

SUMMARY
RNA splicing entails the coordinated interaction of more than 150 proteins in the spliceosome, one of the most complex of the cell’s molecular machines. We previously discovered that the RNA-binding motif protein 17 (RBM17), a component of the spliceosome, is essential for survival and cell maintenance. Here, we find that it interacts with the spliceosomal factors U2SURP and CHERP and that they reciprocally regulate each other’s stability, both in mouse and in human cells. Individual knockdown of each of the three proteins induces overlapping changes in splicing and gene expression of transcripts enriched for RNA-processing factors. Our results elucidate the function of RBM17, U2SURP, and CHERP and link the activity of the spliceosome to the regulation of downstream RNA-binding proteins. These data support the hypothesis that, beyond driving constitutive splicing, spliceosomal factors can regulate alternative splicing of specific targets.
Graphical Abstract
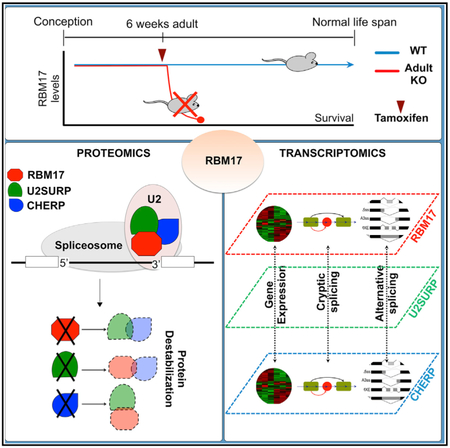
In Brief
De Maio et al. find that the splicing factor RBM17 establishes a physical and functional relation with U2SURP and CHERP. Knockdown of these U2 snRNP-associated spliceosomal components reveals their synergistic activity toward regulation of a given set of transcripts rather than a more predictable transcriptome-wide inhibition of splicing.
INTRODUCTION
Our enormous repertoire of proteins, which vastly exceeds the size of our genome, is generated by RNA splicing, which edits precursor mRNA to remove specific non-coding regions (introns) and join the remaining exons to produce various mature mRNA molecules (Berget et al., 1977; Chow et al., 1977). Some exons are constitutively spliced into mature isoforms, whereas others are alternatively spliced, to be included or excluded depending on the tissue or developmental stage (Fu and Ares, 2014; Luco and Misteli, 2011; Witten and Ule, 2011; Yap and Makeyev, 2013).
This editing of the pre-mRNA transcript into mature mRNA typically occurs within a dynamic macro-protein complex called the spliceosome, whose catalytic core is formed by five small nuclear ribonucleoproteins (snRNPs) (U1 snRNP, U2 snRNP, U4 snRNP, U5 snRNP, and U6 snRNP). The spliceosomal machinery and the control of its activity are quite complex, but the actual splicing reaction is basically a cut-and-paste excision of the unwanted intron and rejoining of the remaining pre-mRNA ends. The basic structure of each snRNP includes a specific small nuclear RNA (snRNA) and a protein ring made of seven Sm proteins (Wahl et al., 2009; Will and Lührmann, 2011). The spliceosome identifies introns by the short, highly conserved consensus sequences that act as docking sites for the snRNPs: a 5′ splice site (5′SS; GU), a 3′ splice site (3′SS; AG), the branchpoint (BP; YNYURAC, with Y as pyrimidine and R as purine), and a polypyrimidine tract. The snRNPs assemble in an orderly fashion on these consensus sequences (Papasaikas and Valcárcel, 2016; Wahl et al., 2009; Will and Lührmann, 2011), and the snRNAs within each snRNP use high-fidelity base pairing with other snRNAs or the mRNA to structurally alter the pre-mRNA. These changes enable the two transesterification reactions that excise the selected intron. At the same time, core and ancillary protein components of each snRNP, including several RNA helicases, coordinate assembly and disassembly of the dynamic spliceosomal machinery (Papasaikas and Valcárcel, 2016; Wahl et al., 2009; Will and Lührmann, 2011).
The splicing cycle is divided into steps that correspond to the formation of five intermediate complexes, E, A, B, B*, and C, that make up the catalytically active spliceosome. In addition to the 150-odd proteins contained in these various iterations of the spliceosome, many other factors contribute to the spatio-temporal regulation of splicing by promoting recruitment of the spliceosome to (or its exclusion from) a specific site (Fu and Ares, 2014; Luco and Misteli, 2011; Witten and Ule, 2011; Yap and Makeyev, 2013). Mutations of RNA splicing sequences or splicing factors that impinge on proper progression of the splicing cycle at any step can therefore cause severe pathology (Cooper et al., 2009; Scotti and Swanson, 2016), if not lethality (Möröy and Heyd, 2007). Increasing evidence, however, indicates that the spliceosome can adapt to alterations in the levels or functioning of specific components (Hsu et al., 2015; Papasaikas et al., 2015). Moreover, limited functioning of core spliceosomal factors does not necessarily cause a generalized reduction of splicing efficiency but can also regulate alternative splicing (Clark et al., 2002; Papasaikas et al., 2015; Park et al., 2004; Pleiss et al., 2007; Saltzman et al., 2011).
We became interested in the splicing factor RNA-binding motif protein (RBM) 17 (also known as splicing factor 45 [SPF45]) when we discovered it is essential for embryonic survival and Purkinje cell maintenance and functions as a repressor of cryptic splicing (Tan et al., 2016). Several cell-based mass spectrometry studies, aimed at characterizing the composition of the spliceosome or of its sub-complexes, found that RBM17 copurifies with the core spliceosomal complex U2 snRNP (Agafonov et al., 2011; Hegele et al., 2012; Neubauer et al., 1998; Papasaikas et al., 2015; Will et al., 2002) and is abundant in the intermediate complex A (Agafonov et al., 2011). Here we perform in vivo studies to delve further into the role of RBM17 within the spliceosome, providing insight into the implications of dynamic interactions among the spliceosomal components.
RESULTS
RBM17 Binds to Spliceosomal Proteins In Vivo
Upon discovering that RBM17 represses cryptic splicing in Purkinje cells, we wanted to gain further insight into its function as a splicing factor in vivo, so we started with an unbiased biochemical analysis of the RBM17 protein interactors. We immunoprecipitated RBM17 from mouse cerebellar protein extracts (Figure S1A) and analyzed the coimmunoprecipitated proteins by mass spectrometry (MS). We identified 156 putative interactors of RBM17 (Table S1) and evaluated them using functional annotation clustering. “Synapse and cell junction,“ “Spliceosome and mRNA processing,” and “Stress fiber” (Figure 1A) were the three most enriched biological categories. “Spliceosome and mRNA processing” was the only cluster that included RBM17, accounting for 19 proteins, of which 14 are factors associated with the spliceosome, and of those, 9 are core or related members (Hegele et al., 2012) of the 17S-U2 snRNP splicing complex (Figure 1B) according to the Spliceosome database (Cvitkovic and Jurica, 2013) (http://spliceosomedb.ucsc.edu/proteins).
Figure 1. RBM17 Binds to Spliceosomal Proteins In Vivo.

(A) Top-ranking hits obtained by IP-MS were analyzed using the DAVID GO functional annotation clustering tool. The bar graph represents the most enriched functional clusters (FDR < 0.05) plotted by their enrichment score. On the right of each bar is indicated the number of genes included in the specific cluster.
(B) Detailed composition of the “Spliceosome and mRNA processing” cluster. Interactors labeled in blue are components of the spliceosome, diamond-shaped interactors are members of the U2 snRNP spliceosomal complex, and gray interactors are proteins involved in RNA metabolism but are not components of the spliceosome.
(C) Representative western blot showing results for the immunoblot analysis of the input, RBM17 IP, and IgG control samples for the validated RBM17 protein interactors.
(D) Reciprocal IP and western blot of the validated interactors to confirm their binding to RBM17.
(E) Representative western blot of the reciprocal IP of the six validated interactors of RBM17 with and without RNase treatment, showing that RNase does not interfere with any of the interactions.
Dashed lines indicate that the image of the membrane has been modified to remove lanes irrelevant to the result. See also Figure S1 and Table S1.
To validate the interactions between RBM17 and its putative spliceosomal partners, we used in vivo coimmunoprecipitation (coIP) followed by immunoblotting (IB). Limited by the availability of verified antibodies, we could test only 11 of the 14 putative interactors, and we validated the reciprocal interactions between RBM17 and six of the spliceosomal factors: SF3B1, SF3B2, DHX15, U2SURP, CHERP, and SNRNP200 (Figures 1C and 1D). Binding between RBM17 and DDX42, DDX46, and EFTUD2 (Figures S1B and S1C) was confirmed in one direction, but not in reciprocal experiments, either because of interference from the antibody or because the interaction is transient or weak; we could not validate the interaction with SF3B3 (no signal) and PRPF6 (non-specific signal in the immunoglobulin G [IgG] lane) (Figures S1B and S1C). One of the confirmed interactors, SNRNP200, is a member of the U5 snRNP complex (Hegele et al., 2012; Wahl et al., 2009), but the remaining five are associated with the 17S-U2 snRNP, supporting the notion that RBM17 associates with this complex in vivo as well.
Interactions among RNA-binding proteins can depend on the protein partners binding closely on RNA sequences. To address this possibility for our confirmed interactions, we immunoprecipitated the six validated protein partners of RBM17 with and without RNase treatment (Figure 1E), which degrades the RNA that could hold together RNA-binding proteins. The RNase treatment did not interfere with RBM17 binding to the bona fide interactors, which indicates that these interactions rely on protein-protein contact.
Rbm17 Deletion in Adult Mice Is Lethal, and Knockdown Alters Levels of Its Interactors
Interactions among two or more proteins can be critical for their stability and function. To determine whether RBM17 influences the levels of the six validated interactors, we measured their abundance after RBM17 loss of function. Because constitutive ablation of Rbm17 causes embryonic lethality and conditional removal of the gene from the developing cerebellum or post-mitotic cerebellar Purkinje cells leads to perinatal lethality and cell death (Tan et al., 2016), respectively, we decided to ablate Rbm17 in adult mice (adult conditional knockout [aCKO]). We used a tamoxifen-inducible ubiquitous Cre (CAGG-CreER) (Hayashi and McMahon, 2002) line and Rbm17flox/− mice (Tan et al., 2016). Sixteen days from the start of tamoxifen administration (Figure S2A), the protein was markedly reduced (Figure S2B), but the mice became moribund (weight loss, reduced activity, and muscle weakness leading to kyphosis) (Figure S2C).
To obtain tissue from healthier animals, we examined the aCKOs at an earlier time point, 14 days after the beginning of tamoxifen treatment (Figure S2D). At this time, RBM17 was reduced by about 70% (Figure S2E). The mice had kyphosis and weight loss, yet most were still alive and in fair condition. By analyzing the cerebellar protein extracts from aCKO mice, we found that the levels of five of six RBM17 interactors were significantly altered when Rbm17 was knocked down (Figure 2A). Specifically, levels of SF3B1, U2SURP, and CHERP decreased, while levels of DHX15 and SNRNP200 increased.
Figure 2. Rbm17 Deletion in Adult Mice Is Lethal, and Knockdown Alters Levels of Its Interactors.

(A) Representative western blots (left) and protein-level quantifications (right) of the six validated interactors of RBM17 in Rbm17 adult conditional knockout (aCKO) mice versus control littermates (n = 6 aCKO and n = 7 control littermates). The area boxed by a dashed line indicates an empty well.
(B) Representative western blots (left) and protein-level quantifications (right) of the six validated interactors of RBM17 in HEK293T treated with siRBM17 or siScramble (n = 6–18 replicates/siRNA). All data were normalized to mouse or human Vinculin (VCL), used here as loading control.
For both panels, bars represent mean ± SEM; p value was calculated by two-tailed Student’s t test, and significance was set at p < 0.05. The 98 kDa mark indicates the relative position of the two bands detected for DHX15 and does not concern any of the other proteins. See also Figure S2.
To evaluate conservation of the functional relationship between RBM17 and its interactors and to rule out the possibility that the effects we observed resulted from the poor health of the aCKO mice, we knocked down RBM17 in HEK293T cells using small interfering RNAs (siRNAs) (siRBM17) and measured the levels of the six interactors (Figure 2B). There was no cell death, and the effect of RBM17 knockdown on the protein levels of SF3B1, U2SURP, CHERP, and DHX15 was conserved.
RBM17, SF3B1, U2SURP, and CHERP Interact with One Another and Regulate Reciprocal Protein Stability
Our finding that RBM17 knockdown led to decreased levels of SF3B1, U2SURP, and CHERP suggests that these factors might interact and reciprocally regulate one another’s stability. To test this hypothesis, we knocked down SF3B1, U2SURP, or CHERP in HEK293T cells to assay the reciprocal effects on protein stability (Figures 3A–3C) and found that loss of function of each of these factors led to reduced levels of RBM17, with the knockdown of U2SURP and CHERP having the strongest effect. U2SURP and CHERP also regulate each other’s levels, while knockdown of U2SURP, but not of CHERP, reduces the abundance of SF3B1 (Figures 3B and 3C). To determine whether the reciprocal regulation among RBM17, U2SURP, CHERP, and SF3B1 affects levels of transcripts or only proteins, we knocked down each protein individually and measured the changes in mRNA levels of the other three by qRT-PCR (Figures S3A–S3D). We found that RBM17 mRNA levels did not change unless cells were treated with siRBM17. U2SURP expression was not reduced by CHERP knockdown (Figure S3D), and CHERP expression was not affected by U2SURP knockdown (Figure S3C); however, U2SURP mRNA was upregulated upon CHERP knockdown (Figure S3D), and CHERP mRNA was upregulated in siRBM17-treated cells (Figure S3A). These results point toward possible compensatory mechanisms that may be activated after loss of function of RBM17 or CHERP. However, RBM17 knockdown reduced SF3B1 levels by 15% (Figure S3A), and SF3B1 knockdown decreased U2SURP expression by 50% (Figure S3B). The reciprocal regulation among RBM17, U2SURP, and CHERP thus seems to occur post-translationally, while direct or indirect RNA regulation could explain the effect of RBM17 knockdown on SF3B1 protein and the impact of the latter on the other three factors.
Figure 3. RBM17, SF3B1, U2SURP, and CHERP Interact with One Another and Regulate Reciprocal Protein Stability.

(A–C) Representative western blots (left) and protein-level quantifications (right) of SF3B1, U2SURP, CHERP, and RBM17 in HEK293T cells treated with siSF3B1 (A), siU2SURP (B), or siCHERP (C) and compared to cells treated with control siScramble (n = 6–18 replicates/siRNA). All data were normalized to VCL, used here as loading control. Bars represent mean ± SEM; p value was calculated by two-tailed Student’s t test, and significance was set at p < 0.05. The presence of a dashed line between Scramble and siRNA samples indicates that the image of the membrane has been modified to eliminate a marker lane between the two sets of samples. See also Figures S3 and S4.
To gain further insight into the relationships among these factors, we tested the reciprocal interaction between SF3B1, U2SURP, and CHERP using immunoprecipitation (IP)-western blot (WB) and found that these proteins bind to one another beyond interacting with RBM17 (Figures S3E–S3G). Moreover, even though western blot analysis of fractions from wild-type brain lysate obtained by size exclusion gel chromatography revealed that RBM17, SF3B1, U2SURP, and CHERP have a similar elution profile (Figures S3H and S3I), the pattern of SF3B1 is slightly shifted toward larger complexes. These data corroborate the hypothesis that RBM17, SF3B1, U2SURP, and CHERP function within similarly composed complexes but strengthen the interpretation of RBM17, U2SURP, and CHERP as having a closer relationship. If this conclusion is accurate, it is plausible to postulate that the interaction among RBM17, U2SURP, and CHERP should be direct. We therefore set out to perform in vitro IP. We were unable to purify U2SURP, a highly disordered protein, but we did have access to recombinant RBM17 and CHERP proteins. By in vitro IP, we showed that CHERP and RBM17 directly bind each other (Figure S3J).
We also evaluated the mutual dependence among RBM17, U2SURP, and CHERP protein levels by measuring the abundance of U2SURP and CHERP proteins in response to a gradient of RBM17 protein obtained by treating HEK293T cells with different concentrations of siRBM17 (Figures S4A and S4B). Levels of U2SURP and CHERP directly correlate to the amount of RBM17 at siRBM17 concentrations of 10 nM and lower (Figure S4B), doses at which we could establish a clear and detectable RBM17 protein gradient.
Lastly, even though reduced RBM17 protein levels do not seem to destabilize all the proteins that we validated as interactors (Figures 2A and 2B), we decided to explore the possibility that RBM17 reduction could affect the stability of the entire U2 snRNP spliceosomal complex. We measured the levels of seven additional factors that the Spliceosome database (Cvitkovic and Jurica, 2013) defines as either core or associated with the 17S-U2 snRNP complex (Figures 4A and 4B); five of these were not detected in our IP-MS, and the remaining two, DDX42 and DDX46, did not validate by IP-WB as RBM17 interactors. This experiment showed that decreased levels of RBM17 in HEK293T cells slightly reduce the levels of these factors, but the effect size is smaller than what we observed for U2SURP and CHERP, with only U2AF1 showing about 30% reduction (Figures 4A and 4B).
Figure 4. Knockdown of RBM17 Only Slightly Affects the Levels of U2 snRNP Components It Does Not Bind.

(A and B) Representative western blots (A) and protein-level quantifications (B) of several U2 snRNP-associated proteins in HEK293T cells treated with siRBM17 and compared to cells treated with control siScramble (n = 12–24 replicates/siRNA). For U2AF2, we quantified both the band of the predicted molecular weight (54 kDa) and an additional band that the antibody manufacturer indicated as specific (65 kDa). All data were normalized to VCL, used here as loading control. Bars represent mean ± SEM; p value was calculated by two-tailed Student’s t test, and significance was set at p < 0.05.
These results suggest that RBM17, U2SURP, and CHERP form a minimal module of proteins functioning together and associating with the U2 snRNP complex.
Individual Knockdowns of RBM17, U2SURP, and CHERP Exhibit Overlapping Molecular Changes
The evidence of a tight reciprocal regulation and interaction among RBM17, U2SURP, and CHERP led us to postulate that they function together to mediate a specific activity, so reducing the levels of any one of them might cause overlapping transcriptional changes. We profiled the gene expression and splicing patterns from total mRNA of HEK293T cells treated with siRBM17, siU2SURP, siCHERP, or siScramble (control). When we compared the differentially expressed genes (DEGs) from the three datasets, the expression changes overlapped by 30%–47% with respect to the background dataset (fold change [FC] > 20% and false discovery rate [FDR] < 0.05 were used as the cutoff in each dataset) (Figure 5A; Table S2), and we validated selected changes using qRT-PCR (Figure 5B). Functional analysis of the overlapping DEGs revealed enrichment for multiple RNA splicing and RNA processing, as well as cell-cycle-related clusters (Figure 5C; Table S3). This suggested that RBM17, U2SURP, and CHERP cooperate to influence RNA homeostasis by modulating other RNA-binding proteins and possibly triggering a quality control feedback system in cell-cycle progression.
Figure 5. Knockdown of RBM17, U2SURP, or CHERP Alters Expression of RNA-Processing Factors.

(A) Plot showing the overlap pattern of differentially expressed genes across the knockdown models of RBM17, U2SURP, or CHERP (n = 3 replicates/siRNA, including siScramble). The red and blue bars represent all genes common to the three datasets (from the three interacting proteins)and consistently altered in the same direction: downregulated (red) and upregulated (blue). The black dots beneath the x axis indicate which proteins have been knocked down for each dataset and the direction of the resulting gene expression changes. The percentages at the top of the graph describe the size of the group of overlapping changes relatively to the biggest and the smallest of the three datasets.
(B) Validation of selected gene expression changes by qRT-PCR. The levels of selected transcripts were measured using qRT-PCR on samples from HEK293T cells treated with siRBM17, siU2SURP, or siCHERP and compared to cells treated with siScramble (n = 4 replicates/siRNA run in technical triplicates). Bars represent mean ± SEM; p value was calculated by two-tailed Student’s t test and significance was set at p < 0.05.
(C) Network plot of functionally enriched categories in overlapping differentially expressed genes. Each node is a functional category. The width of the connector between nodes is directly proportional to the number of genes shared between respective categories.
We next investigated the consequences of loss of RBM17, U2SURP, or CHERP function on both annotated alternative splicing (AS) (Figure 6A; Tables S4 and S5) and cryptic splicing (Figure 6B; Table S4). Knockdown of each of the three proteins caused the alteration of several hundred AS events (Figure 6A) and the appearance of thousands of cryptic junctions (Figure 6B) (both categories of splicing alterations had a cutoff of FDR < 0.05 and inclusion level difference [ILD] ≥ 10%, where ILD is an index of the strength of the splicing event). This suggests that RBM17, U2SURP, and CHERP also share a role in repressing cryptic events. Even the splicing alterations showed considerable overlap across the three datasets, with a stronger effect on the exclusion of the alternative 5′SS and the inclusion of alternative 3′SS (Figure 6A; Table S5); this particular observation is partially consistent with the overall role of the U2 snRNP complex within the splicing cycle and with previously published literature about RBM17 regulation of 3′SS selection in the AS of the Drosophila gene Sex Lethal (Lallena et al., 2002). Using RT-PCR, we validated selected alternative and cryptic splicing alterations (Figures S5 and S6; Table S6). Common mis-spliced transcripts are mostly enriched for RNA metabolism and cell-cycle-regulating factors (Figure 6C; Table S7), in line with what we observed for the gene expression changes. These results support our hypothesis that the three splicing factors regulate a network of RNA-binding proteins involved in RNA homeostasis and directly or indirectly influence cell-cycle progression.
Figure 6. Knockdown of RBM17, U2SURP, or CHERP Reveals Shared Splicing Changes Affecting Numerous RNA-Processing Factors.

(A) Number of exclusion and inclusion splicing events per RNA sequencing dataset relative to the five categories of AS evaluated by rMATS (Shen et al., 2014). The fourth column describes the size of the list of shared events (exclusions and inclusions) across the three datasets calculated as absolute number and as relative percentage of the dataset with the smallest number of events.
(B) Number of cryptic junction gains and losses per RNA sequencing dataset, calculated using CrypSplice. The last column indicates the size of the list of shared events across the three datasets calculated as absolute number and as relative percentage of the dataset with the smallest number of events.
(C) Network plot of functionally enriched categories in overlapping differentially spliced genes. Each node is a functional category. The width of the connector between nodes is directly proportional to the number of genes shared between respective categories.
Next, we analyzed the splicing alterations, annotated and cryptic, shared by the cells with reduced levels of RBM17, U2SURP, or CHERP by looking at their distribution over the ILD (Figures S7A and S7B) and found that most splicing changes associated with loss of function of our proteins of interest have ILD < 0.2 (inclusions) or ILD > −0.2 (exclusion) (Figures S7A and S7B), indicating a large number of mild events. To obtain a more reliable, in-depth analysis of cryptic events, we extended our CrypSplice algorithm (Tan et al., 2016) (see STAR Methods) to classify cryptic alterations into four sub-categories (Figure S7C) based on which portion of the splicing consensus sequences is novel: novel acceptor, novel donor, novel junction, or novel combination between previously annotated exons. Furthermore, the tool distinguishes canonical from non-canonical cryptic junctions according to the presence of the conserved consensus (GU-AG) or a degenerated sequence at the splicing acceptor and/or splicing donor. When we looked at the cumulative distribution function over strength of the four categories of canonical or non-canonical events (Figure S7E), we found that novel donor (ND) (novel 5′SS) events are the strongest among the canonical cryptic junctions and the most abundant within the cryptic junction gains (Figure S7D). Within non-canonical alterations, the novel acceptor (NA) (novel 3′SS) events are the ones with the highest average strength (Figure S7E), but these changes are not extremely numerous (Figure S7D) among neither gain nor loss events.
We next investigated the relationship between gene expression and splicing changes shared across the knockdown of RBM17, U2SURP, and CHERP in cells. About 20% of the genes harboring splicing defects also exhibited transcript-level changes (Figures S7F–S7I), suggesting that most of the splicing alterations we detected do not directly affect the stability of the transcript. We asked which proportion of the genes whose splicing pattern is mutated by knockdown of our proteins of interest carries a premature termination codon (PTC) and might undergo non-sense-mediated decay (NMD) (Figure S7I). About 20% of the genes with aberrant alternative or cryptic splicing carry a PTC or PTCs, and of this fraction, about 10% (2% of the total number of mis-spliced genes) show downregulated (meeting the 20% downregulation cutoff) (Figures S7G–S7I) transcripts potentially ascribable to NMD.
The Molecular Signature of the Functional Unit Formed by RBM17, U2SURP, and CHERP Is Specific
To address the concern that the high overlap in transcriptional alterations observed upon knockdown of RBM17, U2SURP, or CHERP might simply be due to a general impairment of spliceosomal activity, we turned to datasets available through the Encyclopedia of DNA Elements (ENCODE) project. We looked for gene expression data resulting from loss of function of spliceosomal factors not known to interact with RBM17 and belonging to complexes other than U2. Using these criteria, we compared an RNA sequencing dataset from K562 cells treated with short hairpin RNA (shRNA) against RBM17 to those obtained from the same cells treated with shRBM22 (PRP19 related) (Hegele et al., 2012) or shPRP8 (U5 snRNP) (Hegele et al., 2012) (Table S6). This comparison returned far fewer overlapping gene expression changes and almost no common splicing changes (Figures 7A and 7B). We then repeated the analysis comparing the RNA sequencing dataset from K562 cells treated with shRBM17 to those obtained from the same cells treated with shU2AF1 or shU2AF2 (Figures 7C and 7D; Table S6). U2AF1 and U2AF2 form a well-characterized heterodimer that modulates U2 snRNP recruitment to 3′SS and are defined as U2-related factors (Hegele et al., 2012), similar to RBM17, U2SURP, and CHERP; the levels of both U2AF1 and U2AF2 are mildly affected by reduction of RBM17 (Figures 4A and 4B) even though neither of them was identified as a putative RBM17 interactor in our IP-MS experiment. Through this comparison, we intend to rule out the possibility that broad similarities in molecular alterations associated with knockdown of RBM17, U2SURP, or CHERP were not specific to a functional connection among the three proteins but were instead related to their common association with the U2 snRNP complex. We found that the transcriptome of cells expressing reduced levels of RBM17 has little in common with the transcriptome of cells that underwent RNAi against U2AF1 and U2AF2, regardless of the mild effect of RBM17 reduction on U2AF1 and U2AF2 protein levels. Thus, the broad molecular overlap associated with RBM17, U2SURP, or CHERP reduction is specific to their interaction and implies the existence of a functional module associated with the spliceosome. This functional unit controls the expression of a set of transcripts enriched for RNA processing and cell-cycle regulatory factors, likely by fine-tuning of the general U2 snRNP activity.
Figure 7. The Molecular Signature of the Functional Unit Formed by RBM17, U2SURP, and CHERP Is Specific.

(A) Overlap of gene expression changes across cells treated with shRNA against RBM17, RBM22, or PRPF8 in K562 cells. The red and blue bars represent all genes common to the three datasets and consistently altered in the same direction: downregulated (red) or upregulated (blue).
(B) Number of exclusion and inclusion splicing events relative to the five categories of AS evaluated by rMATS and caused by RNAi of RBM17, RBM22, or PRPF8 in K562 cells. The fourth column describes the size of the list of shared events across the three conditions.
(C) Overlap of gene expression changes across cells treated with shRNA against RBM17, U2AF1, or U2AF2 in K562 cells. The red and blue bars represent all genes common to the three datasets and consistently altered in the same direction: downregulated (red) or upregulated (blue).
(D) Number of exclusion and inclusion splicing events relative to the five categories of AS evaluated by rMATS and caused by RNAi of RBM17, U2AF1, or U2AF2. The fourth column describes the size of the list of shared events across the three conditions.
Each of the ENCODE RNA sequencing datasets we reanalyzed has n = 2 replicates/shRNA. SE, skipped exons; A5SS, alternative 5′ splice site; A3SS, alternative 3′ splice site; MXE, mutually exclusive exons; RI, retained introns.
DISCUSSION
In this study, we showed that RBM17 is essential for survival at any stage of life, overcoming the limitations of the conditional knockout models used by Tan et al. (2016) that restricted Rbm17 deletion to early development (since embryonic day 9 [E9]) in the cerebellum and midbrain primordial or to a specific cell type (Purkinje cell). Our previous study profiled the actively translated mRNA of Purkinje cells lacking Rbm17 to investigate its splicing function, but this approach could not examine nuclear mRNA. Here we analyzed total mRNA from HEK293T cells treated with siRBM17 and found that RBM17 loss of function causes extensive alterations of both annotated and cryptic splicing, along with significant gene expression changes.
The in vivo, unbiased proteomic analysis we carried out to define the RBM17 interactome allowed us to identify more than 100 putative protein partners and to validate RBM17 association with the U2 snRNP in mouse; moreover, we were able to confirm in vivo the in vitro findings from high-throughput studies based on protein overexpression that previously suggested RBM17 directly binds to SF3B1, SF3B2, DHX15, U2SURP, and CHERP (Corsini et al., 2007; Hegele et al., 2012; Huttlin et al., 2017). Our data also revealed that RBM17 binds to SNRNP200, confirmed in vivo the interaction between U2SURP and CHERP (Lin-Moshier et al., 2013), and revealed the interaction of both these factors with SF3B1.
Little has been known about U2SURP and CHERP beyond their copurification with the U2 snRNP complex and abundance in the intermediate complex A, which led to their definition as spliceosomal factors (Agafonov et al., 2011; Hegele et al., 2012; Papasaikas et al., 2015; Will et al., 2002). When Lin-Moshieret al. (2013) investigated CHERP interactors in HEK293 cells using MS, their top 26 hits included U2SURP, SF3B1, and SF3B2, all RBM17 partners we validated in vivo and associated with 17S-U2 snRNP. These results are consistent with our finding that the RBM17-U2SURP-CHERP associates with the U2 snRNP complex in vivo.
More importantly, we showed here the specific functional cooperation of RBM17, CHERP, and U2SURP at the molecular level, which is conserved from mouse to human and occurs post-translationally. A possible relationship among these three proteins had been proposed previously by an approach that knocked down 270 splicing factors and tracked the effects on a defined set of 35 AS events (Papasaikas et al., 2015). Our strategy, centered on RBM17, provides a biochemical explanation for their relationship and an unbiased transcrip-tome-wide description of the effect of this functional group on gene expression. The interaction network identified by our IP-MS also suggests that RBM17 links the U2 and the U5 snRNP complexes, because it strongly interacts with the essential U5sn RNP component SNRNP200, a critical player in the step of catalytic activation of the whole spliceosome. This is in line with a previous study that, using high-throughput yeast-two-hybrid screens, observed binding between SF3B2 (U2 snRNP) and SNRNP200 (U5 snRNP) (Hegele et al., 2012; Lin-Moshier et al., 2013).
Analysis of our RNA sequencing data provide several insights into the workings of the spliceosome. First, and surprisingly, altering the levels of abundant spliceosomal components does not necessarily alter constitutive splicing but modulates AS of specific networks of transcripts. Second, spliceosomal factors associated with the U2 snRNP actively control homeostasis of RNA-binding proteins by establishing a feedback loop that links basic splicing and its effectors to modulators of alternative splicing and RNA processing. These results are consistent with previous work describing an active role of the core spliceosome on AS in yeast (Clark et al., 2002; Pleiss et al., 2007), flies (Park et al., 2004), and human cells (Saltzman et al., 2008, 2011).
Lastly, the most notable conclusion of our analysis is the specificity of the common molecular signature associated with RBM17, U2SURP, and CHERP. The knockdown of RBM17 shares almost no molecular changes with the knockdown of other spliceosomal factors (Figures 7A–7D) we tested, irrespective of whether these are related to the U2 snRNP. This evidence strongly suggests that RBM17, U2SURP, and CHERP act as a functional unit that associates with the spliceosome, and in particular the U2 snRNP, to extend the activity of this macro-protein complex beyond regulation of constitutive splicing.
Our analysis also revealed that the gene expression changes linked to RBM17, U2SURP, and CHERP outnumber the genes carrying splicing alterations, letting us infer that in most cases, the observed splicing changes do not alter transcript levels. Because “RNA processing factors” was the most enriched category among the genes with shared splicing changes after RBM17, U2SURP, or CHERP knockdown, it could be that these misregulated RNA-binding proteins are driving most observed gene expression changes. This list includes 83 aberrantly spliced RNA processing factors, 16 of which also exhibited altered transcript abundance. These 16 factors include increased PA-POLA (poly(A) polymerase alpha), necessary for the synthesis of poly(A) tails of mRNA and 3′ end processing (Kaufmann et al., 2004), and downregulated METTL3 (Methyltransferase like 3), which deposits methyl groups on adenosine residues of target mRNAs with consequences on regulation of splicing, RNA processing, translation efficiency, editing, and mRNA stability (Roundtree et al., 2017). Among the remaining 67 RNA processing factors that are aberrantly spliced, but not differentially expressed, are several RBM proteins, RNA helicases, heterogeneous nuclear ribonucleoproteins (hnRNPs), and serine-arginine (SR)-rich proteins.
It is also possible that RBM17-U2SURP-CHERP has an additional role in regulation of transcript stability, but this hypothesis would require further studies to investigate the ability of these three proteins to bind different regions within the transcripts. The “spliceosome and mRNA processing” category (Figure 1A), which revealed the RBM17 interactors we validated, also contained four proteins related to mRNA 3′ end processing and poly(A) tail binding: cleavage stimulation factor subunit 1 (CSTF1), cleavage stimulation factor subunit 3 (CSTF3), poly(A)-binding protein nuclear 1 (PABPN1), and poly(A)-binding protein cytoplasmic 1 (PABPC1). In vivo validation of these interactions and analysis of RBM17’s relationship with these factors could unveil a role for RBM17 in 3′ end processing and poly(A) polymerization and stability.
In addition to providing a deeper understanding of the activity of U2 snRNP-related factors, this study advances computational approaches in decoding specific splicing patterns and their functional consequences. Existing computational approaches explore NMD at the whole transcript level, making it difficult to pinpoint a specific causal splicing error. In this study, we devised an approach to systematically annotate the NMD outcome of specific alternative and cryptic splicing errors. Moreover, we extended the CrypSplice algorithm (Tan et al., 2016) to further categorize the identified splicing alterations (Figures S7C–S7E), enable deeper understanding of their potential functional outcomes, and provide a useful tool for any splicing-centric study.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding author and lead contact, Huda Y. Zoghbi (hzoghbi@bcm.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse handling
All animals were housed in a Level 3, AALAS-certified facility on a 12hr light cycle. Husbandry, housing, euthanasia, and experimental guidelines were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Baylor College of Medicine. Tail tissue was used for PCR genotyping.
Mouse lines
Rbm17flox/+ and Rbm17−/+ animals were previously generated in our laboratory and maintained on a C57BL/6J background. Genotyping protocol and primers used are the same previously described by Tan et al. (2016). Primers are listed in Table S6. The CAGG-CreER line used to generate our adult conditional knockout mice was obtained from the Jackson Lab (stock number 004682). Rbm17−/+;CAGGCreEr/+ animals were bred to Rbm17flox/+ mice to obtain Rbm17flox/−;CAGG-CreER/+ conditional mutants (aCKO) and the control genotypes, Rbm17−/+;CAGG-CreER/+, Rbm17+/+;CAGG-CreER/+, Rbm17flox/+;CAGG-CreER/+, Rbm17−/+;+/+, Rbm17+/+;+/+, Rbm17flox/++/+, Rbm17flox/−;+/+. An n number (indicated in the specific figure legend) of Rbm17flox/−;CAGG-CreER/+ and Rbm17flox/+;+/+ mice of both sexes have been sacrificed to obtain the lysates used for the biochemical experiments described in Figures 2 and S2. 12 weeks old, wild-type, males and females C57BL/6J mice were sacrificed to perform the biochemical experiments described in Figures 1, S1, and S3E–S3G. About 20 weeks old, wild-type, males C57BL/6J mice were used for the biochemical experiments described in Figure S3H–S3I.
Cell lines
HEK293T cells were cultured in DMEM (Thermo Fisher Scientific-Fisher, Waltham, MA USA, MT10013CV) medium supplemented with 10% FBS at 37°C in 5% CO2.
METHOD DETAILS
Tamoxifen treatment
Tamoxifen (Sigma-Aldrich, St. Louis, MO, USA, T5648) was dissolved to 20 mg/ml in peanut oil (Sigma-Aldrich, P2144-1L), aliquoted and frozen at −20°C until use. Peanut oil was also used as vehicle. Tamoxifen or vehicle-only was injected intraperitoneally at a dose of 100 mg/kg, every other week-day for a total of 5 injections. The mice were sacrificed for tissue collection at day 16 or, according to the paradigm, 14 days after the first injection (as described in Figures S2A and S2D).
RNAi in HEK293T cells by siRNAs
HEK293T cells were seeded in 24-well plate. 24h after seeding the cells were transfected with 40nM (unless specified differently) of target siRNAs (Dharmacon, GE-Healthcare, Lafayette, CO, USA, RBM17: SMARTpool: ON-TARGETplus RBM17 siRNA L-005158-01-0005; SF3B1: SMARTpool: ON-TARGETplus SF3B1 siRNA, L-020061-01-0005; U2SURP: SMARTpool: ON-TARGETplus U2SURP siRNA, L-023607-02-0005; CHERP: SMARTpool: ON-TARGETplus CHERP siRNA, L-016176-02-0005) or scramble Control siRNAs (Dharmacon, GE-Healthcare, Scramble: ON-TARGET plus Non-targeting siRNA #3, D-001810-03-20). Transfection was performed using the jetPRIME DNA and siRNA Transfection Reagent-(Polyplus-transfection) (VWR-Summus, Radnor, PA, USA, 89129-922) and the cells were cultured for 72 hours before harvesting for protein or RNA preparation.
Protein lysates and immunoblot analysis
HEK293T cells or mouse cerebellar tissue were homogenized in RIPA buffer (25 mM Tris-HCL pH 7.6, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with Xpert Protease and Phosphatase Inhibitor Cocktail Solutions (100X) (GenDepot, Barker, TX, USA, P3100-100, P3200-020) using, respectively, pipetting or syringe trituration. After homogenization the samples were placed for 30 minutes on ice followed by centrifugation at 13,000 rpm at 4°C for 30 minutes to separate debris from the supernatant protein extract. Proteins were quantified by Pierce BCA Protein Assay Kit (FISHER, PI23225), resolved by high resolution Nupage 4%–12% Bis-Tris Gel (Thermo Fisher Scientific-Invitrogen, NP0336BOX, NP0335BOX) according to the manufacturer’s instructions and analyzed by western blotting. Densitometry was performed using ImageJ software.
All the antibodies used in this paper are listed in the Key Resources Table. For detection of RBM17 and SF3B1 protein by immunoblot the primary antibodies used are the following: Mouse Anti-RBM17 (Neuro mab, Davis, CA, USA, clone N219/5)(Dilution 1:2500); Mouse Anti-SF3B1 (MBL, Woburn, MA, USA, Clone 1A5 D138-3)(Dilution 1:1000). Upon RBM17-IP the RBM17 signal was detected by western blot using the above primary antibody and the following secondary antibody: Secondary Goat anti-Mouse light chain specific HRP conjugated (Jackson ImmunoResearch Labs 115035174) (Dilution 1:10000)
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-RBM17 | Neuromab | Cat# clone N219/5; RRID:AB_10672987 |
| Rabbit polyclonal anti-RBM17 | Bethyl Laboratories Inc | Cat# A310-909A; RRID: AB_1966060 and 1966059 |
| Mouse monoclonal anti-GAPDH | Advanced Immunochemical Inc | Cat# 2-RGM2 |
| Mouse monoclonal anti-VINCULIN | Sigma-Aldrich | Cat#V9131; RRID:AB_477629 |
| Mouse monoclonal anti-SF3B155 | MBL | Cat# D138-3; RRID:AB_592713 |
| Rabbit polyclonal anti-SF3B155 | Bethyl Laboratories Inc | Cat# A300 997A; RRID: AB_2186516 |
| Rabbit polyclonal anti-SF3B2 | Bethyl Laboratories Inc | Cat# A310-569A; RRID: AB_1078826 and 1078828 |
| Rabbit polyclonal anti-DHX15 | Bethyl Laboratories Inc | Cat# A300-390A; RRID:AB_2091992 |
| Rabbit polyclonal anti-DDX42 | Bethyl Laboratories Inc | Cat# A303-354A; RRID:AB_10953655 |
| Rabbit polyclonal anti-DDX46 | Bethyl Laboratories Inc | Cat# A310-342A; RRID:AB_873028 |
| Rabbit polyclonal anti-EFTUD2 | Bethyl Laboratories Inc | Cat# A300-957A; RRID:AB_805780 |
| Rabbit polyclonal anti-CHERP | Bethyl Laboratories Inc | Cat# A304-621A; RRID:AB_2620816 |
| Rabbit polyclonal anti-U2SURP | Bethyl Laboratories Inc | Cat# A304-616A; RRID:AB_2620811 |
| Rabbit polyclonal anti-SNRNP200 | Bethyl Laboratories Inc | Cat# A311-273A; RRID:AB_10953013 |
| Rabbit polyclonal anti-SNRPA1 | Bethyl Laboratories Inc | Cat# A303-948A; RRID:AB_2620297 |
| Rabbit polyclonal anti-Prpf6 | Bethyl Laboratories Inc | Cat# A311-017A; RRID:AB_10630423 |
| Rabbit polyclonal anti-U2AF1 | Bethyl Laboratories Inc | Cat# A310-734A; RRID: AB_1604295 and AB_1604296 |
| Rabbit polyclonal anti-U2AF2 | Abcam | Cat# ab37530; RRID:AB_883336 |
| Rabbit polyclonal anti-SF3A1 | Bethyl Laboratories Inc | Cat# A301-604A; RRID:AB_1078823 |
| Rabbit polyclonal anti-SF3B4 | Bethyl Laboratories Inc | Cat# A303-950A; RRID:AB_2620299 |
| Goat anti-Rabbit HRP conjugated | Biorad | Cat# 170-5046; RRID:AB_11125757 |
| Donkey anti-Mouse HRP conjugated | Jackson ImmunoResearch Lab | Cat# 715035150; RRID:AB_2340770 |
| Goat anti-Mouse IgG light chain specific HRP | Jackson ImmunoResearch Lab | Cat# 115035174; RRID:AB_2338512 |
| Normal Rabbit IgG | Millipore | Cat# 12370; RRID:AB_145841 |
| Rabbit Anti-GST | Bethyl Laboratories Inc | Cat # A190-222A; RRID:AB_67420 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tamoxifen | Sigma-Aldrich | Cat# T5648-5G |
| CHERP (NM_006387) Human Recombinant Protein | Origene | Cat # TP 761205 |
| RBM17 (Human) Recombinant Protein (P01) | Fisher/Abnova | Cat # H00084991- P01 |
| Critical Commercial Assays | ||
| Aurum Total RNA Fatty and Fibrous Tissue Kit | Biorad | Cat# 7326870 |
| Pierce BCA Protein Assay Kit | Fisher | Cat# PI23225 |
| Quantitect Reverse Transcription kit | QIAGEN | Cat# 205311 |
| TruSeq Stranded mRNA Library Preparation Kit | Illumina | RS-122-2101 |
| Agencourt AMPure XP Kit | Beckman Coulter | A63881 |
| SuperScript II Reverse Transcriptase | Thermofisher | 18064-014 |
| Qubit RNA HS Assay Kit | Thermofisher | Q32852 |
| Bioanalyzer HS DNA Kit | Agilent Technologies | 5067-4626 |
| Qubit dsDNA HS Assay Kit | Thermofisher | Q32851 |
| KAPA DNA Standards | KAPA Biosystem (now Roche) | KK4903 |
| Library Quantification Kit-Illumina/Universal | KAPA Biosystem (now Roche) | KK4824 |
| HiSeq PE Cluster Kit v4 cBot | Illumina | PE-401-4001 |
| HiSeq SBS Kit v4 (250 cycles) | Illumina | FC-401-4003 |
| Deposited Data | ||
| Raw RNA-seq data | This paper | GEO: GSE107648 |
| Mass spectrometry data | This paper | MASSIVE: PXD008363 |
| Experimental Models: Organisms/Strains | ||
| Rbm17Flox/− | Tan et al., 2016 | JAX: Cat# 030716 and 030715 |
| CAGG-CreER | Hayashi and McMahon, 2002 | JAX: Cat# 004682 |
| Oligonucleotides | ||
| Primers used for qRT-PCR, sqPCR and genotyping see Table S6 | This paper | N/A |
| SMARTpool: ON-TARGETplus RBM17 siRNA | Dharmacon, GE Healthcare | Cat# L-005158-01-0005 |
| SMARTpool: ON-TARGETplus SF3B1 siRNA | Dharmacon, GE Healthcare | Cat# L-020061-01-0005 |
| SMARTpool: ON-TARGETplus U2SURP siRNA | Dharmacon, GE Healthcare | Cat# L-023607-02-0005 |
| SMARTpool: ON-TARGETplus CHERP siRNA | Dharmacon, GE Healthcare | Cat# L-016176-02-0005 |
| ON-TARGET plus Non-targeting siRNA #3 | Dharmacon, GE Healthcare | Cat# D-001810-03-20 |
| Software and Algorithms | ||
| FastQC v0.10.1 | Andrews, 2010 | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| STAR 2.5.3a | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| rMATS | Shen et al., 2014 | http://rnaseq-mats.sourceforge.net/ |
| Crypesplice see STAR Methods | This paper | N/A |
| ImageJ | https://imagej.nih.gov/ij/ | RRID:SCR_003070 |
| OmniGraffle 6 | Omni Group | N/A |
| GraphPad Prism 6.0c | http://www.graphpad.com/ | RRID:SCR_002798 |
| Spliceosome database | Cvitkovic and Jurica, 2013 | N/A |
| Xcalibur Software | ThermoFisher Scientific | RRID:SCR_014593 |
| Proteome Discoverer | ThermoFisher Scientific | RRID:SCR_014477 |
| Mascot | http://www.matrixscience.com/server.html | RRID:SCR_014322 |
| Other | ||
| Scripts for analysis of sequencing data | This paper | http://www.liuzlab.org/U2complex/Scripts_U2.zip |
| Mass spectrometry data | This paper | http://proteomecentral.proteomexchange.org |
| RNA-sequencing data | This paper | https://www.ncbi.nlm.nih.gov/geo/ |
| ENCODE-datasets | ENCODE | http://www.liuzlab.org/U2complex/ENCODE Data_ links.xlsx |
Immunoprecipitations, IP
Mouse cerebellar tissue was processed with Dounce homogenizer in NETN buffer (50mM Tris ph 7.5, 0.5% NP40, 150mM NaCl, 1mM EDTA) supplemented with Xpert Protease and Phosphatase Inhibitor Cocktail Solutions (100X) (GenDepot, P3100-100, P3200-020). After homogenization the samples were placed for 30 minutes on ice followed by centrifugation at 13,000 rpm at 4°C for 30 minutes to separate debris from the supernatant protein extract. The extract was then diluted 1:5 in lysis buffer and 500 μL were used for each immunoprecipitation. Lysates were initially incubated overnight with antibodies at 4°C on an end-to-end rotator. The day after 40 μL of Protein G plus agarose (Fisher/Pierce, 22851) beads slurry was added to each sample and incubated for 4h at 4°C on an end-to-end rotator. The beads slurry was prepared performing two initial washes with 1 mL of lysis buffer, blocked with 1 mL of 1%BSA (Sigma-Aldrich, 3116964001) solution in lysis buffer for 30 minutes at 4°C on an end-to-end rotator, and finally washed three times in lysis buffer before resuspension of the compact volume of beads in an equal amount of lysis buffer. After incubation with the beads the samples were centrifuged at 5050 rpm for one minute to compact the beads and the supernatant was harvested as post-IP sample. The beads were then washed four times with lysis buffer and eventually resuspended in 40 μL elution buffer (Lysis buffer, Nupage 10X Reducing Agent [Invitrogen, NP0009], Nupage LDS sample buffer 4X [Invitrogen, NP0007]) and boiled at 95°C for 10 minutes before loading the samples in 10 wells Nupage 4%–12% Bis-Tris Gels for further resolution and western blot analysis.
If the IP required RNase treatment the beads were resuspended in 400 μL of Lysis buffer after three final washes and split in two different Eppendorf tubes, 200 μL each. One of the tubes was treated with 4 μL of RNase I (Invitrogen, EN0602) and incubated 15 minutes at 37°C on an end-to-end rotator. For consistency even the samples that were not treated with RNase I were incubated 15 minutes at 37°C. After incubation all the samples were washed one last time with 500 μL of lysis buffer and then eluted in 20 μL of elution buffer.
The immunoprecipitation protocol followed before submitting the samples for Mass spectrometry analysis was the following: we dissected cerebella of twelve C57BL/6J male mice on ice, pulled them together in groups of two to use three final samples for the RBM17 IP and three for the IgG control. After weighing the starting material we added 3 volumes of NENT lysis buffer (see above) supplemented with protease and phosphatase inhibitors (Xpert GenDepot).The tissue was then sonicated using a GE (GE505) sonicator; the sonication paradigm used lasted 3 minutes with 30 s ON pulses and 59 s OFF pulses and amplitude of 20%. The homogenate was spun at 4°C for 5 minutes at 10,000 g. The supernatant was then moved to an ultra-centrifuge tube (Beckman microfuge tube 357448) and spun down in a Beckman (Optima Max XP) ultra-centrifuge at 4°C for 20 minutes at 100,000 g (60,000rpm), with acceleration 1 and deceleration 1. Immediately after spinning an equal amount of supernatant was withdrawn from each sample and moved to a fresh ultra-centrifuge tube. Before proceeding further, protein concentration was measured by Bradford assay. An optimal concentration of at least 10 mg/ml was required for our mass spectrometry analysis. At this point we saved 10 μL of each sample to prepare Input for western blot analysis and then added to each sample 5 μg of RBM17 antibody mixture (Bethyl A302-497A and A302-498A). We incubated the lysates with the antibody for 2h at 4°C on a rotator. Upon incubation we ultra-centrifuged them at 4°C for 20 minutes at 100,000 g (60,000rpm), and then took the supernatant and moved it to a fresh regular Eppendorf tube. We added to the lysate 20 μL of Protein A-Sepharose CL-4B (Fisher, 45-000-143) beads slurry in PBS 1X. We incubated the mix for 2h at 4°C on a rotator and then spun them down at 4°C for 1 minute at 500xg. We removed the supernatant and saved it to prepare post-IP samples for later western blot analysis. We then washed the beads with 500 μL of NETN three times, spinning them down each time by centrifugation at 4°C for 1 minute at 500xg. After the last wash, we took all the supernatant out and re-suspended the beads in 20 μL of eluition buffer (see above). We boiled them at 95°C for 5 minutes and then performed Mass spectrometry analysis. All the antibodies used in this paper are listed in the Key Resources Table. For the IP of RBM17 and SF3B1 the primary antibodies used are the following: Rabbit Anti-RBM17 (Bethyl Lab. A302-497A and A302-498A)(2.5 μg each); Rabbit Anti-SF3B1 (Bethyl Lab. A300-997A)(4 μg).
HPLC-MS/MS
Immunopurified samples were subjected to 4%–20% Tris/Glycine SDS-PAGE (Novex Gel, Invitrogen). The resolved proteins were visualized with Coomassie Brilliant blue-stain and excised into 4 gel pieces according to molecular size. The gel pieces were destained and subjected to in-gel digestion using trypsin (GenDepot T9600). The tryptic peptide was dried under vacuum and was resuspended in 10 μL of loading solution (5% methanol containing 0.1% formic acid). A half of suspended peptide was injected a nano-HPLC 1000 system (Thermo Scientific) coupled to LTQ Orbitrap Elite(Thermo Scientific) mass spectrometer. The peptides were loaded onto an in-house Reprosil-Pur Basic C18 (3 μm, Dr.Maisch GmbH, Germany) trap column, which was 2 cm × 100 μm size. Then the trap column was washed with loading solution and switched in-line with an in-house 5 cm × 150 um column packed with Reprosil-Pur Basic C18 equilibrated in 0.1% formic acid/water. The peptides were separated with a 75 min discontinuous gradient of 2%–24% acetonitrile/0.1% formic acid at a flow rate of 800 nl/min. Separated peptides were directly electro-sprayed into the mass spectrometer. The instrument was operated in data-dependent mode, acquiring fragmentation spectra of the top 25 strongest ions and under direct control of Xcalibur software (Thermo Scientific). Parent MS spectrum was acquired in the Orbitrap with a full MS range of 375-1300 m/z in the resolution of 240,000. CID fragmented MS/MS spectrum was acquired in ion-trap with rapid scan mode. Obtained MS/MS spectra were searched against target-decoy Mouse Ref-seq database (release June 2015, containing 58549 entries) in Proteome Discoverer 1.4 interface (Thermo Fisher) with Mascot algorithm (Mascot 2.4, Matrix Science). Variable modification of hydroxyethyl disulfide for cysteine, oxidation of methionine and protein n-terminal acetylation was permitted. The precursor mass tolerance was confined within 20 ppm with fragment mass tolerance of 0.5 Dalton and a maximum of two missed cleavages was allowed. Assigned peptides were filtered with 1% false discovery rate (FDR) and subject to manual verifications. iBAQ algorithm was used to calculate protein abundance to compare relative amount between different proteins in the sample using in house data processing algorithm (Jung et al., 2017). iBAQ was calculated based on normalization of summed peptide intensity divided by the number of theoretically observable tryptic peptides of a certain protein.
Analysis of Mass Spectrometry results
Spectral counts were filtered by number of average unique peptides (Up), using an arbitrary cut off of ≤ 2Up for the IgG samples and ≥ 2 Up for the RBM17 IP samples. Then, the ratios between averages RBM17 IP iBAQ and IgG iBAQ was calculated and the list filtered by an arbitrary cut off for this ratio of ≥ 2.
In Vitro immunoprecipitation
Full-length Human RBM17 recombinant protein with an N-terminal GST tag was obtained from Abnova (Novus Biologicals, LLC, 8100 Southpark Way, A-8,Littleton, CO 80120, USA)(cat #: H00084991) and full-length Human recombinant CHERP with an N-terminal HIS was obtained from Origene (OriGene Technologies, Inc. 9620 Medical Center Drive, Suite 200, Rockville, MD 20850, USA) (cat #: TP761205). 10 μg purified CHERP was incubated with 60 μL pre-washed TALON metal affinity resin (Clontech,1290 Terra Bella Ave. Mountain View, CA 94043, USA) (cat #: 635502), slurry in 500 μL of binding buffer (6 M urea 20 mM BME 0.5 M NaCl 30 mM Imidazole 50 mM NaPO4 pH 7.4) for 45 minutes at room temperature. Beads were carefully resuspended in immunoprecipitation (IP) buffer (75 mM NaCl 50 mM Tris pH 8.0 0.5% Triton X-100) to a final volume of 1 ml, incubated 10 minutes and spun down on a tabletop centrifuge. Buffer was removed from beads and replaced with 300 μL chilled IP buffer. 1 μg purified RBM17 was added and 10% input was collected. The mixture was rotated at 4°C for 2 hours. 10% post-IP was collected and supernatant was removed. Beads were washed with 300 μL chilled IP buffer 3 times then boiled 10 minutes in 1X SDS buffer supplemented with 500 mM imidazole. A negative control (minus the presence of CHERP) was included to ensure GST-tagged RBM17 did not bind nonspecifically to the TALON metal affinity resin.
Size exclusion gel chromatography
Cerebellar extracts for size exclusion chromatography (SEC) were prepared fresh by dounce homogenization of two cerebella from age-matched mice (~20 weeks) in protein extraction buffer (0.5% Triton X-100, 50mM Tris-HCl pH 8.0, 75mM NaCl) supplemented with complete protease inhibitor cocktail (Fritz Hoffmann-La Roche, Basel, Switzerland), phosphatase inhibitors, and PMSF. 3.5 mg of the total soluble extract were loaded onto a Superose 6 GL300 column using the AKTA purifier UPC10 system from GE. The column buffer was identical to the protein extraction buffer except that Triton X-100 was reduced to 0.1% and NaCl was 50mM. SEC was performed as described previously (Lam et al., 2006; Lim et al., 2008). In short, The flow rate was set at 0.4 ml/min and 1 mL fractions were collected, column void volume was 7.7 ml. Thyroglobulin (669kDa), ADH (150kDa), and Cytochrome C (12.4kDa) were used as gel-filtration standards.
RNA extraction
Total RNA was obtained from HEK293T cells (see Cell Lines) or mouse cerebellar tissue (see Mouse Lines) using the Aurum Total RNA Fatty and Fibrous Tissue Kit (Biorad, 7326870) according to the manufacturer’s instructions. RNA was quantified using the NanoDrop 1000 (Thermo Fisher). Quality of RNA was assessed by gel electrophoresis. cDNA was synthesized using Quantitect Reverse Transcription kit (QIAGEN, Hilden, Germany, 205311) starting from 1 μg of DNase-treated RNA.
Quantitative real time PCR
qRT-PCR experiments were performed using the CFX96 Touch Real-Time PCR Detection System (BioRad Laboratories) with Powerup SYBR master mix (Fisher, A25777). Real-time PCR results were analyzed using the comparative Ct method (Vandesompele et al., 2002) and normalized against the housekeeping gene Hs-TBP or mm-S16 (see Table S6).
Semiquantitative reverse transcription-PCR
sqRT-PCR experiments were performed using EconoTaq PLUS 2X GREEN mastermix (Lucigen, 2905 Parmenter Street Middleton, WI 53562) and analyzed on 1.5% agarose gels in TBE 0.5X. Percentage Spliced In (PSI) and Relative Inclusion (RI) were calculated using densitometry measure of the bands identified by electrophoresis using ImageJ software. PSI = ([inclusion band]/[inclusion + exclusion band])*100; RI = ([band of interest in siRNA treated cells]/[TBP])/([band of interest in Scramble treated cells]/[TBP]). Validation of gene expression changes and splicing alterations identified by RNA-sequencing was carried out on newly generated and independent samples.
Primers
In order to unambiguously distinguish spliced cDNA from genomic DNA contamination, specific exon primers were designed to amplify across introns of the genes tested. All primers were previously tested by reverse transcription (RT)-PCR and –RT controls reactions were performed. The primers for all target genes tested were designed with Primer3 v.0.4.0 (Koressaar and Remm, 2007) and ordered to Sigma-Aldrich.
Primer sequences
The sequences of the primers used for qRT-PCR, for sqRT-PCR and genotyping are listed in Table S6.
RNA–sequencing
Three RNA samples from HEK293T cells treated with each of the on-Target siRNAs (siRBM17, siU2SURP or siCHERP) or scramble control siRNAs (see Cell Lines and RNAi in HEK293T Cells by siRNA), for a total of 12 samples, were quality controlled and processed by the Genomic and RNA Profiling Core at Baylor College of Medicine. The Core first conducted Sample Quality checks using the NanoDrop spectrophotometer and Agilent (Santa Clara, CA, USA) Bioanalyzer 2100. They then used Illumina (San Diego, CA, USA) TruSeq Stranded mRNA library preparation protocol (p/n 15031047, rev. E) along with ThermoFisher’s ERCC RNA Spike-In Control Mixes Protocol (publication number 4455352, rev. D) to generate the actual sequence. The protocol is as follows: A double-stranded DNA library was created using 250ng of total RNA (measured by picogreen), preparing the fragments for hybridization onto a flow cell. ERCC RNA Spike-In Controls were added to each sample according to the manufacturer’s protocol. First, cDNA was created using the fragmented 3′ poly(A) selected portion of total RNA and random primers. During second strand synthesis, dTTP was replaced with dUTP, which quenches the second strand during amplification, thereby achieving strand specificity. Libraries were created from the cDNA by first blunt ending the fragments, attaching an adenosine to the 3′ end and finally ligating unique adapters to the ends (For more information on this process, see below). The ligated products were then amplified using 15 cycles of PCR. The resulting libraries were quantitated using the NanoDrop spectrophotometer and fragment size assessed with the Agilent Bioanalyzer. A qPCR quantitation was performed on the libraries to determine the concentration of adapte-ligated fragments using Applied Biosystems (Thermo Fisher Scientific) ViiA7 Real-Time PCR System and a KAPA Library Quant Kit.
Cluster Generation by Bridge Amplification
Using the concentration from the ViiA7 qPCR machine above, 29pM of library was loaded onto a high output v4 flowcell and amplified by bridge amplification using the Illumina cBot machine. A paired-end 100 cycle run was used to sequence the flowcell on a HiSeq 2500 Sequencing System.
RNASeq data analysis
For each sample lane-wise raw reads were obtained in zipped fastq format. Multiple lane reads of each sample were pooled by concatenating respective fastq files. Sequencing quality and any adaptor contamination are assessed using FastQC v0.10.1 (Andrews, 2010).
Read alignment
Pooled reads were aligned to human reference genome GRCh38 using STAR 2.5.3a (Dobin et al., 2013). Raw FASTA sequence and annotations of genome build GRCh38 were downloaded from UCSC genome browser portal (http://hgdownload.soe.ucsc.edu/downloads.html#human). Raw genome is indexed by setting–runMode to genomeGenerate in STAR. To reliably align splice junction, the raw reads were aligned to the indexed genome by setting an anchor length (splice junction) of 5 and-outSAMstrandField to in-tronMotif. Sample-wise alignments were saved as coordinate sorted binary format (BAM) files.
Differential Gene Expression, DEG
Gene expression values from each sample were quantified as the number of reads mapped (to a specific gene) by setting–quantMode to GeneCounts in STAR. Genes with an average read count < 50 across the samples were considered not expressed and were excluded from the DEG analysis. Raw read counts were normalized and then tested for differential expression using DESeq2 (Love et al., 2014). A false discovery rate (FDR) cutoff of 0.05 and a fold change cutoff of 20% (−0.263 ≤ log2(FC) ≥ +0.263) were used to call DEGs. Sample clustering was assessed by principle component analysis (PCA) on normalized read counts (Yalamanchili et al., 2017). If multiple gene Ids mapped to a same gene symbol, the gene Id with maximum read counts was considered representative.
Splicing analysis
Alternative splicing events were quantified and classified using rMATS. Alignment files (BAM) and reference annotations (GTF) from UCSC genome browser were passed to rMATS. Insert length is computed as average fragment size (400 bp) - (2*read length). rMATS classifies splicing events into 5 categories, skipped exons, retained introns, mutually exclusive exons, alternative 5′ and 3′ splice sites. An FDR cutoff of 0.05 and an inclusion level difference cutoff of less than −0.1 or greater than 0.1 were used to screen for statistically significant changes.
Unannotated or Cryptic splicing changes were quantified using CrypSplice (Tan et al., 2016). However, the method was extended to give deeper insights into the identified cryptic junctions. Based on junction anchor each of the cryptic junction is classified as NJ (novel donor and novel acceptor), NC (novel donor and acceptor combination), ND (novel donor only/Alternative 5′SS) and NA (novel acceptor only/ Alternative 3′SS). Furthermore, each splice site is classified as canonical (GT-AG) or non-canonical (non-GT-AG) sites. All non-annotated junctions are considered cryptic and are labeled Gains and Losses based on respective junction strength difference. Junction strength difference is computed as the difference of junction strength between knock-down and control samples. Junctions with FDR less than 0.05 and junction strength difference ≤ −0.1 and ≥ +0.1 were called significant junction losses and gains respectively. A noise cutoff (minimum reads supporting a junction) of 10 was applied. Junctions spanning more than one gene are assigned to the gene of origin.
Gene Ontology analysis
Functional enrichment analyses were performed on gene lists of interest using WebGestalt (WEB-based GEne SeT AnaLysis Toolkit) (Zhang et al., 2005) with an FDR cutoff of 0.05. Minimum number of genes per category was set to 5. Extracted enrichments were then visualized using GoPlot (Walter et al., 2015) and Cytoscape (Shannon et al., 2003).
Nonsense-mediated decay analysis
De-novo transcripts were assembled from RBM17 knock down samples using cufflinks (Trapnell et al., 2010) and reference GRCh38 annotations (GTF) from UCSC genome browser. Replicate assemblies were merged using cuffmerge and differential analysis was performed using cuffdiff. SpliceR was used to identify premature stop codons (PTC) in the assembled transcripts with the 50nt rule (Vitting-Seerup et al., 2014). Putative splicing-induced NMD-targets were identified mapping splicing events to PTC harboring transcripts.
Splicing strength density plots
Density for each of the splicing category is computed and plotted using geom_density and ggplot (Wickham, 2009) in R. Cumulative density plots for canonical (GT-AG) and non-canonical (non GT-AG) splice sites are computed and plotted using ecdf function in R.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were done with GraphPad-Prism 6.0c and the significance was set at p < 0.05. All the data requiring statistical testing have been analyzed using two-tailed Student’s t test or one-way ANOVA followed by Tukey’s multiple comparisons test. Figure legends and figures contain all statistical details of the experiments described, including the statistical tests performed, the p values, the exact number of n, the nature of the values represented in every plot and the errors calculated. No data point was ever excluded from the quantifications.
DATA AND SOFTWARE AVAILABILITY
The accession number for the sequencing data reported in this paper is GEO: GSE107648. The accession number for the mass-spectrometry data reported in this paper is ProteomeXchange Consortium: PXD008363 (http://proteomecentral.proteomexchange.org).
Scripts and pipelines used in this study are available at: http://www.liuzlab.org/U2complex/Scripts_U2.zip. ENCODE datasets used in this study are listed in Table S6.
Supplementary Material
Highlights.
RBM17 is essential at any stage of life
RBM17, U2SURP, and CHERP regulate reciprocal protein stability
Knockdown of RBM17, U2SURP, or CHERP does not inhibit splicing
RBM17, U2SURP, and CHERP together regulate a specific set of transcripts
ACKNOWLEDGMENTS
We thank the members of the Zoghbi and Liu laboratories for helpful suggestions and discussions and C.E. Alcott, Q. Tan, T.A. Cooper, and V.L. Brandt for valuable advice on the manuscript. V.A.G. is supported by the National Ataxia Foundation (Young Investigator Research Grant 2017), United States. J.Q. and S.Y.J. are supported by the NIH (P30CA125123, NCI center grant), United States. This project was supported by the Howard Hughes Medical Institute, United States; the NIH/National Institute of Neurological Disorders and Stroke (NIH/NINDS) (2R37NS027699), United States; the Baylor College of Medicine IDDRC (U54HD083092, administrative core), United States; the Cancer Prevention Research Institute of Texas (RP170387), Houston endowment, Huffington foundation (Z.L.), United States; the NIH/National Institute of Aging (NIH/NIA) (AG05733902), United States; the NIH/National Institute of General Medical Sciences (NIH/NIGMS) (GM12003302), United States; a P30 Digestive Disease Center support grant (NIDDK-DK56338, genomic and RNA profiling core), United States; and a P30 Cancer Center support grant (NCI-CA125123) at Baylor College of Medicine, United States.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and seven tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.09.041.
REFERENCES
- Agafonov DE, Deckert J, Wolf E, Odenwälder P, Bessonov S, Will CL, Urlaub H, and Lührmann R (2011). Semiquantitative proteomic analysis of the human spliceosome via a novel two-dimensional gel electrophoresis method. Mol. Cell. Biol. 31, 2667–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S (2010). FastQC: a quality control tool for high throughput sequence data. www.bioinformatics.babraham.ac.uk/projects/fastqc. [Google Scholar]
- Berget SM, Moore C, and Sharp PA (1977). Spliced segments at the 5′ terminus of adenovirus 2 late mRNA. Proc. Natl. Acad. Sci. USA 74, 3171¬3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow LT, Gelinas RE, Broker TR, and Roberts RJ (1977). An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell 12, 1–8. [DOI] [PubMed] [Google Scholar]
- Clark TA, Sugnet CW, and Ares M Jr. (2002). Genomewide analysis of mRNA processing in yeast using splicing-specific microarrays. Science 296, 907–910. [DOI] [PubMed] [Google Scholar]
- Cooper TA, Wan L, and Dreyfuss G (2009). RNA and disease. Cell 136, 777–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsini L, Bonnal S, Basquin J, Hothorn M, Scheffzek K, Valcárcel J, and Sattler M (2007). U2AF-homology motif interactions are required for alternative splicing regulation by SPF45. Nat. Struct. Mol. Biol. 14, 620–629. [DOI] [PubMed] [Google Scholar]
- Cvitkovic I, and Jurica MS (2013). Spliceosome database: a tool for tracking components of the spliceosome. Nucleic Acids Res. 41, D132–D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu XD, and Ares M Jr. (2014). Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 15, 689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, and McMahon AP (2002). Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre:a tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol. 244, 305–318. [DOI] [PubMed] [Google Scholar]
- Hegele A, Kamburov A, Grossmann A, Sourlis C, Wowro S, Weimann M, Will CL, Pena V, Lührmann R, and Stelzl U (2012). Dynamic protein-protein interaction wiring of the human spliceosome. Mol. Cell 45, 567–580. [DOI] [PubMed] [Google Scholar]
- Hsu TY, Simon LM, Neill NJ, Marcotte R, Sayad A, Bland CS, Echeverria GV, Sun T, Kurley SJ, Tyagi S, et al. (2015). The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature 525, 384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, et al. (2017). Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung SY, Choi JM, Rousseaux MW, Malovannaya A, Kim JJ, Kutzera J, Wang Y, Huang Y, Zhu W, Maity S, et al. (2017). An anatomically resolved mouse brain proteome reveals Parkinson disease-relevant pathways. Mol. Cell. Proteomics 16, 581–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann I, Martin G, Friedlein A, Langen H, and Keller W (2004). Human Fip1 is a subunit of CPSF that binds to U-rich RNA elements and stimulates poly(A) polymerase. EMBO J. 23, 616–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koressaar T, and Remm M (2007). Enhancements and modifications of primer design program Primer3. Bioinformatics 23, 1289–1291. [DOI] [PubMed] [Google Scholar]
- Lallena MJ, Chalmers KJ, Llamazares S, Lamond AI, and Valcárcel J (2002). Splicing regulation at the second catalytic step by Sex-lethal involves 3′ splice site recognition by SPF45. Cell 109, 285–296. [DOI] [PubMed] [Google Scholar]
- Lam YC, Bowman AB, Jafar-Nejad P, Lim J, Richman R, Fryer JD, Hyun ED, Duvick LA, Orr HT, Botas J, and Zoghbi HY (2006). ATAXIN-1 interacts with the repressor Capicua in its native complex to cause SCA1 neuropathology. Cell 127, 1335–1347. [DOI] [PubMed] [Google Scholar]
- Lim J, Crespo-Barreto J, Jafar-Nejad P, Bowman AB, Richman R, Hill DE, Orr HT, and Zoghbi HY (2008). Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature 452, 713–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin-Moshier Y, Sebastian PJ, Higgins L, Sampson ND, Hewitt JE, and Marchant JS (2013). Re-evaluation of the role of calcium homeostasis endoplasmic reticulum protein (CHERP) in cellular calcium signaling. J. Biol. Chem 288, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco RF, and Misteli T (2011). More than a splicing code: integrating the role of RNA, chromatin and non-coding RNA in alternative splicing regulation. Curr. Opin. Genet. Dev 21, 366–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möröy T, and Heyd F (2007). The impact of alternative splicing in vivo: mouse models show the way. RNA 13,1155–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubauer G, King A, Rappsilber J, Calvio C, Watson M, Ajuh P, Sleeman J, Lamond A, and Mann M (1998). Mass spectrometry and EST-database searching allows characterization of the multi-protein spliceosome complex. Nat. Genet 20, 46–50. [DOI] [PubMed] [Google Scholar]
- Papasaikas P, and Valcárcel J (2016). The spliceosome: the ultimate RNA chaperone and sculptor. Trends Biochem. Sci 41, 33–45. [DOI] [PubMed] [Google Scholar]
- Papasaikas P, Tejedor JR, Vigevani L, and Valcárcel J (2015). Functional splicing network reveals extensive regulatory potential of the core spliceosomal machinery. Mol. Cell 57, 7–22. [DOI] [PubMed] [Google Scholar]
- Park JW, Parisky K, Celotto AM, Reenan RA, and Graveley BR (2004). Identification of alternative splicing regulators by RNA interference in Drosophila. Proc. Natl. Acad. Sci. USA 101, 15974–15979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleiss JA, Whitworth GB, Bergkessel M, and Guthrie C (2007). Transcript specificity in yeast pre-mRNA splicing revealed by mutations in core spliceosomal components. PLoS Biol. 5, e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roundtree IA, Evans ME, Pan T, and He C (2017). Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltzman AL, Kim YK, Pan Q, Fagnani MM, Maquat LE, and Blencowe BJ (2008). Regulation of multiple core spliceosomal proteins by alternative splicing-coupled nonsense-mediated mRNA decay. Mol. Cell. Biol 28, 4320–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltzman AL, Pan Q, and Blencowe BJ (2011). Regulation of alternative splicing by the core spliceosomal machinery. Genes Dev. 25, 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotti MM, and Swanson MS (2016). RNA mis-splicing in disease. Nat. Rev. Genet 17, 19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, and Xing Y (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. USA 111, E5593–55601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Q, Yalamanchili HK, Park J, De Maio A, Lu HC, Wan YW, White JJ, Bondar VV, Sayegh LS, Liu X, et al. (2016). Extensive cryptic splicing upon loss of RBM17 and TDP43 in neurodegeneration models. Hum. Mol. Genet 25, 5083–5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, and Speleman F (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitting-Seerup K, Porse BT, Sandelin A, and Waage J (2014). spliceR: an R package for classification of alternative splicing and prediction of coding potential from RNA-seq data. BMC Bioinformatics 15, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, and Lührmann R (2009). The spliceosome: design principles of a dynamic RNP machine. Cell 136, 701–718. [DOI] [PubMed] [Google Scholar]
- Walter W, Sánchez-Cabo F, and Ricote M (2015). GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics 31, 2912–2914. [DOI] [PubMed] [Google Scholar]
- Wickham H (2009). Ggplot2: Elegant Graphics for Data Analysis (Springer; ). [Google Scholar]
- Will CL, and Lührmann R (2011). Spliceosome structure and function. Cold Spring Harb. Perspect. Biol 3, a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will CL, Urlaub H, Achsel T, Gentzel M, Wilm M, and Lührmann R (2002). Characterization of novel SF3b and 17S U2 snRNP proteins, including a human Prp5p homologue and an SF3b DEAD-box protein. EMBO J. 21, 4978–4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witten JT, and Ule J (2011). Understanding splicing regulation through RNA splicing maps. Trends Genet. 27, 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalamanchili HK, Wan YW, and Liu Z (2017). Data analysis pipeline for RNA-seq experiments: from differential expression to cryptic splicing. Curr. Protoc. Bioinformatics 59, 11.15.1–11.15.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap K, and Makeyev EV (2013). Regulation of gene expression in mammalian nervous system through alternative pre-mRNA splicing coupled with RNA quality control mechanisms. Mol. Cell. Neurosci 56, 420–428. [DOI] [PubMed] [Google Scholar]
- Zhang B, Kirov S, and Snoddy J (2005). WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 33, W741–W748. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.