

Abstract
Objectives
Clinical trials for progressive neurodegenerative disorders such as Alzheimer's Disease and Amyotrophic Lateral Sclerosis have been hindered due to the absence of effective pharmacodynamics markers to assay target engagement. We tested whether measurements of new protein production would be a viable pharmacodynamics tool for RNA‐targeted therapies.
Methods
Transgenic animal models expressing human proteins implicated in neurodegenerative disorders – microtubule‐associated protein tau (hTau) or superoxide dismutase‐1 (hSOD1) – were treated with antisense oligonucleotides (ASOs) delivered to the central nervous system to target these human mRNA transcripts. Simultaneously, animals were administered 13C6‐leucine via drinking water to measure new protein synthesis after ASO treatment. Measures of new protein synthesis and protein concentration were assayed at designated time points after ASO treatment using targeted proteomics.
Results
ASO treatment lowered hTau mRNA and protein production (measured by 13C6‐leucine‐labeled hTau protein) earlier than total hTau protein concentration in transgenic mouse cortex. In the CSF of hSOD1 transgenic rats, ASO treatment lowered newly generated hSOD1 protein driven by decreases in newly synthesized hSOD1 protein, not overall protein concentration, 30 days after treatment. At later time points, decreases in newly generated protein were still observed after mRNA lowering reached a steady state after ASO treatment.
Interpretation
Measures of newly generated protein show earlier pharmacodynamics changes for RNA‐lowering therapeutics compared with total protein concentration. Early in ASO treatment, decreases in newly generated protein are driven by changes in newly synthesized protein. Measuring new protein production in CSF may be a promising early pharmacodynamics marker for RNA‐targeted therapeutics.
Introduction
The number of persons aged 65 and older globally is projected to triple by the year 2050,1 increasing the number of people living with adult‐onset, progressive neurodegenerative diseases in the population. There is a dire need to develop novel therapeutics for these conditions, as current FDA‐approved drugs are not adequately effective, and no treatments exist to reverse disease pathology. Despite promise of new therapies in preclinical models over the past 30 years, these compounds have failed in Amyotrophic Lateral Sclerosis (ALS) and Alzheimer's Disease (AD) clinical trials at rates greater than 95% and 99%, respectively.2, 3 One important way to improve the current treatment landscape is to focus on the development of novel methods to determine whether a therapeutic has engaged the intended target in human clinical trials.
A central hypothesis in neurodegenerative disease is that the accumulation of misfolded proteins over time imparts selective toxicity to specific neuronal populations, causing progressive neurodegeneration and manifestation of symptoms. This pathological misfolding of proteins in AD,4 ALS,5, 6 Parkinson's Disease (PD),7 and Huntington's Disease (HD) 8 suggests that an approach to inhibit the production of toxic proteins within the cell may be an effective therapy. One such approach includes lowering levels of RNA to decrease protein translation using molecules such as antisense oligonucleotides (ASOs) or small interfering RNA (siRNA). ASOs are short, DNA‐like molecules that can be designed to selectively bind mRNA transcripts and cause the catalytic destruction of particular mRNAs.9 ASOs that lower mRNA levels and thus decrease translation of proteins prone to misfolding show great promise in preclinical models of tauopathy,10 ALS,11, 12, 13 and HD,14 and are relatively safe in humans.15 These promising data have led to the launch of multiple clinical trials for ASOs in neurodegenerative diseases (NCT03225846, NCT02623699, NCT02519036, NCT03186989).
For RNA‐targeted therapeutics, it is crucial to establish measurements that validate compound engagement with its desired biological target, mRNA.16 However, it is challenging to isolate mRNA from cerebrospinal fluid (CSF), the primary biofluid available to assay target engagement in the human central nervous system (CNS). Because mRNA translation leads to the production of new protein, protein‐based biomarkers in CSF represent an attractive avenue to pursue target engagement for RNA‐targeted therapeutics. In this study, we used nonradioactive, stable isotope labeling with an essential amino acid 17, 18 to identify inhibition of new protein production after ASO treatment. Using two preclinical models of ASO targets in clinical trials for neurodegenerative disease, we tested the hypotheses that stable isotope labeling with 13C6‐Leucine will: (1) identify pharmacodynamics at an earlier time point than protein concentration‐based measures and (2) show measurable effects in CSF to support translation of this assay approach from animal models to human CNS. These results would support the use of new protein synthesis measurements in clinical trials for RNA‐lowering therapies in neurodegenerative diseases.
Methods
Transgenic animals
Transgenic mice expressing human tau (hTau) 19 (Jackson Laboratories, Bar Harbor, ME) were maintained through in‐house breeding on a C57/Bl6 background. Transgenic rats expressing human superoxide dismutase‐1 (hSOD1WT) 20 were provided by Pak Chan (Stanford University) and maintained through in‐house breeding on a Sprague‐Dawley background. Genotyping was performed by PCR amplification of tail DNA (Table S1). All breeding and experimental protocols were approved by the Institutional Animal Care and Use Committee at Washington University and conducted according to the NIH Guide for Care and Use of Laboratory Animals.
Antisense oligonucleotides (ASOs)
ASOs that recruit RNase‐H to degrade target mRNA and an inactive, control ASO with no sequence specificity were provided by Ionis Pharmaceuticals 10, 21 (Fig. 1). All ASOs were diluted in artificial CSF before use.
Figure 1.

Antisense Oligonucleotides. Oligonucleotides containing phosphothioate backbone modifications: unmodified phosphodiester linkage (red o), 2′‐O‐methoxyethylribose, (orange) and constrained ethyl (blue) groups. All cytosine residues are 5′methylcytosine.
Surgical procedures
All animals were anesthetized by 2%–5% inhalant isoflurane, received constant isoflurane flow during the procedure, and were given an injection of bupivacaine prior to incision. Following the surgical procedure, animals received carprofen and were placed on a 37°C heating pad for recovery.
For intracerebroventricular injections, 60‐day‐old hTau transgenic mice received 300 μg of ASO at −1.0 mm M/L, −0.3 mm A/P, −3.0 mm D/V from Bregma.22 Similarly, 120‐day‐old hSOD1 transgenic rats received 1000 μg of ASO injected at −1.4 mm M/L, −0.4 mm A/P, −3.5 mm D/V from Bregma. For intrathecal ASO injection, 120‐day‐old hSOD1 transgenic rats received 1000 μg of ASO between the L3 and L5 vertebrae.23 Both male and female transgenic animals were used in these studies, and researchers were blinded to the identity of treatment throughout the duration of the experiment.
13C6‐Leucine oral labeling and tissue collection
Methods for oral labeling in rodents with 13C6‐Leucine were described previously using unlabeled L‐leucine or labeled, 13C6‐L‐Leucine (Cambridge Isotope Laboratories, Cambridge, MA) dissolved in water at 5 mg/mL.24 The amount of 13C6‐Leucine found in plasma, the precursor for leucine found in the central nervous system,24 was consistent in all experiments between groups (Figure S1).
At indicated time points after surgery, animals were anesthetized with isoflurane and perfused with cold phosphate buffered saline (PBS) containing 0.03% heparin. Before perfusion, CSF was extracted from rat cisterna magna via a 23x¾” winged infusion butterfly needle (Terumo, Somerset, NJ). Blood was collected from mechanically ruptured vena cavae at the beginning of perfusion, and the brain and spinal cord were harvested after perfusion. All samples were flash frozen in liquid nitrogen and stored at −80°C before use.
mRNA isolation and analysis
mRNA isolation from flash‐frozen tissues was performed using the RNeasy® Mini Kit (Qiagen, Venlo, Netherlands). The EXPRESS One‐Step Superscript qRT‐PCR Universal Kit (Invitrogen, Carlsbad, CA) was used for reverse transcription and amplification of SOD1 and MAPT mRNA transcripts. Comparative analysis by the ΔΔCt method, with GAPDH mRNA transcripts as an internal control, was performed with the ABI PRISM 7500 Fast Real‐Time System (Applied Biosystems, Waltham, MA) (Table S1).
N15 Recombinant Protein Standards
N15‐labeled, recombinant hTau protein (isoform 2N4R 411) was a generous gift from Drs. Isabelle Huvent and Guy Lippens (Université des Sciences et Technologie de Lille 1, France).
Recombinant hSOD1 was produced in Rosetta 2 E Coli (Novagen) using an hSOD1 cDNA construct with an N‐terminal GST tag subcloned into a pGEX4T‐1 backbone. Bacterial cultures were inoculated overnight in 15N‐Celtone Complete Medium (Cambridge Isotope Laboratories, Inc) with shaking at 37°C and induced with 1 mmol/L IPTG for hSOD1 production. Protein was isolated by incubation with 600 μL of glutathione sepharose 4B beads (GE Healthcare) for 30 min, 1 unit of thrombin (Thrombin Cleavage Capture Kit, Millipore) for 20 h, and 30 μL of streptavidin agarose for 30 min, performed at room temperature. Samples were spun at 1000xg for 5 min, and supernatant containing N15‐labeled, hSOD1 recombinant protein was collected.
Tissue homogenization, protein isolation, and mass spectrometric analysis
hTau transgenic mouse brain tissue was homogenized in PBS plus protease inhibitors (P8340; Sigma‐Aldrich, St. Louis, MO). Protein concentration of brain lysate was determined by Pierce BCA assay (ThermoFisher Scientific, Waltham, MA). Total hTau was immunoprecipitated from 100 μg of tissue lysate using 30 μL of CNBr‐activated Sepharose beads (GE Healthcare, Chicago, IL) crosslinked to anti‐hTau antibody (Tau 1 monoclonal, produced by Nicholas M. Kanaan, Michigan State University) at 3 mg antibody/g of beads in 1 mL PBS plus 1% NP‐40, 5 mmol/L guanidine, and protease inhibitors for 1.5 h at room temperature. Before immunoprecipitation, 5 ng of N15‐lableled hTau was added to each sample. hTau was digested using 400 ng of sequencing‐grade endoproteinase Trypsin (Promega, Madison, WI) for 16 h at 37°C. Samples were lyophilized and reconstituted in 25 μL of 2% acetonitrile/0.1% formic acid solution prior to liquid chromatography‐ quadrupole‐orbitrap mass spectrometry analysis (OrbiTrap Fusion, ThermoFisher).
hSOD1 was isolated from transgenic rat tissue or CSF as described previously.24 Before immunoprecipitation, 200 ng of N15‐labeled hSOD1 was added to each brain or spinal cord sample, and 50 ng of N15 SOD1 was added to CSF samples. Isolated hSOD1 was digested with 600 ng of sequencing‐grade endoproteinase GluC in 25 μL of 50 mmol/L NaHCO3 buffer (pH 8.8) for 17 h at room temperature. After solid phase extraction, samples were lyophilized and resuspended in 25 μL 2% acetonitrile/0.1% formic acid solution prior to liquid chromatography‐triple quadrupole mass spectrometry analysis (Xevo TQ‐S, Waters).
Newly synthesized protein rate was quantified by analyzing the incorporation of 13C6‐Leucine into target proteins, indicated by the tracer‐to‐tracee ratio (TTR), by comparing the area under the chromatogram curve for SOD1 or tau peptides containing 13C6‐Leucine or 12C6‐Leucine. Relative protein concentration was quantified by measuring the ratio between the area under the curve of protein immunoprecipitated from animal tissue and N15‐labeled recombinant protein (Tables S2 and S3). To account for non‐steady state conditions due to mRNA lowering in this experiments, newly generated protein was also analyzed by multiplying the relative protein concentration by protein TTR at each time point. For figure presentation, data for hTau is represented by the TPSLPTPPTR peptide and data for hSOD1 is represented by the GLHGFHVHE peptide, as all peptides measured showed strong correlations within each sample (Fig. S2 and S3).
Plasma‐free 13C6‐Leucine abundance was analyzed from TCA precipitation of 100 μL rat or mouse plasma via capillary gas chromatography‐negative chemical ionization‐quadrupole mass spectrometry (Agilent) as described previously.24
Statistical analysis
All statistical analyses were performed using Graphpad Prism 7.0 (GraphPad Software). All hypothesis tests performed were two‐sided assuming a normal distribution of data. P < 0.05 was determined significant. Based on previous results,10, 21 a cutoff of 50% target mRNA reduction relative to the mean mRNA levels in inactive ASO‐treated animals was used as inclusion criteria for subsequent analysis. In CSF analysis, samples were excluded if blood contamination was visibly present or if the volume of CSF collected was less than 75 μL. For relative quantification, samples were normalized to a single sample in each experiment from the inactive ASO treatment group. Quantitative results are expressed as mean difference and 95% confidence interval.
Results
Lowering new hTau production precedes protein concentration changes after ASO treatment
We performed ICV injection of an ASO targeting human tau mRNA 10 in transgenic mice expressing the human tau transgene (hTau mice) to understand lowering of tau mRNA and total protein over time. To analyze newly generated hTau protein, we administered 13C6‐Leucine in the drinking water at designated time points (Fig. 2A). We organized the timing of ASO treatment, 13C6‐Leucine administration, and euthanasia to observe consistent, maximum label incorporation of proteins within the CNS at each time point analyzed. Preliminary studies showed adequate labeling of hTau with 4 days of 13C6‐Leucine labeling (Fig. S4).
Figure 2.
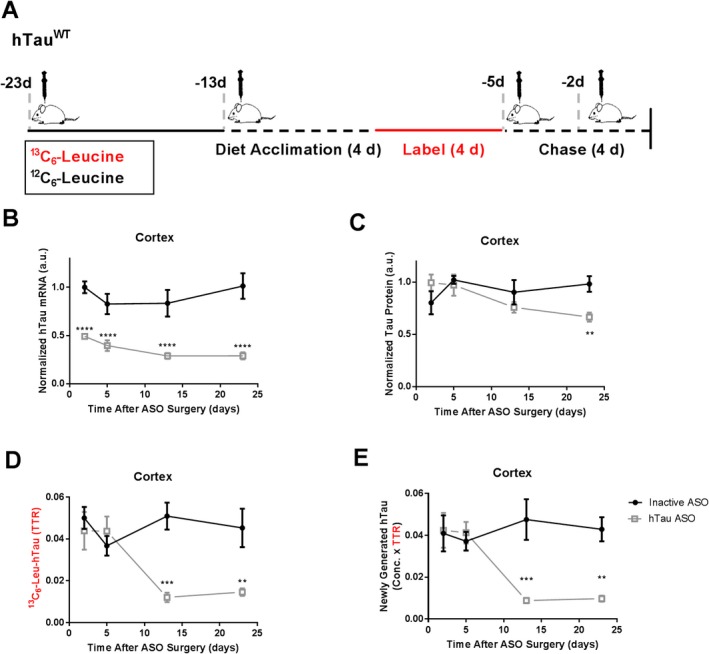
Lowered hTau protein synthesis precedes protein lowering after ASO treatment in hTau transgenic mice. (A) Experimental Design. hTau transgenic mice received 300 μg of either hTau ASO or inactive ASO via intracerebroventricular injection and were labeled with13C6‐Leucine (13C6‐Leu) at designated time points. (B) hTau mRNA levels in temporal cortex by qPCR. Compared to inactive ASO control, hTau ASO treatment lowered hTau mRNA over time (2‐Way ANOVA F 3,24 = 10.04, P = 0.0145). hTau mRNA levels are significantly lowered as early as 2 days after ASO treatment by 50% (95% CI: 34.8%, 66.9%), and remain lowered as late as 23 days post treatment by 72% (55.5%, 87.2%). (C) hTau protein levels relative to N15‐labeled recombinant standard in temporal cortex by LC‐MS. hTau ASO treatment lowers hSOD1 protein over time (2‐Way ANOVA F 3,24 = 3.374, P = 0.0349). This effect is observed at 23 days post‐ASO treatment by 32% (0.05%, 64.0%). (D) Newly synthesized 13C6‐Leu‐labeled hTau in temporal cortex by LC‐MS. Values are measured as the tracer to trace ratio (TTR) of 13C6‐Leu‐labeled:12C6‐Leu‐unlabeled hTau. Lowering of 13C6‐Leu‐labeled hTau is observed over time (2‐Way ANOVA, F 3,24 = 5.748, P = 0.0041), and as early as 13 days post‐ASO treatment by 71% (95% CI: 29.4%, 123%). (E) Newly generated hTau protein measured by multiplying hTau protein concentration and TTR to account for non‐steady state conditions. **** P < 0.0001, *** P < 0.001, ** P < 0.01. n = 4 per treatment group per time point. Error bars in figures represent standard error of the mean (SEM).
hTau ASO treatment lowered hTau mRNA in the cerebral cortex over time (Fig. 2B). As early as 2 days post‐injection, hTau ASO treatment lowered hTau mRNA levels by ~50% relative to inactive ASO, with further change of hTau mRNA by ~72% by 23 days post‐injection. hTau ASO treatment also decreased hTau protein levels over time (Fig. 2C), with significant effects of ~32% observed at 23 days. The discrepancy between overall mRNA lowering and protein lowering is consistent with tau as a long‐lived protein in the CNS.25
Using percent of 13C6‐Leucine incorporation (TTR) as a measure of new protein synthesis rate, hTau ASO treatment lowered the rate of newly synthesized hTau protein over time (Fig. 2D). We observed lowering of newly generated hTau protein at 13 days after ASO treatment by ~71%. Combined, these data suggest that newly generated hTau protein is a reliable measure of hTau ASO pharmacodynamics in central nervous system tissue, with newly generated hTau showing an earlier (13 vs. 23 days) and greater magnitude of effect (71% vs. 32%) compared to hTau protein concentrations (Fig. 2E).
Lowering of 13C6‐Leucine‐labeled hSOD1 occurs in CSF after ASO treatment
hSOD1WT rats were treated with an ICV injection of 1000 μg hSOD1 ASO in the lateral ventricle and administered 13C6‐Leucine in drinking water (Fig. 3A). Based on previous measurements of hSOD1 half‐life in these rats,24 and that lowering of CSF SOD1 after ASO treatment in the G93A SOD1 transgenic rat model was observed at 42 days post‐ASO treatment,26 we euthanized animals 30 days after treatment to assay hSOD1 total protein levels and new hSOD1 protein production to identify early ASO pharmacodynamics. hSOD1 ASO treatment lowered hSOD1 mRNA globally within the CNS (Fig. 3B), leading to overall hSOD1 protein lowering by ~62% in cortex and ~40% in lumbar spinal cord (Fig. 3C). Similarly, we observed a decrease in new hSOD1 protein synthesis rate by ~34% in cortex and ~50% in lumbar spinal cord (Fig. 3D). At this time point, lowering of both hSOD1 protein concentration and new hSOD1 protein synthesis rate drive overall decreases in newly generated hSOD1 protein (Fig. 3E).
Figure 3.
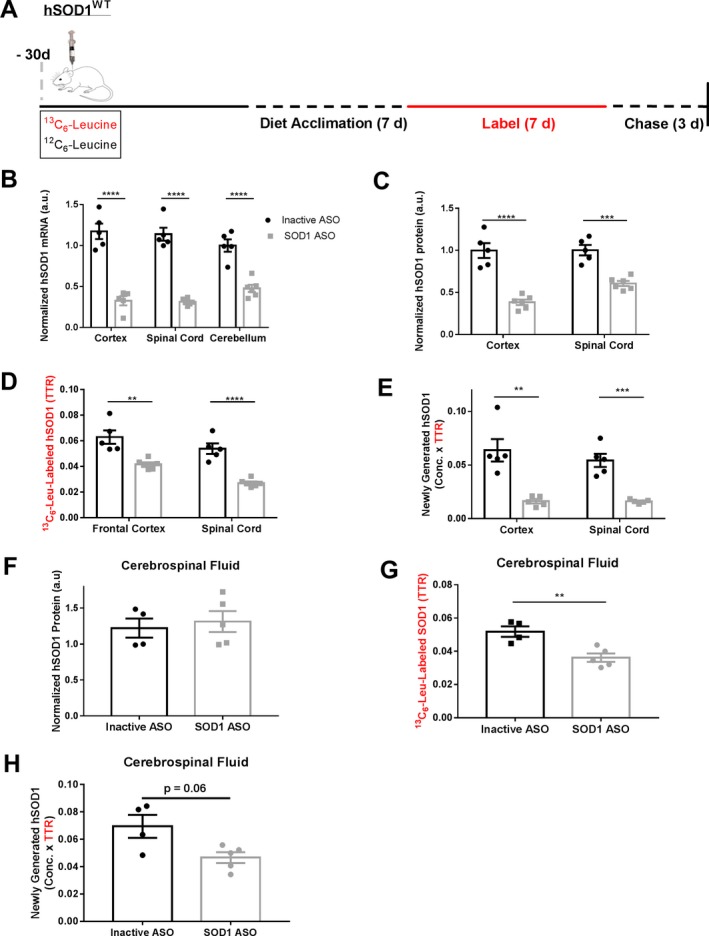
Lowered 13C6‐Leucine‐labeled hSOD1 observed in cerebrospinal fluid after ASO treatment in hSOD1 transgenic rats. (A) Experimental Design. hSOD1 transgenic rats received 1000 μg of either hSOD1 ASO or inactive ASO via intracerebroventricular injection at designated time points. (B) hSOD1 mRNA levels in the CNS by qPCR. hSOD1 mRNA levels are lowered globally in the CNS, with mRNA lowering of ~70% in cortex and lumbar spinal cord and ~55% in cerebellum. (C) hSOD1 protein levels relative to N15‐labeled recombinant standard in CNS cortex by LC‐MS. Lowering of hSOD1 protein is observed in both cortex at ~61% (95% CI: 41.9%, 81.3%) and lumbar spinal cord at ~40% (24.6%, 54.3%). (D) 13C6‐Leucine (13C6‐Leu) labeled hSOD1 in CNS by LC‐MS. Values are measured as the tracer to trace ratio (TTR) of 13C6‐Leu‐labeled:12C6‐Leu‐unlabeled hSOD1. Significant lowering of 13C6‐Leu‐labeled hSOD1 is observed in both cortex at ~34% (15.5%, 51.9%) and lumbar spinal cord at ~50% (33.3%, 66.1%). (E) Newly generated hSOD1 protein measured by multiplying hSOD1 protein concentration and TTR in tissues. (F) hSOD1 protein levels relative to N15‐labeled recombinant standard in CSF by LC‐MS. No changes in CSF hSOD1 protein levels were observed (Student's T‐Test t = 0.448, P = 0.67). (G) 13C6‐Leu labeled hSOD1 in CSF by LC‐MS. Significant lowering of 13C6‐Leu‐labeled hSOD1 by ~30% (95% CI: 12.0%, 48.4%) observed relative to inactive ASO control (Student's T Test t = 3.923, P = 0.0057). (H) Newly generated hSOD1 protein measured by multiplying hSOD1 protein concentration and TTR in CSF. One sample in inactive ASO group was excluded in CSF analysis due to blood contamination during CSF collection. ****P < 0.0001, ***P < 0.001, **P < 0.01. Error bars in figures represent SEM.
In CSF, hSOD1 treatment failed to lower hSOD1 protein concentration (Fig. 3F). However, SOD1 ASO treatment decreased 13C6‐Leucine‐labeled hSOD1 synthesis in CSF by ~30% relative to the inactive ASO treatment group (Fig. 3G). This effect of lowered 13C6‐Leucine‐labeled hSOD1 is specific to CSF, as we did not measure any differences in peripheral hSOD1 protein in plasma (Fig. S5). To test SOD1 protein concentration at a later time point, we treated a separate cohort of animals without 13C6‐Leucine labeling using the same ASO treatment paradigm and analyzed CSF hSOD1 concentration at 50 days post‐surgery (Fig. S6). Although we observed lowering in hSOD1 mRNA throughout the CNS 50 days post‐ASO treatment (Fig. S6A), we did not observe decreases in CSF hSOD1 protein concentration (Fig. S6B), suggesting an even longer incubation time may be required to observe protein lowering in CSF. Taken together, these data suggest changes in newly synthesized, 13C6‐Leucine‐labeled target proteins drive early pharmacodynamics biomarker effects in the CSF after ASO treatment (Fig. 3H).
A therapeutic window exists for newly synthesized protein pharmacodynamics
To enhance the translational relevance of our findings, we performed similar experiments in hSOD1 rats using intrathecal ASO injection, the method of drug delivery used in current clinical trials. We analyzed CNS tissue 10, 30, and 60 days after ASO treatment compared to inactive ASO control to better understand longitudinal changes in hSOD1 protein (Fig. 4A). Contrary to hSOD1 ASO delivery by ICV injection, we observed differential effects of hSOD1 mRNA lowering in CNS tissues after intrathecal ASO injection. We observed a significant hSOD1 mRNA decrease in the lumbar spinal cord over time, with an average hSOD1 mRNA lowering at 60 days by ~66%. However, hSOD1 treatment did not significantly lower hSOD1 mRNA in the cortex or cerebellum over time (Fig. 4B). Because it is likely that SOD1 mRNA will need to be lowered globally in the CNS to affect SOD1 CSF levels, we focused on protein measurements in the spinal cord, where we confidently documented mRNA lowering.
Figure 4.
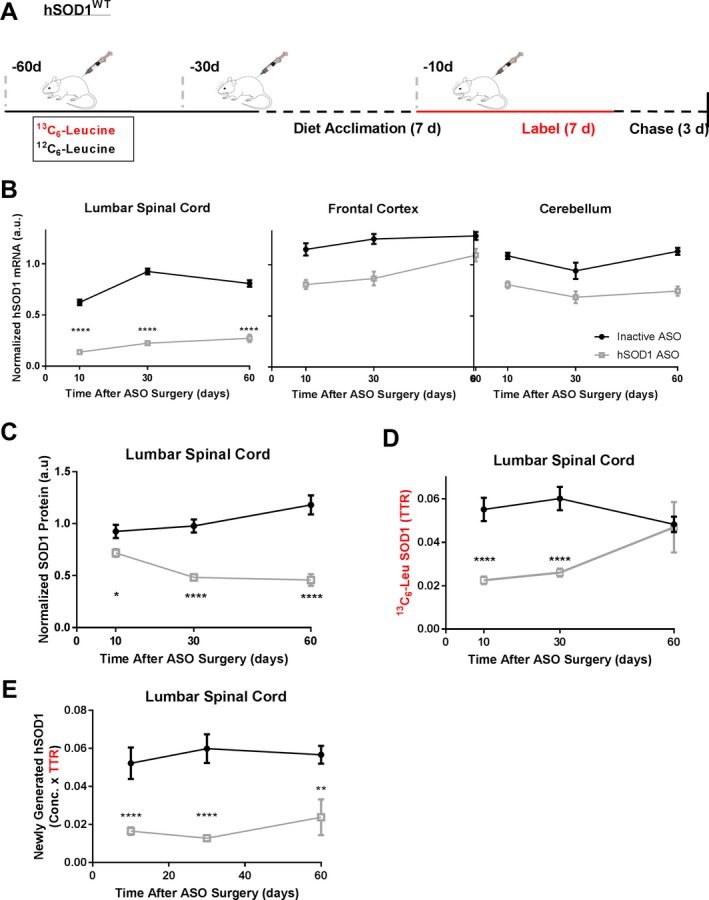
A therapeutic window exists to observe lowered protein synthesis pharmacodynamics in the central nervous system. (A) Experimental Design. hSOD1 transgenic rats received 1000 μg of either hSOD1 ASO or inactive ASO at designated time points via intrathecal injection. (B) hSOD1 mRNA levels in the CNS by qPCR. hSOD1 mRNA levels are significantly lowered in spinal cord after hSOD1 ASO treatment (2 Way ANOVA F 2,39 = 10.91, P = 0.002), with a maximum lowering of ~66% (95% CI: 54.6%, 78.2%) 60 days post‐treatment. No significant hSOD1 mRNA lowering was observed in frontal cortex (two‐Way ANOVA F 2,40 = 1.48, P = 0.24) or cerebellum. (C) hSOD1 protein levels relative to N15‐labeled recombinant standard in lumbar spinal cord by LC‐MS. Lowering of hSOD1 protein is observed over time (two‐Way ANOVA F 2,36 = 10.04, P = 0.0004), with a maximum lowering by ~50% (95% CI: 42.5%, 79.8%) at Day 60. (D) 13C6‐Leu labeled hSOD1 in CNS by LC‐MS. Values are measured as the tracer to trace ratio (TTR) of 13C6‐Leu‐labeled:12C6‐Leu‐unlabeled hSOD1. Significant lowering of newly synthesized hSOD1 observed after hSOD1 ASO Treatment (2 Way ANOVA F 2,36 = 6.269, P = 0.0046). Lowering of 13C6‐Leu‐labeled hSOD1 is observed 10 and 30 days after ASO treatment by ~40–45% (30%, 88%). 60 days post ASO treatment, there was no difference in 13C6‐Leu‐labeled hSOD1 between groups (adjusted P value = 0.9976). (E) Newly generated hSOD1 protein measured by multiplying hSOD1 protein concentration and TTR in tissues. ****P < 0.0001, ***P < 0.001, *P < 0.05. hSOD1 ASO: n = 5–10 per time point, Inactive ASO: n = 5–7 per time point. Error bars in figures represent SEM.
To further understand the relationship between protein lowering and lowering of hSOD1 synthesis rate after ASO treatment, we analyzed hSOD1 protein in lumbar spinal cord. hSOD1 ASO treatment decreased hSOD1 protein levels across all time points measured, and treatment effect was greatest at day 60, with 50% lowering of hSOD1 protein (Fig. 4C). hSOD1 ASO treatment significantly lowered hSOD1 synthesis rate compared to inactive ASO‐treated animals (Fig. 4D). Interestingly, ASO treatment decreased the ratio of newly synthesized hSOD1 at 10 and 30 days by 40% and 45%, but there was no difference in 13C6‐Leucine‐labeled hSOD1 between groups at day 60 despite lowering of total hSOD1 protein concentration. Therefore, at later time points, decreases in newly generated hSOD1 protein by ASO treatment are driven by overall lowering of protein concentration (Fig. 4E).
Discussion
Our data show that, following treatment with a mRNA lowering therapeutic, the lowering of newly synthesized protein occurs before lowering of total protein concentration in tissue. Further, our findings indicate that labeled, newly generated protein measures direct effects of protein translation in the source tissues, which are not seen in CSF protein concentrations at early time points. Given the direct measurement of protein translation, labeling protein production has higher sensitivity than protein concentration‐based measures for mRNA‐lowering therapies. These studies suggest that identifying newly generated protein after ASO treatment is an effective protein‐based biomarker for assaying ASO target engagement (Fig. 5).
Figure 5.

A model of the interplay between new protein production and protein concentration pharmacodynamics in RNA‐lowering treatment strategies. (A) Normal protein turnover at steady state synthesis includes production (top arrow) and clearance (bottom arrow) rates in the CNS. After ASO treatment, target ASO binds to target mRNA transcripts in the central nervous system. (B) Early after treatment, lowering in new protein synthesis is observed due to lowering of target mRNA by ASO treatment. (C) In addition to lowering of new protein synthesis, proteins present before 13C6‐labeling are cleared at the rate of turnover or half‐life. (D) mRNA lowering reaches steady state, resulting in a consistent lowered protein synthesis rate. Protein clearance remains constant and faster than production, leading to a new lowered steady state protein concentration detected at a later time and less magnitude.
Previous work supports the use of CSF protein concentration as a promising pharmacodynamics biomarker for ASO therapy,26, 27 and CSF SOD1 concentration is a secondary outcome measurement in an ongoing Phase I clinical trial (NCT0262399). However, a key limitation in the use of CSF protein concentration as a measure of ASO efficacy is the indirect mechanism informed by this assay. ASOs inhibit the production of new protein, and measures of protein concentration also rely on transport to CSF and the clearance of existing protein which may be unaffected by ASO treatment. Especially in a rapidly progressing neurodegenerative disease such as ALS,28 it is important to develop pharmacodynamics tools that identify direct mechanisms of action at early time points that reflect the actual drug effect in the tissue of interest.
Our study design is translatable to humans, as stable isotope labeling is safe and effective in measuring the kinetics of proteins implicated in neurodegenerative disease within human CSF.17, 18, 24, 25, 29 Previous studies used labeling with 13C6‐Leucine to measure inhibition of amyloid beta peptide synthesis by a gamma secretase inhibitor in human participants.30 However, these experiments were performed over a 36‐hour time period, as the half‐life of amyloid beta peptide is ~9 h in CSF.18 To our knowledge, the results presented here are the first observations measuring new protein production of long‐lived protein targets in the CNS after therapeutic intervention. Our approach can be directly applied to multiple RNA‐directed strategies, such as siRNAs 31 and ASOs,32, 33 that target long‐lived proteins in neurodegeneration for clinical trials. Besides its lowered protein production readout, stable isotope labeling could be a powerful tool to examine increased protein production for therapeutic strategies such as gene therapy 34 and splicing ASO treatments.35, 36 Moreover, an understanding of protein kinetics using stable isotope labeling can also inform on assays for therapies that target protein turnover, such as approaches that activate protein clearance mechanisms 37 or promote proper protein folding.38
Another advantage of our mass spectrometry‐based assay is the ability to specifically isolate mutant from wild‐type protein in patient populations. Many familial forms of neurodegenerative diseases are caused by autosomal dominant mutations 31 and, thus, exhibit both mutant and normal protein forms. However, it is currently unknown if mutant proteins display altered protein kinetics compared with their wild type counterparts in humans. Future studies in symptomatic, mutation‐carrying patients are needed to characterize the half‐life of these target proteins in human CSF. Protein half‐life measures will be crucial to determine the timing required to observe pharmacodynamics effects in humans when designing randomized‐control clinical trials.
One limitation in this study is the inability to identify target protein lowering in CSF after SOD1 ASO treatment. Without this effect, the relationship between lowered protein production and total protein concentration cannot be determined in the biological fluid important for human clinical trials. Previous data suggest this limitation may be due to the drug delivery paradigm in these experiments. The current study used a single bolus injection of ASO in rodent models while previous studies that observed total target protein lowering in CSF after ASO treatment in nonhuman primate models were performed with multiple intrathecal ASO injections.10, 21 Additionally, both transgenic animal models used here express wild‐type forms of the target protein. In previous experiments, the G93A SOD1 rat model was used to demonstrate hSOD1 protein lowering after ASO treatment 42 days after treatment.26 Our group 24 and others 39 have shown that mutant hSOD1 exhibits a shorter protein half‐life in the CNS and CSF compared to WT hSOD1. Taken together, these data suggest that CSF WT hSOD1 protein concentration will occur at an even later time point than 50 days after ASO treatment (Fig. S6). Future investigation should also address these questions in the presence of mutant protein implicated in disease, as no pathological misfolding of either hSOD1 or hTau is observed in the models employed here.19, 20 Therefore, continuing experiments in transgenic rat models or nonhuman primates should examine the relationship between CSF protein half‐life, protein concentration lowering, and changes in new protein production over time after ASO treatment, in addition to understanding how new protein production in CSF changes in the presence of misfolded proteins within the CNS.
In sum, the approach described here provides a framework for the development of assays that isolate the direct mechanism of action of target engagement in neurodegenerative disease therapies, with the hope that such data will lead to a higher success in clinical trial outcomes.
Author Contributions
WKS, KMS, TMM, and RJB contributed to study conception and design of experiments. WKS, KMS, NJB, JGB, JA, and CS contributed to data acquisition and analysis. WKS, KMS, CS, HBK, TC, ES, RJB, and TMM contributed to the drafting of the text and editing of the manuscript.
Conflict of Interest
TMM has served on medical advisory boards for Biogen and Ionis Pharmaceuticals and is a consultant for Cytokinetics. TMM/RJB/Washington University have a licensing agreement with C2N Diagnostics. Washington University and RJB have equity ownership interest in C2N Diagnostics and may receive royalty income based on technology licensed by Washington University to C2N Diagnostics. RJB receives income from C2N Diagnostics for serving on its scientific advisory board. Washington University has submitted nonprovisional patent applications for “Methods for measuring the metabolism of CNS derived biomolecules in vivo” (RJB) and “SOD1 kinetics measurements” (RJB, TMM). TC, HBK, and ES are employees of Ionis Pharmaceuticals.
Supporting information
Figure S1. No differences in 13C6‐Leucine oral labeling between treatment groups in all experiments performed. Plasma 13C6‐Leucine values were analyzed in all samples analyzed and compared between groups. A. hTau ICV ASO plasma 13C6‐Leucine at Day 23 post surgery, as seen in Figure 2. No differences were observed (Student's T‐test, P = 0.4496). B. hSOD1 ICV ASO plasma 13C6‐Leucine at Day 30. No differences were observed (Student's‐T test) C. hSOD1 IT ASO plasma 13C6‐Leucine at all time points assessed. No differences were observed (2‐Way ANOVA). Error bars in figures represent SEM.
Figure S2. Correlation between all hTau peptides measured. Linear regression analysis comparing peptides shows strong correlations exist between all three peptide measurements. All p values < 0.0001 for each analysis.
Figure S3. Correlation between all hSOD1 peptides measured. Linear regression analysis comparing peptides shows strong correlations exist between all peptide measurements. All P values < 0.0001 for each analysis.
Figure S4. Characterizing 13C6‐Leucine incorporation into hTau protein in hTauWT transgenic mice. hTau mice were acclimated to leucine‐free chow for 7 days, and labeled with 5 mg/ml 13C6‐leucine for 7 days. Mice were euthanized at 6 (n = 2), 10 (n = 3), 14 (n = 3), and 17 (n = 2) days post‐end of label. Because of the high label incorporation (nearly 15% TTR) and delayed appearance of higher label incorporation after the end of the labeling period 3‐10 days after the end of labeling, subsequent experiments reduced the label time to 4 days. Given maximal label incorporation at 6‐10 days post‐end of label, all mice in the subsequent experiments were sacrificed at 5 days post‐end of label. Error bars in figures represent SEM.
Figure S5. No changes in peripheral hSOD1 after 1000 ug hSOD1 ASO intracerebroventricular injection. A. hSOD1 protein levels relative to N15‐labeled recombinant standard by LC‐MS in plasma. No changes in plasma hSOD1 protein levels were observed (Student's T‐test). B. 13C6‐Leu labeled hSOD1 in plasma by LC‐MS. No changes in plasma 13C6‐Leu labeled hSOD1 was observed (Student's T‐Test). Error bars in figures represent SEM.
Figure S6. No protein concentration pharmacodynamics observed in CSF after 1000 ug hSOD1 ASO intracerebroventricular injection at 50 days post treatment. A. hSOD1 mRNA levels in the CNS by qPCR. hSOD1 mRNA levels are significantly lowered in spinal cord and frontal cortex by ~70% and 50%, respectively, after ASO treatment relative to inactive ASO controls (Student's T‐test). B. hSOD1 protein levels relative to N15‐labeled recombinant standard by LC‐MS. No changes in CSF hSOD1 protein levels were observed (Student's T‐test). n = 5 for each treatment group, with one CSF sample excluded from each group due to blood contamination. **** P < 0.0001. Error bars in figures represent SEM.
Table S1. Primers and probes used for genotyping and qPCR analysis of hTau (MAPT), hSOD1 (SOD1) and mouse GAPDH (GAPDH) DNA.
Table S2. Transition ions used for hSOD1 tandem LC‐MS/MS. GLHGFHVHE peptide measurements were used as representative data in figures.
Table S3. Transition ions used for hTau tandem LC‐MS/MS. TPSLPTPPTR peptide measurements were used as representative data in figures.
Acknowledgments
We thank Drs. Cheryl Frankfater and John Turk of the Biomedical Mass Spectrometry Core at Washington University in St. Louis for assistance with gas chromatography‐mass spectrometry analysis. Research reported in this publication was supported by the Washington University Institute of Clinical and Translational Sciences grant UL1TR002345, sub‐award TL1TR002344 from the National Center for Advancing Translational Sciences (NCATS), the National Institute of Neurological Disorders and Stroke grant U01NS084870, grant R01NS098716 (TM Miller, PI) and R01NS095773 (RJ Bateman, PI) from the National Institutes of Health, as well as the MetLife Foundation Award (RJ Bateman, PI) (NIH). The content is solely the responsibility of the authors and does not necessarily represent the official view of NIH.
Funding Information
Research reported in this publication was supported by the Washington University Institute of Clinical and Translational Sciences grant UL1TR002345, sub‐award TL1TR002344 from the National Center for Advancing Translational Sciences (NCATS), the National Institute of Neurological Disorders and Stroke grant U01NS084870, grant R01NS098716 (TM Miller, PI), and R01NS095773 (RJ Bateman, PI) from the National Institutes of Health, as well as the MetLife Foundation Award (RJ Bateman, PI) (NIH). The content is solely the responsibility of the authors and does not necessarily represent the official view of NIH.
Funding Statement
This work was funded by National Institutes of Health grants R‐01‐NS095773 and R‐01‐NS098716‐01; Institute of Clinical and Translational Sciences grant UL1TR002345; National Center for Advancing Translational Sciences grant Subaward TL1TR002344; National Institute of Neurological Disorders and Stroke grant U01NS084870.
Contributor Information
Randall J. Bateman, Email: batemanr@wustl.edu
Timothy M. Miller, Email: miller.t@wustl.edu
References
- 1. WHO . Dementia: A public health priority World Health Organization 2012.
- 2. Cummings JL, Morstorf T, Zhong K. Alzheimer's disease drug‐development pipeline: few candidates, frequent failures. Alzheimer's Res Ther 2014;6:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Petrov D, Mansfield C, Moussy A, Hermine O. ALS clinical trials review: 20 years of failure. are we any closer to registering a new treatment? Front Aging Neurosci 2017;9:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Braak H, Braak E. Staging of Alzheimer's disease‐related neurofibrillary changes. Neurobiol Aging 1995;16:271–278. [DOI] [PubMed] [Google Scholar]
- 5. Shibata N, Hirano A, Kobayashi M, et al. Intense superoxide dismutase‐1 immunoreactivity in intracytoplasmic hyaline inclusions of familial amyotrophic lateral sclerosis with posterior column involvement. J Neuropathol Exp Neurol 1996;55:481–490. [DOI] [PubMed] [Google Scholar]
- 6. Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006;314:130–133. [DOI] [PubMed] [Google Scholar]
- 7. Bethlem J, Den Hartog Jager WA. The incidence and characteristics of Lewy bodies in idiopathic paralysis agitans (Parkinson's disease). J Neurol Neurosurg Psychiatry 1960;23:74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. DiFiglia M, Sapp E, Chase KO, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997;277:1990–1993. [DOI] [PubMed] [Google Scholar]
- 9. DeVos SL, Miller TM. Antisense oligonucleotides: treating neurodegeneration at the level of RNA. Neurotherapeutics 2013;10:486–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. DeVos SL, Miller RL, Schoch KM, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med 2017;9:pii eaag0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smith RA, Miller TM, Yamanaka K, et al. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Invest 2006;116:2290–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Donnelly CJ, Zhang PW, Pham JT, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 2013;80:415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Becker LA, Huang B, Bieri G, et al. Therapeutic reduction of ataxin‐2 extends lifespan and reduces pathology in TDP‐43 mice. Nature 2017;544:367–371. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McBride JL, Pitzer MR, Boudreau RL, et al. Preclinical safety of RNAi‐mediated HTT suppression in the rhesus macaque as a potential therapy for huntington's disease. Mol Ther 2011;19:2152–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Miller TM, Pestronk A, David W, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first‐in‐man study. Lancet Neurol 2013;12:435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mitsumoto H, Brooks BR, Silani V. Clinical trials in amyotrophic lateral sclerosis: why so many negative trials and how can trials be improved? Lancet Neurol 2014;13:1127–1138. [DOI] [PubMed] [Google Scholar]
- 17. Bateman RJ, Munsell LY, Chen X, et al. Stable isotope labeling tandem mass spectrometry (SILT) to quantify protein production and clearance rates. J Am Soc Mass Spectrom 2007;18:997–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bateman RJ, Munsell LY, Morris JC, et al. Human amyloid‐beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med 2006;12:856–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Andorfer C, Kress Y, Espinoza M, et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem 2003;86:582–590. [DOI] [PubMed] [Google Scholar]
- 20. Chan PH, Kawase M, Murakami K, et al. Overexpression of SOD1 in transgenic rats protects vulnerable neurons against ischemic damage after global cerebral ischemia and reperfusion. J Neurosci 1998;18:8292–8299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McCampbell A, Cole T, Wegener AJ, et al. New SOD1 Antisense Oligonucelotides markedly extend survival and reverse muscle denervation in SOD1 ALS rodent models. J Clin Invest 2018;128:3558–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. DeVos SL, Miller TM. Direct intraventricular delivery of drugs to the rodent central nervous system. J Vis Exp 2013;12:50326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mazur C, Fitzsimmons B, Kamme F, et al. Development of a simple, rapid, and robust intrathecal catheterization method in the rat. J Neurosci Methods 2017;280:36–46. [DOI] [PubMed] [Google Scholar]
- 24. Crisp MJ, Mawuenyega KG, Patterson BW, et al. In vivo kinetic approach reveals slow SOD1 turnover in the CNS. J Clin Invest 2015;125:2772–2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sato C, Barthélemy NR, Mawuenyega KG, et al. Tau kinetics in neurons and the human central nervous system. Neuron 2008;97:1284–1298.e1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Winer L, Srinivasan D, Chun S, et al. SOD1 in cerebral spinal fluid as a pharmacodynamic marker for antisense oligonucleotide therapy. JAMA Neurol 2013;70:201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gendron TF, Chew J, Stankowski JN, et al. Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72‐associated amyotrophic lateral sclerosis. Sci Transl Med 2017;9:eaai7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bali T, Self W, Liu J, et al. Defining SOD1 ALS natural history to guide therapeutic clinical trial design. J Neurol Neurosurg Psychiatry 2017;88:99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bertram L, Tanzi RE. The genetic epidemiology of neurodegenerative disease. J Clin Invest 2005;115:1449–1457. 10.1172/JCI24761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bateman RJ, Siemers ER, Mawuenyega KG, et al. A gamma‐secretase inhibitor decreases amyloid‐beta production in the central nervous system. Ann Neurol 2009;66:48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pfister EL, Kennington L, Straubhaar J, et al. Five siRNAs targeting three SNPs in Huntingtin may provide therapy for three‐quarters of Huntington's disease patients. Curr Biol 2009;19:774–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhao HT, John N, Delic V, et al. LRRK2 antisense oligonucleotides ameliorate alpha‐synuclein inclusion formation in a parkinson's disease mouse model. Mol Ther Nucleic Acids 2017;8:508–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huynh TV, Liao F, Francis CM, et al. Age‐dependent effects of apoe reduction using antisense oligonucleotides in a model of beta‐amyloidosis. Neuron 2017;96:1013–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tuszynski MH, Yang JH, Barba D, et al. Nerve growth factor gene therapy: activation of neuronal responses in Alzheimer disease. JAMA Neurol 2015;72:1139–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus sham control in infantile‐onset spinal muscular atrophy. N Engl J Med 2017;377:1723–1732. [DOI] [PubMed] [Google Scholar]
- 36. Schoch KM, DeVos SL, Miller RL, et al. Increased 4R‐tau induces pathological changes in a human‐tau mouse model. Neuron 2016;90:941–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee BH, Lee MJ, Park S, et al. Enhancement of proteasome activity by a small‐molecule inhibitor of USP14. Nature 2010;467:179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Das I, Krzyzosiak A, Schneider K, et al. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 2015;348:239–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Farr GW, Ying Z, Fenton WA, Horwich AL. Hydrogen‐deuterium exchange in vivo to measure turnover of an ALS‐associated mutant SOD1 protein in spinal cord of mice. Protein Sci 2011;20:1692–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. No differences in 13C6‐Leucine oral labeling between treatment groups in all experiments performed. Plasma 13C6‐Leucine values were analyzed in all samples analyzed and compared between groups. A. hTau ICV ASO plasma 13C6‐Leucine at Day 23 post surgery, as seen in Figure 2. No differences were observed (Student's T‐test, P = 0.4496). B. hSOD1 ICV ASO plasma 13C6‐Leucine at Day 30. No differences were observed (Student's‐T test) C. hSOD1 IT ASO plasma 13C6‐Leucine at all time points assessed. No differences were observed (2‐Way ANOVA). Error bars in figures represent SEM.
Figure S2. Correlation between all hTau peptides measured. Linear regression analysis comparing peptides shows strong correlations exist between all three peptide measurements. All p values < 0.0001 for each analysis.
Figure S3. Correlation between all hSOD1 peptides measured. Linear regression analysis comparing peptides shows strong correlations exist between all peptide measurements. All P values < 0.0001 for each analysis.
Figure S4. Characterizing 13C6‐Leucine incorporation into hTau protein in hTauWT transgenic mice. hTau mice were acclimated to leucine‐free chow for 7 days, and labeled with 5 mg/ml 13C6‐leucine for 7 days. Mice were euthanized at 6 (n = 2), 10 (n = 3), 14 (n = 3), and 17 (n = 2) days post‐end of label. Because of the high label incorporation (nearly 15% TTR) and delayed appearance of higher label incorporation after the end of the labeling period 3‐10 days after the end of labeling, subsequent experiments reduced the label time to 4 days. Given maximal label incorporation at 6‐10 days post‐end of label, all mice in the subsequent experiments were sacrificed at 5 days post‐end of label. Error bars in figures represent SEM.
Figure S5. No changes in peripheral hSOD1 after 1000 ug hSOD1 ASO intracerebroventricular injection. A. hSOD1 protein levels relative to N15‐labeled recombinant standard by LC‐MS in plasma. No changes in plasma hSOD1 protein levels were observed (Student's T‐test). B. 13C6‐Leu labeled hSOD1 in plasma by LC‐MS. No changes in plasma 13C6‐Leu labeled hSOD1 was observed (Student's T‐Test). Error bars in figures represent SEM.
Figure S6. No protein concentration pharmacodynamics observed in CSF after 1000 ug hSOD1 ASO intracerebroventricular injection at 50 days post treatment. A. hSOD1 mRNA levels in the CNS by qPCR. hSOD1 mRNA levels are significantly lowered in spinal cord and frontal cortex by ~70% and 50%, respectively, after ASO treatment relative to inactive ASO controls (Student's T‐test). B. hSOD1 protein levels relative to N15‐labeled recombinant standard by LC‐MS. No changes in CSF hSOD1 protein levels were observed (Student's T‐test). n = 5 for each treatment group, with one CSF sample excluded from each group due to blood contamination. **** P < 0.0001. Error bars in figures represent SEM.
Table S1. Primers and probes used for genotyping and qPCR analysis of hTau (MAPT), hSOD1 (SOD1) and mouse GAPDH (GAPDH) DNA.
Table S2. Transition ions used for hSOD1 tandem LC‐MS/MS. GLHGFHVHE peptide measurements were used as representative data in figures.
Table S3. Transition ions used for hTau tandem LC‐MS/MS. TPSLPTPPTR peptide measurements were used as representative data in figures.