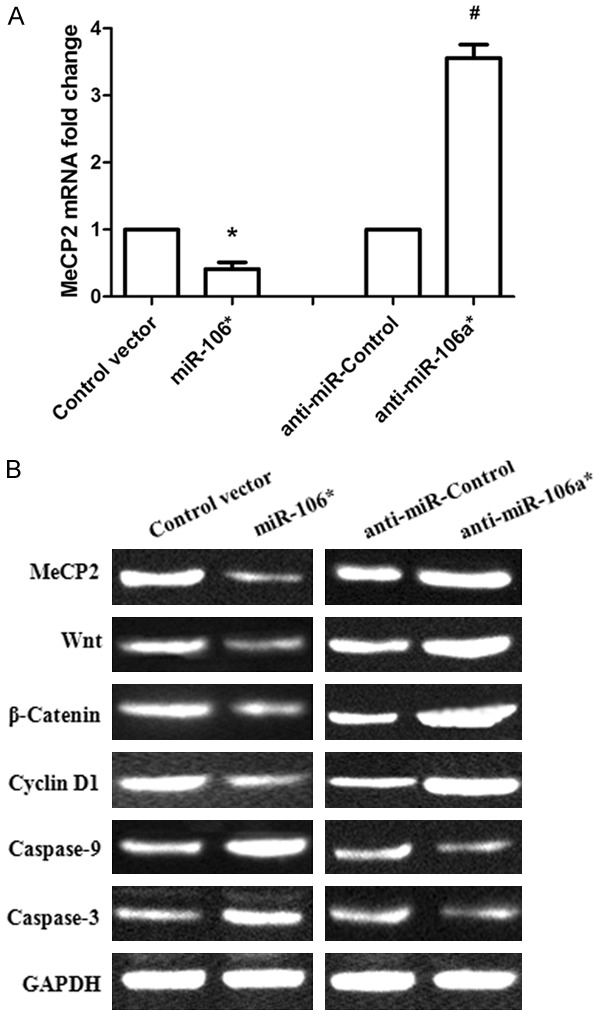
MiR-106a* regulated the Wnt/β-Catenin signaling pathway in human OSCC cells by targeting MeCP2. A. The MeCP2 mRNA levels were determined in OSCC Tca-8113 cells after inducing the overexpression of miR-106a* or treatment with anti-miR-106a*. *P < 0.001, compared with that of the group transfected with Control vector; #P < 0.001, compared with that of the anti-miR-Control group. B. miR-106a* regulated the protein expression of MeCP2, Wnt, β-Catenin, Cyclin D1, and Caspase 3/9 in OSCC Tca-8113 cells.
Overexpression of MeCP2 eliminated the effects of miR-106a* in OSCC Tca-8113 cells
To further demonstrate that miR-106a* exhibited tumor suppressor activity via MeCP2, we constructed an MeCP2 overexpression vector and cotransfected it with miR-106a* into Tca-8113 cells. The overexpression of MeCP2 in Tca-8113 cells rescued the MeCP2 mRNA expression levels that were reduced by miR-106a* (Figure 5A). Following the cotransfection of Tca-8113 cells with the miR-106a* and MeCP2 vectors, we found that the overexpression of MeCP2 counterbalanced the tumor suppressor effects of miR-106a* on cell proliferation (Figure 5B, 5C). The effect of MeCP2 overexpression on cell cycle progression was examined using flow cytometry. It was found that overexpression of MeCP2 disposed the Tca-8113 cells to re-enter S and G2/M phases (Figure 5D). Furthermore, overexpression of MeCP2 eliminated the impact of miR-106a* on Tca-8113 cell apoptosis (Figure 5E). Further analysis revealed that the overexpression of MeCP2 upregulated Wnt, β-Catenin, and Cyclin D1 protein expression and downregulated Caspase 3/9 protein expression compared with that of miR-106a* overexpression (Figure 5F). These results further demonstrated that miR-106a* exhibited a tumor suppressor role through the Wnt/β-Catenin signaling pathway by targeting MeCP2.
Figure 5.
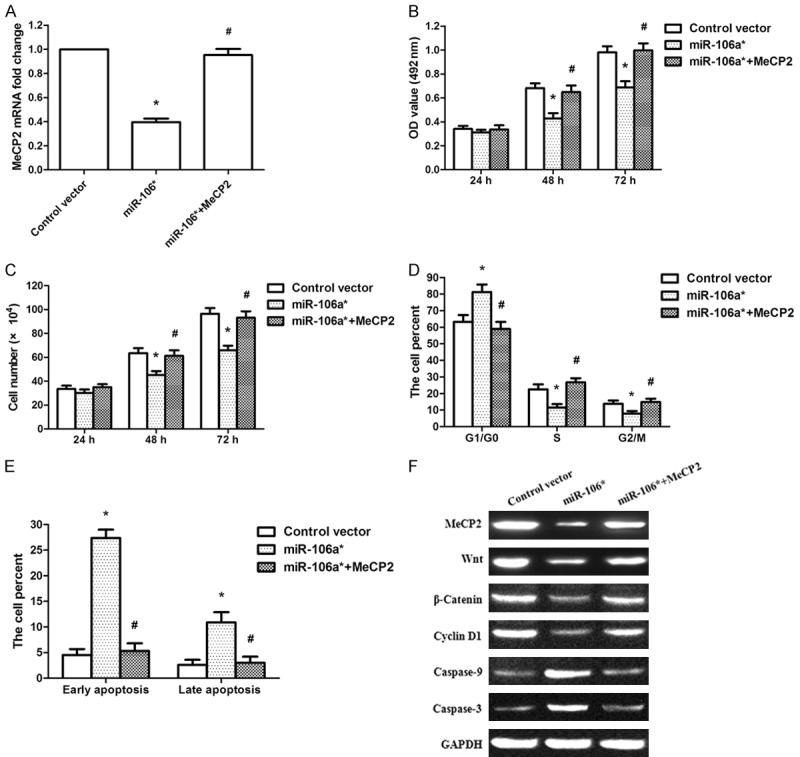
The overexpression of MeCP2 rescued miR-106a*-induced cellular phenotypes in OSCC cells. A. The overexpression of MeCP2 rescued MeCP2 mRNA expression levels reduced by miR-106a*. B. MTT assays showed that the overexpression of MeCP2 counterbalanced the tumor suppressor effects of miR-106a* on cell activity. C. Cell counting assays revealed that the overexpression of MeCP2 counterbalanced the tumor suppressor effects of miR-106a* on cell proliferation. D. Cell cycle indicated that overexpression of MeCP2 disposed the Tca-8113 cells to re-enter S and G2/M phases. E. Apoptosis assays showed that overexpression of MeCP2 eliminated the impact of miR-106a* on Tca-8113 cell apoptosis. F. Expression of MeCP2, Wnt, β-Catenin, Cyclin D1, and Caspase 3/9 proteins were examined after cotransfection with plasmids expressing MeCP2 and miR-106a*. *P < 0.001, compared with that of the group transfected with Control vector group; #P < 0.001, compared with that of the miR-106a* overexpression group, n = 3.
Knockdown of MeCP2 inhibited OSCC Tca-8113 cell proliferation
Because miR-106a* regulated proliferation and apoptosis in OSCC cells and MeCP2 was confirmed as a direct target of miR-106a*, we knocked down MeCP2 expression in OSCC cells using RNA interference to confirm its involvement in the tumor suppressor functions of miR-106a*. The results showed that MeCP2 mRNA expression was specifically suppressed in Tca-8113 cells by the siRNA (Figure 6A). Silencing MeCP2 expression remarkably decreased cellular activity at 48 and 72 h post transfection (Figure 6B). Cell counting assays also showed that silencing MeCP2 significantly inhibited Tca-8113 cell-proliferation (Figure 6C). Silencing of MeCP2 in Tca-8113 cells increased G1/G0 phase populations and decreased S and G2/M phase populations (Figure 6D). Moreover, silencing MeCP2 induced apoptosis in Tca-8113 cells (Figure 6E). These results were similar to those caused by miR-106a* overexpression, indicating a similar effect of MeCP2 knockdown and miR-106a* overexpression. In addition, we analyzed the knockdown efficiency of MeCP2 siRNA at the protein level. The protein expression of MeCP2 was significantly decreased in the siRNA-treated group compared with that of the NC-siRNA-treated group. The Wnt, β-Catenin, and Cyclin D1 protein expression levels were also reduced. However, Caspase 3/9 protein expression increased in the siRNA-treated group compared with that of the NC-siRNA-treated group (Figure 6F).
Figure 6.
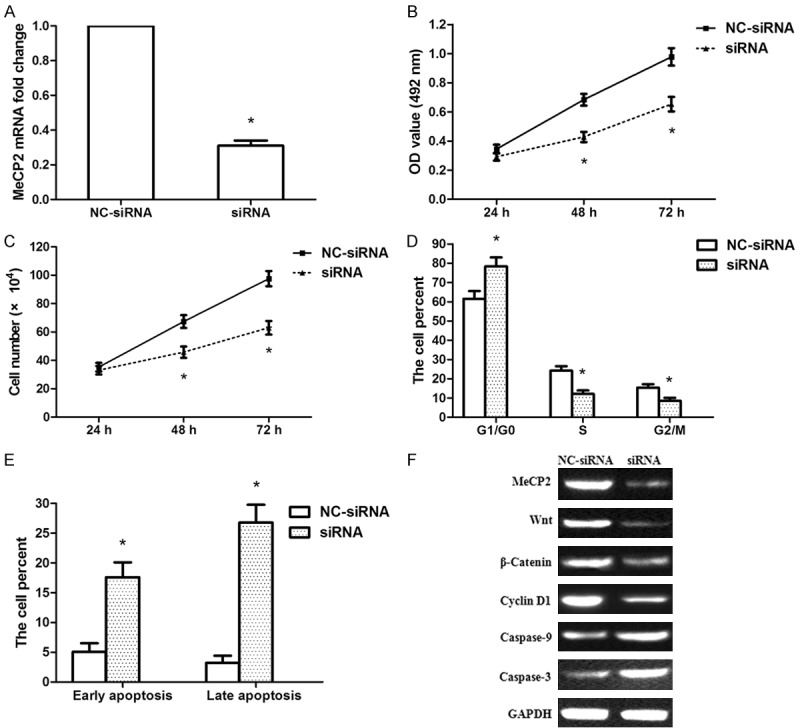
MeCP2 siRNA suppressed the proliferation of human OSCC Tca-8113 cells. A. Results from qRT-PCR showed the knockdown efficiency of MeCP2 siRNA in Tca-8113 cells. B. MTT assays showed that MeCP2 siRNA decreased the activity of Tca-8113 cells at 48 and 72 h post transfection. C. Cell counting assays showed that MeCP2 siRNA inhibited Tca-8113 cell proliferation. D. Flow cytometry analysis revealed the percentage of Tca-8113 cells in G1/G0, S, and G2/M phases. The percentage of G1/G0 phase cells increased following treatment with MeCP2 siRNA and the percentages of S and G2/M phase cells decreased. E. The data showed the percentage of early and late apoptotic cells following treatment with MeCP2 siRNA. F. The expression of MeCP2, Wnt, β-Catenin, Cyclin D1, and Caspase 3/9 proteins were evaluated following treatment with MeCP2 siRNA. *P < 0.01, compared with that of the NC-siRNA-treated group, n = 3.
Discussion
Accumulating evidence demonstrates the importance of miRNAs in the biological characteristics of OSCC, including cell proliferation, survival, differentiation, apoptosis, invasion, and migration [21-24]. Although the clinical significance of miRNAs in OSCC has been well characterized, the roles and underlying molecular mechanism of abnormal miRNAs remain unknown. Therefore, identifying miRNAs and elucidating their biological characteristics in OSCC will help in the search for new targets and the diagnosis and therapy of OSCC. It has been reported that the miR-106a* expression is decreased in follicular lymphoma [25]. MiR-106a* is found to be a tumor suppressor gene in esophageal carcinoma through the targeting of CDK2-associated Cullin 1 [14]. In addition, miR-106a* also inhibits the proliferation of renal carcinoma cells by targeting IRS-2 [15]. However, the clinical significance and function of miR-106a* in the development of OSCC remain unclear. In the current study, we found that miR-106a* expression was significantly downregulated in both OSCC clinical specimens and cell lines. We also analyzed the clinicopathological significance of miR-106a* expression. Our findings showed that low miR-106a* levels were significantly associated with poor tumor histology in OSCC. This study also demonstrated that miR-106a* remarkably inhibited OSCC cell proliferation by inducing G1-S phase arrest and cell apoptosis. The results indicate that miR-106a* played an important role in the development and progression of OSCC.
Our miR-106a* target analysis identified MeCP2 as a direct target of miR-106a*. The role of MeCP2 in cancer has not been well studied. Emerging evidence suggests that MeCP2 as a critical oncogene in several cancers [18,26,27]. In our study, we found that MeCP2 was over expressed in OSCC tissues compared with that in normal tissues and we showed an inverse correlation between MeCP2 expression and miR-106a* expression in OSCC tissues. The results implied that miR-106a* may affect the progression of OSCC by targeting MeCP2. Additional bioinformatics analysis showed that there was an miR-106a*-binding site at nucleotides 9613-9645 of the MeCP2 3’-UTR. Dual-luciferase reporter assays demonstrated that miR-106a* directly targeted MeCP2 by recognizing the 3’-UTR of MeCP2 mRNA and inhibited MeCP2 translation. It has been found that MeCP2 regulates carcinogenesis and progression of neuroblastoma and osteosarcoma [28,29]. Another study detected remarkable upregulation of MeCP2 expression in human hepatocellular carcinoma and gastric cancer tissues and the ability of MeCP2 to promote hepatocellular carcinoma and gastric cancer cell proliferation [18,27]. Our results demonstrated that the overexpression of MeCP2 counterbalanced the tumor suppressor effects of miR-106a* in OSCC cells. In addition, we found that the knockdown of MeCP2 suppressed OSCC cell proliferation by inducing G1-S phase arrest and facilitated cell apoptosis. These findings further verify that miR-106a* functions as a tumor suppressor by repressing MeCP2 expression.
Zhao et al. reported that MeCP2 is able to promote gastric cancer cell proliferation by activating the Wnt/β-Catenin signaling pathway, a key pathway regulating cancer progression [27,30]. The Wnt/β-Catenin downstream-gene Cyclin D1 is a crucial regulatory of the G0/G1 phase [31]. Cyclin D1 controls cell cycle transition from G1 phase to S node [32]. In mammals, cell cycle regulatory gene Cyclin D1 is involved in the growth of various cells. It has been found that Cyclin D1 is upregulated in many types of tumors and that it acts as an oncogene related to tumor occurrence. Furthermore, cell transformation requires amplification of cyclin D1. Our results demonstrated that miR-106a* overexpression and MeCP2 siRNA inhibited the expression of Cyclin D1 and induced G1-S phase arrest through suppressing the Wnt/β-Catenin signaling pathway. In contrast, treatment with anti-miR-106a* and overexpression of MeCP2 promoted the expression of Cyclin D1 and drove more cells into the S phase by activating the Wnt/β-Catenin signaling pathway. Our findings suggest that miR-106a* inhibited G1-S phase transition through suppression of the Wnt/β-Catenin signaling pathway by targeting MeCP2.
The overall proliferation rate of cancer cells is determined by a balance between cell proliferation and cell apoptosis. The Caspase family is the executor of the process of cell apoptosis. Apoptosis signaling cascades are involved in many pathways of the death receptors, mitochondria, and endoplasmic reticulum. In particular, endoplasmic reticulum stress causes a specific cascade including caspase-9 and caspase-3 [33,34]. Caspase-3, a key enzyme in the apoptotic pathway, activates poly (ADP-ribose) polymerase resulting in subsequent apoptotic events and is located downstream of a series of cascades [35,36]. In these cascades, caspase-3 plays an important role. Activated caspase-3 is able to snip DNA and inactivate the relevant proteases in DNA damage repair, thus leading to apoptosis. A previous study found that MeCP2 suppresses gastric cancer cell-apoptosis by inhibiting the caspase-3 signaling pathway [27]. In the current study, we also provided evidence that miR-106a* induced OSCC cell apoptosis via activation of the caspase-3 signaling pathway by targeting MeCP2.
In summary, results from our study demonstrated that miR-106a* functioned as a tumor suppressor gene in OSCC. We determined that miR-106a* was downregulated and associated with clinicopathological characteristics of OSCC patients. We also demonstrated that miR-106a* inhibited OSCC cell proliferation via suppression of the Wnt/β-Catenin signaling pathway and induced apoptosis through activation of the caspase-3 signaling pathway by targeting MeCP2. These findings suggest that miR-106a* plays a significant role in OSCC progression and may serve as a novel potential target for the diagnosis and treatment of OSCC.
Acknowledgements
This study was supported by Shaanxi Province Social Development Scientific and Technological Project Fund (No. 2016SF229).
Disclosure of conflict of interest
None.
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 3.Lu H, Wu B, Ma G, Zheng D, Song R, Huang E, Mao M, Lu B. Melatonin represses oral squamous cell carcinoma metastasis by inhibiting tumor-associated neutrophils. Am J Transl Res. 2017;9:5361–5374. [PMC free article] [PubMed] [Google Scholar]
- 4.Hui Y, Li Y, Jing Y, Feng JQ, Ding Y. miRNA-101 acts as a tumor suppressor in oral squamous cell carcinoma by targeting CX chemokine receptor 7. Am J Transl Res. 2016;8:4902–4911. [PMC free article] [PubMed] [Google Scholar]
- 5.Kong XP, Yao J, Luo W, Feng FK, Ma JT, Ren YP, Wang DL, Bu RF. The expression and functional role of a FOXC1 related mRNA-lncRNA pair in oral squamous cell carcinoma. Mol Cell Biochem. 2014;394:177–186. doi: 10.1007/s11010-014-2093-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer. 2015;15:321–333. doi: 10.1038/nrc3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lai YH, Liu H, Chiang WF, Chen TW, Chu LJ, Yu JS, Chen SJ, Chen HC, Tan BC. MiR-31-5p-ACOX1 axis enhances tumorigenic fitness in oral squamous cell carcinoma via the promigratory prostaglandin E2. Theranostics. 2018;8:486–504. doi: 10.7150/thno.22059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li H, Liu H, Pei J, Wang H, Lv H. miR5423p overexpression is associated with enhanced osteosarcoma cell proliferation and migration ability by targeting Van Goghlike 2. Mol Med Rep. 2015;11:851–856. doi: 10.3892/mmr.2014.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loboda A, Sobczak M, Jozkowicz A, Dulak J. TGF-beta1/Smads and miR-21 in renal fibrosis and inflammation. Mediators Inflamm. 2016;2016:8319283. doi: 10.1155/2016/8319283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao LY, Yao Y, Han J, Yang J, Wang XF, Tong DD, Song TS, Huang C, Shao Y. miR-638 suppresses cell proliferation in gastric cancer by targeting Sp2. Dig Dis Sci. 2014;59:1743–1753. doi: 10.1007/s10620-014-3087-5. [DOI] [PubMed] [Google Scholar]
- 11.Yue H, Tang B, Zhao Y, Niu Y, Yin P, Yang W, Zhang Z, Yu P. MIR-519d suppresses the gastric cancer epithelial-mesenchymal transition via Twist1 and inhibits Wnt/β-catenin signaling pathway. Am J Transl Res. 2017;9:3654–3664. [PMC free article] [PubMed] [Google Scholar]
- 12.Guo H, Zhang X, Chen Q, Bao Y, Dong C, Wang X. miR-132 suppresses the migration and invasion of lung cancer cells by blocking USP9X-induced epithelial-mesenchymal transition. Am J Transl Res. 2018;10:224–234. [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Lee CG. MicroRNA and cancer--focus on apoptosis. J Cell Mol Med. 2009;13:12–23. doi: 10.1111/j.1582-4934.2008.00510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma HL, Wen XP, Zhang XZ, Wang XL, Zhao DL, Che SM, Dang CX. miR-106a* inhibits the proliferation of esophageal carcinoma cells by targeting CDK2-associated Cullin 1 (CACUL1) Cell Mol Biol (Noisy-le-grand) 2015;61:56–62. [PubMed] [Google Scholar]
- 15.Ma Y, Zhang H, He X, Song H, Qiang Y, Li Y, Gao J, Wang Z. miR-106a* inhibits the proliferation of renal carcinoma cells by targeting IRS-2. Tumour Biol. 2015;36:8389–8398. doi: 10.1007/s13277-015-3605-x. [DOI] [PubMed] [Google Scholar]
- 16.Vieira JP, Lopes F, Silva-Fernandes A, Sousa MV, Moura S, Sousa S, Costa BM, Barbosa M, Ylstra B, Temudo T, Lourenco T, Maciel P. Variant Rett syndrome in a girl with a pericentric X-chromosome inversion leading to epigenetic changes and overexpression of the MECP2 gene. Int J Dev Neurosci. 2015;46:82–87. doi: 10.1016/j.ijdevneu.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 17.Mellén M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417–1430. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao LY, Zhang J, Guo B, Yang J, Han J, Zhao XG, Wang XF, Liu LY, Li ZF, Song TS, Huang C. MECP2 promotes cell proliferation by activation ERK1/2 and inhibiting p38 activity in human hepatocellular carcinoma HEPG2 cells. Cell Mol Biol (Noisy-le-grand) 2013;59:OL1876–1881. [PubMed] [Google Scholar]
- 19.Song N, Li K, Wang Y, Chen Z, Shi L. Lentivirus-mediated knockdown of MeCP2 inhibits the growth of colorectal cancer cells in vitro. Mol Med Rep. 2016;13:860–866. doi: 10.3892/mmr.2015.4612. [DOI] [PubMed] [Google Scholar]
- 20.Zhao LY, Tong DD, Xue M, Ma HL, Liu SY, Yang J, Liu YX, Guo B, Ni L, Liu LY, Qin YN, Wang LM, Zhao XG, Huang C. MeCP2, a target of miR-638, facilitates gastric cancer cell proliferation through activation of the MEK1/2-ERK1/2 signaling pathway by upregulating GIT1. Oncogenesis. 2017;6:e368. doi: 10.1038/oncsis.2017.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang X, Ruan H, Hu X, Cao A, Song L. miR-381-3p suppresses the proliferation of oral squamous cell carcinoma cells by directly targeting FGFR2. Am J Cancer Res. 2017;7:913–922. [PMC free article] [PubMed] [Google Scholar]
- 22.Lin J, Lin Y, Fan L, Kuang W, Zheng L, Wu J, Shang P, Wang Q, Tan J. miR-203 inhibits cell proliferation and promotes cisplatin induced cell death in tongue squamous cancer. Biochem Biophys Res Commun. 2016;473:382–387. doi: 10.1016/j.bbrc.2016.02.105. [DOI] [PubMed] [Google Scholar]
- 23.Zeng Q, Tao X, Huang F, Wu T, Wang J, Jiang X, Kuang Z, Cheng B. Overexpression of miR-155 promotes the proliferation and invasion of oral squamous carcinoma cells by regulating BCL6/cyclin D2. Int J Mol Med. 2016;37:1274–1280. doi: 10.3892/ijmm.2016.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu J, Xu JF, Ge WL. MiR-497 enhances metastasis of oral squamous cell carcinoma through SMAD7 suppression. Am J Transl Res. 2016;8:3023–3031. [PMC free article] [PubMed] [Google Scholar]
- 25.Wang W, Corrigan-Cummins M, Hudson J, Maric I, Simakova O, Neelapu SS, Kwak LW, Janik JE, Gause B, Jaffe ES, Calvo KR. MicroRNA profiling of follicular lymphoma identifies microRNAs related to cell proliferation and tumor response. Haematologica. 2012;97:586–594. doi: 10.3324/haematol.2011.048132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neupane M, Clark AP, Landini S, Birkbak NJ, Eklund AC, Lim E, Culhane AC, Barry WT, Schumacher SE, Beroukhim R, Szallasi Z, Vidal M, Hill DE, Silver DP. MECP2 is a frequently amplified oncogene with a novel epigenetic mechanism that mimics the role of activated RAS in malignancy. Cancer Discov. 2016;6:45–58. doi: 10.1158/2159-8290.CD-15-0341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao L, Liu Y, Tong D, Qin Y, Yang J, Xue M, Du N, Liu L, Guo B, Hou N, Han J, Liu S, Liu N, Zhao X, Wang L, Chen Y, Huang C. MeCP2 promotes gastric cancer progression through regulating FOXF1/Wnt5a/beta-Catenin and MYOD1/Caspase-3 signaling pathways. Ebio-Medicine. 2017;16:87–100. doi: 10.1016/j.ebiom.2017.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meng G, Lv Y, Dai H, Zhang X, Guo QN. Epigenetic silencing of methyl-CpG-binding protein 2 gene affects proliferation, invasion, migration, and apoptosis of human osteosarcoma cells. Tumour Biol. 2014;35:11819–11827. doi: 10.1007/s13277-014-2336-8. [DOI] [PubMed] [Google Scholar]
- 29.Murphy DM, Buckley PG, Das S, Watters KM, Bryan K, Stallings RL. Colocalization of the oncogenic transcription factor MYCN and the DNA methyl binding protein MeCP2 at genomic sites in neuroblastoma. PLoS One. 2011;6:e21436. doi: 10.1371/journal.pone.0021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li S, Yang F, Wang M, Cao W, Yang Z. miR-378 functions as an onco-miRNA by targeting the ST7L/Wnt/β-catenin pathway in cervicalcancer. Int J Mol Med. 2017;40:1047–1056. doi: 10.3892/ijmm.2017.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Y, Rao X, Huang K, Jiang X, Wang H, Teng L. FH535 inhibits proliferation and motility of colon cancer cells by targeting Wnt/β-catenin signaling pathway. J Cancer. 2017;8:3142–3153. doi: 10.7150/jca.19273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang K, Liang Q, Li X, Tsoi H, Zhang J, Wang H, Go MY, Chiu PW, Ng EK, Sung JJ, Yu J. MDGA2 is a novel tumour suppressor cooperating with DMAP1 in gastric cancer and is associated with disease outcome. Gut. 2016;65:1619–1631. doi: 10.1136/gutjnl-2015-309276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma J, Zou C, Guo L, Seneviratne DS, Tan X, Kwon YK, An J, Bowser R, DeFrances MC, Zarnegar R. Novel death defying domain in met entraps the active site of caspase-3 and blocks apoptosis in hepatocytes. Hepatology. 2014;59:2010–2021. doi: 10.1002/hep.26769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chai WS, Zhu XM, Li SH, Fan JX, Chen BY. Role of Bcl-2 family members in caspase-3/9-dependent apoptosis during Pseudomonas aeruginosa infection in U937 cells. Apoptosis. 2008;13:833–843. doi: 10.1007/s10495-008-0197-6. [DOI] [PubMed] [Google Scholar]
- 35.Zhang DX, Zhao PT, Xia L, Liu LL, Liang J, Zhai HH, Zhang HB, Guo XG, Wu KC, Xu YM, Jia LT, Yang AG, Chen SY, Fan DM. Potent inhibition of human gastric cancer by HER2-directed induction of apoptosis with anti-HER2 antibody and caspase-3 fusion protein. Gut. 2010;59:292–299. doi: 10.1136/gut.2008.155226. [DOI] [PubMed] [Google Scholar]
- 36.Ling Y, Li X, Yu L, Liang Q, Lin X, Yang X, Wang H, Zhang Y. Sevoflurane exposure in postnatal rats induced long-term cognitive impairment through upregulating caspase-3/cleaved-poly (ADP-ribose) polymerase pathway. Exp Ther Med. 2017;14:3824–3830. doi: 10.3892/etm.2017.5004. [DOI] [PMC free article] [PubMed] [Google Scholar]