

Abstract
Background-
Plasma lipid levels are heritable and genetically associated with risk of coronary artery disease (CAD). However, genome-wide association studies (GWAS) routinely analyze these traits independently of one another. Joint GWAS for two related phenotypes can lead to a higher-powered analysis to detect variants contributing to both traits.
Methods-
We performed a bivariate GWAS to discover novel loci associated with both heart disease, using a CAD Meta-Analysis (122,733 cases and 424,528 controls), and lipid traits, using results from the Global Lipid Genetics Consortium (188,577 subjects).
Results-
We identified six previously unreported loci at genome-wide significance (P < 5 × 10−8), three which were associated with Triglycerides and CAD, two which were associated with LDL cholesterol and CAD, and one associated with Total Cholesterol and CAD. At several of our loci, the GWAS signals jointly localize with genetic variants associated with expression level changes for one or more neighboring genes, indicating that these loci may be affecting disease risk through regulatory activity.
Conclusions-
We discovered six novel variants individually associated with both lipids and coronary artery disease.
Keywords: Genome Wide Association Study, genomics, coronary artery disease, lipids, Genetic, Association Studies, Coronary Artery Disease, Lipids and Cholesterol
Introduction
Genome-wide association studies (GWAS) for coronary artery disease (CAD) have identified numerous susceptibility loci associated with the disease.1–3 These loci account for only a fraction of the total heritability for CAD, indicating that additional loci remain to be discovered.4 In addition, many CAD-associated regions lack an obvious mechanistic connection to heart disease. A recent trend to identify novel mechanistic connections is to utilize data integration, whereby associations for a phenotypic endpoint (e.g. CAD) are statistically analyzed jointly with additional data types to better understand the underlying phenotypic association.
The community is poised to make substantial progress in the understanding of CAD through integration of diverse, publicly available genome-wide datasets This is in part due to a basic understanding of risk factors for CAD. A recent study that looked for association across genome-wide significant loci for CAD and lipid traits found 25 shared loci, indicating a shared genetic basis for the most significantly associated loci.5 Cross-trait LD score regression, a method using the genome-wide correlation in effects on two traits to measure shared heritability, estimates positive genetic correlation between LDL-Cholesterol (LDL-C), Triglycerides (TG), and Total Cholesterol (TC) and CAD (0.25, 0.32, and 0.24 respectively), and negative correlation between HDL-Cholesterol (HDL-C) and CAD (−0.25), indicating that we should expect to find numerous loci shared between lipid traits and CAD.6
Not only are lipid levels and CAD risk genetically associated, but evidence supports a causal relationship between these traits. LDL-C, TG and TC are associated with CAD incidence in longitudinal studies, and statins have a well-established mechanism of action of lowering CAD risk through lowering LDL-C levels.7,8 There is additional evidence of causality between genetically determined lipid levels and CAD. Mendelian randomization adjusted for the pleiotropic effects of lipid loci has demonstrated causal effects of both LDL-C and TG on CAD, independent of one another, and has raised doubts about the independent effects of HDL on CAD,9,10 consistent with the lack of efficacy of HDL-raising therapeutics in clinical trials.11
Because serum lipid levels are heritable, large-scale association scans for these traits are available, and thus can be readily integrated with CAD association data.12,13 In addition, large-scale studies have now emerged which map genetic variation associated with changes in gene expression (i.e., eQTLs) across major human organs and tissues.14 These data provide an opportunity to link a CAD association signal to a putative causal gene and potentially a tissue of action.
Several new statistical approaches have emerged to integrate various data types. Multivariate studies that combine association data from multiple traits can help interpret the underlying associations at existing loci and have higher power for novel locus discovery than univariate studies.15 We and others have demonstrated the utility of such approaches through their use in identifying novel loci affecting CAD and type 2 diabetes,15 or loci affecting fasting insulin, triglyceride levels and HDL cholesterol levels.16 A key advantage of this approach is the potential for direct mechanistic insight into how a locus may affect the phenotypic endpoint of interest. For example, a locus associated with both cholesterol and heart disease might suggest a mechanism of action and therapeutic hypothesis to lower disease risk. However, coincidental associations of two traits at a genomic locus may not reflect a unified etiology. Thus, new methods have been developed which quantify the evidence that an overlapping association pattern across two datasets reflects a single underlying association (i.e., statistical colocalization), instead of coincidental overlap due to proximate, but different, causal variants.17
Here, we report a bivariate GWAS scan to discover novel loci associated with CAD and four lipid-related traits: LDL cholesterol, HDL cholesterol, Total Cholesterol and Triglyceride levels. To focus on a set of loci that potentially point to a shared causal variant and cognate gene of interest across traits, we applied statistical colocalization for the lipid and CAD association signals, both with each other and with gene expression datasets. We report six novel genome-wide significant loci where the GWAS association signals statistically colocalize: three affecting CAD and TG levels, two affecting CAD and LDL-C levels and one affecting CAD and TC. Of these loci, four signals also colocalized with eQTL signals from GTEx,14 indicating gene expression may be influencing disease risk at these loci.
Methods
The data, analytic methods, and study materials have been made available to other researchers for purposes of reproducing the results or replicating the procedure. Genome-wide summary statistics for our bivariate GWAS are available at http://coruscant.itmat.upenn.edu/data/SiewertEA_bivarStats.tar.gz. Methods are available as supplementary data. This study used data from the UK Biobank, CARDIOGRAM+C4D consortium and the GLGC. For each of these studies, appropriate approval by an institutional review committee was obtained, and all subjects gave informed consent, as detailed in the corresponding papers.2,3,13
Results
Overview of Bivariate Scan
Using a bivariate GWAS scan approach, we identified six novel loci associated with lipid levels and heart disease. As input into this scan, we used data from a meta-analysis of coronary artery disease, which combined results from the CARDIOGRAM+C4D consortium (88,192 CAD cases and 162,544 controls)2 and the UK BioBank (34,541 cases and 261,984 controls),3 and GWAS results for each of four lipid traits: HDL-C, LDL-C, Total Cholesterol, and Triglycerides, from the Global Lipid Genetics Consortium (188,577 individuals).13 Our trait covariances and bivariate p-values appeared calibrated (Supplementary Table 2, Supplementary Figure 1) and had a strong correlation with single-trait P-values, as would be expected (Supplementary Figures 2 and 3). In addition, the top hits from our scan included established lipid and CAD associated loci, including PCSK9 (significant for CAD paired with LDL-C and TC), APOE (CAD paired with all four lipids traits), and LPL (CAD paired with HDL-C and TG).
We performed several filtering steps to identify variants with non-trivial associations (P < 5 × 10−8) not yet established from individual trait scans. We first removed loci from our set with prior reported associations with either CAD or the lipid trait under consideration.1,12,18–20 Next, we narrowed our set of variants to those that were nominally associated with both traits from single-trait association data (P < 5 × 10−3 for the lipid trait and CAD). To focus on sites where we could hypothesize that the same causal variant contributes to both the lipid and CAD signals, we filtered out variants where the patterns of association for both traits did not statistically overlap. To determine this, we used the coloc software, which implements a Bayesian test for colocalization of two associations using summary statistics for two traits, to calculate the probabilities that the two traits are associated at a locus and share the same single underlying causal variant (PP4) and the probability that they do not share the causal variant (PP3). 17 We retained variants where the probability of the same causal variant was higher than that of different variants (PP4/(PP3+PP4) > 0.5, Methods, Supplementary Table 3). After these filters, we observed two loci associated with LDL-C and CAD, three with CAD and Triglycerides and one with TC and CAD (Table 1). Sentinel SNPs at these loci were not strongly linked to a nonsynonymous variant (Methods), consistent with a gene regulatory mechanistic hypothesis to explain phenotypic variability. We subsequently focus on associations with eQTLs or relevant prior phenotypic connections. One additional lead SNP, without an obvious link to an eQTL, rs7033354, is associated with LDL-C and CAD (P = 1.16 × 10−8, Table 1, Supplementary Figure 4).
Table 1:
Significant loci by bivariate scan with colocalization of the CAD and lipid trait GWAS signals
| Variant | chr | Position (Mb) | Lipid Trait | EA/OA | EAF | Bivariate P | Lipid Beta |
Lipid s.e. |
Lipid P |
CAD OR | CAD 95% CI | CAD P |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs895953 | 12 | 122.2 | TG† | T/G | 0.81 | 2.4×10−9 | 0.022 | 0.0043 | 8.6×10−8 | 0.98 | 0.97–0.99 | 9.1×10−4 |
| rs3741782 | 12 | 109.0 | LDL | A/G | 0.69 | 3.2×10−9 | −0.020 | 0.0039 | 6.3×10−7 | 0.97 | 0.96–0.99 | 3.3×10−6 |
| rs7033354 | 9 | 16.9 | LDL | T/C | 0.63 | 1.2×10−8 | −0.019 | 0.0038 | 1.4×10−6 | 0.98 | 0.97–0.99 | 7.4×10−6 |
| rs12423664 | 12 | 133.0 | TG | A/G | 0.13 | 1.2×10−8 | 0.026 | 0.005 | 8.2×10−8 | 1.03 | 1.01–1.05 | 5.2×10−4 |
| rs2908290 | 7 | 44.2 | TG | A/G | 0.38 | 1.6×10−8 | 0.017 | 0.0034 | 1.5×10−6 | 1.02 | 1.01–1.03 | 1.4×10−5 |
| rs1378942 | 15 | 75.0 | TC | A/C | 0.62 | 2.2×10−8 | 0.012 | 0.0037 | 9.1×10−6 | 0.98 | 0.97–0.99 | 1.8×10−5 |
EA: Effect Allele, OA: Other Allele, OR: Odds Ratio, CI: Confidence Interval, EAF: Effect Allele. Position is aligned to hg19. Frequency (EUR in 1000 Genomes).
In addition to the TG association, this SNP is nominally associated with HDL (Bivariate P = 8.78 × 10−8)
The monogenic diabetes gene glucokinase is associated with TG and CAD
Our first variant, rs2908290, is associated with TG and CAD (Bivariate P = 1.6 × 10−8) and is in an intron of the GCK (glucokinase) gene (Table 1, Figure 1). Although this SNP is not an eQTL in any assayed tissue, prior evidence supports GCK as the causal gene at this locus. Glucokinase phosphorylates glucose as the first step of glucose metabolism and is an established gene for maturity-onset diabetes of the young (MODY), a monogenic disorder causing early onset diabetes.21 It has been robustly associated with metabolic phenotypes in GWAS, including fasting glucose, glycated hemoglobin and type 2 diabetes (T2D).15,19,22,23 A mutation in the promoter of GCK was previously found to be associated with CAD in a candidate gene analysis,24 however, their variant is in very low linkage with our lead variant (European LD r2=0.05).
Figure 1:
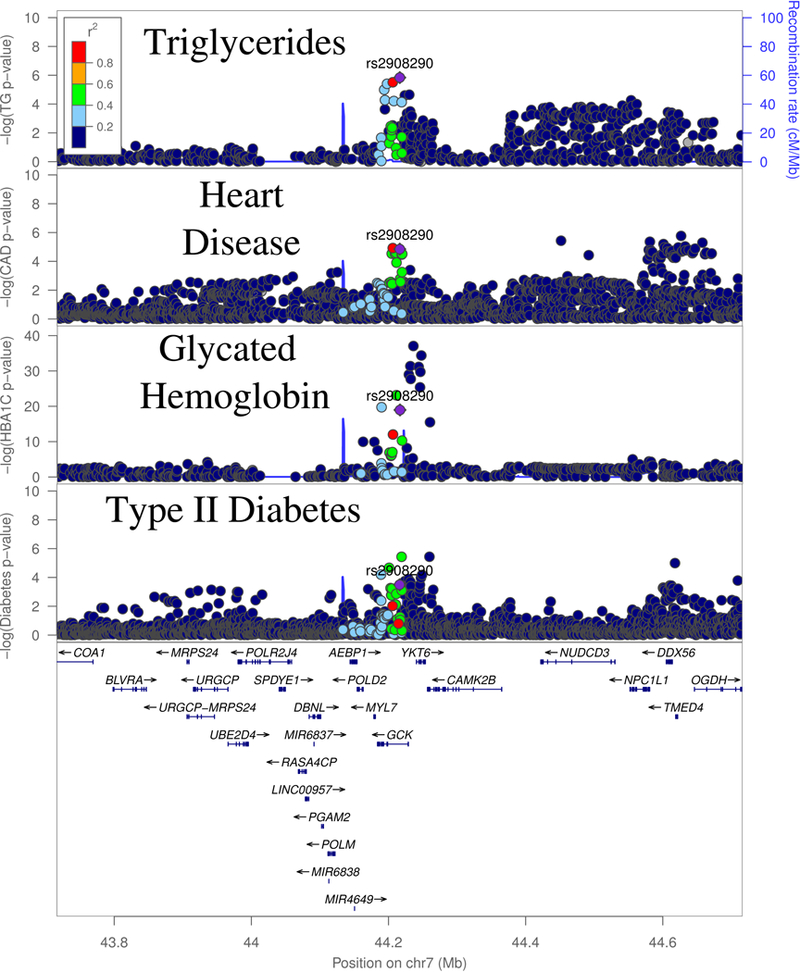
GWAS signals for Triglycerides (TG), CAD and T2D at the GCK locus.
The association signals at this locus are complex and suggest the possibility of several causal variants. Bien et al. found evidence of two independent associations at this locus for fasting glucose, with rs2908290 as the lead SNP of the second association.25 In line with this, we found evidence of three independent genome-wide significant causal variants for glycated hemoglobin, a marker of long-term elevated glucose, within 100kb of our lead SNP using approximate conditional analysis (Methods).23 However, none of the three lead variants are strongly linked with rs2908290, indicating the possibility of many different casual variants for various metabolic traits at this locus. To examine if the TG and CAD signal at rs2908290 statistically overlap, we performed a colocalization analysis on a window 100kb to either side of this SNP, to exclude a nominally significant SNP several hundred kb upstream of GCK. The TG and CAD GWAS showed strong statistical evidence of overlapping (COLOC PP4 = 0.87). Long-term overexpression of GCK has been shown to cause both hyperinsulinemia and hypertriglyceridemia in mice,26 which is broadly consistent with the pattern of genetic associations observed at this site (Supplementary Table 1).
As it is known that GCKR (glucokinase regulator) inhibits the activity of GCK, we next investigated the CAD association at a common variant in GCKR, Pro446Leu. Previous functional studies have shown that this variant diminishes the capability of GCKR to inhibit GCK, which results in increased glycolic flux and liver metabolites that may promote de novo lipogenesis,27 providing a mechanism for the human genetic associations of decreased fasting glucose and T2D risk but increased triglyceride levels seen at this locus.13,15,22,27 In addition to these established associations, we observed a modest association of higher CAD for carriers of the triglyceride-increasing allele (OR = 1.018, P = 1.9 × 10−3, Supplementary Table 1). This observed effect is consistent with the predicted combined effect of elevated triglycerides and lower fasting glucose levels on CAD risk (Predicted OR = 1.022, Methods). These data cumulatively suggest that genetic disruption of GCK influences diabetes and cardiovascular disease risk through known, causal intermediates.
Locus associated with TG and HDL-C may regulate genes affecting tyrosine catabolism or platelet traits
The second locus is associated in our bivariate scan with TG and CAD (Bivariate P = 2.35 × 10−9) and HDL-C and CAD (Bivariate P = 8.78 × 10−8, Figure 2), and is also an eQTL for several nearby genes. The lead variant, rs895953, is in an intron of the SET1B gene and, to our knowledge, has no prior established GWAS associations for related traits. Our sentinel SNP is an eQTL for three genes in the region in multiple tissues (Supplementary Table 4) including RHOF (Ras Homolog Family Member F, Filopodia Associated) and HPD (4-Hydroxyphenylpyruvate Dioxygenase). In contrast to epidemiological expectation, the major allele of this variant is associated with an increase in Triglycerides, but a reduced risk of CAD.
Figure 2.

GWAS and eQTL associations at the SET1B locus. Triglyceride, HDL-C and CAD signals are the GWAS signal corresponding to the respective traits. eQTL associations are for expression of the gene in Adipose Subcutaneous.
There is strong evidence of colocalization between RHOF and HPD expression and the TG signal (Supplementary Table 4). The CAD colocalization was weaker due to an unlinked CAD signal nearby (European LD r2=0.0, Figure 2, 3rd panel denoted by the arrow) which would violate the assumptions of the approach used to quantify colocalization. To confirm that the CAD signal overlapping the lipids and eQTL signals was not due to this secondary signal, we performed an approximate conditional analysis (Methods), conditioning on the top SNP from the nearby peak (rs10849885). The association strength for our sentinel SNP improved slightly (Punconditional = 9.1 × 10−4 to Pconditional = 3.6 × 10−4) upon conditioning, consistent with the lack of linkage disequilibrium between our sentinel SNP and the secondary CAD signal. Furthermore, the probability of colocalization between the eQTLs for RHOF, HPD, and each GWAS signal, as well as between the lipid and CAD GWAS signals themselves, also increased when we performed a colocalization analysis using a 200kb window in this region, which excludes the secondary CAD signal (Supplementary Table 3, Supplementary Table 4). These results are consistent with the hypothesis that a single causal variant underlies the GWAS signals and the expression association for RHOF and HPD at this locus.
Figure 3:
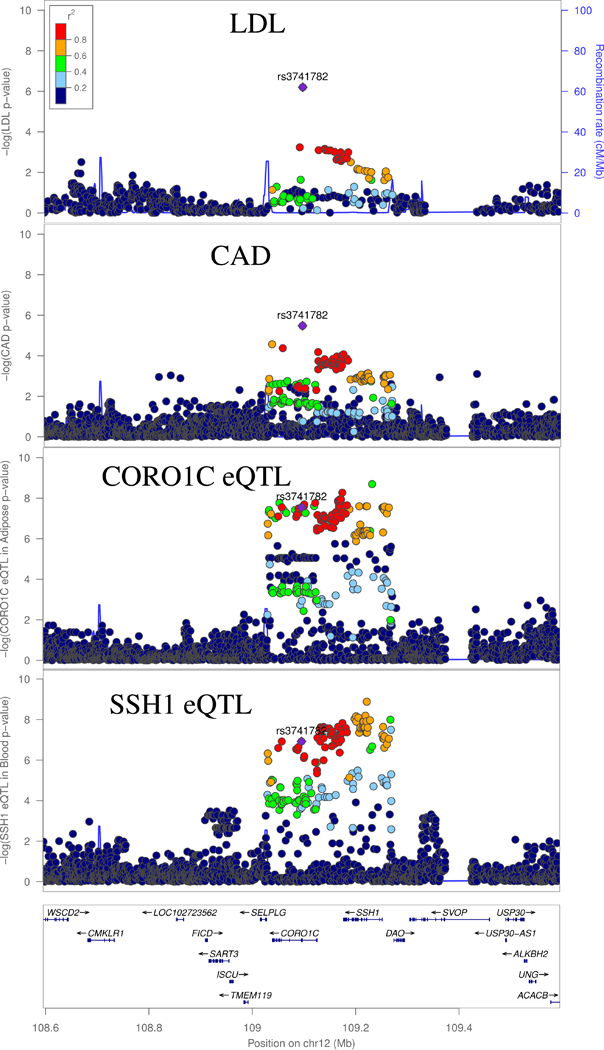
GWAS and eQTL Associations at the CORO1C locus. LDL-C and CAD signals are the GWAS signal corresponding to the respective traits. CORO1C eQTL signal is for expression in adipose, and SSH1 is for expression in whole blood.
Review of the literature suggests that one or both genes are plausible candidate genes for the CAD and lipid associations. HPD is involved in the catabolism of tyrosine, and has been associated with 2-Hydroxyisobutyrate levels, though this previous variant is not strongly linked with ours (European LD r2=0.03)28. 2-Hydroxyisobutyrate concentration is a known biomarker for insulin resistance and adiposity.29 However, literature may also support the hypothesis that RHOF is causal. This gene lies in a GWAS association for platelet count, platelet volume, and reticulocyte fraction of red cells.30 Platelet biology has been implicated in lipid metabolism and atherosclerosis, suggesting rs895953 could be affecting CAD risk through platelet-related pathways.31
Locus associated with LDL-C and CAD may affect actin remodeling
A locus tagged by the sentinel SNP rs3741782 was associated with LDL-C and CAD (Bivariate P = 3.2 × 10−9) and fell within an intron of the CORO1C gene (i.e., Coronin, Actin Binding Protein 1C, Figure 3). This SNP is an eQTL for CORO1C in visceral adipose tissue and in the adrenal gland, and for SSH1 (i.e., Slingshot Protein Phosphatase 1) in whole blood, with strong evidence of colocalization of these eQTL peaks with the CAD and LDL-C GWAS signals (Supplementary Table 4). Both SSH1 and CORO1C regulate actin reorganization.32,33 It has been demonstrated that uptake and degradation of lipoproteins by macrophages requires an actin cytoskeleton,34 and oxidization of the associated lipids in macrophages can result in foam cells, which contribute to atherosclerosis.35 However, to our knowledge, it is unknown whether CORO1C or SSH1 participate in this mechanism.
Locus associated with TG and CAD may regulate expression of FBRSL1
A fourth locus, tagged by sentinel SNP rs12423664 (Bivariate P = 1.2 × 10−8), is located in an intron of FBRSL1 (i.e., Fibrosin Like 1) (Figure 4). Evidence has shown that the protein encoded by this gene may be a member of the polycomb repressive complex 1 (PRC1), which regulates gene expression via epigenetic markers.36 Our lead SNP is an eQTL for this gene in both pancreas and whole blood and is partially linked to a previous GWAS association with red blood cell count (European LD r2=0.52 between rs34390795 and our sentinel SNP).30
Figure 4:
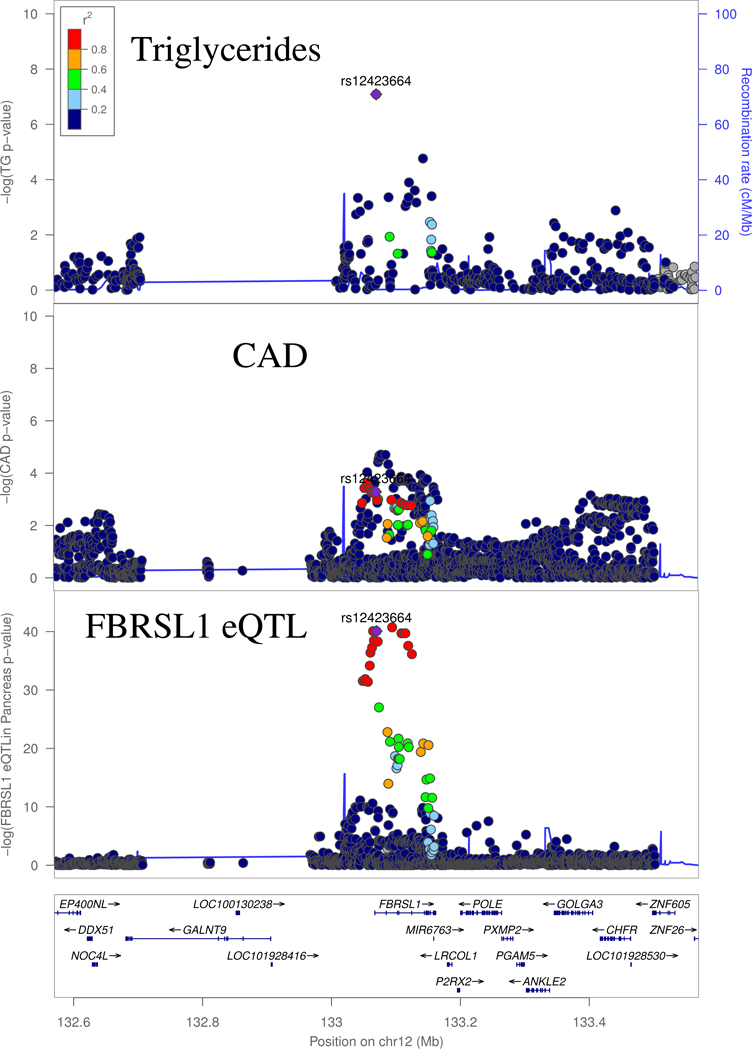
GWAS and eQTL Associations at the FBRSL1 locus. Triglyceride (TG) and CAD signals are the GWAS signal corresponding to the respective traits. FBRSL1 eQTL is signal is for expression of the gene in pancreas.
Available evidence suggests that the eQTL association may underlie the lipid and CAD association. Colocalization analysis supports a shared underlying causal variant between the lipid and CAD signal, and the eQTL (Supplementary Table 3, Supplementary Table 4). That being said, the underlying lipid GWAS was not as density genotyped as the CAD and eQTL studies (Figure 3). Indeed, the lead GWAS variant for CAD (European LD r2=0.08 between rs4883525 and rs12423664), along with several other top CAD variants, were not assayed in the lipid GWAS data. We expect larger-scale imputation studies for the lipids scan should enable more well-powered colocalization analysis at this locus.
The Fibrosin-Like 1 protein has been found to have lower expression in human hearts with dilated cardiomyopathy, relative to control hearts.37 In the eQTL signal colocalizing with our bivariate peak, the alternative allele (A) of our lead SNP is associated with lower expression of this gene, though this association was observed in pancreas, not heart.14 This same allele is associated with increased lipid levels and an elevated risk of heart disease. These results indicate that Fibrosin Like 1 may play a role in healthy heart processes, and thus reduced expression of this gene may increase risk of heart disease. However, the diverse biological processes that PRC1 may influence make it difficult to interpret how this gene may be affecting triglycerides and heart disease.
Variant affecting blood pressure, CAD and Total cholesterol
Our final variant, rs1378942, is associated in our bivariate scan with CAD and total cholesterol (Bivariate P = 2.22 × 10−8), and also nominally associated with LDL-C (Figure 5). The lipid traits strongly colocalize with each other and moderately colocalize with the CAD locus (Supplementary Table 4). In addition, this index SNP has an established association with systolic blood pressure (P = 6 × 10−23),19 with moderate to strong evidence of colocalization between the two lipid traits, CAD and SBP (Supplementary Table 5). The ‘A’ allele of this SNP increases LDL and TC while decreasing SBP and CAD, pointing to SBP as the directionally consistent causal phenotype for the CAD association.
Figure 5:
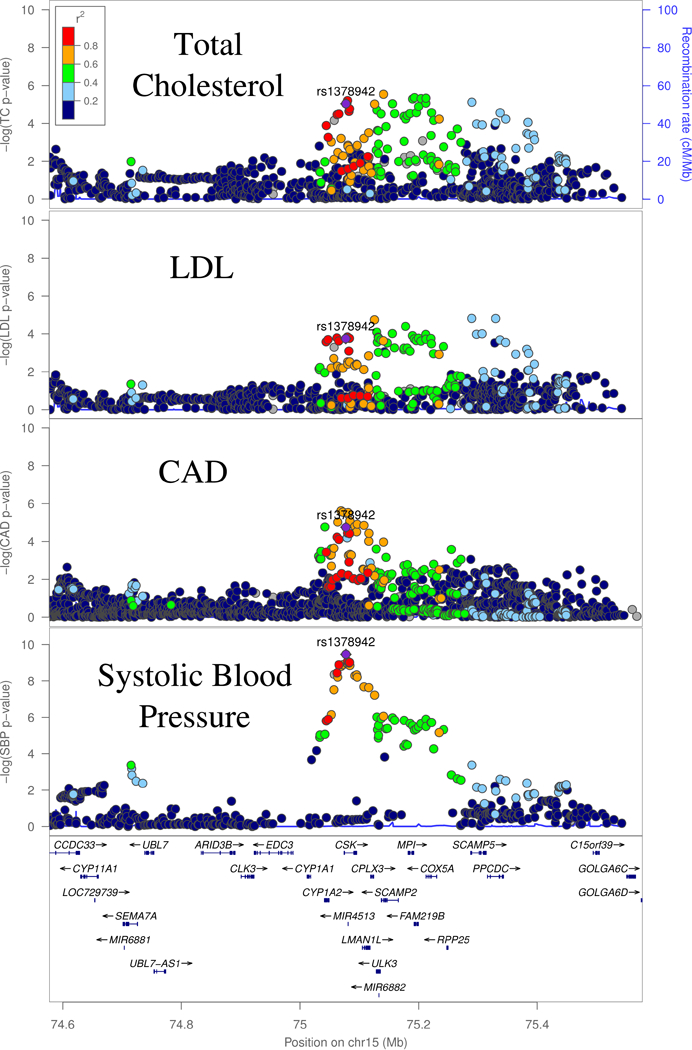
GWAS Associations at the CSK locus. Total Cholesterol (TC), LDL-C and SBP (systolic blood pressure) and CAD signals are the GWAS signal corresponding to the respective traits.
rs1378942 is an eQTL for CSK (C-terminal Src Kinase), in both subcutaneous adipose and the left ventricle of the heart, and CYP1A1 (Cytochrome P450 Family 1 Subfamily A Member 1) in Subcutaneous Adipose and Muscle Skeletal, among several other genes.14 The eQTL signals for these two genes have moderate to strong evidence of colocalization with the CAD and TC GWAS peaks, although visual inspection shows that there may be more than one causal eQTL variant for the CSK eQTL association (Supplementary Figure 5, Supplementary Table 4). siRNA knockdown of CSK was found to increase blood pressure, suggesting it is as a possible causal gene for SBP at this locus.38 However, CYP1A1 is another possible causal gene. Although the substrate of the gene is not firmly established, members of the cytochrome p450 family are known to affect cardiovascular disease risk through effects on a diverse set of processes, including lipid metabolism and vasoconstriction.39
Discussion
We identified six novel variants associated with both CAD and one or more lipid traits using a bivariate approach, with potential candidate genes identified via integration with gene expression data. This demonstrates the power of data integrative approaches to discover novel loci influencing phenotypes of interest. These loci are particularly interesting, because they suggest that CAD risk is influenced through lipid associations at each locus. However, other scenarios are also possible: namely, a variant may not affect CAD directly through lipid levels and may instead influence other processes by which lipid and CAD risk levels are altered independently of one another.
One limitation of the method we applied is that our test may be conservative under the null hypothesis. Cohort overlap, as is seen between the CARDIoGRAM+C4D and Global Lipid Genetics cohorts is expected to increase the estimated covariance of CAD and each lipid trait by increasing risk allele concordance under the null. This results in a reduction of the chi-squared test statistic.6 We also note that while the CAD UK Biobank and lipid GWAS are composed almost entirely of individuals of European ancestry, the meta-analysis for the CARDIoGRAM+CAD is comprised of individuals from both European (~76%) and non-European ancestries. Population differences could decrease the measured covariance between two correlated traits, resulting in increased chi-squared statistics and more significant p-values at a locus that is causal for both traits in the studied populations. To determine the overall impact of these two competing effects, we performed an analysis of these two GWAS studies using cross-trait LD-score regression.6 We observed that the y-intercept departed from zero in the direction of the correlation (data not shown), indicating that the covariance term we estimated is larger than necessary, resulting in a conservative test.
Our results speak to the complexity of assigning functional mechanisms to associated variants. Three of our loci suggest the possibility of several different regulatory targets, indicating these loci may not affect CAD and lipids through a single regulatory mechanism. Instead, these loci may harbor regulatory variants for several genes, which independently affect lipid levels and therefore CAD. Alternatively, as shown in the case of rs3741782, these regulatory targets may be involved in the same biological pathway. Furthermore, conditional analyses reveal that several of the GWAS and eQTL signals may harbor more than one causal variant, as has been previously demonstrated for several metabolic-related loci.40,41
The combination of lipid and heart disease associations and their colocalization with eQTLs for genes involved with cardiometabolic process supports the potential therapeutic potential of the loci found in our bivariate scan. Further functional experimental work will be required to verify and elucidate these candidates’ association with lipid and CAD pathways.
Supplementary Material
Acknowledgments
Sources of Funding: This work was supported by a grant from the National Institutes of Health NIDDK R01DK101478 to B.F.V.
Footnotes
Disclosures: B.F.V. currently serves as an associate editor at Circulation: Genomics and Precision Medicine. There are no additional conflicts of interest to declare.
References:
- 1.Nelson CP, et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet. 2017;49:1385–1391. [DOI] [PubMed] [Google Scholar]
- 2.Nikpay M, et al. A comprehensive 1000 Genomes–based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Harst P, et al. Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ Res. 2018;122:433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McPherson R, et al. Genetics of Coronary Artery Disease. Circ Res. 2016;118:564–78. [DOI] [PubMed] [Google Scholar]
- 5.van der Laan SW, et al. From lipid locus to drug target through human genomics. Cardiovasc Res. 2018;114:1258–1270. [DOI] [PubMed] [Google Scholar]
- 6.Bulik-Sullivan B, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47:1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.The Emerging Risk Factors Collaboration* et al. Major Lipids, Apolipoproteins, and Risk of Vascular Disease. JAMA. 2009;302:1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steinberg D Thematic review series: the pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy, part V: the discovery of the statins and the end of the controversy. J Lipid Res. 2006;47:1339–51. [DOI] [PubMed] [Google Scholar]
- 9.Burgess S, et al. Multivariable Mendelian Randomization: The Use of Pleiotropic Genetic Variants to Estimate Causal Effects. Am J Epidemiol. 2015;181:251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holmes MV, et al. Mendelian randomization of blood lipids for coronary heart disease. Eur Heart J. 2015;36:539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Group THC. Effects of Anacetrapib in Patients with Atherosclerotic Vascular Disease. N Engl J Med. 2017;377:1217–1227. [DOI] [PubMed] [Google Scholar]
- 12.Liu DJ, et al. Exome-wide association study of plasma lipids in >300,000 individuals. Nat Genet. 2017;49:1758–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Willer CJ, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aguet F, et al. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao W, et al. Identification of new susceptibility loci for type 2 diabetes and shared etiological pathways with coronary heart disease. Nat Genet. 2017;49:1450–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lotta LA, et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet. 2016;49:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giambartolomei C, et al. Bayesian Test for Colocalisation between Pairs of Genetic Association Studies Using Summary Statistics. Williams SM, ed. PLoS Genet. 2014;10:e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu X, et al. Exome chip meta-analysis identifies novel loci and East Asian–specific coding variants that contribute to lipid levels and coronary artery disease. Nat Genet. 2017;49:1722–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.MacArthur J, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017;45:D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klarin D, et al. Genetic analysis in UK Biobank links insulin resistance and transendothelial migration pathways to coronary artery disease. Nat Genet. 2017;49:1392–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Froguel P, et al. Familial Hyperglycemia Due to Mutations in Glucokinase -- Definition of a Subtype of Diabetes Mellitus. N Engl J Med. 1993;328:697–702. [DOI] [PubMed] [Google Scholar]
- 22.Manning AK, et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet. 2012;44:659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wheeler E, et al. Impact of common genetic determinants of Hemoglobin A1c on type 2 diabetes risk and diagnosis in ancestrally diverse populations: A transethnic genome-wide meta-analysis. Gregg E, ed. PLOS Med. 2017;14:e1002383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marz W, et al. G(−30)A Polymorphism in the Pancreatic Promoter of the Glucokinase Gene Associated With Angiographic Coronary Artery Disease and Type 2 Diabetes Mellitus. Circulation. 2004;109:2844–2849. [DOI] [PubMed] [Google Scholar]
- 25.Bien SA, et al. Transethnic insight into the genetics of glycaemic traits: fine-mapping results from the Population Architecture using Genomics and Epidemiology (PAGE) consortium. Diabetologia. 2017;60:2384–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferre T, et al. Long-term overexpression of glucokinase in the liver of transgenic mice leads to insulin resistance. Diabetologia. 2003;46:1662–1668. [DOI] [PubMed] [Google Scholar]
- 27.Beer NL, et al. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum Mol Genet. 2009;18:4081–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raffler J, et al. Genome-Wide Association Study with Targeted and Non-targeted NMR Metabolomics Identifies 15 Novel Loci of Urinary Human Metabolic Individuality. PLoS Genet. 2015;11:e1005487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferrannini E, et al. Early metabolic markers of the development of dysglycemia and type 2 diabetes and their physiological significance. Diabetes. 2013;62:1730–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Astle WJ, et al. The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell. 2016;167:1415–1429.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahmadsei M, et al. Immune-mediated and lipid-mediated platelet function in atherosclerosis. Curr Opin Lipidol. 2015;26:438–448. [DOI] [PubMed] [Google Scholar]
- 32.Mizuno K Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cell Signal. 2013;25:457–469. [DOI] [PubMed] [Google Scholar]
- 33.Xavier C-P, et al. Phosphorylation of CRN2 by CK2 regulates F-actin and Arp2/3 interaction and inhibits cell migration. Sci Rep. 2012;2:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hölttä-Vuori M, et al. Endosomal actin remodeling by coronin-1A controls lipoprotein uptake and degradation in macrophages. Circ Res. 2012;110:450–5. [DOI] [PubMed] [Google Scholar]
- 35.Moore KJ, et al. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13:709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boyer LA, et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006;441:349–353. [DOI] [PubMed] [Google Scholar]
- 37.Creemers EE, et al. Genome-Wide Polyadenylation Maps Reveal Dynamic mRNA 3’-End Formation in the Failing Human Heart. Circ Res. 2016;118:433–8. [DOI] [PubMed] [Google Scholar]
- 38.Lee H-J, et al. Gene Silencing and Haploinsufficiency of Csk Increase Blood Pressure. Jia Z, ed. PLoS One. 2016;11:e0146841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elbekai RH, et al. Cytochrome P450 enzymes: Central players in cardiovascular health and disease. Pharmacol Ther. 2006;112:564–587. [DOI] [PubMed] [Google Scholar]
- 40.Roman TS, et al. Multiple Hepatic Regulatory Variants at the GALNT2 GWAS Locus Associated with High-Density Lipoprotein Cholesterol. Am J Hum Genet. 2015;97:801–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smemo S, et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.