

Summary
Emicizumab, a humanised bispecific antibody recognising factors (F) IX/IXa and X/Xa, can accelerate FIXa-catalysed FX activation by bridging FIXa and FX in a manner similar to FVIIIa. However, details of the emicizumab–antigen interactions have not been reported so far. In this study, we first showed by surface plasmon resonance analysis that emicizumab bound FIX, FIXa, FX, and FXa with moderate affinities ( K D = 1.58, 1.52, 1.85, and 0.978 μM, respectively). We next showed by immunoblotting analysis that emicizumab recognised the antigens’ epidermal growth factor (EGF)-like domains. We then performed K D -based simulation of equilibrium states in plasma for quantitatively predicting the ways that emicizumab would interact with the antigens. The simulation predicted that only a small part of plasma FIX, FX, and emicizumab would form antigen-bridging FIX–emicizumab–FX ternary complex, of which concentration would form a bell-shaped relationship with emicizumab concentration. The bell-shaped concentration dependency was reproduced by plasma thrombin generation assays, suggesting that the plasma concentration of the ternary complex would correlate with emicizumab’s cofactor activity. The simulation also predicted that at 10.0–100 μg/ml of emicizumab–levels shown in a previous study to be clinically effective–the majority of plasma FIX, FX, and emicizumab would exist as monomers. In conclusion, emicizumab binds FIX/FIXa and FX/FXa with micromolar affinities at their EGF-like domains. The K D -based simulation predicted that the antigen-bridging ternary complex formed in circulating plasma would correlate with emicizumab’s cofactor activity, and the majority of FIX and FX would be free and available for other coagulation reactions.
Institution where the work was carried out: Research Division, Chugai Pharmaceutical Co., Ltd.
Supplementary Material to this article is available online at www.thrombosis-online.com.
Keywords: Coagulation factors, drug design, factor VIII, haemophilia therapy
Introduction
Routine administration of exogenous factor (F) VIII for bleeding prophylaxis in patients with haemophilia A–a practice widespread mainly in developed countries–improves patients’ quality of life by reducing bleeding episodes and bleeding-related complications ( 1 , 2 ). However, the treatment still has some drawbacks, including imperfect control of bleeding in patients who develop alloantibodies to FVIII and the necessity of administration via frequent intravenous injections ( 2 , 3 ). To overcome such drawbacks, we explored the idea of creating an antibody functioning as a FVIII cofactor, which we believed would be a promising approach because IgG antibodies generally have a long half-life, high subcutaneous bioavailability, and a molecular structure or antigenicity different from that of FVIII ( 4 ). In the 1990s, several pioneering researchers revealed the ways that FVIIIa interacts with both FIXa and FX, which would facilitate acceleration of FIXa-catalysed FX activation ( 5 – 7 ). These interactions were further unveiled through the 2000s and 2010s ( 8 – 10 ), and consequently FVIIIa is now known to have multiple contacts with both FIXa and FX (► Figure 1A ). The idea behind our FVIIIa-mimetic cofactor antibody was to create an anti-FIXa/FX bispecific antibody that places the two antigens(FIXa and FX) in a spatially appropriate position to accelerate FIXa-catalysed FX activation in the same manner that FVIIIa should do ( 4 ). After long research, we eventually identified a potent humanised bispecific antibody, termed ACE910 or emicizumab ( 11 ). As we had intended, emicizumab exerted a potent FVIIIa-mimetic cofactor activity in vitro and in vivo ( 11 – 13 ), and a bleeding preventive effect at around 10–100 µg/ml of plasma emicizumab in the patient part of the phase 1 clinical study ( 14 ).
Figure 1 Schematic illustrations of the interactions of FVIIIa or emicizumab with FIX/FIXa and FX/FXa.
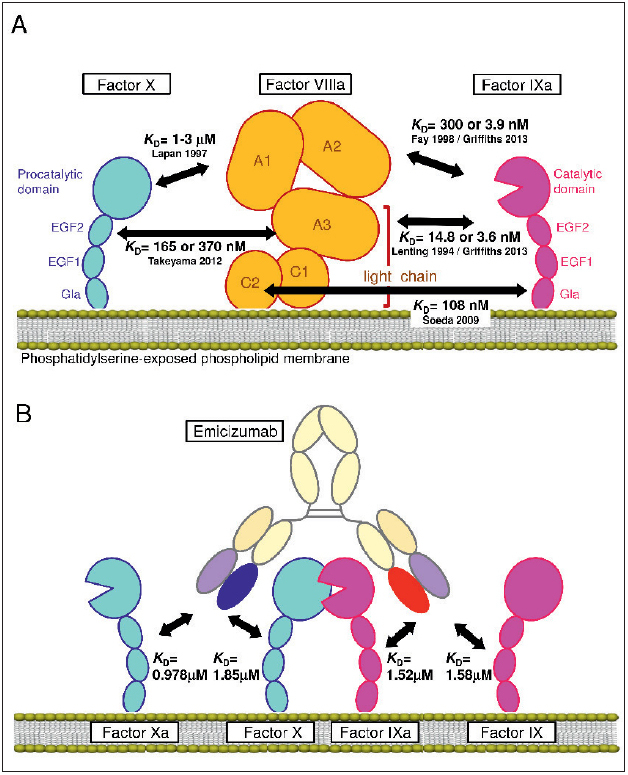
. A) Interactions of FVIIIa with FIXa and FX reported previously (5–10). B) Interactions of emicizumab with FIX/FIXa and FX/FXa. The illustrations do not necessarily indicate precise molecular structures.
As is obvious, however, emicizumab is not the same as FVIII/ FVIIIa and thus has some characteristics different from those of FVIII/FVIIIa. We considered the difference in their binding properties as one of the non-negligible points to be taken into account. In this study, we elucidated emicizumab’s binding affinities ( K D ) to the both antigens by using surface plasmon resonance (SPR) analysis and explored epitopes on the antigens by using immunoblotting analysis. We then used the determined K D values to simulate equilibrium states in plasma in order to predict the ways that emicizumab would interact with the antigens quantitatively, and to discuss how emicizumab and the antigens would behave.
Materials and methods
Antibodies and coagulation factors
Emicizumab (a humanised bispecific IgG 4 antibody recognising FIX/FIXa and FX/FXa) was recombinantly produced from a Chinese hamster ovary cell line ( 15 ). Anti-FIX/FIXa or anti-FX/FXa monospecific two-armed IgG 4 antibodies each having one of the antigen binding fragments of emicizumab (emicizumab Fab) were transiently expressed in HEK293 cells and purified ( 11 ). Except for FVIII used in Suppl. Table 1 (available online at www.thrombosis-online.com ), all coagulation factors used were human plasma-derived (Enzyme Research Laboratories, South Bend, IN, USA).
Table 1.
Antigen-binding affinities of emicizumab .
| Analyte | Arm of emicizumab | ka (/M s) | kd (/s) | KD (μM) |
|---|---|---|---|---|
| Factor IX | Anti-FIX/FIXa | 1.63 × 10 4 | 2.56 × 10– 2 | 1.58 |
| Factor IXa | Anti-FIX/FIXa | 4.14 × 10 4 | 6.14 × 10– 2 | 1.52 |
| Factor X | Anti-FX/FXa | 2.15 × 10 4 | 3.97 × 10– 2 | 1.85 |
| Factor Xa | Anti-FX/FXa | 2.76 × 10 4 | 2.70 × 10– 2 | 0.978 |
The k a k d and K D were analysed with respect to each of two separate experiments and then averaged.
SPR analysis
We analysed the interactions of FIX, FIXa, FX, and FXa with the corresponding variable region of emicizumab by using a Biacore T200 SPR system (GE Healthcare, Uppsala, Sweden). First, we immobilised MabSelect SuRe Ligand (recombinant Protein A; GE Healthcare) onto a CM4 sensor chip (GE Healthcare) that had been pre-activated with NHS and ECD, and pre-deactivated with ethanolamine-HCl using an Amine Coupling Kit (GE Healthcare). To capture the test antibodies on the sensor chip, we injected the anti-FIX/FIXa or anti-FX/FXa monospecific two-armed IgG 4 antibody having either of the emicizumab Fab into flow cell 2, and natalizumab (Biogen, Cambridge, MA, USA) as control humanised IgG 4 antibody into flow cell 1. We next injected each analyte (0 as baseline, 80, 160, 320, 640, or 1280 nM of FIX, FIXa, FX, or FXa), which had been dissolved in running buffer (10 mM HEPES, 150 mM NaCl, 0.05 vol % Surfactant P20, 2.5 mM CaCl 2 [pH 7.4]), into both flow cells on the sensor surface at a flow rate of 30 µl/minute (min) to monitor the association phase for 120 seconds (s) and then the dissociation phase for 30 s with the running buffer. The data were analysed by the 1:1 binding model in the Biacore T200 Evaluation software (version 1.0, GE Healthcare).
Immunoblotting analysis
We expressed recombinant Fc-fusion forms of the first and second epidermal growth factor (EGF)-like domains (EGF1,2), the first EGF-like domain (EGF1), and the second EGF-like domain (EGF2) of FIX and FX (rFIX_EGF1,2–Fc, rFIX_EGF1–Fc, rFIX_EGF2–Fc, rFX_EGF1,2–Fc, rFX_EGF1–Fc, and rFX_EGF2–Fc) in HEK293 cells and purified them with recombinant Protein A. The amino acid sequences of these recombinant proteins consist of the sequences of the respective domain of human FIX or FX, and the hinge and Fc of human immunoglobulin γ1 constant region (► Figure 2A, C ). With rFIX_EGF1,2–Fc and rFIX_EGF2–Fc, we replaced cysteine at position 132 with serine to avoid the possibility of mis-folding by free cysteine. See Suppl. Material (available online at www.thrombosis-online.com ) for the detailed amino acid sequences of the above recombinant proteins. For Coomassie Brilliant Blue (CBB) staining or immunoblotting, we applied 20 pmol or 5 pmol of the above recombinant proteins, as well as 40 pmol or 10 pmol of FIXa and FX, respectively, to gradient acrylamide gels (4 %–20 %) and performed SDS-PAGE in a non-reduced condition. For immunoblotting analyses, we used the anti-FIX/FIXa or anti-FX/FXa monospecific two-armed IgG 4 antibodies having either of the emicizumab Fabs as the test antibody, and detected the antigen–antibody binding by horse radish peroxidase-labelled recombinant Protein L (Actigen, Cambridge, UK) and a peroxidase immunoblotting substrate (Nacalai Tesque, Kyoto, Japan).
Figure 2: Antigen domain recognised by emicizumab and emicizumab’s specificity to the antigens.

. A) FIX/FIXa-related proteins used. B) The electrophoretic pattern of FIX/FIXa-related proteins stained by CBB in a non-reduced condition and the results of immunoblotting with the anti-FIX/FIXa arm of emicizumab. Bands above 100 kDa are considered to be the aggregated multimers. C) FX/FXa-related proteins used. D) The electrophoretic pattern of FX/FXa-related proteins stained by CBB in a non-reduced condition and the results of immunoblotting with the anti-FX/FXa arm of emicizumab. Bands above 100 kDa are considered to be the aggregated multimers. E) The results of ELISA to detect binding of emicizumab to immobilised FVII, FIX, FX, FXII, and Protein C. Data are shown as mean ± SD (n = 3).
ELISA
We coated wells of 96-well immunoplates (Thermo Fisher Scientific, Waltham, MA, USA) with FVII, FIX, FX, FXII, or Protein C (Enzyme Research Laboratories) at 1 µg/ml in phosphate buffered saline. After washing the wells with tris buffered saline containing 1 mM CaCl 2 and 0.05 vol % Tween® 20 (Bio-Rad, Hercules, CA, USA) (TBS-Ca/tween), we blocked the wells with 30 mg/ml bovine serum albumin (BSA) dissolved in TBS-Ca/tween (TBS-Ca/tween/BSA). After the blocking, we applied varied concentrations of emicizumab diluted with TBS-Ca/tween/BSA to the wells for the reaction of the coated test protein-emicizumab interaction. After washing the wells with TBS-Ca/tween, we applied an alkaline phosphatase-labelled mouse human-γ 4 -chain-specific antibody (Southern Biotech, Birmingham, AL, USA) diluted with TBS-Ca/tween/BSA. After washing the wells again with TBS-Ca/tween, we applied phosphatase substrate (Kirkegaard & Perry Laboratories, Gaithersburg, MD, USA) to the wells, and then measured absorbance at 595 nm for quantitating the complex of the coated test protein-emicizumab bound to the alkaline phosphatase-labelled mouse human-γ 4 -chain-specific antibody. The data were collected in triplicate.
Simulation of equilibrium state in plasma containing emicizumab
The fraction of emicizumab’s anti-FIX/FIXa arm bound by FIX ( fb FIX ) was calculated based on the following formula:
where C FIX , C Mab , and K D,F IX represent the total FIX concentration, the total emicizumab concentration, and the K D value to FIX, respectively. The fraction of emicizumab’s anti-FX/FXa arm bound by FX ( fb FX ) was calculated in a similar manner. Concentrations of FIX-emicizumab-FX ternary complex ( C FIX–Emi–FX ), FIX–emicizumab binary complex ( C FIX-Emi ), emicizumab-FX binary complex ( C Emi–FX ), free FIX ( C FIXfree ), and free FX ( C FXfree ) were calculated using the formulae below.
 |
Thrombin generation assay
Thrombin generation (TG) in FVIII-deficient plasma (George King Bio-Medical, Overland Park, KS, USA) was measured by calibrated automated thrombography using a 96-well plate fluorometer (Thermo Fisher Scientific) as described previously ( 12 ). Each well in a 96-well plate was dispensed with 80 µl of FVIII-deficient plasma containing emicizumab, to which was then added 20 µl of an intrinsic triggering solution consisting of 0.16 nM human FXIa (Enzyme Research Laboratories) and 20 µM synthetic phospholipid (10 % phosphatidylserine, 60 % phosphatidyl-choline, and 30 % phosphatidylethanolamine). The synthetic phos-pholipid was prepared as previously described ( 4 ). For calibration, 20 µl of Thrombin Calibrator (Thrombinoscope BV, Maastricht, the Netherlands) was added instead of the triggering solution. To initiate the reaction, 20 µl of FluCa reagent prepared from FluCa kit (Thrombinoscope BV) was dispensed by the instrument as programmed. We analysed the thrombograms and Peak Height by the instrument’s software. Data were collected in triplicate.
Statistical analysis
We presented the averaged values of two sets of measurement for each SPR parameter, and the mean ± SD of three sets of measurement for absorbance in ELISA and a TG parameter.
Results
Antigen-binding affinities of emicizumab
First, we performed SPR analysis in which FIX, FIXa, FX, and FXa were used as analytes to be bound by either anti-FIX/FIXa or anti-FX/FXa monospecific two-armed IgG 4 antibody each having one of the emicizumab Fabs. Consequently, we determined the K D values of emicizumab to FIX, FIXa, FX, and FXa to be 1.58, 1.52, 1.85, and 0.978 µM, respectively, all of which indicated moderate-strength interactions (► Table 1 , ► Figure 1B ). These K D values were much larger than those of typical therapeutic antibodies with antagonistic action ( K D = single digit nM or lower) ( 16 – 20 ). The results also indicated that neither of the emicizumab Fabs clearly discriminated between the activated form and the precursor form of the respective antigens. These observations were aligned with the data of hBS23 (a humanised bispecific antibody recognising FIX/FIXa and FX/FXa; a precursor of emicizumab) reported previously ( 4 ).
Antigens’ domains recognised by emicizumab
Next, we determined by immunoblotting analysis the domain in each antigen that emicizumab recognises. Since hBS23 had recognised the light chains of FIX/FIXa and FX/FXa ( 4 ), we focused on the respective light chains. For FIX/FIXa, we applied plasma-derived FIXa and recombinant Fc-fusion forms of EGF1,2, EGF1, and EGF2 of FIX (rFIX_EGF1,2–Fc, rFIX_EGF1–Fc, and rFIX_EGF2–Fc, respectively; ► Figure 2A ) to immunoblotting analysis with the anti-FIX/FIXa arm of emicizumab. Consequently, we found that FIXa, rFIX_EGF1,2–Fc, and rFIX_EGF1–Fc were detected by the anti-FIX/FIXa arm of emicizumab (► Figure 2B ). We also examined FX/FXa in a similar way, and found that FX and recombinant Fc-fusion forms of EGF1,2 and EGF2 of FX (rFX_EGF1,2–Fc and rFX_EGF2–Fc) were detected by the anti-FX/FXa arm of emicizumab (► Figure 2C, D ). These results indicated that emicizumab recognised the EGF1 of FIX/FIXa with one arm and the EGF2 of FX/FXa with the other arm. An “EGF-like domain” is contained in various proteins including some other coagulation-related proteins. Just to make sure, we examined the binding specificity of emicizumab by ELISA using immunoplates coated with FVII, FIX, FX, FXII, or Protein C. Emicizumab bound to neither FVII, FXII, nor Protein C (► Figure 2E ), underwriting that emicizumab specifically binds to FIX/FIXa and FX/FXa.
Simulated equilibrium states in plasma containing emicizumab
Based on the K D values determined, we simulated equilibrium states in plasma containing varied concentrations of emicizumab. For the simulation, we employed 90 and 135 nM as standard plasma concentrations of FIX and FX, respectively ( 21 ). First, we simulated concentrations of the antigen-bridging (FIX–emicizumab–FX) ternary complex in plasma containing varied concentrations of emicizumab. Plotting concentration of the FIX–emicizumab–FX ternary complex against the concentration of emicizumab formed a bell-shaped curve (► Figure 3A ). At the peak of the bell-shaped curve, the predicted concentration of emicizumab and the FIX–emicizumab–FX ternary complex would be 1820 nM (265 µg/ml) and as low as 1.72 nM, respectively. When the plasma concentration of FIX or FX in the simulation was changed to 200 %, 50 % or 20 % of the standard, the predicted emicizumab concentrations at the peak of FIX–emicizumab–FX ternary complex changed little (Suppl. Figures 1A and Figure 2A , available online at www.thrombosis-online.com ).
Figure 3: KD -based simulation of an equilibrium state in the presence of the standard plasma concentrations of FIX and FX .

. Concentrations of FIX–emicizumab–FX ternary complex (A), and the percent ratio of monomer, binary complex, and ternary complex of FIX to total FIX (B) or those of FX to total FX (C), which were simulated on the basis of the K D values at varied concentrations of emicizumab.
Next, we examined emicizumab concentration-dependency by a plasma TG assay in order to elucidate whether plasma concentrations of FIX–emicizumab–FX ternary complex would correlate with thrombin burst in FVIII-deficient plasma. Without phos-phatidylserine-exposed phospholipid membrane, emicizumab was unable to accelerate FIXa-catalysed FX activation (Suppl. Figure 3 , available online at www.thrombosis-online.com ). Thus, circulating FIX–emicizumab–FX ternary complex would not directly promote coagulation, but we assumed that the amount of circulating FIX–emicizumab–FX ternary complex should correlate with the amount of FIX/FIXa–emicizumab–FX/FXa ternary complex on the phosphatidylserine-exposed phospholipid membrane at a haemostatic site when bleeding occurred. As we anticipated, a TG parameter, Peak Height, also showed a bell-shaped dependency when plotted against emicizumab concentration (► Figure 4 ), similar to that predicted in the above simulation (► Figure 3A ).
Figure 4: Emicizumab concentration-dependency of a parameter of an intrinsic pathway-triggered TG assay in FVIII-deficient plasma.
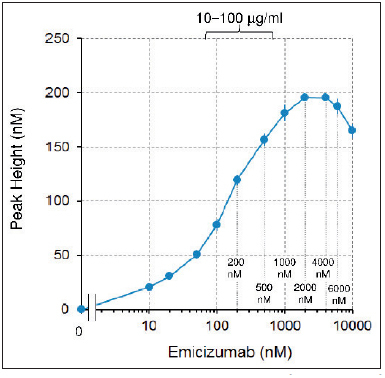
. Peak Height analysed from the intrinsic pathway-triggered TG assay at varied concentrations of emicizumab in FVIII-deficient plasma. Data are shown as mean ± SD (n = 3).
We also simulated concentrations of FIX monomer and FX monomer in plasma containing varied concentrations of emicizumab. Since emicizumab’s binding epitopes on FIX/FIXa and FX/ FXa existed in the EGF-like domains, there may arise a concern that binding of emicizumab may restrict the availability of FIX/ FIXa and FX/FXa for the other reactions in the coagulation cascade. The simulation predicted that, at plasma emicizumab concentrations shown in a previous study to be clinically effective (around 10.0–100 µg/ml or 68.7–687 nM) ( 14 ), the majority of FIX and FX would exist as monomers (► Figure 3B, C ). Even when the plasma concentration of FIX or FX in the simulation changed to 200 %, 50 % or 20 % of the standard, the majority of FIX and FX would exist as monomers at the above range of emicizumab (Suppl. Figures 1B – D and 2B–D, available online at www.thrombo sis-online.com). These results suggested that the influence of emicizumab on the other reactions in the coagulation cascade would be small, if any, in clinical settings.
Discussion
In this study, we found that emicizumab binds FIX/FIXa and FX/ FXa with micromolar affinities, which indicates moderate-strength interactions. These K D values were much larger than those of typical antagonistic therapeutic antibodies ( K D = single digit nM or lower) ( 16 – 20 ). In the case of antagonistic antibodies, a higher antigen-binding affinity is obviously desirable in order not to allow the antigen to escape from the antibody. In the case of agonistic antibodies, however, repeated attachment and detachment of the antibody may contribute to a higher activity by the rapid turnover of action or signalling. A previous report on antierythropoietin receptor agonistic antibodies presented an example where antibody variants with faster off-rate (larger k d or k off ) were more effective than ones with slower off-rate ( 22 ). As for emicizumab, it should be also theoretically better for the generated FXa to be released quickly from the enzyme–emicizumab–substrate ternary complex and supplied to the downstream of the coagulation cascade. Actually, the binding affinity of FVIIIa to FX is moderate (► Figure 1A ), and emicizumab showed faster off-rates ( k d = several 10 –2 /s) than typical antagonistic therapeutic antibodies –the k d ( k off ) values of tocilizumab, bevacizumab, omalizumab, adalimumab, infliximab, and palivizumab range in order of magnitude from 10 –3 /s to 10 –5 /s ( 16 – 20 ).
Antibodies to a soluble antigen sometimes cause antibody-dependent antigen accumulation, because the plasma half-life of an antigen–antibody complex is often much longer than that of the antigen alone. A previous report showed that an anti-FIX antibody accumulated FIX–antibody complex in vivo to a level 10 times that of the baseline concentration of FIX ( 23 ). We recognise that such an accumulation should be a possible thrombophilic concern, because our bispecific antibody is not a neutralising one. In the clinical study of emicizumab, plasma concentrations of FIX and FX, which included both monomeric and complexed forms, did not increase, although molecule-based concentration of plasma emicizumab reached several times the baseline levels of FIX and FX in the cohort receiving the highest doses of emicizumab ( 14 ). We assume that the micromolar affinities of emicizumab contributed to avoiding antibody-dependent antigen accumulation by allowing the antigens to escape from emicizumab.
We also found that emicizumab recognised the EGF1 of FIX/ FIXa and the EGF2 of FX/FXa. Although other coagulation-related factors, including FVII, FIX, FX, FXII, and Protein C, also have EGF-like domains, we showed that emicizumab bound neither of these factors by ELISA while emicizumab bound to FIX and FX. In the ELISA, the signals for FIX were unexpectedly lower than those for FX (► Figure 2E ), although FIX and FX have a similar molecular weight and similar binding affinities to emicizumab. We have not revealed the reason for the discrepancy yet, but assume that it may attribute to a difference of the immobilised state of the respective antigens on immunoplates and/or a difference of the binding epitope recognised by emicizumab.
Each of EGF1 in FIX/FIXa and FX/FXa has an ability to bind Ca 2+ , which is necessary for proper orientation of Gla and EGF modules in each molecule ( 24 ). Moreover, the binding of Ca 2+ to the EGF1 of FIX/FIXa promotes its binding to the light chain of FVIIIa and also the enzyme activity of FIXa ( 25 ). Thus, the effect of Ca 2+ on emicizumab’s binding to the antigens should be one of the curious points of view. In the SPR analyses and the ELISA assay shown in ► Table 1 and ► Figure 2E , respectively, we used Ca 2+ -containing buffer for the binding reaction and also the dissociation phase or the washing procedure as described in Materials and methods . Although we do not show the detailed data here, we also performed ELISA using Ca 2+ -free buffer and found that emicizumab similarly bound to FIX and FX even in the Ca 2+ -free solution. Post-translational modifications of the EGF domains of FIX/FIXa and FX/FXa, including glycosylation and β-hydroxy-as-partate, have been also reported so far ( 26 , 27 ). Their involvement in emicizumab’s binding to the antigens should be one of the other curious points of view. Although we have no answer to it at the moment, we anticipate that further structural analyses of emicizumab’s binding to the antigens will give a definite answer in the future.
The K D -based simulation predicted that the concentration of the antigen-bridging FIX–emicizumab–FX ternary complex formed would follow a bell-shaped curve dependent on the concentration of emicizumab. The reason for such a bell-shaped concentration-dependency is that, in the presence of too high a concentration of emicizumab, formation of binary complexes (FIX–emicizumab and emicizumab–FX) would be dominant and would impede the formation of FIX–emicizumab–FX ternary complex.
This bell-shaped concentration-dependency also means that the plasma concentration of FIX–emicizumab–FX ternary complex would not be necessarily proportional to the plasma concentration of emicizumab (Suppl. Figure 4 , available online at www. thrombosis-online.com). The concentration of FIXa–FVIIIa–FX ternary complex formed should be nearly proportional to plasma FVIII concentration at the therapeutic range, since the standard plasma concentration of FVIII is far lower than that of FIX or FX (0.3 nM vs 90 or 135 nM) ( 21 ). These considerations suggest that equivalent FVIII cofactor activity of emicizumab would relate to, but not be necessarily proportional to, plasma concentration of emicizumab.
The plasma TG assay parameter, Peak Height, indicating the intensity of thrombin burst also presented a bell-shaped concentration-dependency (► Figure 4 ) similar to that observed with the simulated FIX–emicizumab–FX ternary complex formation (► Figure 3A ). We used the intrinsic pathway trigger for the TG assay in this study, but the supplementary appendix of a previous paper ( 14 ) presented such a bell-shaped concentration-dependency also in the TG assay using the extrinsic pathway trigger. Therefore, it should not be specific for the pathway to trigger the coagulation reactions. Because the intensity of thrombin burst is a key process that determines the extent of a haemostatic plug ( 28 ), we hypothesised that the concentrations of circulating FIX–emicizumab–FX ternary complex would possibly correlate with the amount of FIXa–emicizumab–FX (enzyme–cofactor–substrate) ternary complex formed at a haemostatic site and with an in vivo haemostatic activity. Apart from it, we had previously hypothesised from the animal study that the factor for conversion of emicizumab concentration (µg/ml) to equivalent FVIII haemostatic activity (IU/dl or %) would be around 0.3 at an emicizumab concentration of around 36 to 61 µg/ml (247 to 419 nM) ( 12 , 14 ). We wondered if these two independent hypotheses were jointly concordant with the enzymatic kinetics.
Based on the in vivo conversion factor, 48.5 µg/ml (average of 36 and 61 µg/ml) or 333 nM of emicizumab would exert 14.6 % (48.5 µg/ml multiplied by 0.3 % per µg/ml) of FVIII-equivalent haemostatic activity. At this state, the K D -based simulation predicts that 0.886 nM of FIX–emicizumab–FX ternary complex would exist in circulating plasma. For FVIII to exert the same cofactor activity, 0.0438 nM (0.3 nM [standard plasma level of FVIII] multiplied by 14.6 %) has to exist in circulating plasma. Thus, 20-fold (0.886 nM divided by 0.0438 nM) molecular-based concentration of FIX–emicizumab–FX ternary complex should be required than that of FVIII to exert 14.6 % of FVIII:C or the equivalent. From a viewpoint of enzymatic kinetics, the turnover rate ( k cat ) of enzyme (FIXa)–substrate (FX) ternary complex was 2.88 /min or 126 /min in the presence of emicizumab or FVIIIa as cofactor, respectively (Suppl. Table 1 , available online at www. thrombosis-online.com). Thus, the speed of FXa generation from the FIXa–emicizumab–FX ternary complex is 1/44 (2.88 /min divided by 126 /min) of that from the FIXa–FVIIIa–FX ternary complex. As discussed later, we have not found any solution to measure the amount of enzyme (FIXa)–cofactor (emicizumab or FVIIIa)–substrate (FX) ternary complex on phosphatidylserine-exposed phospholipid membrane. Therefore, there remains a jump in logic, but we roughly assume that plasma emicizumab at the clinical doses would allow the formation of a several tens fold amount of enzyme–cofactor–substrate ternary complex on phos-phatidylserine-exposed phospholipid membrane than would plasma FVIII, compensating for the several tens fold lower speed of FXa generation from FIXa–emicizumab–FX ternary complex than that from FIXa–FVIIIa–FX ternary complex.
The K D -based simulation also predicted that the majority of FIX and FX would exist as monomers at the plasma concentrations of emicizumab that were effective in the phase 1 clinical study (around 10.0–100 µg/ml or 68.7–687 nM) (► Figure 3B, C ). Even when plasma concentration of FIX or FX in the simulation changed to 200 %, 50 % or 20 % of the standard concentrations, similar simulation results were also obtained (Suppl. Figure 1B – D and 2B–D, available online at www.thrombosis-online.com ). These results suggest that the influence of emicizumab on the other coagulation reactions would be small, if any. This will be one of the preferred features of emicizumab.
What is known about this topic?
Emicizumab, a humanised bispecific monoclonal IgG 4 antibody designed to bridge factors (F) IXa and X, exerted a FVIIIa-mimetic cofactor activity regardless of the presence of FVIII inhibitors.
Emicizumab had a high subcutaneous bioavailability in non-human primate and a long half-life (∼ 1 month) in human.
Emicizumab exerted a potent haemostatic activity in non-human primate models of acquired haemophilia A and in severe haemophilia A patients in a clinical study.
What does this paper add?
In this paper, we started with the analyses of emicizumab’s binding properties to FIX/FIXa and FX/FXa: moderate affinities at their EGF-like domains.
The K D -based simulation predicted that amount of antigen-bridging FIX-emicizumab-FX ternary complex in plasma would correlate with the cofactor activity.
The simulation also predicted that the majority of plasma FIX and FX would exist as monomer at clinically effective doses.
The K D values that we determined are quite large. It caused a technical issue on obtaining specific binding signals in the SPR. To overcome it, we immobolised the monospecific two-armed antibody having one of emicizumab Fabs instead of emicizumab itself for increasing a signal intensity, and also refined the detailed conditions so as to minimise non-specific signals. Despite these, we cannot deny that some discrepancy between our determined K D values and the “true” ones may possibly exist. If emicizumab’s K D values to FIX and FX both changed, for example, to one-third, half, double or triple, the predicted concentration of FIX–emicizumab–FX ternary complex would change to 0.585, 0.873, 3.33 or 4.85 nM from 1.72 nM, respectively, and the predicted concentration of emicizumab at the peak of the bell-shaped curve would change to 681, 966, 3530 or 5240 nM from 1820 nM, respectively. As for the TG parameter, however, the concentration of emicizumab at the peak of the bell-shaped curve was 2000 nM among the employed emicizumab concentration points (0, …, 500, 1000, 2000, 4000, 6000 and 10000 nM, ► Figure 4 ). It exactly corresponded to our determined K D -based simulation, moderately suggesting that our determined K D values should be fairly accurate and the discrepancy from the “true” K D values should be within a few folds, if any. Difficulty in measuring the equilibrium state of FIX/FIXa, FX/ FXa, emicizumab and their complexes on phosphatidylserine-exposed phospholipid membrane is another technical issue. It should be affected by various factors including ratio of FIXa to FIX, condensing of FIX/FIXa and FX/FXa to the membrane via their Gla domains, an avidity effect of emicizumab’s binding both to membrane-bound FIX/FIXa and FX/FXa, and a state of thrombus, all of which would change time to time. As we were unable to reach such a high level of analyses in this study, the above two technical issues have to be carefully considered. We emphasise that there still remains a certain possibility to come to different consequences at settings in patients.
In conclusion, we found that emicizumab binds FIX/FIXa and FX/FXa at their respective EGF-like domains with micromolar affinities. The simulation using the K D values provided several insights. For example, the plasma concentration of the antigen-bridging FIX–emicizumab–FX ternary complex would form a bell-shaped relationship with emicizumab’s concentration as a plasma TG parameter forms, suggesting that the ternary complex formed in circulating plasma would correlate with emicizumab’s cofactor activity. Only a small part of FIX, FX, and emicizumab in circulating plasma would form the ternary complex, and the majority of plasma FIX and FX would exist as monomers at the effective plasma concentrations of emicizumab, suggesting that undesired interference of emicizumab with the other reactions in the coagulation cascade would be avoided.
Acknowledgements
We thank our colleagues at Chugai Pharmaceutical Co., Ltd. (Chugai): S. Suzuki, Y. Higuchi, A. Ueyama, T. Toyoda and M. Saito for their assistance with the in vitro experiments; and T. Wakabayashi, E. Tanaka, Y. Kikuchi, A. Sakamoto, M. Wada, and M. Goto for preparing the test items.
Funding Statement
Financial Support: This study was supported by Chugai Pharmaceutical Co., Ltd.
Footnotes
Conflicts of interest T. K., K. E., Y. K., H. T., K. H., T. T., S. I., T. S., A. M., and T. I. are employees of Chugai and the former 5 persons own Chugai stock. K.N. and M. S. have received research support from Chugai, are engaged in clinical studies sponsored by Chugai and F. Hoffmann-La Roche, and have received consulting honoraria from these companies. T.K. T.S., A. M., T.I, T. H., K. N., M. S., and K. H. are inventors of the patents relating to anti-FIXa/FX bispecific antibodies.
Online Supplementary Material (PDF)
Online Zusatzmaterial (PDF)
References
- 1.Srivastava A, Brewer AK, Mauser-Bunschoten EP et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19:e1–e47.. doi: 10.1111/j.1365-2516.2012.02909.x. [DOI] [PubMed] [Google Scholar]
- 2.Peyvandi F, Garagiola I, Young G. The past and future of haemophilia: diagnosis, treatments, and its complications. Lancet. 2016;388:187–197. doi: 10.1016/S0140-6736(15)01123-X. [DOI] [PubMed] [Google Scholar]
- 3.Leissinger CA. Advances in the clinical management of inhibitors in hemophilia A and B. Semin Hematol. 2016;53:20–27. doi: 10.1053/j.seminhematol.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 4.Kitazawa T, Igawa T, Sampei Z et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat Med. 2012;18:1570–1574. doi: 10.1038/nm.2942. [DOI] [PubMed] [Google Scholar]
- 5.Lenting PJ, Donath MJ, van Mourik JA et al. Identification of a binding site for blood coagulation factor IXa on the light chain of human factor VIII. J Biol Chem. 1994;269:7150–7155. [PubMed] [Google Scholar]
- 6.Lapan KA, Fay PJ. Localization of a factor X interactive site in the A1 subunit of factor VIIIa. J Biol Chem. 1997;272:2082–2088. doi: 10.1074/jbc.272.4.2082. [DOI] [PubMed] [Google Scholar]
- 7.Fay PJ, Koshibu K. The A2 subunit of factor VIIIa modulates the active site of factor IXa. J Biol Chem. 1998;273:19049–19054. doi: 10.1074/jbc.273.30.19049. [DOI] [PubMed] [Google Scholar]
- 8.Soeda T, Nogami K, Nishiya K et al. The factor VIIIa C2 domain (residues 2228–2240) interacts with the factor IXa Gla domain in the factor Xase complex. J Biol Chem. 2009;284:3379–3388. doi: 10.1074/jbc.M804955200. [DOI] [PubMed] [Google Scholar]
- 9.Takeyama M, Wakabayashi H, Fay PJ. Factor VIII light chain contains a binding site for factor X that contributes to the catalytic efficiency of factor Xase. Biochemistry. 2012;51:820–828. doi: 10.1021/bi201731p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Griffiths AE, Rydkin I, Fay PJ. Factor VIIIa A2 subunit shows a high affinity interaction with factor IXa: contribution of A2 subunit residues 707–714 to the interaction with factor IXa. J Biol Chem. 2013;288:15057–15064. doi: 10.1074/jbc.M113.456467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sampei Z, Igawa T, Soeda T et al. Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activity. PLoS One. 2013;8:e57479.. doi: 10.1371/journal.pone.0057479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muto A, Yoshihashi K, Takeda M et al. Anti-factor IXa/X bispecific antibody (ACE910): hemostatic potency against ongoing bleeds in a hemophilia A model and the possibility of routine supplementation. J Thromb Haemost. 2014;12:206–213. [PubMed] [Google Scholar]
- 13.Muto A, Yoshihashi K, Takeda M et al. Anti-factor IXa/X bispecific antibody ACE910 prevents joint bleeds in a long-term primate model of acquired hemophilia A. Blood. 2014;124:3165–3171. doi: 10.1182/blood-2014-07-585737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shima M, Hanabusa H, Taki M et al. Factor VIII-mimetic function of humanized bispecific antibody in hemophilia A. N Engl J Med. 2016;374:2044–2053. doi: 10.1056/NEJMoa1511769. [DOI] [PubMed] [Google Scholar]
- 15.Uchida N, Sambe T, Yoneyama K et al. A first-in-human phase 1 study of ACE910, a novel factor VIII-mimetic bispecific antibody, in healthy subjects. Blood. 2016;127:1633–1641. doi: 10.1182/blood-2015-06-650226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Igawa T, Ishii S, Tachibana T et al. Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nat Biotechnol. 2010;28:1203–1207. doi: 10.1038/nbt.1691. [DOI] [PubMed] [Google Scholar]
- 17.Presta LG, Chen H, O’Connor SJ et al. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997;57:4593–4599. [PubMed] [Google Scholar]
- 18.Hamilton RG, Saini SS, MacGlashan D. Surface plasmon resonance analysis of free IgE in allergic patients receiving omalizumab (Xolair) J Immunol Methods. 2012;383:54–59. doi: 10.1016/j.jim.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 19.Kaymakcalan Z, Sakorafas P, Bose S et al. Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin Immunol. 2009;131:308–316. doi: 10.1016/j.clim.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 20.Wu H, Pfarr DS, Johnson S et al. Development of motavizumab, an ultra-potent antibody for the prevention of respiratory syncytial virus infection in the upper and lower respiratory tract. J Mol Biol. 2007;368:652–665. doi: 10.1016/j.jmb.2007.02.024. [DOI] [PubMed] [Google Scholar]
- 21.Coagulation sequence and structure database (CoagBase)Available athttp://www.isth.org/?page=CoagBase.Accessed January 15, 2017.
- 22.Lacy SE, DeVries PJ, Xie N et al. The potency of erythropoietin-mimic antibodies correlates inversely with affinity. J Immunol. 2008;181:1282–1287. doi: 10.4049/jimmunol.181.2.1282. [DOI] [PubMed] [Google Scholar]
- 23.Davis CB, Tobia LP, Kwok DC et al. Accumulation of antibody-target complexes and the pharmacodynamics of clotting after single intravenous administration of humanized anti-factor IX monoclonal antibody to rats. Drug Delivery. 1999;6:171–179. [Google Scholar]
- 24.Sunnerhagen M, Drakenberg T, Forsen S et al. Effect of Ca 2+ on the structure of vitamin K–dependent coagulation factors . Haemostasis. 1996;26 01:45–53. doi: 10.1159/000217240. [DOI] [PubMed] [Google Scholar]
- 25.Lenting PJ, Christophe OD, Maat H et al. Ca 2+ binding to the first epidermal growth factor-like domain of human blood coagulation factor IX promotes enzyme activity and factor VIII light chain binding . J Biol Chem. 1996;271:25332–25337. doi: 10.1074/jbc.271.41.25332. [DOI] [PubMed] [Google Scholar]
- 26.Makino Y, Omichi K, Kuraya N et al. Structural analysis of N-linked sugar chains of human blood clotting factor IX. J Biochem. 2000;128:175–180. doi: 10.1093/oxfordjournals.jbchem.a022738. [DOI] [PubMed] [Google Scholar]
- 27.Chevreux G, Tilly N, Faid V et al. Mass spectrometry based analysis of human plasma-derived factor X revealed novel post-translational modifications. Protein Sci. 2015;24:1640–1648. doi: 10.1002/pro.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salvagno GL, Berntorp E. Thrombin generation testing for monitoring hemophilia treatment: a clinical perspective. Semin Thromb Hemost. 2010;36:780–790. doi: 10.1055/s-0030-1265295. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Online Supplementary Material (PDF)
Online Zusatzmaterial (PDF)