

Abstract
Background
Cancer develops when pathways controlling cell survival, cell fate or genome maintenance are disrupted by the somatic alteration of key driver genes. Understanding how pathway disruption is driven by somatic alterations is thus essential for an accurate characterization of cancer biology and identification of therapeutic targets. Unfortunately, current cancer pathway analysis methods fail to fully model the relationship between somatic alterations and pathway activity.
Results
To address these limitations, we developed a multi-omics method for identifying biologically important pathways and genes in human cancer. Our approach combines single-sample pathway analysis with multi-stage, lasso-penalized regression to find pathways whose gene expression can be explained largely in terms of gene-level somatic alterations in the tumor. Importantly, this method can analyze case-only data sets, does not require information regarding pathway topology and supports personalized pathway analysis using just somatic alteration data for a limited number of cancer-associated genes. The practical effectiveness of this technique is illustrated through an analysis of data from The Cancer Genome Atlas using gene sets from the Molecular Signatures Database.
Conclusions
Novel insights into the pathophysiology of human cancer can be obtained from statistical models that predict expression-based pathway activity in terms of non-silent somatic mutations and copy number variation. These models enable the identification of biologically important pathways and genes and support personalized pathway analysis in cases where gene expression data is unavailable.
Electronic supplementary material
The online version of this article (10.1186/s12859-018-2476-8) contains supplementary material, which is available to authorized users.
Keywords: Gene set testing, Pathway analysis, Cancer genomics, Driver mutations
Background
High-throughput genomic assays have revolutionized our understanding of cancer. Projects such as The Cancer Genome Atlas (TCGA) [1] and the Catalog of Somatic Mutations in Cancer (COSMIC) [2] have collected detailed measurements of DNA sequence, mRNA expression and methylation for thousands of individual tumors across multiple cancer types. Leveraging this data, researchers have identified hundreds of genes whose somatic alterations drive human cancer [3]. Although the discovery of cancer associated mutations and genes has enabled significant advances in cancer care through the identification of new therapeutic targets and support for personalized treatment, a strictly gene or mutation-level analysis fails to capture many important aspects of cancer biology. While a small number of cancer-associated genes are commonly mutated, i.e., present in more than 10%, of tumors, and can therefore be more easily studied, there exists a much larger set of rarely mutated cancer genes spread across the human genome [4]. Adding to this complexity, the activity of cancer-associated genes can be impacted in a variety of ways including copy number variation, somatic mutations and methylation changes [5]. Capturing all of these mechanisms requires the measurement and joint analysis of multiple types of omics data. To address the complexity of the cancer genomics landscape, researchers have turned to pathway analysis methods that combine the somatic alterations or expression of multiple, functionally related, genes [6]. Cancer is fundamentally a disease of disrupted pathways in which a population of cells develops a selective growth advantage due to the altered function of pathways controlling cell survival, cell fate or genome maintenance [7]. To fully elucidate the mechanisms driving cancer, it is thus critical that researchers understand how the somatic alterations present in each tumor collectively impact these pathways.
To interpret cancer genomic data at the level of biological pathways, researchers have developed a large number of pathway analysis methods, many specifically customized for cancer data [6]. For these methods, a pathway, or gene set, refers to a group of genes whose products share a common biological function. A number of large and well maintained repositories of such gene sets now exist with some, e.g., the Gene Ontology (GO) [8], holding simple unordered genes sets and others, e.g., Reactome [9], defining pathways in terms of a complex topology of molecular interactions. In this paper, the terms pathway and gene set are used synonymously and it is assumed that topological information is unavailable. Given a collection of such gene sets, pathway analysis methods aim to identify statistically significant associations between the activity of pathway members and a phenotype of interest, e.g., cancer type or case/control status [10]. Although most pathway methods focus on population-level associations, a number of recent approaches provide pathway enrichment results at the single-subject level [11–14].
Although the pathways most commonly impacted by somatic alterations in cancer have been identified [7] and significant progress has been made developing cancer-specific pathway analysis methods [6, 11, 14–21], existing approaches have several important limitations. Many current methods focus on either gene expression data [11] or mutation data [21] and therefore fail to capture the important association between the two in cancer. The utility of methods that use only expression data is also impacted by the lack of gene expression data for many tumor samples. For methods that just use mutation data, performance is hindered by the limited number of genomic regions sequenced in many clinical settings, the overall sparsity of somatic alterations and the fact that pathways are often defined in the context of protein activity/abundance, which may not be closely linked to the mutational status of the underlying gene. Most methods that jointly analyze multiple types of omics data ignore pathway information and instead aim to identify specific mutations or mutated genes that are associated with alterations in gene expression [22, 23] or perform unsupervised clustering [24, 25]. Among the few existing pathway methods that do combine mutations with expression [14–16, 19, 20], none of them support both population-level and single subject analyzes, few provide information on both cancer-associated pathways and genes, all of them rank pathways or genes according to measures of statistical association rather than predictive performance, and most require knowledge of pathway topology, which is unavailable for many gene sets of interest, e.g., those from GO [8]. A further limitation of most existing methods is the dependence on data from matched controls, which is very limited for certain data sets [1]. Collectively, these limitations make it difficult to catalog the full set of pathways impacted in cancer along with the genes whose somatic alteration drives pathway dsyregulation. Especially challenging is the identification of the small number of pathways with potential therapeutic value from among the larger set of pathways with altered activity in human cancer.
To address these limitations, we have developed a new multi-omics pathway analysis method for cancer genomic data that aims to:
Identify pathways that play an important role in the pathophysiology of human cancers.
Identify genes whose somatic alterations are significantly associated with pathway activity.
Support personalized pathway analysis using only somatic alteration data for known cancer genes.
To achieve these aims, our method jointly analyzes gene expression and somatic alteration data from human tumors to build statistical models that predict the subject-level pathway activity in terms of the somatic alterations of known cancer genes. Importantly, this method does not require data from matched controls and can analyze pathways lacking topological information. This design is motivated by our hypothesis that biologically important pathways are those for which expression-level activity of the genes in the pathway relative to other cancers of the same type can be well predicted by the somatic alterations present in the tumor. To realize our method, we leveraged three important advances in cancer genomics and biostatistics, namely the development of large cancer genomics data sets that combine gene expression and somatic alteration data, e.g., TCGA [1], the creation of effective single-sample pathway analysis methods [11–14], and the development of computationally efficient estimation algorithms [26] for penalized regression models such as the LASSO [27]. The novelty of our approach lies in the combination of these three advances to build regression models that explain the variation of pathway activity within a single cancer type using gene-level measures of somatic alteration.
Methods
Our approach, illustrated in Fig. 1, finds pathways whose expression-level activity within a single cancer type is well predicted by somatic alterations. For a detailed description of the method, including data source details and relevant mathematics, please see the Additional file 1. The computational approach and code implementing this approach along with a description of its logic is provided at http://www.dartmouth.edu/hrfrost/MutPath/. This website also contains detailed information on the regression models fit using this approach. Reflecting the data sources used to evaluate our method, Fig. 1 shows TCGA as the source of cancer genomic data, the Molecular Signatures Database (MSigDB) [28] as the source of pathway definitions and the COSMIC cancer gene census [29] as the source of known cancer genes, however, the proposed method can be used with any appropriate source of tumor genomic data, any desired collection of gene sets and any relevant list of cancer-associated genes. Our approach is comprised by the following high-level steps (the step numbers match the numbered blue boxes in Fig. 1):
Fig. 1.

Illustration of the proposed pathway analysis method. Green cylinders represent data sources, grey boxes are intermediate data structures, blue boxes are processing steps, and orange boxes represent the analysis results associated with each of the primary aims. The numbers in each blue box correspond to one of the processing steps detailed in the “Methods” section
Step 1. Estimate single-sample pathway activity
Our method first determines the activity of each candidate pathway within each tumor relative to other tumors of the same cancer type. This step is performed using the single-sample pathway analysis method gene set variation analysis (GSVA) [12] which takes as inputs a set of pathway definitions (i.e., a mapping of genes to pathways) and a matrix of gene expression data measured on multiple tumors of the same cancer type. We specifically utilize the variant of GSVA that identifies gene sets whose members are primarily up-regulated or primarily down-regulated. Using this data, the GSVA method generates a matrix holding pathway scores for each tumor that capture the extent to which the expression of pathway genes in that tumor deviates from the mean pathway gene expression measured in all tumor samples for that cancer type. Our motivation for performing single sample pathway analysis on just the data from a single cancer type is based on findings that variation in the expression of genes among just cancer cases is a better predictor of cancer driver genes than differential expression between cancer cases and non-cancer controls [30]. Focusing on just one cancer type also enables case-only analyses, which is important for data sets, such as TCGA, that contain little data from matched controls. It should be noted an alternate single sample gene set testing method (or variation of GSVA) can be used with our approach if such a method better captures the features of pathway activity of interest for a specific analysis (see Section 1.3. of the Additional file 1 for more details; comparative results from GSVA and ssGSEA [13] for pancreatic cancer are included in Additional file 1: Tables S24 and S25).
Step 2. Estimate the association between pathway activity and somatic alterations
Our approach next determines how well the expression-based activity of each pathway can be predicted from gene-level somatic alterations. This step is performed via the regression of the single-sample pathway scores computed in Step 1 on gene-level indicators of non-silent somatic mutations and copy number variation (CNV) values. These models are estimated using LASSO-penalized multiple linear regression [31] with the penalty threshold and predictive performance computed via cross-validation. In particular, the predictive performance is represented by the proportion of null deviance explained by the model on the test data, which is equivalent to the predicted coefficient of determination in this case. LASSO penalization is used both to identify a parsimonious set of uncorrelated predictors and to support the analysis of data sets where the number of tumor samples for a given cancer type is less than the number of predictor variables. To obtain non-shrunken coefficient estimates and the approximate statistical significance of each predictor, the penalized regression is followed by an unpenalized multiple linear regression using only those predictors with non-zero coefficients in the LASSO fit. For each pathway and cancer type combination, we fit two different regression models using this procedure. The first model uses as predictor variables non-silent somatic mutation indicators and CNV values for all genes captured in the TCGA data for the target cancer type. The second model uses somatic alteration values for the subset of TCGA genes that also belong to the COSMIC cancer gene census [2]. By comparing the models fit using only consensus cancer genes with the models fit using all available genes, it is possible to assess whether the models are capturing cancer-specific phenomena and potentially identify novel cancer-associated pathways and genes. Please see Sections 1.2 and 1.3 in the Additional file 1 for a detailed mathematical description of these regression models and the estimation procedure.
Step 3. Interpret regression models to identify important pathways and genes
Using the regression models estimated in Step 2, one or more of the primary aims can be addressed:
Aim 1. To identify biologically important pathways, the pathways are ranked according to the mean from cross-validation of the LASSO-penalized models fit using just consensus cancer gene predictors. The pathways whose activity can be well predicted by somatic alterations in cancer associated genes are deemed to be biologically important, and have potential therapeutic value, for the analyzed cancer type.
Aim 2. To identify genes whose somatic alteration is associated with pathway activity for a specific cancer type, the estimated coefficients for the mutation and CNV predictors in the unpenalized pathway regression models are inspected. If somatic alteration of a gene is retained as a significant predictor in the model for a specific pathway, the gene is deemed to be a potential driver for that pathway in the analyzed cancer type. A list of inferred driver genes for each cancer type can be generated by summarizing predictor significance across all pathway models while taking into account model predictive performance.
Aim 3. The regression models estimated in the second step enable personalized pathway analysis using just somatic alteration data for a limited number of cancer-associated genes. Specifically, given tumor-specific mutational status and CNV values for the genes with non-zero coefficients in the LASSO-penalized models, it is possible to predict the activity of each evaluated pathway in that patient. When predictions are based on the unpenalized regression models, an approximate prediction interval can also be computed.
Results
To evaluate our proposed method, we analyzed 20 TCGA cancer types using gene sets from the MSigDB curated canonical (C2.CP) and oncogenic signatures (C6) collections (see the Additional file 1 for details on the TCGA data sets and MSigDB collections). Figure 2 illustrates the predicted R2 values generated for the pathways in the C2.CP collection for each supported TCGA cancer type (Additional file 1: Figure S1 contains a similar heatmap for the C6 collection). Through this analysis, we aimed to answer six questions that address the overall and aim-specific effectiveness of our method:
Do the estimated regression models capture non-random associations?
Do the models capture cancer-specific phenomena?
(Aim 1) Does model predictive performance identify biologically important pathways?
(Aim 2) Can the models be used to identify cancer driver genes?
(Aim 3) Is model predictive performance sufficient for personalized pathway analysis?
Can the models be used to characterize cancer subtypes?
Fig. 2.

Visualization of model predictive performance. Illustration of results from the analysis of TCGA pan cancer data using the MSigDB curated canonical pathways (C2.CP) collection. Rows correspond to the TCGA cohorts and columns to MSigDB gene sets. Each cell represents a pathway and cancer type-specific model colored according to the value. Both the columns and rows are ordered according to the output from hierarchical clustering
The following sections discuss each of these questions, and the relevant analysis results, in more detail.
Do the estimated regression models capture non-random associations?
To answer this question, we compared the values obtained on randomized TCGA somatic alteration data with the values estimated using non-randomized data. Because randomized data should have no predictive power, we expected the values for random data to be very close to zero. In contrast, we expected the values for TCGA data to have a mean value significantly larger than zero. These expectations were confirmed by examining the distribution of values computed using the C2.CP and C6 collections (see Fig. 3 and Additional file 1: Figure S2).
Fig. 3.
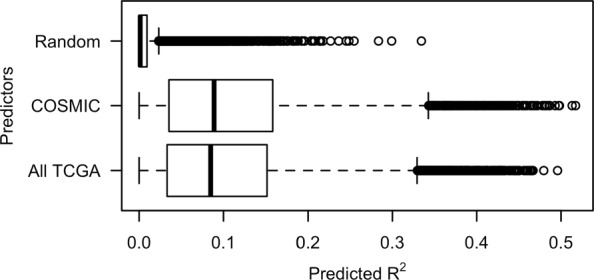
Distribution of values. Box plot illustrating the distribution of values from the analysis of TCGA pan cancer data using the MSigDB curated canonical pathways (C2.CP) collection. Values are shown for three different analyses: COSMIC: estimation of pathway regression models using somatic alteration data for COSMIC consensus cancer genes, All TCGA: estimation of models using somatic alteration data for all genes included in the TCGA datasets, and Random: estimation of models using randomized somatic alteration data for COSMIC consensus cancer genes
Do the models capture cancer-specific phenomena?
To determine if the models correctly capture cancer-specific phenomena, we compared the empirical distribution and rank ordering of values obtained for pathway models fit using two different sets of predictors: 1) the somatic alteration status of genes in the COSMIC cancer gene census or 2) the somatic alteration status of all genes included in the TCGA data. As an expert curated list of mutated human genes that have an experimentally supported association with oncogenesis [29], the COSMIC cancer gene census provides a comprehensive list of known human cancer genes. We expected the empirical distribution of values for these two potential predictor sets to be similar. Although the somatic alteration of non-cancer genes will impact gene expression levels within tumors, we expected that the ability to predict the variation of pathway activity across tumors of a single cancer type would be driven largely by the somatic alteration status of genes with a known cancer association. In other words, a model that included somatic alteration predictors for all TCGA genes would not have predictive performance significantly greater than a model that just included predictors for genes in the COSMIC cancer gene census. Following similar reasoning, we expected the rank ordering of pathways according to to be to similar regardless of whether non-COSMIC genes were included as predictors. As seen in Fig. 3, the empirical distribution is similar for both predictor sets with mean values that are significantly larger than the mean generated using random data, matching our expectation. In fact, the mean for models that include predictors for all TCGA genes is slightly lower than the mean for models that use just COSMIC genes, confirming that the addition of non-COSMIC predictors does not meaningfully improve predictive performance. As seen in Fig. 4, the Spearman rank correlation between the values computed using the two predictor sets increases with sample size and approaches 1 for the largest cohorts. These results are consistent with our expectation that pathway ranking depends primarily on somatic alterations in cancer-associated genes. The lower correlation values for the smaller cohorts reflects an expected increase in variance for the estimates.
Fig. 4.

Correlation of values between models fit using just COSMIC genes and models fit using all TCGA genes. Spearman rank correlation between values for curated canonical pathway (C2.CP) models estimated using just COSMIC consensus cancer genes and the values for C2.CP pathway models estimated using all TCGA genes. The rank correlation values for each TCGA cohort are plotted relative to cohort sample size
Does model predictive performance identify biologically important pathways?
To address this question, we ranked the pathway models for each high-level TCGA cancer type according to values. We expected that the pathways with the largest R2 values would represent biological processes that play an important role in oncogenesis for the analyzed cancer type, would be more likely to identify therapeutic targets and would have activity levels linked to patient prognosis. As an illustrative example, Table 1 lists the top MSigDB C2.CP pathways for the TCGA lung adenocarcinoma cohort (one of the larger cohorts with stable pathway ranking between predictor sets) with separate lists for the two potential predictor sets (i.e., somatic alterations for COSMIC concensus cancer genes or somatic alterations for all available TCGA genes). The Additional file 1 contains similar tables for the other four of the five largest TCGA cohorts (breast cancer, lower grade glioma, head and neck cancer and thyroid cancer) for both the C2.CP and C6 collections (see Additional file 1: Tables S3, S4, S6, S7, S9, S10, S15 and S16). As shown in Table 1, the top ten pathways for lung adenocarcinoma are almost identical for both potential predictor sets with a Spearman rank correlation for the entire C2.CP collection of 0.96. All of the top ten pathways for models built using just the COSMIC consensus cancer genes are related to the cell cycle, which has a well known association with lung adenocarinoma [32, 33]. Importantly, four of the top ten pathways (pathways with ranks 1, 2, 6 and 10) are associated with genes identified as either therapeutic targets for lung adenocarcinoma (Ran [34] and ATR [35, 36]) or as biomarkers of patient prognonsis (CDC6 [37]).
Table 1.
Top pathway models for lung adenocarcinoma
| Consensus cancer genes | All TCGA genes | ||||
|---|---|---|---|---|---|
| # | Gene set | # | Gene set | ||
| 1 | BIOCARTA_RANMS_PATHWAY | 0.487 | 1 | BIOCARTA_RANMS_PATHWAY | 0.465 |
| 2 | REACTOME_ACTIVATION_OF_ATR_IN_RESPO... | 0.486 | 3 | REACTOME_G2_M_CHECKPOINTS | 0.459 |
| 3 | REACTOME_G2_M_CHECKPOINTS | 0.483 | 2 | REACTOME_ACTIVATION_OF_ATR_IN_RESPO... | 0.456 |
| 4 | REACTOME_CELL_CYCLE | 0.472 | 6 | REACTOME_CDC6_ASSOCIATION_WITH_THE_... | 0.450 |
| 5 | REACTOME_MITOTIC_M_M_G1_PHASES | 0.468 | 14 | REACTOME_CHROMOSOME_MAINTENANCE | 0.434 |
| 6 | REACTOME_CDC6_ASSOCIATION_WITH_THE_... | 0.468 | 9 | REACTOME_G0_AND_EARLY_G1 | 0.433 |
| 7 | REACTOME_DNA_REPLICATION | 0.464 | 4 | REACTOME_CELL_CYCLE | 0.431 |
| 8 | REACTOME_CELL_CYCLE_MITOTIC | 0.464 | 8 | REACTOME_CELL_CYCLE_MITOTIC | 0.429 |
| 9 | REACTOME_G0_AND_EARLY_G1 | 0.454 | 5 | REACTOME_MITOTIC_M_M_G1_PHASES | 0.428 |
| 10 | PID_ATR_PATHWAY | 0.450 | 7 | REACTOME_DNA_REPLICATION | 0.427 |
Top ten MSigDB C2.CP pathways ranked according to for regression models constructed using the TCGA lung adenocarcinoma data. Separate rankings are shown for models estimated using both potential predictor sets (i.e., the somatic alterations for either genes in the COSMIC cancer gene census or all genes available in the TCGA data). The "#" columns contain the pathway rank according to the COSMIC models. Note that the complete names of the rank 2 and rank 6 pathways for COSMIC genes are REACTOME_ACTIVATION_OF_ATR_IN_RESPONSE_TO_REPLICATION_STRESS and REACTOME_CDC6_ASSOCIATION_WITH_THE_ORC_ORIGIN_COMPLEX
Can the models be used to identify cancer driver genes?
To ascertain if the models can identify known cancer driver genes, we ranked the somatic alteration predictors for each cancer type according to predictor significance in the pathway models. Specifically, alterations were ranked according to a weight computed as the average across all pathway models in a specific MSigDB collection of the product of predictor significance (i.e., the -log(p-value) for the predictor in the unpenalized model) and model . An example of this ranking is shown in Table 2 for the TCGA lung adenocarcinoma cohort with ranks computed using all four combinations of MSigDB collection and predictor set (the Additional file 1 contains similar tables for the other four of the five largest TCGA cohorts, see Additional file 1: Tables S5, S8, S11, S17). It is important to note the potential bias in these weights caused by pathway overlaps. Specifically, the weights for somatic alterations associated with the expression of genes annotated to multiple pathways will be inflated relative to the weights for somatic alterations that are only associated infrequently annotated genes.
Table 2.
Top predictors for lung adenocarcinoma
| Consensus cancer genes | All TCGA genes | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| C2.CP | C6 | C2.CP | C6 | ||||||||
| # | Predictor | W | # | Predictor | W | # | Predictor | W | # | Predictor | W |
| 1 | TP53 | 0.784 | 1 | TP53 | 0.672 | 1 | KEAP1 | 0.766 | 1 | KEAP1 | 0.942 |
| 2 | SMARCA4 | 0.559 | 3 | MET (CNV) | 0.590 | 2 | TP53 | 0.484 | 2 | TP53 | 0.423 |
| 3 | MET (CNV) | 0.445 | 2 | SMARCA4 | 0.541 | 3 | SMARCA4 | 0.339 | 4 | KRAS | 0.397 |
| 4 | KRAS | 0.374 | 4 | KRAS | 0.507 | 4 | KRAS | 0.249 | 3 | SMARCA4 | 0.298 |
| 5 | SETD2 | 0.268 | 8 | EGFR (CNV) | 0.319 | 5 | STK11 | 0.171 | 8 | *BAGE2 | 0.209 |
| 6 | RBM10 | 0.263 | 25 | EGFR | 0.279 | 6 | *DST | 0.166 | 7 | *FRG1B | 0.148 |
| 7 | STK11 | 0.239 | 9 | FOXA1 (CNV) | 0.262 | 7 | *FRG1B | 0.145 | 13 | MET (CNV) | 0.146 |
| 8 | EGFR (CNV) | 0.220 | 10 | MYC (CNV) | 0.241 | 8 | *BAGE2 | 0.131 | 43 | *PKHD1 | 0.137 |
| 9 | FOXA1 (CNV) | 0.213 | 44 | EP300 (CNV) | 0.229 | 9 | *SPTA1 | 0.127 | 36 | EGFR | 0.130 |
| 10 | MYC (CNV) | 0.209 | 11 | SMARCA4 (CNV) | 0.227 | 10 | *ANK2 | 0.115 | 6 | *DST | 0.127 |
Top ten gene-level somatic alteration predictors from models estimated using the TCGA lung adenocarcinoma cohort. The predictors are ranked according to a weight, W, computed as the average across all pathway models in the MSigDB collection of the product the -log(p-value) for the predictor in the unpenalized model and model . Separate rankings are shown for the C2.CP and C6 collections using both potential sets of predictors. The "#" columns contain the rank of the predictor in the list computed using C2.CP collection and the target predictor set. Predictors marked in bold represent known driver genes for lung adenocarcinoma. Predictors prefixed with a "*" are not in the COSMIC cancer gene census
We expected that the top ranking gene-level somatic alterations would disproportionately represent known driver genes. To evaluate this, we first generated a list of known driver genes for each cancer type using the cancer type associations from the COSMIC cancer gene census (see Additional file 1: Table S1 for details). Although not comprehensive, these represent cancer type relationships that are independent of the TCGA data (i.e., they are not inferred from TCGA somatic alteration data). Enrichment of these known driver genes among the ranked predictors associated with all available TCGA genes was then tested using a Wilcoxon rank sum test and false discovery rate (FDR) q-values were computed using the Benjamini and Hochberg (BH) [38] method. Consistent with our expectations, the enrichment q-values for all but three of the cancer types using the C2.CP models was below 0.07 with the majority below 0.01 (see Fig. 5 and S8). For the lung cancer example, a careful examination of the top 10 COSMIC predictors for the C2.CP collection (see Table 2) reveals that all ten in fact have a known association with lung adenocarcinoma (only six are in the list of known driver genes) with most also serving as therapeutic targets and/or prognostic indicators (TP53 [32], SMARCA4 [39], MET [40, 41], KRAS [42, 43], SETD2 [44], RBM10 [45], STK11 [46, 47], EGFR [43, 48], FOXA1 [49] and MYC [43]).
Fig. 5.

Enrichment of known cancer driver genes. The false discovery rate (FDR) q-values (as computed using the Benjamini and Hochberg (BH) [38] method) for the Wilcox rank sum tests of enrichment of known driver genes among the ranks of all predictors for the MSigDB C2.CP models using all TCGA genes. The q-value for each cancer type cohort is plotted relative to the cohort sample size. All but one cancer type has a q-value of < 0.1 with a general association between significance and sample size
We also expected that the predictor ranking would be insensitive to the pathway collection on which it was computed, i.e., the C2.CP and C6 rankings would be similar. As seen in Fig. 6, the Spearman rank correlation between the predictor weights computed using the C2.CP or C6 pathway models increases from ∼ 0.5 for the smallest cohorts to ∼ 0.8 for the largest cohorts. For the lung cancer example shown in Table 2, seven of the top ten predictors for the C2.CP collection are also in the top ten list for the C6 collection with this magnitude overlap holding for both predictor sets. These results are consistent with our expectation that predictor ranking is identifying true driver genes and provides additional evidence that pathways with large values are associated with important aspects of cancer biology.
Fig. 6.

Correlation of predictor weights between C2.CP and C6 regression models. Spearman rank correlation between the weights for COSMIC consensus cancer gene predictors computed for the MSigDB curated canonical pathways (C2.CP) and oncogenic signatures (C6) collections. The correlation values for each TCGA cohort and are plotted relative to cohort sample size
Can the models be used to identify novel driver genes?
To determine if the models are effective at identifying novel driver genes, we examined the high-ranking somatic alteration predictors for genes not included in the COSMIC cancer gene census. In particular, we examined non-COSMIC predictors whose high-ranking was replicated across both the C2.CP and C6 collections. Because these genes do not have a well-established role in cancer but are significant predictors in models associated with different pathway collections, we hypothesized that they could represent novel drivers for the analyzed cancer type. For the lung adenocarcinoma cohort, the predictors prefixed with an * in the right two columns of Table 2 are associated with non-COSMIC genes. Importantly, three of the non-COSMIC predictors included in the top ten for the C2.CP collection (DST, FRG1B and BAGE2) are also in the top ten for the C6 collection and none of the three has an established association with lung adenocarcinoma. DST, FRG1B and BAGE2 are thus candidate driver genes for lung adenocarcinoma and good targets for follow-on experiments.
Is model predictive performance sufficient for personalized pathway analysis?
The values computed via CV on the TCGA data sets reflect the predictive performance that can be expected for personalized pathway analysis. Although more research is needed to determine the true utility of these models for personalized pathway analysis, we believe that the current results are encouraging, especially for the larger TCGA cohorts and pathways models that have values around ∼ 0.5. These models, when combined with model uncertainty to generate a prediction interval, may provide useful information regarding pathway dysregulation within a single tumor. The prediction of expression-based activity from somatic alteration status may be especially useful for cases where gene expression data is unavailable. Appropriately, this approach to personalized pathway analysis is strongly influenced by somatic alterations of common cancer driver genes, i.e., the somatic alteration of a gene like p53 will impact the predicted aberrant activity of many pathways whose expression-based activity is closely associated with somatic alterations in the tumor. This follows from the criteria used by our method to identify candidate driver genes, i.e., the somatic alteration predictors that are significant in highly predictive regression models are assumed to represent potential cancer driver genes. Improvements in predictive performance of this approach can likely be achieved with larger sample sizes (see Additional file 1: Figures S4 and S5 for the association between mean and cohort size) or with the inclusion of additional predictors such as methylation values.
Can the models be used to characterize cancer subtypes?
Although the TCGA cohorts provide a useful high-level grouping of cancers, significant heterogeneity often exists within each type of cancer. For example. breast cancers are often sub-divided according to gene expression or mutational profiles [50] into two (e.g., luminal-like and basal-like) or more (e.g., luminal A, luminal B, HER2 enriched, basal-like) subtypes. To determine if the pathway models fit for the high-level TCGA cancer types could be used to characterize the features of cancer subtypes, we explored the differences in predicted pathway activity and somatic mutation predictors for the TCGA BRCA subjects assigned to either the basal or luminal PANCAN cluster-of-cluster assignments [51] (see Section 1.4 of the Additional file 1 for analysis details). As illustrated in the Additional file 1 (Tables S18-S23), the differences in predicated pathway activity and important somatic alterations are consistent with known differences between luminal and basal breast cancer types subtypes.
Discussion
We have described a novel approach for jointly analyzing gene expression and somatic alteration data through the lens of biological pathways. Our approach combines single sample pathway analysis on gene expression data with LASSO-penalized regression to build statistical models that use the somatic alterations present in each tumor to predict the deviation of pathway activity from the expected activity for the associated cancer type. Because this method can analyze case-only data and does not require information regarding pathway topology, it allows researchers to explore data sets that cannot be analyzed by existing cancer pathway analysis techniques. These models can be used to achieve our three primary aims:
Aim 1: Identify pathways that play an important role in the pathophysiology of human cancers. By ranking the pathway-specific regression models according to predictive performance, it is possible identify pathways whose activity is driven by somatic alterations in the tumor. These pathways can be expected to play a key role in the pathophysiology of each cancer type and are thus strong candidates for therapeutic intervention. As shown through our analysis of TCGA data using MSigDB pathways, the regression models are capturing real cancer-specific phenomena. A qualitative analysis of the models with the largest predictive performance indicates that the associated pathways have a clear relationship with the cancer type and, in many cases, identify therapeutic targets.
Aim 2: Identify genes whose somatic alteration is significantly associated with pathway activity. By ranking the gene-level somatic alteration predictors according to predictor significance and model predictive performance, it is possible to identify the genes whose somatic alteration drives pathway activity for each cancer type. As demonstrated by the analysis of TCGA data and MSigDB pathways, the predictor rankings can be replicated across disjoint pathway collections and are significantly enriched for genes known to be associated with each cancer type. By fitting regression models using somatic alterations for genes without a known cancer association, our approach also supports the discovery of novel driver genes.
Aim 3: Support personalized pathway analysis using only somatic alteration data. The predictive performance estimated for the MSigDB pathway models on the TCGA data reflects the expected performance for personalized pathway analysis. Although additional software engineering will be required to create a tool that can be easily used by other researchers for personalized pathway analysis, the current results are promising and motivate future work in this direction. For cases in which gene expression data is unavailable and somatic alteration data may be limited to known cancer driver genes, these models can provide useful information on the activity of pathways within a patient’s tumor; information that may help assess prognosis or guide treatment.
Limitations
Important limitations of our method and the reported results include the scope and quality of the data drawn from the TCGA, COSMIC and MSigDB, limitations of the statistical models and estimation approaches employed for pathway analysis, and limitations associated with the subjective interpretation of pathways associated with highly predictive regression models. Limitations associated with the TCGA data include the small number of samples for many of the analyzed cohorts, the heterogeneity of tumors within each cohort (e.g., the major subtypes of breast cancer), errors in the gene-level estimates of non-silent somatic mutations and copy number variation, sparsity of the somatic alteration data, and the fact that the employed non-silent mutation indicators fail to distinguish between gain-of-function and loss-of-function mutations. An additional limitation associated with the leveraged TCGA data is the fact that somatic alteration data types like methylation, mutations of non-protein coding genes and structure features such as fusions and translations are not included as predictors in the regression models. Although the COSMIC cancer gene census provides a comprehensive list of genes with a known cancer association, the census does not quantify the degree or direction of association, provides only approximate cancer type associations for each gene, and likely misses many genes that have a true link to cancer. Limitations of the gene set collections in MSigDB include the variable quality of annotations, bias in pathway annotations (i.e., more annotations will exist for well studied genes and pathways), the fact that the analyzed MSigDB pathways do not represent all potential cancer-related pathways, and overlaps between the members of many pathways. An important implication of our use of curated pathways from MSigDB is that our method is unlikely to identify truly novel pathway-cancer associations. The statistical model used to predict pathway activity only includes copy number alterations and indicators of non-silent somatic alterations as predictors; other genomic features that are known to impact gene expression such as epigenetic changes (e.g., methylation), translocations, gene fusions and mutation of non-protein coding genes are ignored. Given the unknown marginal and joint distribution of the values, a formal statistical test was not performed on individual values or comparing the different distributions shown in Fig. 3. Other analytical limitations include the fact that the scores generated by GSVA only approximate pathway activity within each tumor, and the stochastic nature of LASSO-penalized estimation. An important limitation of the evaluation results is the qualitative and subjective analysis used to ascertain the cancer relevance and therapeutic value of identified pathways and genes. While the estimated regression models provide useful insight into the somatic alterations can drive pathway dysregulation in the analyzed TCGA cancer types, model performance has not been evaluated on non-TCGA data and the current models and implementation logic do not support the direct use of these models for personalized pathway analysis on new patient tumor data.
Future directions
Possible extensions to this method include support for additional data types, modifications to the statistical model, exploration of cancer subtypes, validation on other cancer genomics data sets and creation of tools to support personalized pathway analysis on new tumor genomic data. To more accurately model the somatic alterations that drive gene expression, the current approach can be expanded to include epigenetic modifications (e.g., methylation), mutations of non-protein coding genes, and features such as gene fusions and translocations as predictors in the regression model. An important issue that must be address in future efforts to integration additional predictor variables will be the limited available of many of these additional genomic data types. Potential enhancements of the statistical model include the addition of interaction terms, predictor weighting based on prior knowledge regarding the role of specific genes in cancer and modification of predictor weights to account for the overlap between pathways. To support formal statistical analysis of the values computed for each pathway model, resampling approaches could be used (i.e., generate multiple bootstrap resampled versions of the TCGA data and estimate pathway regression models for each resampled data set). Alternative approaches for computing the single sample pathway scores can also be investigated, e.g., generate GSVA statistics that can identify gene sets with both up and down-regulated members, base single sample scores on gene expression relative to controls or another cancer type, etc. Evaluation of this approach can be expanded to include the analysis of other cancer types or subtypes of the analyzed cohorts (e.g., extend the analysis of breast cancer subtypes to other cancers), and the analysis of data from other cancer genomics repositories. A particularly important topic for future research involves the use of more a objective and systematic approach for evaluating identified pathways and genes with experimental confirmation of any novel findings.
Conclusions
We have developed a new approach for the pathway-based analysis of multi-omics cancer data. Our approach combines single-sample pathway analysis with multi-stage, lasso-penalized regression to find pathways whose gene expression can be explained largely in terms of the non-silent somatic mutations and copy number variations present in the tumor. This method enables the identification of biologically important pathways and genes and can be used for personalized pathway analysis in cases where gene expression data is unavailable. Importantly, this method can be used on case-only data sets and does not require information regarding pathway topology. An analysis of 20 human cancer types using TCGA genomic data and MSigDB gene sets illustrates the effectiveness of our technique. These analysis results also provide cancer researchers with ranked lists of pathways and genes that likely play a key role in the etiology of these cancer types, information that can be used to generate hypotheses for more detailed experimental exploration of cancer pathways and novel driver genes.
Additional file
Additional results and details on the computational pipeline, analyzed data sets and logic used to generate all tables and figures. (PDF 1772 kb)
Acknowledgments
Funding
National Institutes of Health grants K01LM012426, P20GM103534, P30CA023108, U19CA148127 and U01CA196386.
Availability of data and materials
All data used to generate the results presented in this paper is publicly accessible. The analyzed gene sets were from version 5.2 of the Molecular Signatures Database (MSigDB) [28] (http://software.broadinstitute.org/gsea/downloads.jsp). Information about cancer-associated genes was taken from the Catalog of Somatic Mutations in Cancer (COSMIC) [2] cancer gene census (release v78, 5th September 2016; http://cancer.sanger.ac.uk/cosmic). Cancer genomic data was from The Cancer Genome Atlas (TCGA) [1] as downloaded from the UCSC Cancer Browser [52] pan cancer data set (https://genome-cancer.ucsc.edu/). The computational approach and code implementing this approach along with a description of its logic is provided at http://www.dartmouth.edu/~hrfrost/MutPath/.
Abbreviations
- C2.CP
MSigDB curated canonical pathways collection
- C6
MSigDB oncogenic signatures collection
- CNV
Copy number variation
- COSMIC
Catalog of somatic mutations in cancer
- GO
Gene ontology
- GSVA
Gene set variation analysis
- LASSO
Least absolute shrinkage and selection operator
- MSigDB
Molecular signatures database
- TCGA
The cancer genome atlas
Authors’ contributions
HRF designed and implemented the computational pipeline, performed the real data analysis and drafted the manuscript. CIA participated in the development of the methodology, assisted with the real data analysis and helped draft the manuscript. Both HRF and CIA have read and approve of the final manuscript.
Ethics approval and consent to participate
Not applicable. Although the results contained in this manuscript were generated through the analysis of data collected from human subjects, only previously collected, publicly available and de-identified data sources were be used. Consequently, the proposed research was except from Federal regulations according to category 4 (45 CFR 46.101.b.4) of the Common Rule for the Protection of Human Subjects.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
H. Robert Frost, Email: rob.frost@dartmouth.edu.
Christopher I. Amos, Email: christopher.i.amos@dartmouth.edu
References
- 1.Cancer Genome Atlas Research Network. Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. The cancer genome atlas pan-cancer analysis project. Nat Genet. 2013;45(10):1113–20. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, Teague JW, Stratton MR, McDermott U, Campbell PJ. Cosmic: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43(Database issue):805–11. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng F, Zhao J, Zhao Z. Advances in computational approaches for prioritizing driver mutations and significantly mutated genes in cancer genomes. Brief Bioinform. 2016;17(4):642–56. doi: 10.1093/bib/bbv068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kristensen VN, Lingjærde OC, Russnes HG, Vollan HKM, Frigessi A, Børresen-Dale A-L. Principles and methods of integrative genomic analyses in cancer. Nat Rev Cancer. 2014;14(5):299–313. doi: 10.1038/nrc3721. [DOI] [PubMed] [Google Scholar]
- 6.Mutation Consequences and Pathway Analysis working group of the International Cancer Genome Consortium: Pathway and network analysis of cancer genomes. Nat Methods. 2015; 12(7):615–21. 10.1038/nmeth.3440. [DOI] [PMC free article] [PubMed]
- 7.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz Jr LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–58. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Croft D, Mundo AF, Haw R, Milacic M, Weiser J, Wu G, Caudy M, Garapati P, Gillespie M, Kamdar MR, Jassal B, Jupe S, Matthews L, May B, Palatnik S, Rothfels K, Shamovsky V, Song H, Williams M, Birney E, Hermjakob H, Stein L, D’Eustachio P. The reactome pathway knowledgebase. Nucleic Acids Res. 2014;42(Database issue):472–7. doi: 10.1093/nar/gkt1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khatri P, Sirota M, Butte AJ. Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput Biol. 2012;8(2):1002375. doi: 10.1371/journal.pcbi.1002375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drier Y, Sheffer M, Domany E. Pathway-based personalized analysis of cancer. Proc Natl Acad Sci U S A. 2013;110(16):6388–93. doi: 10.1073/pnas.1219651110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hänzelmann S, Castelo R, Guinney J. Gsva: gene set variation analysis for microarray and rna-seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, Fröhling S, Chan EM, Sos ML, Michel K, Mermel C, Silver SJ, Weir BA, Reiling JH, Sheng Q, Gupta PB, Wadlow RC, Le H, Hoersch S, Wittner BS, Ramaswamy S, Livingston DM, Sabatini DM, Meyerson M, Thomas RK, Lander ES, Mesirov JP, Root DE, Gilliland DG, Jacks T, Hahn WC. Systematic rna interference reveals that oncogenic kras-driven cancers require tbk1. Nature. 2009;462(7269):108–12. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vaske CJ, Benz SC, Sanborn JZ, Earl D, Szeto C, Zhu J, Haussler D, Stuart JM. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using paradigm. Bioinformatics. 2010;26(12):237–45. doi: 10.1093/bioinformatics/btq182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paull EO, Carlin DE, Niepel M, Sorger PK, Haussler D, Stuart JM. Discovering causal pathways linking genomic events to transcriptional states using tied diffusion through interacting events (tiedie) Bioinformatics. 2013;29(21):2757–64. doi: 10.1093/bioinformatics/btt471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng S, Collisson EA, Sokolov A, Goldstein T, Gonzalez-Perez A, Lopez-Bigas N, Benz C, Haussler D, Stuart JM. Paradigm-shift predicts the function of mutations in multiple cancers using pathway impact analysis. Bioinformatics. 2012;28(18):640–6. doi: 10.1093/bioinformatics/bts402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gundem G, Lopez-Bigas N. Sample-level enrichment analysis unravels shared stress phenotypes among multiple cancer types. Genome Med. 2012;4(3):28. doi: 10.1186/gm327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bashashati A, Haffari G, Ding J, Ha G, Lui K, Rosner J, Huntsman DG, Caldas C, Aparicio SA, Shah SP. Drivernet: uncovering the impact of somatic driver mutations on transcriptional networks in cancer. Genome Biol. 2012;13(12):124. doi: 10.1186/gb-2012-13-12-r124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahn T, Park T. Pathway-driven discovery of rare mutational impact on cancer. Biomed Res Int. 2014;2014:171892. doi: 10.1155/2014/171892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding J, McConechy MK, Horlings HM, Ha G, Chun Chan F, Funnell T, Mullaly SC, Reimand J, Bashashati A, Bader GD, Huntsman D, Aparicio S, Condon A, Shah SP. Systematic analysis of somatic mutations impacting gene expression in 12 tumour types. Nat Commun. 2015;6:8554. doi: 10.1038/ncomms9554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uzilov AV, Ding W, Fink MY, Antipin Y, Brohl AS, Davis C, Lau CY, Pandya C, Shah H, Kasai Y, Powell J, Micchelli M, Castellanos R, Zhang Z, Linderman M, Kinoshita Y, Zweig M, Raustad K, Cheung K, Castillo D, Wooten M, Bourzgui I, Newman LC, Deikus G, Mathew B, Zhu J, Glicksberg BS, Moe AS, Liao J, Edelmann L, Dudley JT, Maki RG, Kasarskis A, Holcombe RF, Mahajan M, Hao K, Reva B, Longtine J, Starcevic D, Sebra R, Donovan MJ, Li S, Schadt EE, Chen R. Development and clinical application of an integrative genomic approach to personalized cancer therapy. Genome Med. 2016;8(1):62. doi: 10.1186/s13073-016-0313-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masica DL, Karchin R. Correlation of somatic mutation and expression identifies genes important in human glioblastoma progression and survival. Cancer Res. 2011;71(13):4550–61. doi: 10.1158/0008-5472.CAN-11-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Liang M, Zhang Z. Regression analysis of combined gene expression regulation in acute myeloid leukemia. PLoS Comput Biol. 2014;10(10):1003908. doi: 10.1371/journal.pcbi.1003908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen R, Olshen AB, Ladanyi M. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics. 2009;25(22):2906–12. doi: 10.1093/bioinformatics/btp543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hofree M, Shen JP, Carter H, Gross A, Ideker T. Network-based stratification of tumor mutations. Nat Methods. 2013;10(11):1108–15. doi: 10.1038/nmeth.2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friedman JH, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33(1):1–22. doi: 10.18637/jss.v033.i01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tibshirani R. Regression shrinkage and selection via the Lasso. J R Stat Soc Ser B Stat Methodol. 1996;58(1):267–88. [Google Scholar]
- 28.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (msigdb) 3.0. Bioinformatics. 2011;27(12):1739–40. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat Rev Cancer. 2004;4(3):177–83. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gorlov IP, Yang J-Y, Byun J, Logothetis C, Gorlova OY, Do K-A, Amos C. How to get the most from microarray data: advice from reverse genomics. BMC Genom. 2014;15(1):223. doi: 10.1186/1471-2164-15-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tibshirani R. Regression shrinkage and selection via the lasso: a retrospective. J R Stat Soc Ser B Stat Methodol. 2011;73(Part 3):273–82. doi: 10.1111/j.1467-9868.2011.00771.x. [DOI] [Google Scholar]
- 32.Esposito V, Baldi A, Tonini G, Vincenzi B, Santini M, Ambrogi V, Mineo TC, Persichetti P, Liuzzi G, Montesarchio V, Wolner E, Baldi F, Groeger AM. Analysis of cell cycle regulator proteins in non-small cell lung cancer. J Clin Pathol. 2004;57(1):58–63. doi: 10.1136/jcp.57.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eymin B, Gazzeri S. Role of cell cycle regulators in lung carcinogenesis. Cell Adh Migr. 2010;4(1):114–23. doi: 10.4161/cam.4.1.10977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuen H-F, Chan K-K, Platt-Higgins A, Dakir E-H, Matchett K, Haggag YA, Jithesh P, Habib T, Faheem A, Dean F, Morgan R, Rudland P, El-Tanani M. Ran gtpase promotes cancer progression via met receptormediated downstream signaling. Oncotarget. 2016;7(46):75854–64. doi: 10.18632/oncotarget.12420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanjiv K, Hagenkort A, Calderón-Montaño JM, Koolmeister T, Reaper PM, Mortusewicz O, Jacques SA, Kuiper RV, Schultz N, Scobie M, Charlton PA, Pollard JR, Berglund UW, Altun M, Helleday T. Cancer-specific synthetic lethality between atr and chk1 kinase activities. Cell Rep. 2016;14(2):298–309. doi: 10.1016/j.celrep.2015.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Syljuåsen RG, Hasvold G, Hauge S, Helland Å. Targeting lung cancer through inhibition of checkpoint kinases. Front Genet. 2015;6:70. doi: 10.3389/fgene.2015.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allera-Moreau C, Rouquette I, Lepage B, Oumouhou N, Walschaerts M, Leconte E, Schilling V, Gordien K, Brouchet L, Delisle MB, Mazieres J, Hoffmann JS, Pasero P, Cazaux C. Dna replication stress response involving plk1, cdc6, polq, rad51 and claspin upregulation prognoses the outcome of early/mid-stage non-small cell lung cancer patients. Oncogenesis. 2012;1:30. doi: 10.1038/oncsis.2012.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B. 1995;57(1):289–300. [Google Scholar]
- 39.Orvis T, Hepperla A, Walter V, Song S, Simon J, Parker J, Wilkerson MD, Desai N, Major MB, Hayes DN, Davis IJ, Weissman B. Brg1/smarca4 inactivation promotes non-small cell lung cancer aggressiveness by altering chromatin organization. Cancer Res. 2014;74(22):6486–98. doi: 10.1158/0008-5472.CAN-14-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Landi L, Minuti G, D’Incecco A, Cappuzzo F. Targeting c-met in the battle against advanced nonsmall-cell lung cancer. Curr Opin Oncol. 2013;25(2):130–6. doi: 10.1097/CCO.0b013e32835daf37. [DOI] [PubMed] [Google Scholar]
- 41.Gelsomino F, Facchinetti F, Haspinger ER, Garassino MC, Trusolino L, De Braud F, Tiseo M. Targeting the met gene for the treatment of non-small-cell lung cancer. Crit Rev Oncol Hematol. 2014;89(2):284–99. doi: 10.1016/j.critrevonc.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 42.Westcott PMK, To MD. The genetics and biology of kras in lung cancer. Chin J Cancer. 2013;32(2):63–70. doi: 10.5732/cjc.012.10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McFadden DG, Politi K, Bhutkar A, Chen FK, Song X, Pirun M, Santiago PM, Kim-Kiselak C, Platt JT, Lee E, Hodges E, Rosebrock AP, Bronson RT, Socci ND, Hannon GJ, Jacks T, Varmus H. Mutational landscape of egfr-, myc-, and kras-driven genetically engineered mouse models of lung adenocarcinoma. Proc Natl Acad Sci U S A. 2016;113(42):6409–17. doi: 10.1073/pnas.1613601113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pfister SX, Ahrabi S, Zalmas L-P, Sarkar S, Aymard F, Bachrati CZ, Helleday T, Legube G, La Thangue NB, Porter ACG, Humphrey TC. Setd2-dependent histone h3k36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014;7(6):2006–18. doi: 10.1016/j.celrep.2014.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hernández J, Bechara E, Schlesinger D, Delgado J, Serrano L, Valcárcel J. Tumor suppressor properties of the splicing regulatory factor rbm10. RNA Biol. 2016;13(4):466–72. doi: 10.1080/15476286.2016.1144004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gill RK, Yang S-H, Meerzaman D, Mechanic LE, Bowman ED, Jeon H-S, Roy Chowdhuri S, Shakoori A, Dracheva T, Hong K-M, Fukuoka J, Zhang J-H, Harris CC, Jen J. Frequent homozygous deletion of the lkb1/stk11 gene in non-small cell lung cancer. Oncogene. 2011;30(35):3784–91. doi: 10.1038/onc.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pécuchet N, Laurent-Puig P, Mansuet-Lupo A, Legras A, Alifano M, Pallier K, Didelot A, Gibault L, Danel C, Just P-A, Riquet M, Le Pimpec-Barthes F, Damotte D, Fabre E, Blons H. Different prognostic impact of stk11 mutations in non-squamous non-small-cell lung cancer. Oncotarget. 2015. 10.18632/oncotarget.6379. [DOI] [PMC free article] [PubMed]
- 48.da Cunha Santos G, Shepherd FA, Tsao MS. Egfr mutations and lung cancer. Annu Rev Pathol. 2011;6:49–69. doi: 10.1146/annurev-pathol-011110-130206. [DOI] [PubMed] [Google Scholar]
- 49.Wang H, Meyer CA, Fei T, Wang G, Zhang F, Liu XS. A systematic approach identifies foxa1 as a key factor in the loss of epithelial traits during the epithelial-to-mesenchymal transition in lung cancer. BMC Genom. 2013;14:680. doi: 10.1186/1471-2164-14-680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cancer Genome Atlas Network: Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490(7418):61–70. 10.1038/nature11412. [DOI] [PMC free article] [PubMed]
- 51.Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, Leiserson MDM, Niu B, McLellan MD, Uzunangelov V, Zhang J, Kandoth C, Akbani R, Shen H, Omberg L, Chu A, Margolin AA, Van’t Veer LJ, Lopez-Bigas N, Laird PW, Raphael BJ, Ding L, Robertson AG, Byers LA, Mills GB, Weinstein JN, Van Waes C, Chen Z, Collisson EA, Cancer Genome Atlas Research Network. Benz CC, Perou CM, Stuart JM. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158(4):929–44. doi: 10.1016/j.cell.2014.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goldman M, Craft B, Swatloski T, Cline M, Morozova O, Diekhans M, Haussler D, Zhu J. The ucsc cancer genomics browser: update 2015. Nucleic Acids Res. 2015;43(Database issue):812–7. doi: 10.1093/nar/gku1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional results and details on the computational pipeline, analyzed data sets and logic used to generate all tables and figures. (PDF 1772 kb)
Data Availability Statement
All data used to generate the results presented in this paper is publicly accessible. The analyzed gene sets were from version 5.2 of the Molecular Signatures Database (MSigDB) [28] (http://software.broadinstitute.org/gsea/downloads.jsp). Information about cancer-associated genes was taken from the Catalog of Somatic Mutations in Cancer (COSMIC) [2] cancer gene census (release v78, 5th September 2016; http://cancer.sanger.ac.uk/cosmic). Cancer genomic data was from The Cancer Genome Atlas (TCGA) [1] as downloaded from the UCSC Cancer Browser [52] pan cancer data set (https://genome-cancer.ucsc.edu/). The computational approach and code implementing this approach along with a description of its logic is provided at http://www.dartmouth.edu/~hrfrost/MutPath/.