

Summary
While prasugrel is indicated for the treatment of myocardial infarction, its effects in the most severely affected patients requiring intensive care is unknown, so that we measured the antiplatelet effects and sparse pharmacokinetics of prasugrel in critically ill patients. Twenty-three patients admitted to medical intensive care units, who were treated with 10 mg prasugrel once daily, were included in this prospective trial. Critically ill patients responded poorly to daily prasugrel treatment: adenosine diphosphate (ADP)-induced aggregation in whole blood classified 65 % (95 % confidence intervals (CI) 43–84 %) of patients as having high on treatment platelet reactivity, platelet function under high shear rates even 74 % (95 %CI 52–90 %). There was only limited additional inhibition provided 2 hours after the next dose of prasugrel. In contrast, insufficient inhibition of the target was only seen in 26 % (95 %CI 10–48 %) of patients as measured by the vasodilator-stimulated phosphoprotein phosphorylation (VASP-P) assay. Low effective plasma levels of prasugrel active metabolite were measured at trough [0.5 (quartiles 0.5–1.1) ng/ml at baseline], and 2 hours after intake [5.7 (3.8–9.8) ng/ml], but showed coefficients of variation of ~70 %. In sum, inhibition of platelet aggregation by prasugrel is not uniform but highly variable in critically ill patients, similar to clopidogrel in a general population. The pharmacokinetic measurements indicate that poor absorption/metabolism of prasugrel may partly contribute while inflammation induced heightened intrinsic platelet reactivity may also play a role.
Supplementary Material to this article is available online at www.thrombosis-online.com .
Note: This work was performed at the Medical University of Vienna, Austria.
Keywords: Antiplatelet drugs, intensive care unit, pharmacodynamics, pharmacokinetics, platelet function
Introduction
Critical illness is defined as any clinical condition that requires vital organ support in order to avoid imminent death ( 1 ). While some patients recover quickly, a substantial number of patients develop multiple organ failure with a mortality of up to 50 % ( 1 , 2 ). Frequently, systemic inflammatory responses due to infectious or non-infectious stimuli are either the cause for or complication of intensive care treatment ( 3 ).
Prasugrel is a potent irreversible inhibitor of the P2Y12 ADP-receptor on platelets, which requires metabolic activation ( 4 , 5 ). Treatment is usually initiated with a 60 mg loading dose to achieve quick and sufficient platelet inhibition, which is followed by a daily maintenance dose of 10 mg ( 5 , 6 ). Insufficient platelet inhibition despite antiplatelet therapy is termed high “on-treatment” platelet reactivity (HTPR) ( 4 ). Although prasugrel is thought to be more potent than clopidogrel, studies reported HTPR rates of up to 25 % as measured by the vasodilator-stimulated phosphoprotein phosphorylation (VASP-P) assays ( 4 , 7 ).
Only limited data is available on the antiplatelet effects of prasugrel in critically ill patients. Two studies reported high HTPR rates for patients with acute myocardial infarction, who underwent successful cardiopulmonal resuscitation and therapeutic hypothermia after cardiac arrest, in spite of receiving a 60 mg loading dose of prasugrel ( 8 , 9 ). However, the number of patients with HTPR decreased over time and after 72 hours (h) all patients were found to respond, which may indicate slow and prolonged absorption of prasugrel in these patients ( 9 ). Of note, platelet reactivity in these two studies was assessed by a single assay: the vasodilator stimulated phosphoprotein phosphorylation assay (VASP-P). Noteworthy, HTPR rates vary significantly between various available platelet function assays ( 7 ).
No data is available on the absorption, drug concentrations or metabolisation of prasugrel in patients requiring intensive care treatment. Yet, the pharmacological properties of drugs are significantly altered in critically ill patients due to altered drug absorption, organ dysfunction, altered cytochrome and P-glycoprotein activity, increased volume of distribution and, drug interactions ( 10 ). This may be of importance for treatment with prasugrel, as lower doses were associated with higher HTPR rates in patients with coronary artery disease ( 11 ), opiates such as morphine decreased the maximum concentration of the active metabolite by approximately 30 % even in healthy volunteers receiving 60 mg prasugrel ( 12 ), and grapefruit juice, an inhibitor of cytochrome enzyme activity, reduced the formation of active metabolite by 50 % in healthy volunteers ( 13 ).
Thus, the aim of this trial was to investigate the anti-platelet effects of prasugrel, by means of ADP-induced whole blood aggregometry, VASP-P assay and platelet function under high shear rates, and sparse pharmacokinetics in critically ill patients with established prasugrel therapy.
Materials and methods
The Ethics Committee of the Medical University of Vienna and the National Competent authority approved the study, which complied with the principles set forth in the Good Clinical Practice guideline and the Declaration of Helsinki. The trial was registered in publicly available databases (EudraCT-nr. 2012–002226–76 and https://clinicaltrials.gov/ct2/show/NCT02285751 ).
All conscious patients gave their informed consent before inclusion in the trial. However, because not all critically ill patients were able to give their informed consent before inclusion in the study, the Ethics Committee waived informed consent in these patients.
Study design
Patients >18 years of age, admitted to an intensive care unit (ICU) with prior prasugrel treatment (10 mg prasugrel tablets, Daiichi Sankyo, Tokyo, Japan) were included in the trial. Exclusion criteria included allergies or hypersensitivities to the trial drugs, active bleeding, known coagulation disorders or intake of other antiplatelet drugs except for aspirin.
Blood samples were drawn before patients received the daily dose of 10 mg prasugrel, orally or via a nasogastric tube (crushed and/or dissolved in 0.9 % sodium chloride solution), 2 h and 24 h thereafter.
Platelet function assays
Whole blood aggregation (primary endpoint) was determined using the Multiple Electrode Aggregometry (MEA) on the Multiplate Analyzer (Dynabyte Medical) as previously described (in detail: see Suppl. Material, available online at www.thrombosis-online.com ). To define HTPR we chose the recommended cut-off of >46 U (arbitrary units) for our study ( 14 ). As an external control group, we compared our data to data from stable patients with acute myocardial infarction treated with prasugrel, which were included in a trial run at the same time by us. The vasodilator-stimulated phosphoprotein phosphorylation (VASP-P) assay was measured by an enzyme linked immune assay (see also Suppl. Material, available online at www.thrombosis-online.com ) ( 12 ). Platelet reactivity is expressed as the platelet reactivity index (PRI). A PRI cut-off of 42 % was chosen for HTPR because it predicted ischemic events most sensitively ( 14 ). The collagen ADP closure time (CADP-CT) was measured with the platelet function analyzer (Dade Behring, Marburg, Germany) under high shear rates (see Suppl. Material, available online at www.thrombosis-online.com ) ( 12 , 14 ). The cut-off was defined as 105 seconds based on previous trials ( 14 ).
Pharmacokinetics
Blood samples were drawn into EDTA-anticoagulated blood tubes (Vacuette, Greiner Bio-one, Kremsmünster, Austria) and plasma was harvested after centrifugation at 2000g for 10 minutes (min) at 4 °C, and aliquots (500 µl) stored at –80 °C. Plasma concentrations of prasugrel active metabolite were assessed as described previously ( 12 ) (see Suppl. Material, available online at www.thrombosis-online.com ).
Disease scores
The Sequential Organ Failure Assessment (SOFA) score ( 15 ) and the simplified acute physiology score (SAPS) III were calculated on trial day 1 ( 16 ).
Statistical analysis
Initially we planned to include a minimum of 16 patients to identify five patients with HTPR according to aggregometry, which then could be switched to ticagrelor treatment. We based our sample size calculation on a previous trial reporting that 30 % of patients taking 5 mg prasugrel responded poorly ( 11 ). Although we identified many non-responders, we were not able to switch five patients to ticagrelor, because physicians were reluctant to do so. Thus, we terminated recruitment early into the trial.
Baseline characteristics, demographics, results of platelet function tests and pharmacokinetics are presented by descriptive statistics (medians ± quartiles, unless otherwise stated). Spearman correlations were calculated. Commercially available statistical software was used (IBM, SPSS 22, Microsoft Excel 2010).
Results
Twenty-three (19 male, 4 female) critically ill patients participated in this trial. Baseline data and demographics are presented in Table 1 . Twenty patients underwent successful cardiopulmonary resuscitation, of which 19 patients were treated with therapeutic hypothermia, and three patients suffered from cardiogenic shock after acute myocardial infarction. Patients received the prasugrel loading dose a median 4.5 ( 3 – 13 ) days before inclusion in the trial. Three patients died in the course of the ICU treatment (28-day mortality: 3 of 23, 13 %).
Table 1.
Baseline data and Demographics of 23 critically ill patients are presented . Medians ± IQR, or absolute numbers are presented.
| Parameter | Medians (IQR) |
|---|---|
| Gender m (f) | 19 (4) |
| Age [years] | 52 (47–57) |
| Height [cm] | 180 (172–185) |
| Weight [kg] | 85 (80–100) |
| SAPS 3 score | 63.5 (50–71) |
| SOFA Score | 6.5 (4–10) |
| C-reactive protein [mg/dl] | 12.8 (8.9–16.4) |
| Platelet count [*10 9 /l] | 245 (124–288) |
| Haemoglobin [g/dl] | 10.8 (9.3–11.9) |
| Catecholamine treatment (%) | 8/23 (35%) |
| Mechanical Ventilation (%) | 16/23 (70%) |
Platelet function
Whole blood aggregometry showed that 15 of 23 patients (65 %, 95 % confidence intervals (CI) 43–84 %)) responded poorly to daily prasugrel treatment (► Figure 1 ). ADP-induced platelet aggregation was 65 AU (interquartile range [IQR] 33–70) at baseline, and decreased insignificantly to 55 AU (IQR 32–66) 2h after intake of 10 mg prasugrel and 56 AU (IQR 32–66) after 24 h. ADP-induced platelet aggregometry was significantly higher in critically ill patients compared to stable patients with acute myocardial infarction, who were included a median 4 (IQR 3–5) days after loading dose (p<0.001). MEA results correlated well with platelet function under high shear rates (r=0.67, p=0.001), platelet counts (r=0.66, p=0.001), D-dimer (r=0.71, p=0.001) and notably with the time elapsed after the loading dose (r=0.70, p<0.001, ► Figure 1 ) in critically ill coronary artery disease patients.
Figure 1: Platelet function tests.
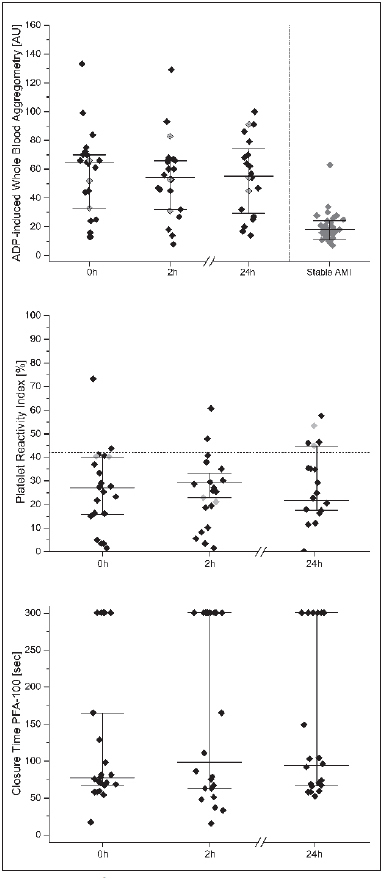
. ADP-induced whole blood aggregometry (upper panel), platelet reactivity index (middle panel), and closure times of platelet function under high shear rates (PFA-100, ADP/collagen coated cartridges) (lower panel) at baseline, 2 and 24 h after intake of 10 mg prasugrel. Presented are individual values and medians ± quartiles (n=23). For whole blood aggregometry data from stable patients with acute myocardial infarction (AMI) are presented (right column, n=35, unpublished data).
According to the VASP assay six patients (26 %) had a PRI>42 % (► Figure 1 ). The median PRI was 25 % (IQR 16–36 %). Intake of 10 mg prasugrel did not significantly reduce the PRI after 2 h. The overall coefficient of variation was 53 %. A high PRI correlated with C-reactive protein (r=0.53, p=0.011), and with a low haemoglobin level (r=-0.62, p=0.002).
Seventeen patients (74 %) had a CADP-CT<106 seconds and therefore HTPR. A short CADP-CT correlated with high fibrinogen (r=-0.49, p=0.02), D-dimer (r=-0.58, p=0.01) and time after the loading dose (► Figure 3 , r=-0.55, p=0.01). Noteworthy, the time after the loading dose, which also reflects the time spent in the ICU, also correlated with platelet counts (r=0.47, p=0.02).
Figure 3: Correlations of ADP-induced whole blood aggregometry and time after loading dose (in days) (left upper panel); Prasugrel active metabolite and time after loading dose (right upper panel); Platelet reactivity index and haemoglobin level (left lower panel); Prasugrel active metabolite and C-reactive protein levels (right lower panel).
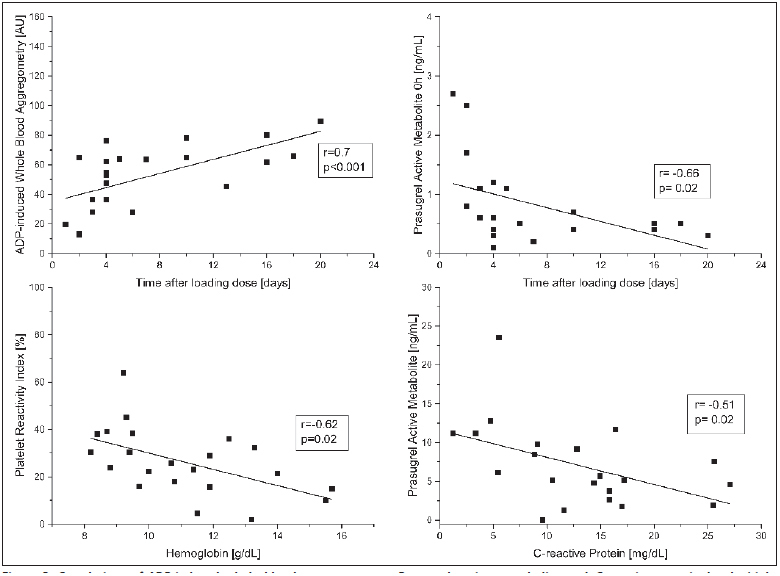
. Presented are individual values and results of non-parametric Spearman correlations.
We performed the following exploratory post-hoc subgroup analyses, for which the trial was not powered:
Four patients received unfractionated heparin, whereas 19 patients were treated with low-molecular-weight heparin. Although not significant, there was a trend to higher platelet reactivity with regards to the PRI%, the CADP-CT and ADP-induced whole blood aggregometry in patients treated with unfractionated heparin (Suppl. ► Table 1 , available online at www.thrombosis-online.com ).
Patients treated with high doses of catecholamines (>0.15mcg/ kg/min, n=4) had lower ADP-induced platelet aggregation compared to patients who received low doses of catecholamines (<0.15 µg/kg/min, n=4) or patients who did not receive such treatment (Suppl. Table 2, available online at www.thrombosis-online.com ). These results may be explained by the lower platelet counts in these patients. The PRI% remained similar between groups (Suppl. Table 2, available online at www.thrombosis-online.com ).
Opiate treatment was associated with lower plasma concentrations of the prasugrel active metabolite, but platelet reactivity remained largely unaffected, except for a numerically higher PRI% in the opiate group (Suppl. Table 3, available online at www.thrombosis-online.com ).
Pharmacokinetics
Plasma concentrations of the prasugrel active metabolite were 0.5 (IQR 0.5–1.1) ng/ml at baseline before intake of the next daily dose of 10 mg prasugrel, 5.7 (IQR 3.8–9.8) ng/ml 2 h after intake and, 0.5 (IQR 0.5–0.9) ng/ml before the next consecutive dose at 24 h (► Figure 2 ). The coefficient of variation of the plasma concentration was 78 % at baseline and 75–58 % at 2–24 h after intake. Interestingly, baseline levels of prasugrel active metabolite where higher shortly after the loading dose (r=-0.77, p<0.001). High levels of C-reactive protein (r=-0.51, p=0.02) were inversely associated with lower peak concentrations of prasugrel.
Figure 2: Plasma concentrations of prasugrel active metabolite at baseline, 2 h and 24 h after intake of 10 mg prasugrel.
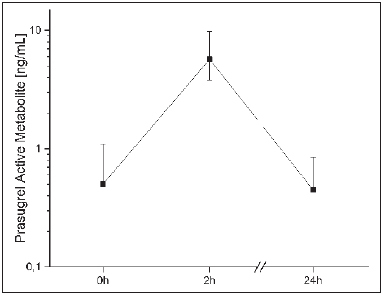
. Presented are medians ± quartiles.
Discussion
In our population of critically ill patients 65 %, 74 % and 26 % responded poorly to daily treatment with 10 mg prasugrel, according to ADP-induced whole blood aggregometry, platelet function under high shear rates and the VASP-assay, respectively.
The chosen platelet function tests are commonly used to assess the response to anti-platelet treatment ( 14 , 17 ). However, no information is available on prasugrel effects in critically ill patients. Thus, one may only speculate on the causes of the divergent results. The VASP-P assay, which is considered a specific indicator of down-stream P2Y12 receptor mediated platelet inhibition ( 14 ), may be biased by low haemoglobin levels in our patients. This may require further validation in similar populations. In contrast, whole blood aggregometry and platelet function under high shear rates are rapid whole blood assays, which may be altered by inflammation. Critically ill patients without antiplatelet treatment, who did not develop sepsis, had higher platelet aggregation compared to controls ( 18 ). However, lower platelet counts in septic patients may have influenced the results of platelet aggregation tests ( 18 ). Consistently, the closure time in PFA was shorter and ADP-induced whole blood aggregometry trendwise higher during experimental endotoxemia, but the effects of prasugrel on the VASP assay were not influenced ( 19 ). In our patients, platelet counts increased over time, which may have affected the results of MEA and PFA. Additionally, ADP may also activate platelets via the P2Y1 receptor, which is not inhibited by prasugrel ( 20 ). Although this receptor usually induces only weak and transient platelet responses ( 20 ), platelets in critically ill patients may differ from other patient populations and therefore activation may contribute to aggregation. For instance, the fraction of reticulated platelets may be larger and activation of other receptors, i. e. toll-like receptors, may contribute to the increased aggregability ( 20 – 22 ). Hence, the VASP-P assay may overestimate the inhibition of platelets by prasugrel in critically ill patients as other pathways of platelet aggregation may gain more importance.
Given these results, in critically ill patients both, specific and P2Y12-receptor unspecific platelet reactivity tests appear to provide complementary information regarding the platelet reactivity in this patient population. Remaining ADP-induced platelet aggregation may not be improved by increasing the prasugrel dose in those critically ill patients who have a low PRI% indicating substantial P2Y12 receptor inhibition. In high-risk patients other strategies to reduce platelet reactivity may be more reasonable, if such is desirable.
A 60 mg loading dose of prasugrel is used to achieve quick and effective platelet inhibition ( 4 ). Interestingly, in our trial, patients who were tested within a few days after the loading dose responded better in both functional tests. Lower platelet counts at admission, which increased over time, may partially explain this finding. This phenomenon may also be triggered by the inflammatory response.
To minimise blood loss in anaemic critically ill patients we only performed sparse blood sampling with three measurements within 24 h, which is a limitation. Concentrations of prasugrel active metabolite appeared ∼50 % lower at 2 h compared to healthy controls ( 13 ) or ∼30 % lower compared to patients after myocardial infarction who received a 60 mg loading dose, assuming dose-proportional pharmacokinetics ( 23 ). Intriguingly, our patients had at least three-fold higher trough plasma concentrations of prasugrel active metabolite, suggesting slow absorption due to gastrointestinal alterations, or impaired metabolic activation by cytochrome enzymes ( 10 ), which affect prasugrel metabolism ( 13 ). Additionally, in our trial high levels of Creactive protein, indicating an inflammatory response, correlated with low peak concentrations of the prasugrel active metabolite. Interestingly, the maximum concentration of the prasugrel active metabolite was approximately six-fold lower in stable patients with acute myocardial infarction compared to healthy volunteers ( 12 , 23 ). Slow absorption or impaired metabolisation may also, at least in part, explain the findings of Flierl et al., who reported decreasing HTPR rates over the first 72 h ( 9 ). Therapeutic hypothermia on top of the inflammatory response may additionally reduce the activity of cytochrome P450 enzymes ( 24 ). Our data in critically ill patients extend these findings and suggest significantly different pharmacokinetics of prasugrel in these differing clinical situations. However, a more extensive analysis of plasma concentrations of prasugrel and its active metabolite is needed to better define its pharmacokinetics in critically ill patients.
What is known about this topic?
Prasugrel is a potent P2Y12-receptor inhibitor. To achieve quick and sufficient platelet inhibition treatment with prasugrel is initiated with a 60 mg loading dose, followed by a daily maintenance dose of 10 mg.
In patients undergoing therapeutic hypothermia after cardiac arrest a high number of patients responded poorly to the initial loading dose, according to the vasodilator-stimulated phosphoprotein phosphorylation. No information on the response to prasugrel treatment in critically ill patients is currently available.
The response rate of patients to platelet inhibitory drugs varies with different platelet function tests and the chosen cutoffs.
What does this paper add?
Critically ill patients respond poorly to daily prasugrel treatment. Whole blood aggregometry classified 65 %, platelet function under high shear rates 74 % and vasodilator-stimulated phosphoprotein phosphorylation 26 % of patients, with high on-treatment platelet reactivity.
Low levels of prasugrel active metabolite were measured, which may be caused by slow absorption of these orally administered drugs, or impaired activation of the prodrug by downregulated cytochrome enzymes. These low levels may contribute to the high variability in platelet inhibition found in critically ill patients.
Note added in proof
The authors made an additional analysis in samples from this manuscript and made a quite interesting observation. In critically ill patients microRNA 130b levels were significantly higher in patients with trough prasugrel active metabolite concentrations >1 ng/ml compared to those with lower or nonmeasurable concentrations (p=0.005). MicroRNA 130b, which is upregulated by inflammatory processes, is a negative regulator of cytochrome enzyme activity. Thus, microRNA 130b may contribute to the partially impaired prasugrel metabolism (or activation) in critically ill patients.
Another limitation is the lack of post-mortem examinations, so that we cannot rule in or out stent thrombosis.
In summary, there is a high variability in plasma concentrations of prasugrel active metabolite and in its ability to inhibit platelet aggregation in critically ill patients.
Acknowledgements
The authors would like to thank Sabine Schranz, Karin Petroczi and Christa Drucker for their invaluable help throughout the trial.
Funding Statement
Financial support: The Austrian Science Funds (FWF) funded this work (grant SFB54-P04).
Footnotes
Conflicts of interest J. Siller-Matula has received lecture fees from AstraZeneca, Daiichi Sankyo, Eli Lilly, and Roche. None of the other authors declares any conflicts of interest.
References
- 1.Sakr Y, Lobo SM, Moreno RP et al. Patterns and early evolution of organ failure in the intensive care unit and their relation to outcome. Crit Care. 2012;16:R222.. doi: 10.1186/cc11868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bone RC. Immunologic dissonance: a continuing evolution in our understanding of the systemic inflammatory response syndrome (SIRS) and the multiple organ dysfunction syndrome (MODS) Ann Intern Med. 1996;125:680–687. doi: 10.7326/0003-4819-125-8-199610150-00009. [DOI] [PubMed] [Google Scholar]
- 3.Krychtiuk KA, Ruhittel S, Hohensinner PJ et al. Mitochondrial DNA and Toll-Like Receptor-9 Are Associated With Mortality in Critically Ill Patients. Crit Care Med. 2015;43:2633–2641. doi: 10.1097/CCM.0000000000001311. [DOI] [PubMed] [Google Scholar]
- 4.Bonello L, Pansieri M, Mancini J et al. High on-treatment platelet reactivity after prasugrel loading dose and cardiovascular events after percutaneous coronary intervention in acute coronary syndromes. J Am Coll Cardiol. 2011;58:467–473. doi: 10.1016/j.jacc.2011.04.017. [DOI] [PubMed] [Google Scholar]
- 5.Mega JL, Simon T. Pharmacology of antithrombotic drugs: an assessment of oral antiplatelet and anticoagulant treatments. Lancet. 2015;386:281–291. doi: 10.1016/S0140-6736(15)60243-4. [DOI] [PubMed] [Google Scholar]
- 6.Roffi M, Patrono C, Collet JP et al. 2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: Task Force for the Management of Acute Coronary Syndromes in Patients Presenting without Persistent ST-Segment Elevation of the European Society of Cardiology (ESC) Eur Heart J. 2016;37:267–315. doi: 10.1093/eurheartj/ehv320. [DOI] [PubMed] [Google Scholar]
- 7.Franchi F, Rollini F, Aggarwal N et al. Pharmacodynamic Comparison of Prasugrel Versus Ticagrelor in Patients With Type 2 Diabetes Mellitus and Coronary Artery Disease: The OPTIMUS (Optimizing Antiplatelet Therapy in Diabetes Mellitus)-4 Study. Circulation. 2016;134:780–792. doi: 10.1161/CIRCULATIONAHA.116.023402. [DOI] [PubMed] [Google Scholar]
- 8.Ibrahim K, Christoph M, Schmeinck S et al. High rates of prasugrel and ticagrelor non-responder in patients treated with therapeutic hypothermia after cardiac arrest. Resuscitation. 2014;85:649–656. doi: 10.1016/j.resuscitation.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 9.Flierl U, Rontgen P, Zauner F et al. Platelet inhibition with prasugrel in patients with acute myocardial infarction undergoing therapeutic hypothermia after cardiopulmonary resuscitation. Thromb Haemost. 2016;115:960–968. doi: 10.1160/TH15-07-0599. [DOI] [PubMed] [Google Scholar]
- 10.Roberts DJ, Hall RI. Drug absorption, distribution, metabolism and excretion considerations in critically ill adults. Expert Opin Drug Metab Toxicol. 2013;09:1067–1084. doi: 10.1517/17425255.2013.799137. [DOI] [PubMed] [Google Scholar]
- 11.Jernberg T, Payne CD, Winters KJ et al. Prasugrel achieves greater inhibition of platelet aggregation and a lower rate of non-responders compared with clopidogrel in aspirin-treated patients with stable coronary artery disease. Eur Heart J. 2006;27:1166–1173. doi: 10.1093/eurheartj/ehi877. [DOI] [PubMed] [Google Scholar]
- 12.Hobl EL, Reiter B, Schoergenhofer C et al. Morphine interaction with prasugrel: a double-blind, cross-over trial in healthy volunteers. Clin Res Cardiol. 2016;105:349–355. doi: 10.1007/s00392-015-0927-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holmberg MT, Tornio A, Hyvarinen H et al. Effect of grapefruit juice on the bioactivation of prasugrel. Br J Clin Pharmacol. 2015;80:139–145. doi: 10.1111/bcp.12581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siller-Matula JM, Trenk D, Schror K et al. Response variability to P2Y12 receptor inhibitors: expectations and reality. JACC Cardiovasc Interv. 2013;06:1111–1128. doi: 10.1016/j.jcin.2013.06.011. [DOI] [PubMed] [Google Scholar]
- 15.Vincent JL, de Mendonca A, Cantraine F et al. Use of the SOFA score to assess the incidence of organ dysfunction/failure in intensive care units: results of a multicenter, prospective study. Working group on “sepsis-related problems” of the European Society of Intensive Care Medicine. Crit Care Med. 1998;26:1793–1800. doi: 10.1097/00003246-199811000-00016. [DOI] [PubMed] [Google Scholar]
- 16.SAPS 3 Score Calculation SheetAvailable at:http://www.saps3.org
- 17.Spiliopoulos S, Pastromas G. Current status of high on-treatment platelet reactivity in patients with coronary or peripheral arterial disease: Mechanisms, evaluation clinical implications. World J Cardiol. 2015;07:912–921. doi: 10.4330/wjc.v7.i12.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davies GR, Mills GM, Lawrence M et al. The role of whole blood impedance aggregometry and its utilisation in the diagnosis and prognosis of patients with systemic inflammatory response syndrome and sepsis in acute critical illness. PLoS One. 2014;09:e108589.. doi: 10.1371/journal.pone.0108589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spiel AO, Derhaschnig U, Schwameis M et al. Effects of prasugrel on platelet inhibition during systemic endotoxaemia: a randomized controlled trial. Clin Sci. 2012;123:591–600. doi: 10.1042/CS20120194. [DOI] [PubMed] [Google Scholar]
- 20.Hechler B, Gachet C. Purinergic Receptors in Thrombosis and Inflammation. Arterioscler Thromb Vasc Biol. 2015;35:2307–2315. doi: 10.1161/ATVBAHA.115.303395. [DOI] [PubMed] [Google Scholar]
- 21.Stratz C, Nuhrenberg T, Amann M et al. Impact of reticulated platelets on antiplatelet response to thienopyridines is independent of platelet turnover. Thromb Haemost. 2016;116:941–948. doi: 10.1160/TH16-03-0191. [DOI] [PubMed] [Google Scholar]
- 22.Wu Q, Ren J, Hu D et al. An elevated percentage of reticulated platelet is associated with increased mortality in septic shock patients. Medicine. 2015;94:e814.. doi: 10.1097/MD.0000000000000814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rollini F, Franchi F, Hu J et al. Crushed Prasugrel Tablets in Patients With STEMI Undergoing Primary Percutaneous Coronary Intervention: The CRUSH Study. J Am Coll Cardiol. 2016;67:1994–2004. doi: 10.1016/j.jacc.2016.02.045. [DOI] [PubMed] [Google Scholar]
- 24.Anderson KB, Poloyac SM, Kochanek PM et al. Effect of Hypothermia and Targeted Temperature Management on Drug Disposition and Response Following Cardiac Arrest: A Comprehensive Review of Preclinical and Clinical Investigations. Ther Hypothermia Temp Manag. 2016;06:169–179. doi: 10.1089/ther.2016.0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.