

Abstract
The KRAS GTPase plays a fundamental role in transducing signals from plasma membrane growth factor receptors to downstream signalling pathways controlling cell proliferation, survival and migration. Activating KRAS mutations are found in 20% of all cancers and in up to 40% of colorectal cancers, where they contribute to dysregulation of cell processes underlying oncogenic transformation. Multiple KRAS-regulated cell functions are also influenced by changes in intracellular Ca2+ levels that are concurrently modified by receptor signalling pathways. Suppression of intracellular Ca2+ release mechanisms can confer a survival advantage in cancer cells, and changes in Ca2+ entry across the plasma membrane modulate cell migration and proliferation. However, inconsistent remodelling of Ca2+ influx and its signalling role has been reported in studies of transformed cells. To isolate the interaction between altered Ca2+ handling and mutated KRAS in colorectal cancer, we have previously employed isogenic cell line pairs, differing by the presence of an oncogenic KRAS allele (encoding KRASG13D), and have shown that reduced Ca2+ release from the ER and mitochondrial Ca2+ uptake contributes to the survival advantage conferred by oncogenic KRAS. Here we show in the same cell lines, that Store-Operated Ca2+ Entry (SOCE) and its underlying current, ICRAC are under the influence of KRASG13D. Specifically, deletion of the oncogenic KRAS allele resulted in enhanced STIM1 expression and greater Ca2+ influx. Consistent with the role of KRAS in the activation of the ERK pathway, MEK inhibition in cells with KRASG13D resulted in increased STIM1 expression. Further, ectopic expression of STIM1 in HCT 116 cells (which possess the KRASG13D mutation) rescued SOCE, demonstrating a fundamental role of STIM1 in suppression of Ca2+ entry downstream of KRASG13D. These results add to the knowledge of how ERK controls cancer cell physiology and highlight STIM1 as an important biomarker in cancerogenesis.
Keywords: Calcium signalling, STIM, KRAS, colorectal cancer cancer, ERK, SOCE/ICRAC
Graphical Abstract
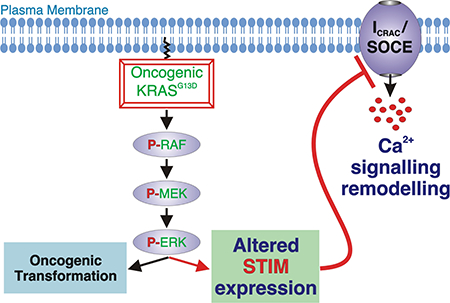
1. Introduction
The small GTPase RAS is a molecular switch that couples receptor tyrosine kinase (RTK) activation with downstream signalling pathways regulating cell proliferation, survival and migration [1]. Somatic mutations at several levels in these pathways, for example in RTKs (EGFR), RAS itself, BRAF and PI3K, are highly prevalent in cancer and are responsible for the constitutive activation of affected pathways observed [2,3]. Notably, underlining its importance in oncogenic transformation, RAS is mutated in 20–30 % of all cancers and up to 40% of colorectal cancer (CRC), which is the second and third most prevalent cancer in women and men, respectively, affecting over one million patients per year globally [4]. Of the three RAS isoforms, KRAS is most frequently mutated in cancer, and as the only essential isoform it has the greatest impact on cell biology [1]. The importance of growth factor signalling pathways in oncogenic transformation has placed them at the centre of cancer drug development efforts. To date however, while drugs targeting RTKs, BRAF, MEK and PI3K are at various stage of development or in use in the clinic, no effective drugs targeting KRAS are available [5].
Constitutive activation of KRAS in cancer is brought about through missense point mutations in the codons encoding Gly12, Gly13 or Gln61 [6,7]. Consequently, the activities of pathways downstream of KRAS are increased. KRAS signals via four main pathways: the RAF/MEK/ERK pathway, the PI3K/AKT pathway, the RAL pathway and the PLCε/PKC/Ca2+ pathway. These pathways do not act in isolation however but signal in concert. Notably, Ca2+ interacts with the KRAS-RAF-MEK-ERK pathway at multiple levels as well as engaging overlapping downstream mediators [8–10]. For example, inhibition of BRAF increases the expression of Ca2+ ATPase isoform 4b (PMCA4) [11] and facilitates endoplasmic reticulum (ER)mitochondrial Ca2+ transfer [12], while opening of canonical transient receptor potential 3 (TRPC3) non-selective cation channels is required for ERK activation in B-lymphocytes [13]. Like KRAS, Ca2+ signals have pleiotropic effects controlling cellular life and death decisions [14–16]. As such, dysregulated Ca2+ signalling pathways contribute to the altered activity of cell processes underlying cancer [14,16–19]. Of particular note, Ca2+ signals are necessary to sustain the cell cycle at specific checkpoints such as G1/S and G2/M transitions [20,21] and mitochondrial Ca2+ overload is a key event in activating the apoptotic cascade [22]. Cell migration and invasion associated with tumour metastasis are also regulated by Ca2+ [23–26].
Ca2+ signals are generated by release from intracellular stores and/or influx across the plasmalemma. While Ca2+ signalling pathways centred on the endoplasmic reticulum (ER) have primarily been invoked in the altered cell physiology of cancer cells, a prominent role for Ca2+ influx across the plasmalemma is now also emerging [17,18]. Indeed, the Ca2+ influx pathway engaged following depletion of the ER Ca2+ store (Store-Operated Ca2+ Entry; SOCE) is considered important in the regulation of cell proliferation, migration and apoptosis [27–29] – cell processes modified in cancer cells [30,31]. Ca2+ influx via SOCE directly affects the proliferation of normal (non cancer cells) as well as cancer cell types [32–34], including melanoma cells [35], renal cell carcinoma [36], cervical cancer cells [37], glioblastoma cells [38–40], HEK 293 cells [41,42] and colorectal cancer [43,44]. As well as acting to replenish a depleted ER Ca2+ store to allow further Ca2+ signalling, changes in SOCE are likely to have broad implications for the phenotype of cancer cells [45–49]. Indeed, the mediators of SOCE, STIM1 and ORAI1, have been found to control G1/S transition in cervical cancer SiHa cells [20]. Moreover, inhibition of STIM1 and ORAI1 via siRNA impairs migration of breast [50,51] and cervical cancer cells [37], whereas overexpression of STIM1 favours invasion [43]. Endothelial cell proliferation and tube formation in renal cell carcinoma underlying tumour angiogenesis is also dependent on SOCE [52].
Despite these advances in identifying altered Ca2+ intracellular Ca2+ regulation as a hallmark of cancer, the link between Ca2+ homeostasis/signalling and cellular transformation induced by a particular oncogenic mutation remains poorly explored. While analysis of tumour-derived and normal cells of the same tissue origin or neighbouring to the tumour has provided important information [44,53], these controls may not adequately reflect the cell type/genetic status of the cell type of origin. Further inconsistencies are observed on analysis of transformed cells from tumours at different stages and origins as well as whether the original tumour or its metastasis are analysed [54–56]. An approach successfully used to circumvent these issues in cellular diversity has been to determine the consequences of genetic deletion of specific mutated oncogenic alleles from a transformed cell [57–59]. Comparison of isogenic cell line pairs thus generated provides specific information regarding the interaction between the mutated allele and the phenotype studied. Given the frequency of KRAS mutations in colorectal cancer, we have used this approach to probe functional interactions between this GTPase and Ca2+ handling. We previously demonstrated that through suppression of inositol 1,4,5-trisphosphate receptor (InsP3R) expression, cells harbouring a mutated KRAS allele exhibited a reduction in both Ca2+ signalling induced by an InsP3-generating agonist and mitochondrial Ca2+ uptake, which together served to protect from death-inducing stimuli [60]. Given the growing body of evidence supporting a role of Ca2+ influx in cell proliferation and cell migration in cancer, including in CRC, we here examined whether Ca2+ entry mechanisms were also a target of activated KRAS. As previously, we analysed CRC cell line pairs that were isogenic albeit for a single copy of oncogenic KRAS (encoding KRASG13D), which was deleted by homologous recombination (Shirasawa et al., 1993). Use of these cell line pairs thus allows the selective analysis of the influence of the oncogenic KRAS on cell phenotype without the confounding effects observed in overexpression studies for example where oncogenic KRAS is expressed at supraphysiological levels [61]. Using biochemical, fluorometric and electrophysiological approaches, we demonstrate that KRASG13D expression in the isogenic CRC cell model was associated with reduced SOCE and ICRAC as well as remodelled expression of STIM proteins. KRASG13D expression was also associated with a reduced sensitivity to cell death induced by activation of SOCE. The lower Ca2+ entry in KRASG13D expressing cells was augmented to the levels in the KRAS-deficient cells by STIM1 overexpression, indicating that STIM1 expression is a direct downstream target of KRASG13D in CRCs. Targeting KRAS/Ca2+ signalling interactions pathway may provide a strategy to intervene in the development of CRC.
2. Methods
2.1. Materials
Salts for physiological recordings were of the highest grade and purchased from SigmaAldrich, Fisher Scientific or BDH. Na-methanesulfonate, Adenosine 5′ -triphosphate magnesium salt and Cs-methanesulfonate and salts for internal solutions were from Sigma (St. Louis, MO, USA). Cs-BAPTA and Ca2+-sensitive fluorescent indicators were from Invitrogen (Eugene, OR, USA). Sources for other reagents used are indicated where described.
2.2. Cell culture
HCT 116 and DLD-1 cells (both KRASG13D/WT) and their respective isogenic derivatives HKH-2 and DKO-4 (both KRAS-/WT) were a kind gift of S. Shirasawa (Fukuoka University, Japan) and have been previously described [57]. Cells were cultured in DMEM (Life Technologies, Carlsbad, CA, USA), containing 10 % heat-inactivated foetal bovine serum (FBS) (Invitrogen), 1 % penicillin/streptomycin solution (5 units penicillin, 55 μg streptomycin) (Sigma, Dorset, UK). Cells were maintained at 37 °C with 5 % CO2 in saturated humidity and passaged upon reaching 80–90 % confluency.
2.3. Imaging of cytosolic Ca2+
Cytosolic Ca2+ was imaged as previously described [62]. Briefly, cells were seeded onto polyL-lysine-coated coverslips at equivalent densities and imaged after 48 h. Imaging was carried out using Ca2+ containing and Ca2+ free HEPES buffered solutions as previously described [62] and included in the supplementary information. Prior to each experiment, coverslips were mounted into stainless steel imaging chambers and loaded with fura-2 AM diluted in Ca2+ containing imaging buffer (Life Technologies; 2 μM for 30 min), followed by de-esterification in Ca2+-free buffer for 30 min during which thapsigargin (Tg, 2 μM; SIGMA) was included for the final 15 min. Coverslips were imaged on the stage of a Nikon Eclipse TE200 inverted epifluorescence microscope equipped with a Nikon PlanFluor 20x/0.75 NA multi immersion objective (Nikon, Kingston Upon Thames, Surrey, UK). Excitation light at 340 and 380 nm was selected using a motorised filter wheel (Sutter Industries, Novato, CA, USA) at a frequency of 1 image pair every 3 s with an exposure of 200 ms and emitted light was selected using a 400 nm dichroic mirror and filtered through a 460 nm long pass filter. Images were captured using a Hamamatsu ORCA ER Charge-Coupled Device (CCD) camera. Three coverslips per cell type were imaged per day on 3 separate days. Ca2+ concentration values were calculated according to published methods [63].
2.4. Whole-cell patch clamp electrophysiology
Whole-cell patch clamp recordings were performed at room temperature using an Axopatch 200B and Digidata 1440A (Axon Instruments) previously published [64,65]. Internal and bath solutions were as previously described [64,65] and as provided in the supplementary information. Clampfit 10.1 software was used for data analysis. Pipettes were pulled from borosilicate glass capillaries (World Precision Instruments, Inc. Sarasota, FL) with a P-1000 Flaming/Brown micropipette puller (Sutter Instrument Company, Novato, CA) and polished with DMF1000 (World Precision Instruments, Inc. Sarasota, FL) to a resistance of 2–4 MΩ when filled with pipette solutions. Immediately before the experiments, cells were washed with bath solution. Only cells with tight seals (>16 GΩ) were selected to break in. Cells were maintained at a 0 mV holding potential during experiments and subjected to voltage ramps from +100 to −140 mV lasting 250 ms every 2 s. “Reverse” ramps were designed to inhibit Na+ channels potentially expressed in these cells. High MgCl2 (8 mM) was included in the patch pipette to inhibit TRPM7 currents. All experiments were performed at room temperature.
2.5. Transient transfection
For expression of STIM1 in HCT 116 cells, plasmids were used encoding YFP-STIM1 (kind gift of Prof S. Muallem, NIDCR, Bethesda, USA) or YFP (purchased from Clontech). Cells were transfected using JetPei (Polyplus Transfection) following the manufacturer’s instructions 24 h post seeding the cells onto glass coverslips. Transfection efficiency was ~50 %.
2.6. Mn2+ quench imaging
Cells were seeded as described for ratiometric imaging and imaged using an Olympus IX81 inverted epifluorescence microscope equipped with an Olympus UPlanSApo 20x/0.75 NA air objective. Wavelengths for excitation of fura-2 in its Ca2+ free (380/10 nm), Ca2+-bound (340/10 nm), and Ca2+ insensitive isosbestic −2 (360/4 nm) forms were selected using a Polychrome V monochromator. Emitted light was filtered through a 400 nm dichroic mirror and an emission filter wheel. Images were captured using a Hamamatsu ORCA ER CCD camera. The imaging system was controlled by the Olympus CellR software. When imaging YFP-transfected cells, a reference image of the YFP-positive cells was captured using 480 nm excitation and filtering through a 520 nm long-pass filter.
2.7. Immunoblotting
Cells were seeded and harvested after 48 h or at the times specified. 15 to 30 μg of protein lysate for each sample, quantitated using a bincinchoninic acid (BCA) protein assay kit (Thermo Scientific), was loaded per lane onto 7 % SDS- polyacrylamide gels or 4–12 % Bis-Tris gradient gels (NuPage). Proteins were transferred from the gels onto polyvinilidene fluoride (PVDF) membranes and probed with the following antibodies: anti-STIM1 (dilution 1:1000, BD Bioscience), anti-STIM2 (dilution 1:1000, Sigma) and anti-calnexin (dilution 1:20000, Sigma).
Detection of bands was carried out after incubation with horseradish peroxidase (HRP)conjugated secondary antibodies (dil. 1:10,000) using Enhanced Chemiluminescence (ECL) reagent (Thermo Scientific) or using fluorescence-based detection (Li-Cor CLx) using secondary antibodies conjugated to fluorophores excited at 680 nm and 800 nm. In the case of Li-Cor detection, multiple primary antibodies conjugated to different species were used at the same time, producing one set of loading control bands.
2.8. Immunofluorescence
Cells were seeded as described for cytosolic Ca2+ imaging. 48 h post-seeding cells were processed for immunofluorescence analysis as previously described [60] Briefly, after washing in PBS, cells were fixed with 2% paraformaldehyde 0.05% glutaraldehyde. Cells were then permeabilised in PBS containing 0.2 % TRITON X-100 after which non-specific sites were blocked with Chemiblocker (5% in PBS/0.1% TRITON X-100; EMD Millipore). Cells were then labelled with primary antibodies against STIM-1 (BD Bioscience; 1:100) and calnexin (SIGMA; 1:500) diluted in blocking buffer overnight at 4 °C. After washing for 1 h, coverslips were incubated with Alexa-Fluor conjugated secondary antibody (dilution 1:500; Thermo Fisher). Coverslips were then mounted onto glass slides with Vectashield mounting agent containing DAPI (Vector laboratories). Cells were imaged using an Olympus FV1000 point-scanning confocal imaging system configured on an Olympus IX81 inverted microscope equipped with an Olympus PlanSApo 60x/1.35 oil objective.
2.9. Flow Cytometry
Analysis of sub-G1 DNA content was performed as previously described [60,66] with minor modifications. Cells in the medium were collected and pooled with cells that remained attached to their substrate that were harvested by trypsinization. After washing in PBS, cells were fixed with 70% ethanol and then treated with RNase and stained with propidium iodide (PI). Stained cells were analysed with a Becton Dickinson Canto II AIG flow cytometer (Oxford, UK). Single cells in suspension were excited at 488 nm by an argon laser and analysed according to the intensity of emitted fluorescence at 572 nm.
2.10. Statistical analysis
Statistical analyses were performed using GraphPad Prism 6. Data represents a minimum of 3 independent biological replicates, sample numbers for each experiment indicated in the figure legend. For testing statistical significance of three or more conditions, the means were compared by analysis of variance (ANOVA) followed by post-hoc Holm-Sidak’s Multiple Comparison test (for testing statistical significance between groups of the same dataset, illustrated with a line between groups). A one-sample Student’s t-test was used for establishing significance between two experimental conditions within a data set. Data was taken as significant when P was less than 0.05, which was represented as *. P-values less than 0.01, 0.001, 0.0001 were represented by **, *** and ****, respectively.
3. Results
3. 1. K-RasG13D suppresses SOCE in colorectal cancer cell lines
Using the HCT 116 colorectal cancer cell line (possessing the oncogenic KRAS allele encoding KRASG13D) and its isogenic derivative HKH-2 (oncogenic allele ablated by homologous recombination), we previously identified an interaction between InsP3-mediated Ca2+ release from intracellular ER stores, mitochondrial Ca2+ uptake and oncogenic KRAS that was protective against apoptotic stimuli [60]. Given the importance of SOCE in cancer cell processes and regulation of ER Ca2+ levels, which are altered in KRAS mutated cells, SOCE was examined in the same HCT 116 and HKH-2 cell pair. To this end, experimental strategies that allowed isolation of SOCE from other Ca2+ signalling mechanisms were employed. SOCE was first investigated by measuring Ca2+ influx across the PM during a period of Ca2+ re-addition following depletion of ER Ca2+ with the irreversible inhibitor of the SERCA pump thapsigargin (Tg). Application of Tg to cells bathed in Ca2+-free imaging buffer induced a typical increase in cytosolic Ca2+ concentration [Ca2+]i due to uncovering of the passive leak of Ca2+ from the ER (Fig. 1A i and as in Pierro et al. 2014). After 10 min treatment with Tg to allow depletion of the ER Ca2+ store, cells were superfused with Ca2+-containing imaging buffer, thereby initiating SOCE. The resulting increase in [Ca2+]i reached a peak amplitude and magnitude (integrated signal) that was no different between the two cell types (Fig. S1A). As the peak amplitude of the entry Ca2+ signal is influenced by feedback mechanisms, Ca2+ buffering and clearance mechanisms, the rate of rise of the Ca2+ entry signal, which provides a more direct measure of Ca2+ flux [67,68], was also determined. To this end, the Ca2+ entry signals in both HCT 116 and HKH-2 cells were normalized to the maximal [Ca2+]i level (Fig 1Aii) and the first derivative was obtained (Fig. 1Aiii and iv). The maximal rate of Ca2+ influx was significantly greater in HKH-2 cells than in HCT 116 cells (Fig. 1Aiv). Consistent with the reduced Ca2+ entry following store depletion with Tg in HCT 116 cells, these cells were protected against cell death resulting from chronic Tg application when compared to HKH-2 cells (Fig S1).
Fig.1. SOCE is greater in KRASG13D null HKH-2 cells than in KRASG13D-expressing HCT 116 cells.

A: i. Overview of the imaging protocol used to assess SOCE. ER Ca2+ stores were depleted by Tg application in Ca2+-free buffer, after which Ca2+-containing buffer was superfused over the cells to induce SOCE. ii. SOCE response normalised to the peak [Ca2+]i level. iii. Traces of the first derivative of the normalised data from ii. plotted over the time course of the experiment. iv. Histogram showing the maximum first derivative of the SOCE signals for HCT 116 and HKH-2 cells. The data represent the mean ± SEM, obtained from four days of experiments, where three coverslips bearing either HCT 116 or HKH-2 cells were imaged on each day. The fields of view chosen for imaging contained at least 60 HCT 116 or HKH-2 cells. *** indicates P < 0.001 (Student’s t-test). B: i. Normalised traces of Mn2+ quench of fura-2 fluorescence during excitation at 360 nm in Tg-treated HCT 116 and HKH-2 cells. ii. First derivative of the normalised Mn2+ quench in i. iii. Histogram showing summary data of the maximum first derivative of the Mn2+ quench. The data represent the mean ± SEM, obtained from three days of experiments, where three coverslips bearing either HCT 116 or HKH-2 cells were imaged on each day. The fields of view chosen for imaging contained at least 100 HCT 116 or HKH-2 cells. *** indicates P < 0.001 (Student’s t-test).
To further probe SOCE in isolation from the Ca2+ homeostatic mechanisms described above, we assessed the rate of manganese (Mn2+) influx following store depletion with Tg. Mn2+ serves as a surrogate for Ca2+ permeating SOCE channels, but is a poor substrate for Ca2+ dependent ATPases, and as such is not efficiently extruded across the plasmalemma. The influx of Mn2+ was monitored through the quenching of fura-2 fluorescence, determined when excited at its Ca2+-independent isosbestic point (~360 nm). ER stores were depleted of Ca2+ with Tg during the de-esterification step of fura-2 loading prior to imaging. Cells were then imaged in Ca2+- and, MnCl2-free imaging buffer to obtain a baseline level of fura-2 fluorescence. To monitor the activity of SOCE, MnCl2 (2 mM) in Ca2+-free imaging buffer was then superfused over the cells. While MnCl2 addition induced a mono-exponential decline in fura-2 fluorescence in both cell types (Fig. 1Bi), the rate of decline, illustrated by the maximal derivative of the fura-2 quench by Mn2+, was significantly greater in HKH-2 cells than in HCT 116 cells (Fig. 1Bii and iii).
Basal Ca2+ influx (i.e. Ca2+ entry occurring in resting HCT 116 and HKH 2 cells with replete Ca2+ stores), and Ca2+ influx following agonist application (partial store depletion) were also assessed using Mn2+ quench of fura-2, and found to be lower in the HCT 116 cells than HKH-2 cells, further confirming an influence of KRASG13D on SOCE (Fig S2B and C).
3.2. KRASG13D suppresses ICRAC in CRC cell lines
In non-excitable cells such as the colorectal cancer cells examined in this study, Ca2+ entry across the plasmalemma occurs via both SOCE dependent and independent pathways. Experiments were therefore carried out to establish that SOCE was specifically affected by loss of mutated KRAS. To this end, the defining and underlying current of SOCE, ICRAC, was measured in HCT 116 and HKH-2 cells by whole cell patch clamp electrophysiology. ICRAC was induced by Ca2+ store depletion with BAPTA (20 mM), included in the pipette solution and cells were bathed in bath solution containing 20 mM Ca2+ to maximize inward Ca2+ currents [40]. The recordings in Fig 2Ai and ii, from HCT 116 and HKH-2 cells respectively, show a slowly developing inward current that was activated within 3–4 min of whole cell configuration being achieved. The current was substantially augmented by replacement of the Ca2+ containing bath solution with divalent free (DVF) solution, which results in Na+ being the principal charge carrier. The recordings also show a rapid loss of potentiation of the Na+ current during the short time windows of exposure to DVF. This loss of potential is experimentally observed as a rapid increase in inward current caused by exposure to DVF (i.e. Na+ influx), followed by a progressive decline in the current during the DVF application. During the first exposure to DVF immediately after break-in, where ICRAC was negligible, a small current which does not depotentiate, likely representing leak, was observed. A second DVF pulse was applied after ICRAC had fully developed resulting in a relatively large Na+ current that showed a rapid depotentiation during the short time windows of exposure to DVF, a defining biophysical signature of ICRAC [69] (Fig 2Ai and ii). The sensitivity of the current recorded in both cell lines to Gd3+ (5 μM) supports the identity of the current as ICRAC (Fig. 2A). Current-voltage relationships for the currents recorded in HCT 116 and HKH-2 cells are also shown in Fig 2Aiii. These data indicate inward rectification of the current and a positive reversal potential (+60 mV), which are known features of ICRAC [70]. Notably, the magnitude of the current recorded at −100 mV was significantly greater in HKH-2 cells than in HCT 116 cells (Fig. 2Aiv).
Fig.2. CRAC currents are increased following KRASG13D ablation in the HCT116/HKH2 and DLD-1/DKO4 cell line pairs.

Ai-ii and Bi-ii. Whole cell CRAC currents activated by 20 mM BAPTA dialysed via patch pipette were measured in extra cellular solution containing 20 mM Ca2+. Na+ CRAC currents were recorded in divalent free (DVF) solution. Current density recordings over time (pA/pF vs t) in HCT 116 (i) and HKH-2 cells (ii) and in DLD-1 (Bi) and DKO-4 (Bii) cells were taken at −100 mV. Na+ CRAC during 3 pulses of DVF solution are shown. The first pulse was applied at break-in to gauge leak current, the second pulse was applied when current was maximal, and the third pulse was applied after current inhibition with 5 μM Gd3+. Note rapid depotentiation of CRAC current during the second DVF pulse, a defining characteristic of ICRAC. Aiii and Biii. I/V relationships of Na+ CRAC in the HCT 116/HKH-2 and DLD-1/DKO-4 cell line pairs respectively. Aiv and Biv. Summary data of Na+ CRAC currents taken at −100 mV in the HCT 116/HKH-2 and DLD-1/DKO-4 cell line pairs respectively. The mean ± SEM and the number of cells are shown (n). * indicates P<0.05 and **P<0.01 determined by Student’s t-test.
To verify whether lesser ICRAC was a common feature of KRASG13D expressing cells, we measured ICRAC in a second KRASG13D-expressing cell line, DLD-1, and its isogenic derivative DKO-4 (in which the oncogenic KRAS allele was also deleted by homologous recombination, [57]) (Fig. 2B). Similar protocols were used for the DLD-1/DKO-4 cell line pair as were used for HCT/HKH-2 in Fig 2A. Although currents were generally greater in both DLD-1 and DKO-4 cells (Fig 2Bi-iii) compared to HCT 116 and HKH-2 cells, characteristic features of ICRAC (i.e. inward rectification, positive reversal potential and depotentiation in DVF solutions) were also observed in DLD-1 and DKO-4 cells. As shown in Fig. 2Biv, ICRAC was lower in the KRASG13D-expressing DLD-1 cells than in the DKO-4 cells (Fig. 2Biv).
Together, the data obtained from fura-2 measurements (Fig. 1) and electrophysiological recordings of ICRAC (Fig. 2) show that loss of the oncogenic KRAS allele in CRC cell lines results in increased SOCE.
3.3. STIM1 and STIM2 expression is regulated by KRASG13D in CRC cell lines
STIM proteins are essential for coupling ER Ca2+ store depletion with Ca2+ entry channels on the plasma membrane. To assess whether the enhanced SOCE observed following deletion of oncogenic KRAS allele was due to altered STIM expression, STIM1 and STIM2 protein and mRNA levels were assessed by immunoblotting and RT-qPCR, respectively, in HCT 116 and HKH-2 cells. STIM1 protein and mRNA levels were significantly greater in HKH-2 compared to HCT 116 cells (Fig. 3Ai-iii). STIM2 protein levels were also differentially expressed between HCT 116 and HKH-2 cells, although STIM2 levels were significantly lower in the absence of KRASG13D (Fig. 3Aiv and v). mRNA levels of STIM2 were however unchanged (Fig. 3Avi).
Fig.3. Expression of STIM1 and STIM2 in isogenic CRC cell lines.

A. Analysis of STIM1 and STIM2 expression in the HCT 116/HKH 2 cell line pair. i. Representative immunoblot of STIM1 in HCT 116 and HKH-2 cells. Calnexin was used a loading control. ii. Fold change in STIM1 protein levels in HKH-2 cells with respect to HCT 116 cells. iii. Fold change of STIM1 mRNA expression in HKH-2 cells with respect to HCT 116 determined by RT-qPCR. iv. Representative immunoblot of STIM2 in HCT 116 and HKH-2 cells. Calnexin was used a loading control. v. Fold change in STIM2 protein level in HKH-2 cells with respect to HCT 116 cells. vi. Fold change in STIM2 mRNA expression in HKH-2 cells with respect to HCT 116 determined by RT-qPCR. B. Analysis of STIM1 and STIM2 expression in the DLD-1/DKO-4 cell line pair. i. Representative immunoblot of STIM1 in DLD-1 and DKO-4 cells. Calnexin was used a loading control. ii. Fold change in STIM1 protein levels in DKO-4 cells with respect to DLD-1 cells. iii. Fold change in STIM1 mRNA expression in DKO-4 cells with respect to DLD-1 measured by RT-qPCR. iv. Fold change in STIM2 protein levels in DKO-4 cells with respect to DLD-1 cells. v. Fold change in STIM2 mRNA expression in DKO-4 cells with respect to DLD-1 measured by RT-qPCR. All bar graphs represent the mean ± SEM of eight experiments for immunoblotting and six experiments for RT-qPCR. *, ** and *** indicate P < 0.05, P < 0.01, and P < 0.001, respectively (one-sample Student’s t-test).
Given the greater ICRAC in DKO-4 cells compared to their isogenic DLD-1 counterparts harbouring an oncogenic KRAS allele, the expression of STIM1 and 2 was also investigated in these cells. The expression profile of STIM1 and 2 in the DLD-1/DKO-4 cell line pair was different than between the HCT 116/HKH-2 cell lines (Fig. 3B). Specifically, STIM1 protein expression was significantly greater in DLD-1 than DKO-4 cells (Fig. 3Bi and ii). In contrast to that observed for HCT 116 and HKH-2 cells, STIM2 protein was more abundant in DKO-4 cells compared to DLD-1 (Fig. 3Bi and iv). mRNA levels of STIM1 and STIM2 were similar between DLD-1 and DKO-4 cell lines (Fig. 3Biii and v).
3.4. YFP-STIM1 expression is sufficient to elevate SOCE in the HCT 116 cells
The correlation between increased STIM1 expression, enhanced SOCE/ICRAC and loss of KRASG13D between HCT 116 and HKH-2 cells suggested a causal role of STIM1 in determining SOCE/ICRAC activity. Whether increasing STIM1 levels was sufficient to bring about an increase in SOCE/ICRAC in HCT 116 was therefore tested. To this end, yellow fluorescent protein (YFP)-tagged STIM1 (YFP-STIM1) was overexpressed in HCT 116 cells and SOCE was measured. HCT 116 cells were transfected with YFP-STIM1 or with YFP alone, which was used as a transfection control (Fig. S3). YFP-STIM1 exhibited the expected ER distribution, co-localising with ER-localised chaperone calnexin (FIG. S2 top row, right panel). SOCE was analysed in YFP-STIM1- and YFP-expressing cells using Mn2+ quench of fura-2, as described above (Fig. 4) i. The maximal derivative of the rate of Mn2+ quench of fura-2 in HCT 116 cells was substantially increased by YFP-STIM1 expression (Fig. 4ii and iii), reaching levels approaching that observed in HKH-2 cells (Fig. 1B.). These data are consistent with the hypothesis that a difference in STIM1 expression underlies the altered SOCE between HCT 116 and HKH-2 cells.
Fig. 4. STIM1 overexpression is sufficient to increase SOCE in HCT 116 cells.

i. Normalised fura-2 signal (when excited at 360 nm) in YFP and YFP-STIM1-transfected HCT 116 cells prior and subsequent to addition of MnCl2. ii. First derivative of the normalised Mn2+ quench of fura-2 fluorescence. iii. Summary data showing the maximum value of the derivative of Mn2+ quench curve from iii. All graphs represent the mean ± SEM of data from three days of experiments, where 3 coverslips per cell type were imaged on each day. Each coverslip contained at least 30 YFP- or YFP-STIM1-positive cells. *** indicates P < 0.001 (Student’s t-test).
3.5. Expression of STIM proteins is regulated by ERK signalling in the HCT 116 CRC cell line
KRASG13D-expressing HCT 116 cells exhibit increased ERK pathway activation compared to their HKH-2 isogenic derivatives [60,71,72]. Given the role of ERK signalling in regulation of gene expression, we next investigated whether this signalling pathway contributed to the regulation of STIM expression in HCT 116 cells. ERK signalling was inhibited in HCT 116 cells by application of the MEK inhibitor (48 h with 5 μM PD184352; an exposure duration and dose determined to be sufficient to elicit an increase in STIM1 expression, Fig. S4). Since MEK lies directly upstream of ERK1/2, inhibition of its activation prevents ERK1/2 activation as well as downstream signalling [73]. PD184352 application resulted in a significant increase in STIM1 protein and mRNA, reaching levels comparable with those expressed by HKH-2 cells (Fig 5Ai-iii). Protein levels of STIM2 in HCT 116 cells were also altered by PD184352 application, so that its expression was reduced to levels comparable to that for HKH-2 cells (Fig. 5B). PD184352 treatment did not influence mRNA abundance of STIM2 in HCT 116 cells.
Fig.5. MEK inhibition with PD184352 results in remodelling of STIM1/2 expression in HCT 116 cells.

Ai. Representative immunoblot of STIM1 expression in HCT 116 cells treated with PD184352 or DMSO (vehicle control), and of untreated HKH2 cells, as indicated. GAPDH was used as loading control. ii. Histogram showing quantification of STIM1 protein expression. iii. mRNA levels of STIM1 in HCT 116 cells treated with DMSO or PD184352 determined by RT-qPCR. Bi. Immunoblotting of STIM2 under the same conditions as in Ai. ii. Quantification of STIM2 protein expression. iii. mRNA levels of STIM2 following treatment of HCT 116 cells with DMSO or PD184352 determined by RT-qPCR. Histograms of protein expression and for RT-qPCR show a mean ± SEM of 6 experiment each. *represents P<0.05 determined by Student’s t-test.
Together, these data indicate that STIM expression and SOCE lie downstream of KRASG13D in CRC cell lines.
4. Discussion
Here we established a functional interaction between oncogenic KRAS and SOCE in two isogenic pairs of CRC cell lines. We determined that the presence of oncogenic KRAS was associated with a suppression of SOCE and its underlying current ICRAC. Consistent with these observations, the expression of the STIM proteins required for transduction of the state of filling of the ER to the plasma membrane to activate Ca2+ entry was remodelled in cancer cells expressing mutated KRAS. Ectopic expression of STIM1 in KRASG13D cells was sufficient to enhance SOCE, demonstrating the role of reduced STIM1 in the Ca2+ handling phenotype of these cells.
SOCE is fundamental to intracellular Ca2+ signalling and homeostasis and as such is emerging as an important contributor to the hallmarks of transformed cells [17,18,74–77]. Indeed, SOCE is augmented and expression of its molecular mediators, STIM1 and ORAI1 are increased in a range of different cancer types [35–37,40,43,50,53,78–82]. Moreover, the expression level of components of the SOCE pathway directly correlates with tumour cell proliferation, aggressiveness and invasiveness in the above-mentioned reports, highlighting a role for changes in expression, and not just activity, of SOCE and its regulators. In contrast to these studies, we found that the presence of the oncogenic KRAS allele was associated with reduced SOCE and its underlying current ICRAC. Given the reported requirement for Ca2+ influx for cell migration and proliferation for example, these observations might seem counterintuitive. However, divergent effects of Ca2+ entry on cell proliferation, tumour invasiveness and metastasis have been reported [83,84]. Possible explanations for these discrepancies included cell type origin/tumour type, inappropriate controls, stage of transformation or whether the original tumour or metastasis is analysed. Indeed, the requirement for Ca2+ signals during oncogenic transformation likely varies according to the stage of differentiation and functional requirements of the cell [14,17,85]. Oncogenic transformation of cancer cells is a multi-stage process. Initially, cells exhibit increased proliferation and invasive capacity: cell processes generally associated with increased Ca2+ entry. During the later stages, cell proliferation is reduced and resistance to death is increased. Consistent with this idea, over the course of transformation, different types of cancers remodel the abundance of SOCE components in different ways, thus altering the ratios between the two STIM isoforms (STIM1 and STIM2) and the three ORAI isoforms (ORAI1, ORAI2 and ORAI3). For example, in prostate cancer, an increase in the expression of ORAI3 switches Ca2+ entry from SOCE to non-SOCE via the sequestration of ORAI1 into ARC channels [86]. In colorectal cancer (CRC), STIM1 was differentially important between colonic and rectal cancers [87].
The data showing repression of Ca2+ influx in the presence of activated KRAS described here are in line with the widely reported anti-apoptotic role of reduced ER-based Ca2+ signalling mechanisms observed in transformed cells. According to this paradigm, reduced Ca2+ release from the ER and lesser Ca2+ transfer to the mitochondria acts as a mechanism to protect transformed cells from apoptotic cell death. Several mechanisms have been proposed to underlie the reduced ER Ca2+ release, including a reduction in ER luminal Ca2+ and altered expression [60] or activity of InsP3Rs, for example by association with Bcl-2 family members [88–90]. Given that SOCE participates in maintaining ER store Ca2+ content, the reduction in SOCE described here could also contribute to reducing the pool of releasable Ca2+ [91–93]. Notably, in a previous study in which we analysed Ca2+ handling in the same isogenic cell line pairs as described here, activated KRAS expression corresponded with reduced ER luminal Ca2+ [60]. Comparison of CRC cells with normal colon mucosa cells [44] also revealed that despite increased SOCE in the cancer counterpart, store content was diminished, consistent with our previous results. Here we find that SOCE contributes to the differences in sensitivity to cell death between the HCT 116 and HKH-2 cells. Cell death induced by Tg, which has previously been shown to induce apoptosis in a SOCE-dependent manner [94] was greater in the HKH-2 cells than in the oncogenic KRAS-expressing HCT 116 cells. A more direct link between Ca2+ entry, mitochondrial Ca2+ uptake and cell survival has also recently been delineated. STIM proteins directly regulate and are themselves regulated by the mitochondrial Ca2+ uniporter MCU. Specifically, expression of MCU that mediates mitochondrial Ca2+ uptake is decreased in cells with reduced STIM1 [95] and in turn SOCE is lower following MCU silencing [93]. Moreover, STIM1 regulates ER Ca2+ signals through interaction with IP3Rs [96]. In addition, Wang and colleagues recently showed that STIM1 inhibition confers a survival advantage in endothelial progenitor cells [97]. Indeed, whether STIM1 acts as tumour suppressor [98–100] or oncogene [37,38,101,102] is debated.
STIM1 and 2 play different roles in regulation of Ca2+ influx. As such, the relative abundance of STIM1 and/or STIM2, as well as their association with ORAI1 [44,86,103], define their effect on SOCE and cell function. Here we found that while STIM1 expression was lower in oncogenic KRAS-expressing HCT 116 cells compared to their mutated KRAS deficient isogenic derivatives, the expression of STIM2 was greater. This raises the possibility that it is the reciprocal regulation of these two STIM isoforms, and not only a change in STIM1, that contributes to the altered SOCE regulation observed. Indeed, differences in the STIM-ORAIactivating regions (SOAR) of STIM1 and 2 are responsible for the differential coupling of the two STIM isoforms with ORAI1, in which STIM1 acts as a full ORAI1 agonist and STIM2 exerts an inhibitory effect [104]. Thus, the lower SOCE observed in HCT 116 compared to HKH-2 cells may be due to not only lower STIM1–mediated ORAI activation but also increased inhibition by STIM2. Overexpression of STIM1 was however sufficient to enhance SOCE in HCT116 cells, indicating the importance of its repressed expression in the phenotype of these cells. In accordance with this notion, STIM2 was recently found to be overexpressed in melanoma [105,106] and in CRC (Aytes et al. 2012), with its presence associated with a less invasive phenotype. It is possible that, in HCT 116 cells and in cancers with a high STIM2/STIM1 ratio, SOCE is decreased to a basal level, which impairs apoptosis yet provides the Ca2+ necessary to sustain the cell cycle [20].
The repressive effect of oncogenic KRAS on SOCE in both DLD-1 and HCT 116 cells suggests reduced SOCE is a feature of CRC cells expressing mutated KRAS. However, a different expression profile of STIM1 and 2 in the HCT 116/HKH-2 and DLD-1/DKO-4 cell line pairs was observed that would possibly indicate different mechanisms for coupling KRAS activity and expression of STIM1/2 between HCT 116 and DLD-1 cells. These differences may in part be explained by the different signalling pathways engaged downstream of activated KRAS in HCT 116 and DLD-1 cells. While the ERK pathway is principally engaged in HCT 116 cells and as shown here to regulate STIM1/2 expression, the PI3K-AKT pathway dominates in DLD-1 cells [71,107–109]. Despite these differences, HCT 116 and DLD-1 cells exhibit properties of the transformed phenotype, including cell proliferation, regulation of gene expression and avoidance of cell death. Consistent with this hypothesis, Schmidt et al. reported that in ovarian cancer cells, STIM1 expression and SOCE were under the control of AKT [110], which is increased in activity in DLD-1 cells.
In conclusion, our findings identify SOCE and its molecular components as targets of oncogenic KRAS in colorectal cancer cells. Whether remodelled SOCE, altered expression of STIM proteins, or other components of the SOCE machinery contribute to transformation of other cancer types in which activated KRAS mutations are expressed remains to be established however. It will also be important to determine the contribution of SOCE remodelling and SOCE regulated processes during the different stages of oncogenesis. Addressing these questions together with the knowledge of the role of SOCE in cancer provided here will provide new STIM/SOCE-specific avenues for the design of cancer therapeutics.
Supplementary Material
Highlights.
Oncogenic KRAS suppresses SOCE/ICRAC in colorectal cancer cell (CRC) lines.
STIM expression is remodeled in CRCs following oncogenic KRAS deletion.
STIM1 expression is sufficient to rescue SOCE in CRCs.
STIM1 expression is regulated by the MEK/ERK pathway in CRCs.
Acknowledgements
C.P. was funded by the Aldobrandini Studentship of St John’s College, Cambridge, UK. The H.L.R. laboratories are supported by the Babraham Institute, the Biotechnology and Biological Sciences Research Council (BBSRC) [Epigenetics and Signalling ISPGs (Institute Strategic Programme Grant)]. HLR also received funding from an Odysseus Award from the Research Foundation Flanders (FWO) and The Royal Society (University Research Fellowship to HLR). MT received funding from National Institutes of Health Grants R01 HL-123364, R01 HL-097111, and R21 AG-050072, and grant NPRP8–110-3–021 from the Qatar National Research Fund (QNRF). We are grateful to Prof Shmuel Muallem (NIDCR, Bethesda, USA) for the YFP-tagged STIM1 construct; Dr Simon Walker (Imaging Facility, Babraham Institute, Cambridge, UK), for assistance with imaging, Senji Shirasawa, (Fukuoka University, Japan) for the cell lines and the Roderick laboratory for assistance throughout this work. We thank Simon Cook for comments on the manuscript and provision of PD184352.
Footnotes
Duality of Interest
The authors declare that they do not have any potential conflicts relevant to this study.
References
- [1].Downward J, Targeting RAS signalling pathways in cancer therapy., Nat. Rev. Cancer 3 (2003) 11–22. doi:10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- [2].Cicenas J, Tamosaitis L, Kvederaviciute K, Tarvydas R, Staniute G, Kalyan K, Meskinyte-Kausiliene E, Stankevicius V, Valius M, KRAS, NRAS and BRAF mutations in colorectal cancer and melanoma., Med. Oncol 34 (2017) 26. doi:10.1007/s12032-016-0879-9. [DOI] [PubMed] [Google Scholar]
- [3].Sugai T, Eizuka M, Takahashi Y, Fukagawa T, Habano W, Yamamoto E, Akasaka R, Otuska K, Matsumoto T, Suzuki H, Molecular subtypes of colorectal cancers determined by PCR-based analysis., Cancer Sci. (2017). doi:10.1111/cas.13164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Arvelo F, Sojo F, Cotte C, Biology of colorectal cancer, (2015) 1–20. doi:10.3332/ecancer.2015.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chuang H-C, Huang P-H, Kulp SK, Chen C-S, Pharmacological Strategies to Target Oncogenic KRAS Signaling in Pancreatic Cancer., Pharmacol. Res (2017). doi:10.1016/j.phrs.2017.01.006. [DOI] [PubMed] [Google Scholar]
- [6].Barbacid M, ras genes., Annu. Rev. Biochem 56 (1987) 779–827. doi:10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- [7].Bollag G, McCormick F, Regulators and effectors of ras proteins., Annu. Rev. Cell Biol 7 (1991) 601–32. doi:10.1146/annurev.cb.07.110191.003125. [DOI] [PubMed] [Google Scholar]
- [8].Cook SJ, Lockyer PJ, Recent advances in Ca(2+)-dependent Ras regulation and cell proliferation., Cell Calcium. 39 (2006) 101–12. doi:10.1016/j.ceca.2005.10.014. [DOI] [PubMed] [Google Scholar]
- [9].Cullen PJ, Lockyer PJ, Integration of calcium and Ras signalling., Nat. Rev. Mol. Cell Biol 3 (2002) 339–48. doi:10.1038/nrm808. [DOI] [PubMed] [Google Scholar]
- [10].Agell N, Bachs O, Rocamora N, Villalonga P, Modulation of the Ras/Raf/MEK/ERK pathway by Ca(2+), and calmodulin., Cell. Signal 14 (2002) 649–54. [DOI] [PubMed] [Google Scholar]
- [11].Hegedũs L, Garay T, Molnár E, Varga K, Bilecz Á, Török S, Padányi R, Pászty K, Wolf M, Grusch M, Kállay E, Döme B, Berger W, Hegedũs B, Enyedi A, The plasma membrane Ca(2+) pump PMCA4b inhibits the migratory and metastatic activity of BRAF mutant melanoma cells., Int. J. Cancer 140 (2017) 2758–2770. doi:10.1002/ijc.30503. [DOI] [PubMed] [Google Scholar]
- [12].Corazao-Rozas P, Guerreschi P, André F, Gabert P-E, Lancel S, Dekiouk S, Fontaine D, Tardivel M, Savina A, Quesnel B, Mortier L, Marchetti P, Kluza J, Mitochondrial oxidative phosphorylation controls cancer cell’s life and death decisions upon exposure to MAPK inhibitors., Oncotarget. 7 (2016) 39473–39485. doi:10.18632/oncotarget.7790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Numaga-Tomita T, Nishida M, Putney JW, Mori Y, TRPC3 amplifies B-cell receptor-induced ERK signalling via protein kinase D-dependent Rap1 activation., Biochem. J 473 (2016) 201–10. doi:10.1042/BJ20150596. [DOI] [PubMed] [Google Scholar]
- [14].Roderick HL, Cook SJ, Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival, Nat. Rev. Cancer 8 (2008) 361–375. doi:10.1038/nrc2374. [DOI] [PubMed] [Google Scholar]
- [15].Berridge MJ, Bootman MD, Lipp P, Calcium--a life and death signal, Nature. 395 (1998) 645–648. doi:10.1038/27094. [DOI] [PubMed] [Google Scholar]
- [16].Ivanova H, Kerkhofs M, La Rovere RM, Bultynck G, Endoplasmic Reticulum– Mitochondrial Ca2+ Fluxes Underlying Cancer Cell Survival, Front. Oncol 7 (2017). doi:10.3389/fonc.2017.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Monteith GR, Prevarskaya N, Roberts-Thomson SJ, The calcium–cancer signalling nexus, Nat. Rev. Cancer (2017). doi:10.1038/nrc.2017.18. [DOI] [PubMed] [Google Scholar]
- [18].Cui C, Merritt R, Fu L, Pan Z, Targeting calcium signaling in cancer therapy, Acta Pharm. Sin. B 7 (2017) 3–17. doi:10.1016/j.apsb.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chen Y-F, Chen Y-T, Chiu W-T, Shen M-R, Remodeling of calcium signaling in tumor progression., J. Biomed. Sci 20 (2013) 23. doi:10.1186/1423-0127-20-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chen Y-W, Chen Y-F, Chen Y-T, Chiu W-T, Shen M-R, The STIM1-Orai1 pathway of store-operated Ca(2+) entry controls the checkpoint in cell cycle G1/S transition., Sci. Rep 6 (2016) 22142. doi:10.1038/srep22142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kahl CR, Means AR, Regulation of Cell Cycle Progression by Calcium/Calmodulin-Dependent Pathways, Endocr. Rev 24 (2003) 719–736. doi:10.1210/er.2003-0008. [DOI] [PubMed] [Google Scholar]
- [22].Vervliet T, Clerix E, Seitaj B, Ivanova H, Monaco G, Bultynck G, Modulation of Ca2+ Signaling by Anti-apoptotic B-Cell Lymphoma 2 Proteins at the Endoplasmic Reticulum–Mitochondrial Interface, Front. Oncol 7 (2017) 1–9. doi:10.3389/fonc.2017.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Fabian A, Fortmann T, Dieterich P, Riethmüller C, Schön P, Mally S, Nilius B, Schwab A, TRPC1 channels regulate directionality of migrating cells., Pflugers Arch. 457 (2008) 475–84. doi:10.1007/s00424-008-0515-4. [DOI] [PubMed] [Google Scholar]
- [24].Witze ES, Connacher MK, Houel S, Schwartz MP, Morphew MK, Reid L, Sacks DB, Anseth KS, Ahn NG, Wnt5a directs polarized calcium gradients by recruiting cortical endoplasmic reticulum to the cell trailing edge, Dev. Cell 26 (2013) 645–657. doi:10.1016/j.devcel.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tsai F, Seki A, Yang HW, Hayer A, Carrasco S, Meyer T, A polarized Ca2+ , diacylglycerol, and STIM1 signaling system regulates directed cell migration, 16 (2014) 133–144. doi:10.1038/ncb2906.A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Prudent J, Popgeorgiev N, Gadet R, Deygas M, Rimokh R, Gillet G, Mitochondrial Ca2+ uptake controls actin cytoskeleton dynamics during cell migration, Sci. Rep 6 (2016) 36570. doi:10.1038/srep36570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chen Y-F, Hsu K-F, Shen M-R, The store-operated Ca(2+) entry-mediated signaling is important for cancer spread., Biochim. Biophys. Acta 1863 (2016) 1427–35. doi:10.1016/j.bbamcr.2015.11.030. [DOI] [PubMed] [Google Scholar]
- [28].Villalobos C, Sobradillo D, Hernández-Morales M, Núñez L, Calcium remodeling in colorectal cancer., Biochim. Biophys. Acta 1864 (2017) 843–849. doi:10.1016/j.bbamcr.2017.01.005. [DOI] [PubMed] [Google Scholar]
- [29].Tanwar J, Motiani RK, Role of SOCE architects STIM and Orai proteins in Cell Death., Cell Calcium. (2017). doi:10.1016/j.ceca.2017.06.002. [DOI] [PubMed] [Google Scholar]
- [30].Hanahan D, Weinberg RA, Hallmarks of cancer: the next generation., Cell. 144 (2011) 646–74. doi:10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- [31].Hanahan D, Weinberg RA, The hallmarks of cancer., Cell. 100 (2000) 57–70. [DOI] [PubMed] [Google Scholar]
- [32].Arredouani A, Yu F, Sun L, Machaca K, Regulation of store-operated Ca2+ entry during the cell cycle., J. Cell Sci 123 (2010) 2155–62. doi:10.1242/jcs.069690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Machaca K, Ca(2+) signaling, genes and the cell cycle., Cell Calcium. 49 (2011) 323–30. [DOI] [PubMed] [Google Scholar]
- [34].Vashisht A, Trebak M, Motiani RK, STIM and Orai Proteins as Novel Targets for Cancer Therapy., Am. J. Physiol. Cell Physiol (2015) ajpcell.00064.2015. doi:10.1152/ajpcell.00064.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Umemura M, Baljinnyam E, Feske S, De Lorenzo MS, Xie LH, Feng X, Oda K, Makino A, Fujita T, Yokoyama U, Iwatsubo M, Chen S, Goydos JS, Ishikawa Y, Iwatsubo K, Store-operated Ca2+ entry (SOCE) regulates melanoma proliferation and cell migration, PLoS One. 9 (2014). doi:10.1371/journal.pone.0089292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kim J-H, Lkhagvadorj S, Lee M-R, Hwang K-H, Chung HC, Jung JH, Cha S-K, Eom M, Orai1 and STIM1 are critical for cell migration and proliferation of clear cell renal cell carcinoma, Biochem. Biophys. Res. Commun 448 (2014) 76–82. doi:10.1016/j.bbrc.2014.04.064. [DOI] [PubMed] [Google Scholar]
- [37].Chen Y-F, Chiu W-T, Chen Y-T, Lin P-Y, Huang H-J, Chou C-Y, Chang H-C, Tang M-J, Shen M-R, Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis., Proc. Natl. Acad. Sci. U. S. A 108 (2011) 15225–30. doi:10.1073/pnas.1103315108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Liu H, Hughes JD, Rollins S, Chen B, Perkins E, Calcium entry via ORAI1 regulates glioblastoma cell proliferation and apoptosis., Exp. Mol. Pathol 91 (2011) 753–60. doi:10.1016/j.yexmp.2011.09.005. [DOI] [PubMed] [Google Scholar]
- [39].Li G, Zhang Z, Wang R, Ma W, Yang Y, Wei J, Wei Y, Suppression of STIM1 inhibits human glioblastoma cell proliferation and induces G0/G1 phase arrest, J. Exp. Clin. Cancer Res 32 (2013) 20. doi:10.1186/1756-9966-32-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Motiani RK, Hyzinski-García MC, Zhang X, Henkel MM, Abdullaev IF, Kuo YH, Matrougui K, Mongin AA, Trebak M, STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion, Pflugers Arch. Eur. J. Physiol 465 (2013) 1249–1260. doi:10.1007/s00424-013-1254-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Feng M, Grice DM, Faddy HM, Nguyen N, Leitch S, Wang Y, Muend S, Kenny PA, Sukumar S, Roberts-Thomson SJ, Monteith GR, Rao R, Store-independent activation of Orai1 by SPCA2 in mammary tumors., Cell. 143 (2010) 84–98. doi:10.1016/j.cell.2010.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].El Boustany C, Katsogiannou M, Delcourt P, Dewailly E, Prevarskaya N, Borowiec A-S, Capiod T, Differential roles of STIM1, STIM2 and Orai1 in the control of cell proliferation and SOCE amplitude in HEK293 cells., Cell Calcium. 47 (2010) 350–9. doi:10.1016/j.ceca.2010.01.006. [DOI] [PubMed] [Google Scholar]
- [43].Wang J-Y, Sun J, Huang M-Y, Wang Y-S, Hou M-F, Sun Y, He H, Krishna N, Chiu S-J, Lin S, Yang S, Chang W-C, STIM1 overexpression promotes colorectal cancer progression, cell motility and COX-2 expression., Oncogene. 34 (2015) 4358–67. doi:10.1038/onc.2014.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sobradillo D, Hernández-Morales M, Ubierna D, Moyer MP, Núñez L, Villalobos C, A reciprocal shift in transient receptor potential channel 1 (TRPC1) and stromal interaction molecule 2 (STIM2) contributes to Ca2+remodeling and cancer hallmarks in colorectal carcinoma cells, J. Biol. Chem 289 (2014) 28765–28782. doi:10.1074/jbc.M114.581678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Courjaret R, Machaca K, STIM and Orai in cellular proliferation and division., Front. Biosci. (Elite Ed) 4 (2012) 331–41. [DOI] [PubMed] [Google Scholar]
- [46].Jardin I, Rosado JA, STIM and calcium channel complexes in cancer., Biochim. Biophys. Acta 1863 (2016) 1418–26. doi:10.1016/j.bbamcr.2015.10.003. [DOI] [PubMed] [Google Scholar]
- [47].Xie J, Pan H, Yao J, Zhou Y, Han W, SOCE and cancer: Recent progress and new perspectives, Int. J. Cancer 138 (2016) 2067–2077. doi:10.1002/ijc.29840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Fiorio Pla A, Kondratska K, Prevarskaya N, STIM and ORAI proteins: crucial roles in hallmarks of cancer, Am. J. Physiol. - Cell Physiol 310 (2016) C509–C519. doi:10.1152/ajpcell.00364.2015. [DOI] [PubMed] [Google Scholar]
- [49].Iamshanova O, Fiorio Pla A, Prevarskaya N, Molecular mechanisms of tumor invasion: regulation by calcium signals, J. Physiol 10 (2017) 3063–3075. doi:10.1113/JP272844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yang N, Tang Y, Wang F, Zhang H, Xu D, Shen Y, Sun S, Yang G, Blockade of store-operated Ca2+ entry inhibits hepatocarcinoma cell migration and invasion by regulating focal adhesion turnover, Cancer Lett. 330 (2013) 163–169. doi:10.1016/j.canlet.2012.11.040. [DOI] [PubMed] [Google Scholar]
- [51].Motiani RK, Abdullaev IF, Trebak M, A novel native store-operated calcium channel encoded by Orai3: Selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells, J. Biol. Chem 285 (2010) 19173–19183. doi:10.1074/jbc.M110.102582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lodola F, Laforenza U, Bonetti E, Lim D, Dragoni S, Bottino C, Ong HL, Guerra G, Ganini C, Massa M, Manzoni M, Ambudkar IS, Genazzani AA, Rosti V, Pedrazzoli P, Tanzi F, Moccia F, Porta C, Store-Operated Ca2+ Entry Is Remodelled and Controls In Vitro Angiogenesis in Endothelial Progenitor Cells Isolated from Tumoral Patients, PLoS One. 7 (2012). doi:10.1371/journal.pone.0042541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Pérez-Riesgo E, Gutiérrez L, Ubierna D, Acedo A, Moyer M, Núñez L, Villalobos C, Transcriptomic Analysis of Calcium Remodeling in Colorectal Cancer, Int. J. Mol. Sci 18 (2017) 922. doi:10.3390/ijms18050922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wang X-T, Nagaba Y, Cross HS, Wrba F, Zhang L, Guggino SE, The mRNA of L-Type Calcium Channel Elevated in Colon Cancer, Am. J. Physiol. - Cell Physiol 157 (2000) 1549–1561. doi:10.1016/S0002-9440(10)64792-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Latour I, Louw DF, Beedle AM, Hamid J, Sutherland GR, Zamponi GW, Expression of T-type calcium channel splice variants in human glioma., Glia. 48 (2004) 112–9. doi:10.1002/glia.20063. [DOI] [PubMed] [Google Scholar]
- [56].Toyota M, Ho C, Ohe-toyota M, Baylin SB, Issa JJ, Advances in Brief Inactivation of CACNA1G, a T-Type Calcium Channel Gene, by Aberrant Methylation of Its 5 CpG Island in Human Tumors 1, (1999) 4535–4541. [PubMed]
- [57].Shirasawa S, Furuse M, Yokoyama N, Sasazuki T, Altered growth of human colon cancer cell lines disrupted at activated Ki-ras., Science. 260 (1993) 85–8. [DOI] [PubMed] [Google Scholar]
- [58].Mou H, Moore J, Malonia SK, Li Y, Ozata DM, Hough S, Song C-Q, Smith JL, Fischer A, Weng Z, Green MR, Xue W, Genetic disruption of oncogenic Kras sensitizes lung cancer cells to Fas receptor-mediated apoptosis., Proc. Natl. Acad. Sci. U. S. A 114 (2017) 3648–3653. doi:10.1073/pnas.1620861114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Fasterius E, Raso C, Kennedy S, Rauch N, Lundin P, Kolch W, Uhlen M, Al-Khalili Szigyarto C, A novel RNA sequencing data analysis method for cell line authentication, PLoS One. 12 (2017) 1–19. doi:10.1371/journal.pone.0171435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Pierro C, Cook SJ, Foets TC, Bootman MD, Roderick HL, Oncogenic K-Ras suppresses IP₃-dependent Ca2+ release through remodelling of the isoform composition of IP₃Rs and ER luminal Ca2+ levels in colorectal cancer cell lines, J Cell Sci. 127 (2014) 1607–1619. doi:10.1242/jcs.141408. [DOI] [PubMed] [Google Scholar]
- [61].Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D, Hingorani SR, Zaks T, King C, Jacobetz MA, Wang L, Bronson RT, Orkin SH, DePinho RA, Jacks T, Endogenous oncogenic K-rasG12D stimulates proliferation and widespread neoplastic and developmental defects, Cancer Cell. 5 (2004) 375–387. doi:10.1016/S1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- [62].Peppiatt CM, Holmes AM, Seo JT, Bootman MD, Collins TJ, McDonald F, Roderick HL, Calmidazolium and arachidonate activate a calcium entry pathway that is distinct from store-operated calcium influx in HeLa cells., Biochem. J 381 (2004) 929–39. doi:10.1042/BJ20040097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Grynkiewicz G, Poenie M, Tsien RY, A new generation of Ca2+ indicators with greatly improved fluorescence properties., J. Biol. Chem 260 (1985) 3440–50. [PubMed] [Google Scholar]
- [64].Zhang X, Zhang W, González-Cobos JC, Jardin I, Romanin C, Matrougui K, Trebak M, Gonzalez-Cobos JC, Jardin I, Romanin C, Matrougui K, Trebak M, Complex role of STIM1 in the activation of store-independent Orai1/3 channels, J. Gen. Physiol 143 (2014) 345–359. doi:10.1085/jgp.201311084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Desai PN, Zhang X, Wu S, Janoshazi A, Bolimuntha S, Putney JW, Trebak M, Multiple types of calcium channels arising from alternative translation initiation of the Orai1 message., Sci. Signal 8 (2015) ra74. doi:10.1126/scisignal.aaa8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hanson CJ, Bootman MD, Distelhorst CW, Maraldi T, Roderick HL, The cellular concentration of Bcl-2 determines its pro- or anti-apoptotic effect, Cell Calcium. 44 (2008) 243–258. doi:10.1016/j.ceca.2007.11.014. [DOI] [PubMed] [Google Scholar]
- [67].Bird G, DeHaven W, Smyth J, Methods for studying store-operated calcium entry, Methods. 46 (2008) 204–212. doi:10.1016/j.ymeth.2008.09.009.Methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Lur G, Sherwood MW, Ebisui E, Haynes L, Feske S, Sutton R, Burgoyne RD, Mikoshiba K, Petersen OH, V Tepikin A, InsP₃receptors and Orai channels in pancreatic acinar cells: co-localization and its consequences., Biochem. J 436 (2011) 231–239. doi:10.1042/BJ20110083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Prakriya M, Lewis RS, Separation and characterization of currents through store-operated CRAC channels and Mg2+-inhibited cation (MIC) channels., J. Gen. Physiol 119 (2002) 487–507. doi:10.1085/jgp.20028551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hoth M, Penner R, Depletion of intracellular calcium stores activates a calcium current in mast cells., Nature. 355 (1992) 353–6. doi:10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- [71].Balmanno K, Cook SJ, Tumour cell survival signalling by the ERK1/2 pathway., Cell Death Differ. 16 (2009) 368–77. doi:10.1038/cdd.2008.148. [DOI] [PubMed] [Google Scholar]
- [72].Vartanian S, Bentley C, Brauer MJ, Li L, Shirasawa S, Sasazuki T, Kim JS, Haverty P, Stawiski E, Modrusan Z, Waldman T, Stokoe D, Identification of mutant KRas-dependent phenotypes using a panel of isogenic cell lines, J. Biol. Chem 288 (2013) 2403–2413. doi:10.1074/jbc.M112.394130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Allen LF, Sebolt-Leopold J, Meyer MB, CI-1040 (PD184352), a targeted signal transduction inhibitor of MEK (MAPKK)., Semin. Oncol 30 (2003) 105–16. [DOI] [PubMed] [Google Scholar]
- [74].Déliot N, Constantin B, Plasma membrane calcium channels in cancer: Alterations and consequences for cell proliferation and migration, Biochim. Biophys. Acta - Biomembr 1848 (2015) 2512–2522. doi:10.1016/j.bbamem.2015.06.009. [DOI] [PubMed] [Google Scholar]
- [75].Wen J, Huang YC, Xiu HH, Shan ZM, Xu KQ, Altered expression of stromal interaction molecule (STIM)-calcium release-activated calcium channel protein (ORAI) and inositol 1,4,5-trisphosphate receptors (IP3Rs) in cancer: Will they become a new battlefield for oncotherapy?, Chin. J. Cancer 35 (2016) 1–9. doi:10.1186/s40880-0160094-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Moccia F, Zuccolo E, Poletto V, Turin I, Guerra G, Pedrazzoli P, Rosti V, Porta C, Montagna D, Targeting Stim and Orai Proteins as an Alternative Approach in Anticancer Therapy., Curr. Med. Chem 23 (2016) 3450–3480. [DOI] [PubMed] [Google Scholar]
- [77].Azimi I, Roberts-Thomson SJ, Monteith GR, Calcium influx pathways in breast cancer: Opportunities for pharmacological intervention, Br. J. Pharmacol 171 (2014) 945–960. doi:10.1111/bph.12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Zhang Z, Liu X, Feng B, Liu N, Wu Q, Han Y, Nie Y, Wu K, Shi Y, Fan D, STIM1, a direct target of microRNA-185, promotes tumor metastasis and is associated with poor prognosis in colorectal cancer, Oncogene. 35 (2015) 6043–6043. doi:10.1038/onc.2016.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Cheng H, Wang S, Feng R, Cheng H, Wang S, Feng R, Huanyi Cheng SWRF, STIM1 plays an important role in TGF-β-induced suppression of breast cancer cell proliferation, Oncotarget. 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Li P, Bian X-Y, Chen Q, Yao X-F, Wang X-D, Zhang W-C, Tao Y-J, Jin R, Zhang L, Blocking of stromal interaction molecule 1 expression influence cell proliferation and promote cell apoptosis in vitro and inhibit tumor growth in vivo in head and neck squamous cell carcinoma., PLoS One. 12 (2017) e0177484. doi:10.1371/journal.pone.0177484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Zhu H, Zhang H, Jin F, Fang M, Huang M, Yang CS, Chen T, Fu L, Pan Z, Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma., Oncotarget. 5 (2014) 3455–71. doi:10.18632/oncotarget.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Liu B, Yu H, Ye H, Luo Z, Xiao F, Effects of stromal interacting molecule�1 gene silencing by short hairpin RNA on the biological behavior of human gastric cancer cells, Mol. Med. Rep (2015) 3047–3054. doi:10.3892/mmr.2015.3778. [DOI] [PubMed] [Google Scholar]
- [83].Vanden Abeele F, Shuba Y, Roudbaraki M, Lemonnier L, Vanoverberghe K, Mariot P, Skryma R, Prevarskaya N, Store-operated Ca2+ channels in prostate cancer epithelial cells: Function, regulation, and role in carcinogenesis, Cell Calcium. 33 (2003) 357–373. doi:10.1016/S0143-4160(03)00049-6. [DOI] [PubMed] [Google Scholar]
- [84].Monteith GR, Davis FM, Roberts-Thomson SJ, Calcium channels and pumps in cancer: Changes and consequences, J. Biol. Chem 287 (2012) 31666–31673. doi:10.1074/jbc.R112.343061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Monteith GR, McAndrew D, Faddy HM, Roberts-Thomson SJ, Calcium and cancer: targeting Ca2+ transport, Nat. Rev. Cancer 7 (2007) 519–530. doi:10.1038/nrc2171. [DOI] [PubMed] [Google Scholar]
- [86].Dubois C, Vanden Abeele F, Lehen’kyi V, Gkika D, Guarmit B, Lepage G, Slomianny C, Borowiec A, Bidaux G, Benahmed M, Shuba Y, Prevarskaya N, Remodeling of Channel-Forming ORAI Proteins Determines an Oncogenic Switch in Prostate Cancer, Cancer Cell. 26 (2014) 19–32. doi:10.1016/j.ccr.2014.04.025. [DOI] [PubMed] [Google Scholar]
- [87].Wong HS-C, Chang W-C, Correlation of clinical features and genetic profiles of stromal interaction molecule 1 (STIM1) in colorectal cancers, Oncotarget. 6 (2015) 42169–42182. doi:10.18632/oncotarget.5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, Berridge MJ, Conway SJ, Holmes AB, Mignery GA, Velez P, Distelhorst CW, Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate, J. Cell Biol 166 (2004) 193–203. doi:10.1083/jcb.200309146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Scorrano L, BAX and BAK Regulation of Endoplasmic Reticulum Ca2+: A Control Point for Apoptosis, Science (80-. ). 300 (2003) 135–139. doi:10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- [90].White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, Foskett JK, The endoplasmic reticulum gateway to apoptosis by Bcl-XL modulation of the InsP3R, Nat. Cell Biol 7 (2005) 1021–1028. doi:10.1038/ncb1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Courjaret R, Machaca K, Mid-range Ca2+ signalling mediated by functional coupling between store-operated Ca2+ entry and IP3-dependent Ca2+ release., Nat. Commun 5 (2014) 3916. doi:10.1038/ncomms4916. [DOI] [PubMed] [Google Scholar]
- [92].Deak a T., Blass, Khan MJ, Groschner LN, Waldeck-Weiermair M, Hallström S, Graier WF, Malli R, Inositol-1,4,5-trisphosphate (IP3)-mediated STIM1 oligomerization requires intact mitochondrial Ca2+ uptake., J. Cell Sci 2 (2014) 2944–2955. doi:10.1242/jcs.149807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Tang S, Wang X, Shen Q, Yang X, Yu C, Cai C, Cai G, Meng X, Zou F, Mitochondrial Ca2+ uniporter is critical for store-operated Ca2+ entry-dependent breast cancer cell migration, Biochem. Biophys. Res. Commun 458 (2015) 186–193. doi:10.1016/j.bbrc.2015.01.092. [DOI] [PubMed] [Google Scholar]
- [94].Flourakis M, Lehen’kyi V, Beck B, Raphaël M, Vandenberghe M, V Abeele F, Roudbaraki M, Lepage G, Mauroy B, Romanin C, Shuba Y, Skryma R, Prevarskaya N, Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells, Cell Death Dis. 1 (2010) e75. doi:10.1038/cddis.2010.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Shanmughapriya S, Rajan S, Hoffman NE, Zhang X, Guo S, Kolesar JE, Hines KJ, Ragheb J, Jog NR, Caricchio R, Baba Y, Zhou Y, Kaufman BA, Cheung JY, Kurosaki T, Gill DL, Madesh M, Ca2+ signals regulate mitochondrial metabolism by stimulating CREB-mediated expression of the mitochondrial Ca2+ uniporter gene MCU., Sci. Signal 8 (2015) ra23. doi:10.1126/scisignal.2005673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Beliveau E, Lessard V, Guillemette G, STIM1 positively regulates the Ca2+ release activity of the inositol 1,4,5-trisphosphate receptor in bovine aortic endothelial cells, PLoS One. 9 (2014) 1–15. doi:10.1371/journal.pone.0114718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Wang YW, Zhang JH, Yu Y, Yu J, Huangame L, Inhibition of store-operated calcium entry protects endothelial progenitor cells from H2O2-induced apoptosis, Biomol. Ther 24 (2016) 371–379. doi:10.4062/biomolther.2015.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Sabbioni S, Barbanti-Brodano G, Croce CM, Negrini M, GOK: a gene at 11p15 involved in rhabdomyosarcoma and rhabdoid tumor development., Cancer Res. 57 (1997) 4493–7. [PubMed] [Google Scholar]
- [99].Sabbioni S, Veronese A, Trubia M, Taramelli R, Barbanti-Brodano G, Croce CM, Negrini M, Exon structure and promoter identification of STIM1 (alias GOK), a human gene causing growth arrest of the human tumor cell lines G401 and RD., Cytogenet. Cell Genet 86 (1999) 214–8. doi:15341. [DOI] [PubMed] [Google Scholar]
- [100].Suyama E, Wadhwa R, Kaur K, Miyagishi M, Kaul SC, Kawasaki H, Taira K, Identification of metastasis-related genes in a mouse model using a library of randomized ribozymes., J. Biol. Chem 279 (2004) 38083–6. doi:10.1074/jbc.C400313200. [DOI] [PubMed] [Google Scholar]
- [101].Abdullaev IF, Bisaillon JM, Potier M, Gonzalez JC, Motiani RK, Trebak M, Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation., Circ. Res 103 (2008) 1289–99. doi:10.1161/01.RES.0000338496.95579.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Yang S, Zhang JJ, Huang XY, Orai1 and STIM1 Are Critical for Breast Tumor Cell Migration and Metastasis, Cancer Cell. 15 (2009) 124–134. doi:10.1016/j.ccr.2008.12.019. [DOI] [PubMed] [Google Scholar]
- [103].Hodeify R, Selvaraj S, Wen J, Arredouani A, Hubrack S, Dib M, Al-Thani SN, McGraw T, Machaca K, A STIM1-dependent “trafficking trap” mechanism regulates Orai1 plasma membrane residence and Ca2+ influx levels., J. Cell Sci 128 (2015) 3143–54. doi:10.1242/jcs.172320. [DOI] [PubMed] [Google Scholar]
- [104].Wang X, Wang Y, Zhou Y, Hendron E, Mancarella S, Andrake MD, Rothberg BS, Soboloff J, Gill DL, Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site., Nat. Commun 5 (2014) 3183. doi:10.1038/ncomms4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Stanisz H, Saul S, Müller CSL, Kappl R, Niemeyer BA, Vogt T, Hoth M, Roesch A, Bogeski I, Inverse regulation of melanoma growth and migration by Orai1/STIM2dependent calcium entry, Pigment Cell Melanoma Res. 27 (2014) 442–453. doi:10.1111/pcmr.12222. [DOI] [PubMed] [Google Scholar]
- [106].Stanisz H, Vultur A, Herlyn M, Roesch A, Bogeski I, The role of Orai-STIM calcium channels in melanocytes and melanoma., J. Physiol 594 (2016) 2825–35. doi:10.1113/JP271141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Keller JW, Franklin JL, Graves-Deal R, Friedman DB, Whitwell CW, Coffey RJ, Oncogenic KRAS provides a uniquely powerful and variable oncogenic contribution among RAS family members in the colonic epithelium., J. Cell. Physiol 210 (2007) 740–9. doi:10.1002/jcp.20898. [DOI] [PubMed] [Google Scholar]
- [108].Carón RW, Yacoub A, Mitchell C, Zhu X, Hong Y, Sasazuki T, Shirasawa S, Hagan MP, Grant S, Dent P, Radiation-stimulated ERK1/2 and JNK1/2 signaling can promote cell cycle progression in human colon cancer cells., Cell Cycle. 4 (2005) 456–64. doi:10.4161/cc.4.3.1249. [DOI] [PubMed] [Google Scholar]
- [109].Carón RW, Yacoub A, Zhu X, Mitchell C, Han SI, Sasazuki T, Shirasawa S, Hagan MP, Grant S, Dent P, H-RAS V12-induced radioresistance in HCT116 colon carcinoma cells is heregulin dependent., Mol. Cancer Ther. 4 (2005) 243–55. [PubMed] [Google Scholar]
- [110].Schmidt S, Liu G, Liu G, Yang W, Honisch S, Pantelakos S, Stournaras C, Hönig A, Lang F, Enhanced Orai1 and STIM1 expression as well as store operated Ca2+ entry in therapy resistant ovary carcinoma cells., Oncotarget. 5 (2014) 4799–810. doi:2035 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.