

Abstract
Label-free mass spectrometry (MS) has developed into an important tool applied in various fields of biological and life sciences. Several software exist to process the raw MS data into quantified protein abundances, including open source and commercial solutions. Each software includes a set of unique algorithms for different tasks of the MS data processing workflow. While many of these algorithms have been compared separately, a thorough and systematic evaluation of their overall performance is missing. Moreover, systematic information is lacking about the amount of missing values produced by the different proteomics software and the capabilities of different data imputation methods to account for them.
In this study, we evaluated the performance of five popular quantitative label-free proteomics software workflows using four different spike-in data sets. Our extensive testing included the number of proteins quantified and the number of missing values produced by each workflow, the accuracy of detecting differential expression and logarithmic fold change and the effect of different imputation and filtering methods on the differential expression results. We found that the Progenesis software performed consistently well in the differential expression analysis and produced few missing values. The missing values produced by the other software decreased their performance, but this difference could be mitigated using proper data filtering or imputation methods. Among the imputation methods, we found that the local least squares (lls) regression imputation consistently increased the performance of the software in the differential expression analysis, and a combination of both data filtering and local least squares imputation increased performance the most in the tested data sets.
Keywords: proteomics, software workflow, differential expression, logarithmic fold change, imputation, evaluation
Introduction
Mass spectrometry (MS)-based proteomics has developed rapidly during the recent decades. High-resolution MS enables modern-day proteomics to identify and quantify tens of thousands of peptides and thousands of proteins in a single run [1]. MS-powered quantitative proteomics has emerged into an important tool applied in various biosciences (e.g. biology, biochemistry and medicine) [2] and is expected to further evolve with regard to resolution, speed and cost-efficiency [1].
MS technologies can be coarsely divided into two categories: label-based and label-free quantification methods [3]. Although label-based quantification methods, such as SILAC (Stable isotope labeling with amino acids in cell culture) [4], provide undisputable accuracy and robustness, they also have their limitations when compared with the more simple label-free methods. For example, SILAC requires the use of live cell cultures, and compared with label-free methods, most of the label-based methods require more steps in sample preparation and higher sample concentration, are more expensive and can only be performed for a limited number of samples [3, 5]. Label-free methods can be used both in shotgun (discovery analysis of the whole proteome) and in targeted (analysis of a specified set of proteins) proteomics experiments [6] and can be applied even when the metabolic labeling of samples is not possible [5]. So far, the shotgun method has been more popular than the targeted method [6]. In recent years, data-independent acquisition (DIA) shotgun proteomics has emerged as a possible solution to the low reproducibility of the traditional data-dependent acquisition (DDA) shotgun proteomics [7–12].
Two main strategies for relative quantification by label-free methods exist: peak intensity-based methods and spectral counting [2, 3]. It has been shown that relative quantification by peptide peak intensities provides more accurate relative quantities than spectral counting if the protein concentrations are high or if the sample complexity changes drastically between samples [13]. Therefore, in this study, we concentrate on relative quantification using peptide peak intensities. After the actual MS measurements, the raw data need to be processed appropriately for accurate results. Label-free proteomics software workflows typically consist of multiple steps, including peptide peak picking, peptide identification, feature finding, matching of the features with peptide identities, alignment of the features between different samples and possibly aggregation of the identified and quantified peptides into protein quantifications [14].
The label-free proteomic software workflows can be divided into two categories based on their structure: modular workflows and non-modular complete workflows [15]. Both commercial and nonprofit solutions exist [15]. Some excellent reviews on existing software are available, such as [13–17]. For instance, in [15], three different software platforms, Progenesis, MaxQuant and Proteios (with and without feature alignment and with features imported from MaxQuant or detected using MsInspect) were compared for peptide-level quantification in shotgun proteomics using a spike-in peptide data set with two different spike-in peptide dilution series. The performance of the software workflows was evaluated with different metrics, including harmonic mean of precision and sensitivity, mean accuracy, coverage and the number of unique peptides found [15]. The comparison suggested that Progenesis performed best, but a noncommercial combination of Proteios with imported features from MaxQuant also performed well [15]. Algorithms, such as peak picking and retention time alignment, usually included within a quantitative shotgun proteomics label-free workflow, have also been evaluated and compared separately [18–21]. While such separate comparisons are interesting and the evaluation by [15] is informative, a thorough comparison of multiple workflows on protein level is still missing, especially in terms of differential expression analysis.
A common and pervasive problem in MS data are missing values [22]. When using peptide peak intensities for relative quantification, missing values are intensity values that are not recorded for a peptide in a sample during the MS measurement [22]. Because protein intensities are aggregated from peptide intensities, enough missing values on peptide-level propagate to missing values on protein level. Missing values can occur because of multiple reasons and are mainly divided into two main categories: abundance-dependent missing values (e.g. the concentration of the peptide is below the detection limit of the instrument) or values missing completely at random (e.g. the identification of the peptide is incorrect) and are discussed in more detail in [22, 23]. Although missing values can severely bias the results [22], so far, the number of missing values produced by different software and their impact on the results has not been systematically examined. Data imputation has been proposed as a solution to missing values in proteomics [22, 24, 25]. A variety of different data imputation methods exist [24, 26], but their effect on performance, especially with regard to differential expression, has not been comprehensively evaluated in proteomics.
In this study, we present a thorough evaluation of five commonly used software workflows for quantitative shotgun label-free proteomics with special attention to missing values. We evaluated the software workflows on protein level using four different spike-in data sets. As the common interest in many proteomics studies is detecting differentially expressed proteins between sample groups, we benchmarked the software in their ability to correctly detect the truly differentially expressed proteins in the spike-in data and in their ability to estimate the known fold changes. Additionally, we measured the number of missing values produced by each software workflow and evaluated the ability of different filtering and imputation methods to counter the effect of missing values in the differential expression analysis.
Material and methods
Data sets
The UPS1 data set
The data set of Pursiheimo et al. [27] consists of 48 Universal Proteomics Standard Set (UPS1) proteins spiked into a yeast proteome digest in five different concentrations: 2, 4, 10, 25 and 50 fmol/μl. An LTQ Orbitrap Velos MS was used to analyze three technical replicates of each concentration. The data set is available from the PRIDE Archive with the identifier PXD002099 (http://www.ebi.ac.uk/pride/archive/projects/PXD002099).
The CPTAC data set
The CPTAC (Study 6) data set [28] includes 48 UPS1 proteins spiked into a yeast proteome digest in five different concentrations: 0.25, 0.74, 2.2, 6.7 and 20 fmol/μl. An LTQ Orbitrap MS was used to analyze three technical replicates of each concentration (at test site 86). The data set is available from the CPTAC Portal (https://cptac-data-portal.georgetown.edu/cptac/dataPublic/list/LTQ-Orbitrap%4086?currentPath=%2FPhase_I_Data%2FStudy6).
The UPS1B data set
The UPS1B data set of Ramus et al. [29] contains 48 UPS1 proteins spiked into yeast proteome digest in nine different concentrations: 0.05, 0.125, 0.25, 0.5, 2.5, 5, 12.5, 25 and 50 fmol/μl. An LTQ Orbitrap Velos MS was used to analyze three technical replicates of each concentration. The data set is available from the PRIDE Archive with the identifier PXD001819 (https://www.ebi.ac.uk/pride/archive/projects/PXD001819).
The Shotgun Standard Set data set (SGSDS)
A profiling standard data set of Bruderer et al. [7] consists of 12 nonhuman proteins spiked into a constant human background [human embryonic kidney (HEK-293)], including eight different sample groups with known concentrations of spike-in proteins in three master mixes. A Q Exactive Orbitrap MS was used to analyze three technical replicates of each sample group both in DDA and DIA modes. In this study, the DDA shotgun proteomics data (referred to here as shotgun standard set, SGSDS) was used. The profiling standard is available from PeptideAtlas: No. PASS00589 (username PASS00589, password WF6554orn).
Software workflows
All of the software workflows were run using the default settings as much as possible. The proper level of instrument resolution was selected (Orbitrap or high resolution in all data sets). A FASTA database of the yeast Saccharomyces cerevisiae protein sequences merged with the Sigma-Aldrich 48 UPS1 protein sequences was used for the UPS1, CPTAC and UPS1B data sets for all software. A FASTA database of the human HEK-293 cell proteins merged with the sequences of the nonhuman spike-in proteins was used for the SGSDS data set for all software. Cysteine carbamidomethylation was set as a fixed modification, and methionine oxidation and acetylation at the N-terminus were used as dynamic modifications. Peptides formed by trypsin digestion were searched. Minimum peptide length to be used was set to 6. Precursor mass tolerance was set to 20 ppm, and fragment mass tolerance to 0.5 Da. Peptides belonging to only one protein (i.e. nonconflicting unique peptides or equivalent setting) were used to calculate protein-level relative abundances in all software.
Progenesis workflow
The default peak-picking settings were used to process the raw MS files in Progenesis QI version 2.0.5387.52102. Peptide identifications were performed using Mascot search engine via Proteome Discoverer version 1.4.1.14. A Mascot score corresponding to a false discovery rate (FDR) of 0.01 was set as a threshold for peptide identifications. FDR of 0.01 was also used for protein-level identifications. Progenesis was allowed to automatically align the runs, and the retention time was entered based on visual inspection of the retention time image. Although Progenesis does not produce missing values per se, it produces some zeros, which can be interpreted as protein abundance being below detection capacity or protein not existing in the sample. The zeros produced by Progenesis were transformed into not available (NA) and treated as missing values in this study.
MaxQuant workflow
The default peak-picking settings were used to process the raw MS files in MaxQuant [30] version 1.5.3.30. Peptide identifications were performed within MaxQuant using its own Andromeda search engine [31]. MaxLFQ was on. Match type was ‘match from and to’. ‘Advanced ratio estimation’, ‘stabilize large LFQ ratios’ and ‘advanced site intensities’ were on. Match time window size was by default 0.7 min, and alignment time window size was 20 min. ‘Require MS/MS for comparisons’ was on, and decoy mode was ‘revert’. FDR of 0.01 was set as a threshold for peptide and protein identifications. MaxQuant automatically aligned the runs.
Proteios workflow
Files were first converted to mgf and mzML formats using MSConvert of ProteoWizard 3.0.9322 with 64 bit binary encoding without any compression. The converted files were imported into a virtual server running Proteios Software Environment version 2.20.0-dev [32]. Peptide identifications were performed on the mgf files using the combined hits of XTandem [33] and MS-GF+ [34] search engines. FDR of 0.01 was set as a threshold for the combined peptide identifications. The native scoring algorithm in XTandem was used for scoring the identifications. The Dinosaur algorithm [35] was used for peptide feature detection on the mzML files. Features were matched with identifications using the default settings. After matching the peptide identities with features, Proteios feature alignment algorithm [36] was used to align the sample runs. Because using protein quantification with nonconflicting peptides was not possible in the Proteios software environment, peptide quantities were exported from Proteios and composed into protein quantities by summing the nonconflicting peptides belonging to a protein in the R statistical analysis programming environment version 3.3.1.
PEAKS workflow
Default settings were used with PEAKS studio proteomics software version 7.5 [37] without merging the scans. Correct precursor was detected using mass only. Peptide identifications were performed within PEAKS using its own search engine PEAKS DB combined with PEAKS de novo sequencing [38]. PEAKS PTM search tool [39] was used to search for peptides with unspecified modifications and mutations, and the SPIDER [40] search tool was used for finding novel peptides that are homologous to peptides in the protein database. The default maximum number of variable posttranslational modifications per peptide was 3, and the number of de novo dependencies was 16. The default de novo score threshold for SPIDER was 15 and Peptide hit score threshold 30. Retention time shift tolerance was 20 min. All the search tools are included in the PEAKS studio software. FDR was estimated with target decoy fusion and set to 0.01. Label-free quantification with PEAKS Q was used. PEAKS was allowed to autodetect the reference sample and automatically align the sample runs. To allow the exporting of complete results, protein significance filter was set to 0, protein fold change filter to 1 and unique peptide filter to 1 in the export settings.
OpenMS workflow
Toppas [41] version 2.0.0 was used to create workflows in the OpenMS [42] software environment. Files were first converted to mzML format using MSConvert of ProteoWizard 3.0.9322 with 64 bit binary encoding without any compression. The converted files were imported into Toppas. Peptide identifications were performed using the combined hits of XTandem [33] and MS-GF+ [34] search engines. The native scoring algorithm in XTandem was used for scoring the identifications. FDR of 0.01 was set as a threshold for the combined peptide identifications using the PeptideIndexer, FalseDiscoveryRate and IDFilter algorithms. Reverse decoys were used to calculate the FDR for the peptide identifications. The PeakPickerHiRes algorithm with parameters for Orbitrap/high-resolution data combined with BaselineFilter algorithm was used for peak picking. The FeatureFinderCentroided algorithm with parameters for Orbitrap/high-resolution data was used for peptide feature finding. Peptide identifications were linked to peptide features using the IDMapper algorithm, and the different sample runs were aligned using the MapAlignerPoseClustering algorithm, allowing the algorithm to automatically select the appropriate reference run. The FeatureLinkerUnlabeledQT algorithm was used to group the features before quantifying the protein abundances with the ProteinQuantifier algorithm. Summing of the peptide intensities belonging to a protein was used to compose the protein-level intensities.
Post-processing
Non-normalized protein intensities (peptide intensities for Proteios) were extracted from all workflows and imported into the R statistical programming software environment version 3.3.1 [43] for post-processing and analysis. All of the downstream data processing and analysis were done in the R environment.
All data were normalized using the variance stabilization normalization (vsn) [44] shown to perform consistently well with proteomics spike-in data [45]. The justvsn function from the vsn package [44] was used for normalization.
Missing values and imputation methods
All missing intensity values were transformed to a uniform NA notation. All missing values were treated uniformly before imputation. No special class was assigned to missing values (e.g. missing completely at random or missing because of being censored) rather they were considered missing because of an unknown reason. Data imputation and filtering were done on protein level after normalization as suggested by [22]. Seven imputation methods and two filtering approaches were tested as detailed below.
Zero imputation (zero)
All missing values (NA) were replaced with zeros.
Background imputation (back)
All missing values were replaced with the lowest detected intensity value of the data set. This imputation simulates the situation where protein values are missing because of having small concentrations in the sample and thus cannot be detected during the MS run. The lowest intensity value detected is therefore imputed for the missing protein values as a representative of the background.
Censored imputation (censor)
If only a single NA for a protein in a sample group was found, it was considered as being ‘missing completely at random’, and no value was imputed for it. If a protein contained more than one missing value in a sample group (consisting of technical replicates), they were considered missing because of being below detection capacity, and the lowest intensity value in the data set was imputed for them.
Bayesian principal component analysis imputation (bpca)
Bayesian principal component analysis (bpca) imputation was first developed for microarray data [46]. It combines an expectation maximization algorithm with principal component regression and Bayesian estimation to calculate the likelihood of an estimate for the missing value [46]. Bpca is based on variational Bayesian framework, which does not force orthogonality between principal components [47]. The bpca imputation was implemented with the principal component analysis function of the pcaMethods package [47]. The number of principal components k used for missing value estimation was set to n-1 as suggested by [46], where n is the number of samples in the data.
Local least squares imputation (lls)
In the local least squares (lls) imputation, k most similar proteins are first selected, and the missing values are then estimated with least squares regression as a linear combination of the values of these k proteins. Similarity is inferred based on protein intensities of other samples than the one being imputed for. A value of 150 for parameter k has been observed to be enough in most cases [48–50] and was also used in this study. The lls imputation was implemented with the llsImpute function of the pcaMethods package [47].
K-nearest neighbor imputation (knn)
The k-nearest neighbor (knn) imputation algorithm finds k most similar proteins (k-nearest neighbors) and uses a weighted average over these k proteins to estimate the missing value [51]. More weight is given to more similar proteins [51]. Similarity is inferred based on intensities of other samples than the one being imputed for. A number of k between 10 and 20 have been suggested to be appropriate for the knn algorithm, and the performance of the algorithm has been observed to be uniform with these values of k [51]. A k value of 10 was used in this study.
Singular value decomposition imputation (svd)
Singular value decomposition is applied to the data to obtain sets of mutually orthogonal expression patterns of all proteins in the data [51]. These expression patterns, which are identical to the principal components of the data, are referred to as eigenproteins following the terminology of [51]. The missing values are estimated as a linear combination of the k most significant eigenproteins by regressing the protein containing the missing value against the eigenproteins [51]. It has been observed that a suitable value for parameter k is 20% of the most significant eigengenes [51], and a k value corresponding to 20% of the most significant eigenproteins was also used in this study.
Basic filtering (filtered)
All proteins with more than one missing value per sample group (consisting of three technical replicates in each data set) were considered as ‘not missing at random’ [22, 52, 53], and were filtered out to enable differential expression analysis between sample groups. Single missing values for a protein per sample group were considered as being ‘missing completely at random’, and no values were imputed for them.
Filtering + local least squares imputation (filtlls)
First, all proteins with more than one missing value per sample group were filtered out as above, and then lls imputation was used to impute values for the remaining missing values.
Evaluation of the software workflows and imputation methods
Differential expression analysis
Differential expression analysis was performed between all possible combinations of two sample groups in each data set. The reproducibility-optimized test statistic (ROTS) [54] has been shown to perform well for label-free proteomics data [27, 45] and was used in this study.
Receiver operating characteristic (ROC) curve analysis was used to measure the performance of different software workflows and imputation methods in detecting the truly differentially expressed spike-in proteins. In the ROC curve analysis, the true-positive rate (or sensitivity) is plotted against the true-negative rate (or specificity). The partial area under the ROC curve (pAUC) was used to rank the software and imputation methods. As generally the interest is in detecting the top differentially expressed proteins for further validation, pAUCs between specificity values 1 and 0.9 were used to focus on the most essential part of the ROC curve. The pAUCs were scaled so that they had a maximal value of 1.0 and a non-discriminant value of 0.5 using the R package pROC [55]. If a P-value for the differential expression of a protein between any two sample groups could not be calculated because of missing values, the protein was assigned a P-value of 1 in that two-group comparison to keep the number of proteins in the ROC analysis unchanged between the different comparisons in each data set and each software. The bootstrap method was used to calculate the significance of the differences between the pAUCs using the roc.test-function of the pROC package [55].
Ranks for each software were calculated in each two-sample comparison; better ranks were assigned to software with higher pAUCs. If pAUCs were equal, they received equal ranks. An overall rank was calculated for each software using the mean ranks of the software for each data set. The Satterthwaite approximation was used to calculate the associated standard error. Similarly, ranks, mean ranks and overall ranks were also calculated for each software after applying the different imputation methods.
Evaluation of the logarithmic fold change
While the ability of a software to correctly detect true differential expression is pivotal, producing accurate estimates for the logarithmic fold changes (LogFCs) can also be of interest. In the spike-in data sets used, the expected LogFCs were known both for the spike-in proteins and for the background proteins (for which the expected LogFC is zero). We evaluated how close the LogFCs estimated by the different software workflows were to the expected LogFCs. For each two-group comparison of each data set, we calculated the mean squared error (MSE) between the estimated and expected LogFCs of proteins.
Results
Missing values and the number of proteins quantified
Progenesis quantified a considerably smaller number of proteins than the other software in all data sets except the SGSDS data set (Table 1). In the SGSDS data set, Progenesis quantified more proteins than OpenMS, while PEAKS and especially MaxQuant and Proteios still quantified considerably larger numbers of proteins.
Table 1.
The total number of proteins quantified, the number of spike-in proteins detected and the proportion of missing values produced by each proteomics software workflow in each data set
| Progenesis |
MaxQuant |
Proteios |
PEAKS |
OpenMS |
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UPS1 | CPTAC | SGSDS | UPS1B | UPS1 | CPTAC | SGSDS | UPS1B | UPS1 | CPTAC | SGSDS | UPS1B | UPS1 | CPTAC | SGSDS | UPS1B | UPS1 | CPTAC | SGSDS | UPS1B | |
| Proteins quantified | 889 | 614 | 2168 | 1033 | 1581 | 1247 | 3487 | 1126 | 1799 | 1383 | 3554 | 1490 | 1632 | 1223 | 3161 | 1581 | 1753 | 1276 | 1401 | 1787 |
| Spike-in proteins detected, n (%) | 47 (98) | 32 (67) | 12 (100) | 47 (98) | 47 (98) | 41 (85) | 12 (100) | 48 (100) | 47 (98) | 42 (88) | 12 (100) | 48 (100) | 48 (100) | 42 (88) | 12 (100) | 47 (98) | 47 (98) | 41 (85) | 11 (92) | 48 (100) |
| Proportion of missing values in the detected spike-in proteins (%) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 29.4 | 4.2 | 35.2 | 0.1 | 20.8 | 0.0 | 19.9 | 1.2 | 21.7 | 0.3 | 21.4 | 1.0 | 23.6 | 2.3 | 13.0 |
| Proportion of missing values in the detected background proteins (%) | 0.4 | 1.2 | 0.0 | 0.1 | 14.5 | 19.1 | 3.4 | 7.4 | 19.0 | 18.0 | 6.4 | 8.1 | 16.4 | 19.5 | 3.2 | 5.9 | 29.8 | 32.1 | 8.8 | 20.0 |
| Total proportion of missing values (%) | 0.0 | 1.0 | 0.0 | 0.0 | 14.0 | 19.0 | 3.0 | 9.0 | 19.0 | 18.0 | 6.0 | 8.0 | 16.0 | 20.0 | 3.0 | 6.0 | 29.0 | 32.0 | 9.0 | 20.0 |
| Mean proportion of missing values (%) | 0.3 | 11.3 | 12.8 | 11.3 | 22.5 | |||||||||||||||
Note: The proportions of missing values are presented separately for the spike-in proteins, background proteins and all proteins combined. The mean proportion of missing values is calculated for each software based on the total proportion of missing values for that software over all the data sets.
In the UPS1, SGSDS and UPS1B data sets, all the software detected most of the spike-in proteins. In the CPTAC data set, Progenesis was able to discover only 67% of the spike-in proteins, while the rest of the software workflows detected a substantially larger proportion of them (85–88%). This might be related to Progenesis dropping out one technical replicate from the 20 fmol sample group when performing the automatic alignment of the MS runs.
In general, the number of missing values was markedly lower with Progenesis than with the other software, being near zero in all data sets (Table 1). With the other software, the number of missing values in the detected spike-in proteins was low in the UPS1 and SGSDS data sets (0–4.2%), but higher in the CPTAC and UPS1B data sets (13.0–35.2%). For instance, in the CPTAC data set, the proportion of missing values in the spike-in proteins with Progenesis was zero, while it was as much as 29.4% with MaxQuant and 20.8–23.6% with the other software. Overall, the number of missing values in the background proteins was much higher with the other software than with Progenesis, being highest in the CPTAC and UPS1 data sets (Table 1).
While the different software quantified different numbers of proteins, the bulk of the protein quantifications was shared (Figure 1). Nearly all proteins identified and quantified by Progenesis in each data set were proteins identified and quantified also by the other software (Figure 1A–D). In the UPS1, CPTAC and SGSDS data sets, MaxQuant had the largest number of unique protein quantifications (Figure 1A–C), whereas OpenMS had the largest number of unique protein quantifications in the UPS1B data set (Figure 1D).
Figure 1.

Venn diagrams of quantified proteins by each software in the (A) UPS1, (B) CPTAC, (C) SGSDS and (D) UPS1B data set.
Differential expression analysis
Progenesis performed systematically well in the differential expression analysis delivering high pAUCs in each data set (Figure 2). While the other software also performed well, Progenesis clearly outperformed them in the CPTAC and UPS1B data sets (Figure 2B, D, F and H). This difference in performance was prominent, regardless of whether investigating the ROC curves drawn over all the two-group comparisons (Figure 2B and D) or the pAUCs of each two-group comparison separately (Figure 2F and H). A closer investigation of the ROC curves revealed differences between the software depending on the data set analyzed.
Figure 2.

Results of the differential expression analysis using different software. ROC curves over all the two-group comparisons in the (A) UPS1 data, (B) CPTAC data (C) SGSD data and (D) UPS1B data, and the pAUCs of each two-group comparison in the (E) UPS1 data, (F) CPTAC data (G) SGSD data and (H) UPS1B data. In A–D, the diagonal grey line represents the identity (no discrimination) line. In E–H, the x-axes denote the two-group comparisons of the sample groups.
In the UPS1 data set, all software performed well. However, when looking at the pAUCs of the two-group comparisons (Figure 2E), MaxQuant, Progenesis and Proteios outperformed PEAKS and OpenMS. Especially, MaxQuant performed well, delivering the highest pAUCs in most of the two-group comparisons, being significantly better than PEAKS or OpenMS in 9 of 10 of the two-group comparisons (P < 0.05).
In the CPTAC data set, Progenesis significantly outperformed all the other software in all but two comparisons (0.25 versus 0.74 fmol and 2.2 versus 6.7 fmol) (P < 0.05) (Figure 2F). However, as was already noted earlier, in this data set, Progenesis discovered only 67% of the spike-in proteins, while the rest of the software discovered 85–88% of the spike-in proteins. In four comparisons (0.25 versus 0.74 fmol, 0.25 versus 2.2 fmol, 0.25 versus 6.7 fmol and 0.25 versus 20 fmol), MaxQuant performed significantly worse than the other software (P < 0.05), whereas in three comparisons (2.2 versus 6.7 fmol, 2.2 versus 20 fmol and 6.7 versus 20 fmol), OpenMS performed worst (P < 0.05). In the other three comparisons, Proteios, PEAKS, OpenMS and MaxQuant performed comparably.
In the SGSDS data set, Proteios and Progenesis performed best delivering the highest pAUCs in most of the two-group comparisons (Figure 2G). The differences in pAUCs between the two software were significant in only one of the comparison pairs (3 versus 7), where Progenesis performed significantly better than Proteios (P = 0.039). PEAKS performed almost on par with Progenesis and Proteios; the differences in pAUCS to Proteios were not significant in any of the two-group comparisons, but Progenesis performed significantly better in two of the comparisons (5 versus 6 and 5 versus 7, P < 0.04).
In the UPS1B data set, Progenesis systematically outperformed the other software in the two-group comparisons (Figure 2H). The differences were significant in 72–89% of all the 36 two-group comparisons (P < 0.05). In this data set, there were many comparisons with low concentrations of spike-in proteins in the samples. When both sample groups had a low concentration of spike-in proteins, and the differences in the concentrations were small, all the software struggled in detecting the truly differentially expressed proteins.
When ranked based on pAUCs of each two-group comparison in each data set, Progenesis received an overall rank of 1.49 with a standard error estimate of 0.35. Proteios was second with an overall rank of 2.79 and a standard error of 0.34, followed by MaxQuant (3.12 ± 0.57), PEAKS (3.51 ± 0.42) and OpenMS (4.09 ± 0.44).
As the number of proteins identified and quantified in a data set was different for each software workflow, we also examined to which extent this affected the results in the differential expression analysis. To investigate this, we composed union data sets, which contained all the proteins identified and quantified by at least one software in that data set. Proteins not originally quantified by a software were treated as missing values in the differential expression analysis. Using the union data sets did not have a major effect; the performance of Progenesis in the CPTAC data set was lowered, but it still outperformed the other software (Supplementary Figure S1). Otherwise, the results of the differential expression analysis using the union data sets were similar to those of the original data sets (Supplementary Figure S1, Supplementary Table S1) and were thus excluded from the main results.
Estimates of LogFC
There were clear differences between the accuracy of the LogFC estimates calculated from the protein intensities produced by the different software workflows. MaxQuant consistently produced low MSEs between the observed LogFCs compared with the expected LogFCs (Figure 3).
Figure 3.

The MSE of the LogFC estimates in the two-group comparisons produced by the software workflows compared with the known expected LogFCs. MSEs of LogFC of the spike-in proteins in the (A) UPS1 data set, (B) CPTAC data set, (C) SGSDS data set and (D) UPS1B data set. MSEs of LogFCs of the background proteins in the (E) UPS1 data set, (F) CPTAC data set, (G) SGSDS data set and (H) UPS1B data set.
For the spike-in proteins, MaxQuant had systematically low median MSE compared with the other software in each data set (Figure 3A–D), being the lowest in the UPS1 and UPS1B data sets. Over all the two-group comparisons, MaxQuant had an overall mean MSE of 3.3 with a standard error of 0.4, followed by Proteios (mean MSE 5.3 ± 0.64), PEAKS (mean MSE 7.36 ± 0.88), Progenesis (mean MSE 8.57 ± 1.32) and OpenMS (mean MSE 8.96 ± 1.45). Also, the distribution of MSEs for the spike-in proteins was generally more compact with MaxQuant than with the other software; MaxQuant had fewer large MSEs than the other software (Figure 3A–D).
While Progenesis produced intensity values that resulted in relatively high MSEs for the spike-in proteins, it estimated the LogFCs of the background proteins most accurately in each data set (Figure 3E–H). Overall, all the software had rather compact distributions of MSEs and low median MSEs in the background proteins. Over all the two-group comparisons, Progenesis had an overall mean MSE of 0.31 with a standard error of 0.01, followed by MaxQuant (mean MSE 0.36 ± 0.06), Proteios (mean MSE 0.61 ± 0.07), OpenMS (mean MSE 0.63 ± 0.08) and PEAKS (mean MSE 0.64 ± 0.08).
As LogFC has been observed to depend also on the normalization method chosen [45], for a comprehensive evaluation of LogFC, we examined whether normalizing the data differently had an effect on the results. We normalized the data from each software workflow using five different normalization methods (vsn, median, quantile, fast LOESS using a reference array and robust linear regression using a reference array) and recalculated the MSEs of the LogFCs. However, the different normalization methods had no major impact on the order or magnitude of the results and were thus excluded from the main results (Supplementary Figure S2).
Effect of imputation on performance
Finally, we examined the effect of filtering and imputation on the performance of the different software workflows (Table 2, Supplementary Table S2, Figure 4, Supplementary Figures S3–5). When missing values were abundant, filtering and imputation improved pAUCs in the differential expression analysis. This effect was most prominent in the CPTAC and UPS1B data sets with MaxQuant, OpenMS, Proteios and PEAKS (Figure 4B, D, F and H, Supplementary Figure S3B, D, F and H). In general, imputation or filtering improved the performance of MaxQuant in the differential expression analysis the most (Supplementary Figure S5, Supplementary Table S2). As expected, imputation or filtering did not have a major effect on the performance of Progenesis, which had only a small number of missing values (Table 2, Supplementary Table S2).
Table 2.
Rankings of the software and imputation method combinations based on partial areas under the ROC-curves (pAUC) of the differential expression analysis.
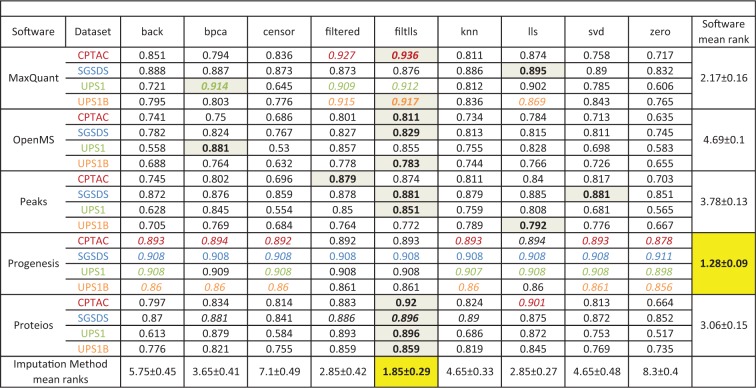 |
Note: The best imputation method for each dataset with each software is highlighted in grey and the value is bolded. Since imputation had little to none effect on the performance of Progenesis, no best imputation method for Progenesis could be selected. The best software in each dataset with each imputation method (found on each column) is colored according to the colors of datasets. The best mean ranking for the imputation methods and software is highlighted in yellow and the value is bolded. The pAUCs were calculated from the ROC-curves over all the two-group comparisons in a dataset.
Figure 4.

Results of the differential expression analysis of different software after using the best ranking filtlls method. ROC curves over all the two-group comparisons in the (A) UPS1 data, (B) CPTAC data (C) SGSD data and (D) UPS1B data, and the pAUCs of each two-group comparison in the (E) UPS1 data, (F) CPTAC data (G) SGSD data and (H) UPS1B data. In A–D, the diagonal gray line represents the identity (no discrimination) line. In E–H, the x-axes denote the two-group comparisons of the sample groups.
The combination of filtering and imputation (filtlls) improved the performances of the software the most (Table2, Supplementary Table S2, Figure 4). Among the pure imputation methods, the lls imputation consistently produced the highest improvement in the pAUCs of the software in different data sets (Table 2, Supplementary Table S2, Supplementary Figure S3). The basic filtering method (filtered) received an equal rank to the lls imputation and consistently improved the performance of the software in the differential expression analysis.
In the SGSDS data set, Progenesis always performed best despite the imputation or filtering method used. Notably, however, when using the filtlls method, MaxQuant performed best in the differential expression analysis in the UPS1, CPTAC and UPS1B data sets (Table 2, Figure 4). The same was true, when using the basic filtering; MaxQuant performed best in the UPS1, CPTAC and UPS1B data sets (Table 2). Accordingly, when using the filtlls or filtered methods, MaxQuant performed best in the differential expression analysis based on the mean ranks of all the two-group comparisons (Supplementary Table S2). Proteios also performed generally well and consistently outperformed PEAKS and OpenMS after filtering or imputation (Figure 4, Supplementary Figure S3, Supplementary Figure S4).
Discussion
In the four spike-in data sets tested in this study, Progenesis performed systematically well in the differential expression analysis. It performed well even in the most challenging CPTAC and UPS1B data sets, in which it clearly outperformed the other software before filtering or imputation. Without filtering or imputation, Proteios performed second best; however, there were no large differences in the performances of OpenMS, PEAKS and Proteios (Figure 2). MaxQuant performed best in the UPS1 data set and almost on par with PEAKS and Proteios in SGSDS data set but was clearly worst in the CPTAC and UPS1B data sets. These results are in agreement with the prior results of [15] on peptide level. However, after basic filtering or combining filtering with the lls imputation, MaxQuant performed best in three of the four tested data sets (Table 2, Supplementary Table S2).
In this study, Progenesis produced a significantly lower number of missing values than the other software in each data set examined (Table 1). We speculate that this is essentially because of the efficient co-detection algorithm, which uses an aggregate ion map for the detection of peaks from each sample. While all the other software perform similar alignment, the feature ions are generally detected separately in each sample run before aligning the runs conversely to the co-detection solution used by Progenesis. However, as was noted in the case of the CPTAC data set, Progenesis is not always successful in aligning all the sample runs, which may be problematic in the case of a large and complex data set with many samples. The fact that Progenesis was unable to automatically align all the runs in the CPTAC data set might have partly contributed to the smaller number of quantified proteins compared with the other software in that data set, including ∼10 fewer spike-in proteins (Table 1).
In addition to the ‘matching between the runs’ alignment algorithm used in this study [56], MaxQuant includes another algorithm, MaxLFQ [5], which is especially useful in studies of large, complex proteomes, where the samples have been fractionated before the MS analysis. While we did not examine every possible algorithm and parameter combination of each software, we decided to explore the effect of MaxLFQ, as it can possibly essentially affect the number of missing values produced by MaxQuant. In the data sets where MaxQuant was previously found to struggle (CPTAC and UPS1B data sets in particular), using MaxLFQ decreased the performance of MaxQuant in the differential expression analysis, whereas no noticeable effect was found with the other data sets (Supplementary Figure S6).
The number of proteins quantified by different software workflows differed greatly in the tested data sets (Table 1). While the use of different search engines understandably creates variation in the number of proteins discovered and quantified, even using the same search engines with the exact same settings in different software workflows can still lead to different quantified proteins, as was the case with OpenMS and Proteios (Table 1). The number of proteins quantified by each software workflow depends on the peptides and proteins identified by the search engine used but also on the successful matching of peptide identifications with the detected features, the alignment of different sample runs and grouping of peptides into proteins by the software. It is hard to estimate the impact of the different search engines on the performance of the software workflows in the differential expressions analysis. However, in most cases, a similar number of spike-in proteins were detected by the different software workflows in each data set (Table 1), indicating that the differences seen in the performance were not necessarily because of differences in identification but rather because of differences in successful quantification of the proteins. This is perhaps even more highlighted with Progenesis detecting only 67% of the spike-in proteins in the CPTAC data set compared with the 85–88% with the other software and still outperforming the other software in that data set (Figure 2, Supplementary Figure S1).
LogFC is a popular measure in proteomics studies to determine the degree of differential expression in detected differentially expressed proteins [27, 57, 58]. Notably in our tests, MaxQuant estimated the LogFCs of the spike-in proteins most accurately having the most compact distribution of MSEs. However, all software estimated the LogFCs of the background proteins fairly accurately.
Finally, we examined the effect of two filtering and seven imputation methods on the performance of the software in the differential expression analysis. Importantly, filtering and imputation had a major impact on increasing the performance of all other software than Progenesis in the differential expression analysis (Figure 4, Supplementary Figure S5, Table 2). Among the imputation methods tested, lls performed best, consistently improving the performance of the software in the differential expression analysis the most in the spike-in data sets tested. Also, bpca ranked well and was able to systematically increase the performance of all other software than Progenesis. These results are in agreement with prior results from DNA microarrays [26, 59] where simple imputation methods such as zero imputation or row average have not performed as well as more advanced methods such as bpca. However, it has recently been suggested that the more simple imputation methods might be more suitable than the advanced methods, when most of the missing values in a data set are among the low-abundance peptides because of the detection limit of the instrument [25]. Furthermore, [25] suggested that performing imputation already on the peptide level improves the performances of the imputation methods in most cases. Be that as it may, as most of the software workflows used in this study aggregate peptide quantities into protein quantities using specialized methods, examining peptide-level imputation was beyond the scope of this study.
For the other software than Progenesis, the best performance in the differential expression analysis was achieved, when data filtering was followed by imputation with the lls method (Table 2). Among the tested software, MaxQuant benefitted most from filtering or imputation. While filtering seemed to be an effective way to increase the performance of a software to detect the truly differentially expressed proteins, it might have resulted in losing some possibly interesting proteins when missing values were abundant. The best approach and method most likely depend on the data and on whether more emphasis is put on finding as many differentially expressed proteins as possible or in finding the most differentially expressed proteins as accurately as possible.
Even though suggestive, with the current setting of our study, our results cannot be generalized to represent absolute differences between the tested software. We tested the software workflows using spike-in data sets, where the number of truly differentially expressed proteins is rather small. While this is representative of many real experimental settings, it might be insufficient in some cases where there might be a considerable number of proteins changing between sample groups. Furthermore, we tested the different software using the default and automated settings as much as possible, which may favor some software workflows and not be optimal in all research settings. The performance of each software depends on the data set, but it also depends on the instrument used and on the complex combination of different software parameters. Each software workflow contains a multitude of parameters that can be altered and which potentially affect the performance.
The effect of parameters or modules on the performance is especially true for modular workflows, such as OpenMS and Proteios, in which even different algorithms can be chosen for a given task, which may change the performance of the workflow completely. An experienced user can possibly optimize the performance of any software workflow by choosing the correct algorithms and parameters based on the data set and prior expert knowledge. In this comparison, based on the default settings of each software, as uniform settings as possible were used, and the software were not optimized for best possible performance. Instead of aiming to compare absolute differences between the tested software, our comparison rather corresponds to the circumstances where the different software workflows are used with more limited amount of prior knowledge about the software algorithms. This setting likely reflects the situation in which many researchers and research groups stand with a more intense focus on the experimental side than data analysis. While selecting the appropriate software can be a demanding task for the average user, careful consideration should be applied to the amount of prior knowledge and the available resources for mastering the attainable software. Most of the complete solutions are easily set up and are ready to be used in just a few steps with good results, whereas the modular solutions generally require more time to familiarize with, while offering extra control (i.e. algorithm selection, the inclusion of multiple search engines) and adjustability for the experienced user. In addition to the software workflows tested in this comparison, a broad range of other software for the analysis of MS data exist, such as The Transproteomic Pipeline [60] or Skyline [61]. The results from this comparison cannot be generalized to different types of MS techniques (such as DIA, targeted MS, labeling-based MS, etc.), which would require testing and evaluation of their own.
Key Points
The performance of five software workflows, MaxQuant, OpenMS, PEAKS, Progenesis and Proteios, in detecting true differential expression was examined, including the number of missing values produced and the effect of seven imputation and two filtering methods on the performance.
Progenesis performed consistently well in the differential expression analysis in every data set tested.
Filtering and proper imputation increased the performance of MaxQuant, OpenMS, PEAKS and Proteios. The combination of filtering and imputation improved the performances the most.
After filtering or using the combination of filtering and imputation, MaxQuant performed best in the differential expression analysis.
The LogFCs of the spike-in proteins were estimated most accurately by MaxQuant. The LogFCs of the background proteins were estimated most accurately by Progenesis.
Supplementary Material
Acknowledgements
The authors gratefully thank Fredrik Levander from the Department of Immunotechnology, University of Lund for his assistance with the Proteios software environment.
Funding
L.L.E. reports grants from the European Research Council (ERC) (677943), European Union's Horizon 2020 research and innovation programme (675395), Academy of Finland (296801 and 304995), Juvenile Diabetes Research Foundation JDRF (2-2013-32), Tekes – the Finnish Funding Agency for Innovation (1877/31/2016) and Sigrid Juselius Foundation, during the conduct of the study.
Biographies
Tommi Välikangas is a PhD student in the Computational Biomedicine Group at the Turku Centre for Biotechnology Finland. He is interested in computational biology and bioinformatics.
Tomi Suomi is a PhD student in the Computational Biomedicine research group at the Turku Centre for Biotechnology Finland. His research interests include scientific computing and bioinformatics.
Laura L. Elo is Adjunct Professor in Biomathematics, Research Director in Bioinformatics and Group Leader in Computational Biomedicine at Turku Centre for Biotechnology, University of Turku, Finland. Her main research interests include computational biomedicine and bioinformatics.
References
- 1. Meissner F, Mann M.. Quantitative shotgun proteomics: considerations for a high-quality workflow in immunology. Nat Immunol 2014;15:112–17. [DOI] [PubMed] [Google Scholar]
- 2. Megger DA, Bracht T, Meyer HE, et al. Label-free quantification in clinical proteomics. Biochim Biophys Acta 2013;1834:1581–90. [DOI] [PubMed] [Google Scholar]
- 3. Zhu W, Smith JW, Huang CM.. Mass spectrometry-based label-free quantitative proteomics. J Biomed Biotechnol 2010;2010:840518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ong S-E, Blagoev B, Kratchmarova I, et al. Stable isotope labeling by Amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 2002;1:376–86. [DOI] [PubMed] [Google Scholar]
- 5. Cox J, Hein MY, Luber C. A, et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 2014;13:2513–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Domon B, Aebersold R.. Options and considerations when selecting a quantitative proteomics strategy. Nat Biotech 2010;28:710–21. [DOI] [PubMed] [Google Scholar]
- 7. Bruderer R, Bernhardt OM, Gandhi T, et al. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen treated 3D liver microtissues. Mol Cell Proteomics 2015;14:1400–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Venable JD, Dong MQ, Wohlschlegel J, et al. Automated approach for quantitative analysis of complex peptide mixtures from tandem mass spectra. Nat Meth 2004;1:39–45. [DOI] [PubMed] [Google Scholar]
- 9. Plumb RS, Johnson KA, Rainville P, et al. UPLC/MS(E); a new approach for generating molecular fragment information for biomarker structure elucidation. Rapid Commun Mass Spectrom 2006;20:1989–94. [DOI] [PubMed] [Google Scholar]
- 10. Distler U, Kuharev J, Navarro P, et al. Drift time-specific collision energies enable deep-coverage data-independent acquisition proteomics. Nat Meth 2014;11:167–70. [DOI] [PubMed] [Google Scholar]
- 11. Moran D, Cross T, Brown LM, et al. Data-independent acquisition (MSE) with ion mobility provides a systematic method for analysis of a bacteriophage structural proteome. J Virol Methods 2014;195:9–17. [DOI] [PubMed] [Google Scholar]
- 12. Egertson JD, Kuehn A, Merrihew GE, et al. Multiplexed MS/MS for improved data-independent acquisition. Nat Meth 2013;10:744–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grossmann J, Roschitzki B, Panse C, et al. Implementation and evaluation of relative and absolute quantification in shotgun proteomics with label-free methods. J Proteomics 2010;73:1740–6. [DOI] [PubMed] [Google Scholar]
- 14. Sandin M, Teleman J, Malmström J, et al. Data processing methods and quality control strategies for label-free LC–MS protein quantification. Biochim Biophys Acta 2014;1844:29–41. [DOI] [PubMed] [Google Scholar]
- 15. Chawade A, Sandin M, Teleman J, et al. Data processing has major impact on the outcome of quantitative label-free LC-MS analysis. J Proteome Res 2015;14:676–87. [DOI] [PubMed] [Google Scholar]
- 16. Navarro P, Kuharev J, Gillet LC, et al. A multicenter study benchmarks software tools for label-free proteome quantification. Nat Biotechnol 2016;34:1130–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Runxuan Z, Barton A, Brittenden J, et al. Evaluation for computational platforms of LC-MS based label-free quantitative proteomics: a global view. J Proteomics Bioinform 2010;3:260–5. [Google Scholar]
- 18. Smith R, Ventura D, Prince JT.. LC-MS alignment in theory and practice: a comprehensive algorithmic review. Brief Bioinform 2015;16:104–17. [DOI] [PubMed] [Google Scholar]
- 19. Lange E, Tautenhahn R, Neumann S, et al. Critical assessment of alignment procedures for LC-MS proteomics and metabolomics measurements. BMC Bioinformatics 2008;9:375.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sandin M, Krogh M, Hansson K, et al. Generic workflow for quality assessment of quantitative label-free LC-MS analysis. Proteomics 2011;11:1114–24. [DOI] [PubMed] [Google Scholar]
- 21. Zhang J, Gonzalez E, Hestilow T, et al. Review of peak detection algorithms in liquid-chromatography-mass spectrometry. Curr Genomics 2009;10:388–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Karpievitch YV, Dabney AR, Smith RD.. Normalization and missing value imputation for label-free LC-MS analysis. BMC Bioinformatics 2012;13:S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Karpievitch YV, Taverner T, Adkins JN, et al. Normalization of peak intensities in bottom-up MS-based proteomics using singular value decomposition. Bioinformatics 2009;25:2573–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Webb-Robertson BJ, Wiberg HK, Matzke MM, et al. Review, evaluation, and discussion of the challenges of missing value imputation for mass spectrometry-based label-free global proteomics. J Proteome Res 2015;14:1993–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lazar C, Gatto L, Ferro M, et al. Accounting for the multiple natures of missing values in label-free quantitative proteomics data sets to compare imputation strategies. J Proteome Res 2016;15:1116–25. [DOI] [PubMed] [Google Scholar]
- 26. Tuikkala J, Elo LL, Nevalainen OS, et al. Missing value imputation improves clustering and interpretation of gene expression microarray data. BMC Bioinformatics 2008;9:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pursiheimo A, Vehmas AP, Afzal S, et al. Optimization of statistical methods impact on quantitative proteomics data. J Proteome Res 2015;14:4118–26. [DOI] [PubMed] [Google Scholar]
- 28. Tabb DL, Vega-Montoto L, Rudnick PA, et al. Repeatability and reproducibility in proteomic identifications by liquid chromatography-tandem mass spectrometry. J Proteome Res 2010;9:761–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ramus C, Hovasse A, Marcellin M, et al. Spiked proteomic standard dataset for testing label-free quantitative software and statistical methods. Data Brief 2016;6:286–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cox J, Mann M.. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 2008;26:1367–72. [DOI] [PubMed] [Google Scholar]
- 31. Cox J, Neuhauser N, Michalski A, et al. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 2011;10:1794–805. [DOI] [PubMed] [Google Scholar]
- 32. Hakkinen J, Vincic G, Mansson O, et al. The Proteios software environment: an extensible multiuser platform for management and analysis of proteomics data. J Proteome Res 2009;8:3037–43. [DOI] [PubMed] [Google Scholar]
- 33. Craig R, Beavis RC.. TANDEM: matching proteins with tandem mass spectra. Bioinformatics 2004;20:1466–7. [DOI] [PubMed] [Google Scholar]
- 34. Kim S, Pevzner PA.. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat Commun 2014;5:5277.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Teleman J, Chawade A, Sandin M, et al. Dinosaur: a refined open-source peptide MS feature detector. J Proteome Res 2016;15:2143–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sandin M, Ali A, Hansson K, et al. An adaptive alignment algorithm for quality-controlled label-free LC-MS. Mol Cell Proteomics 2013;12:1407–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ma B, Zhang K, Hendrie C, et al. PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun Mass Spectrom 2003;17:2337–42. [DOI] [PubMed] [Google Scholar]
- 38. Zhang J, Xin L, Shan B, et al. PEAKS DB: de novo sequencing assisted database search for sensitive and accurate peptide identification. Mol Cell Proteomics 2012;11:M111.010587.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Han X, He L, Xin L, et al. PeaksPTM: mass spectrometry-based identification of peptides with unspecified modifications. J Proteome Res 2011;10:2930–6. [DOI] [PubMed] [Google Scholar]
- 40. Han Y, Ma B, Zhang K.. SPIDER: software for protein identification from sequence tags with de novo sequencing error. J Bioinform Comput Biol 2005;3:697–716. [DOI] [PubMed] [Google Scholar]
- 41. Junker J, Bielow C, Bertsch A, et al. TOPPAS: a graphical workflow editor for the analysis of high-throughput proteomics data. J Proteome Res 2012;11:3914–20. [DOI] [PubMed] [Google Scholar]
- 42. Sturm M, Bertsch A, Gröpl C, et al. OpenMS—an open-source software framework for mass spectrometry. BMC Bioinformatics 2008;9:163.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. R Core Team (2017). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/.
- 44. Huber W, von Heydebreck A, Sültmann H, et al. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 2002;18 (Suppl 1):S96–104. [DOI] [PubMed] [Google Scholar]
- 45. Välikangas T, Suomi T, Elo LL.. A systematic evaluation of normalization methods in quantitative label-free proteomics. Brief Bioinform 2016, DOI: 10.1093/bib/bbw095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Oba S, Sato MA, Takemasa I, et al. A Bayesian missing value estimation method for gene expression profile data. Bioinformatics 2003;19:2088–96. [DOI] [PubMed] [Google Scholar]
- 47. Stacklies W, Redestig H, Scholz M, et al. pcaMethods—a bioconductor package providing PCA methods for incomplete data. Bioinformatics 2007;23:1164–7. [DOI] [PubMed] [Google Scholar]
- 48. Xiang Q, Dai X, Deng Y, et al. Missing value imputation for microarray gene expression data using histone acetylation information. BMC Bioinformatics 2008;9:252.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim H, Golub GH, Park H.. Missing value estimation for DNA microarray gene expression data: local least squares imputation. Bioinformatics 2005;21:187–98. [DOI] [PubMed] [Google Scholar]
- 50. Tuikkala J, Elo L, Nevalainen OS, et al. Improving missing value estimation in microarray data with gene ontology. Bioinformatics 2006;22:566–72. [DOI] [PubMed] [Google Scholar]
- 51. Troyanskaya O, Cantor M, Sherlock G, et al. Missing value estimation methods for DNA microarrays. Bioinformatics 2001;17:520–5. [DOI] [PubMed] [Google Scholar]
- 52. Elo LL, Karjalainen R, Öhman T, et al. Statistical detection of quantitative protein biomarkers provides insights into signaling networks deregulated in acute myeloid leukemia. Proteomics 2014;14:2443–53. [DOI] [PubMed] [Google Scholar]
- 53. Foss EJ, Radulovic D, Stirewalt DL, et al. Proteomic classification of acute leukemias by alignment-based quantitation of LC-MS/MS data sets. J Proteome Res 2012;11:5005–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Elo L, Filén S, Lahesmaa R, et al. Reproducibility-optimized test statistic for ranking genes in microarray studies. IEEE/ACM Trans Comput Biol Bioinform 2008;5:423–31. [DOI] [PubMed] [Google Scholar]
- 55. Robin X, Turck N, Hainard A, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 2011;12:77.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tyanova S, Temu T, Cox J.. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc 2016;11:2301–19. [DOI] [PubMed] [Google Scholar]
- 57. Cho SH, Goodlett D, Franzblau S.. ICAT-based comparative proteomic analysis of non-replicating persistent Mycobacterium tuberculosis. Tuberculosis 2006;86:445–60. [DOI] [PubMed] [Google Scholar]
- 58. Kammers K, Cole RN, Tiengwe C, et al. Detecting significant changes in protein abundance. EuPA Open Proteomics 2015;7:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jörnsten R, Wang HY, Welsh WJ, et al. DNA microarray data imputation and significance analysis of differential expression. Bioinformatics 2005;21:4155–61 [DOI] [PubMed] [Google Scholar]
- 60. Keller A, Eng J, Zhang N, et al. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol Syst Biol 2005;1:2005.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. MacLean B, Tomazela DM, Shulman N, et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010;26:966.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.