

Abstract
Improving feed efficiency in cattle is important because it increases profitability by reducing costs, and it also shrinks the environmental footprint of cattle production by decreasing manure and greenhouse gas emissions. Residual feed intake (RFI) is 1 measurement of feed efficiency and is the difference between actual and predicted feed intake. Residual feed intake is a complex trait with moderate heritability, but the genes and biological processes associated with its variation still need to be found. We explored the variation in expression of genes using RNA sequencing to find genes whose expression was associated with RFI and then investigated the pathways that are enriched for these genes. In this study, we used samples from growing Angus bulls (muscle and liver tissues) and lactating Holstein cows (liver tissue and white blood cells) divergently selected for low and high RFI. Within each breed-tissue combination, the correlation between the expression of genes and RFI phenotypes, as well as GEBV, was calculated to determine the genes whose expression was correlated with RFI. There were 16,039 genes expressed in more than 25% of samples in 1 or more tissues. The expression of 6,143 genes was significantly associated with RFI phenotypes, and expression of 2,343 genes was significantly associated with GEBV for RFI (P < 0.05) in at least 1 tissue. The genes whose expression was correlated with RFI phenotype (or GEBV) within each breed-tissue combination were enriched for 158 (78) biological processes (Fisher Exact Statistics for gene-enrichment analysis, EASE score < 0.1) and associated with 13 (13) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (P < 0.05 and fold enrichment > 2). These biological processes were related to regulation of transcription, translation, energy generation, cell cycling, apoptosis, and proteolysis. However, the direction of the correlation between RFI and gene expression in some cases reversed between tissues. For instance, low levels of proteolysis in muscle were associated with high efficiency in growing bulls, but high levels of proteolysis in white blood cells were associated with efficiency of milk production in lactating cows.
Keywords: Bos taurus, differential gene expression, feed efficiency, residual feed intake, RNA sequencing
INTRODUCTION
Improving feed efficiency in dairy and beef cattle is an important objective to reduce production costs (Beever and Doyle, 2007; Tizioto et al., 2015). Furthermore, efficient cattle produce less methane and manure, which reduces the environmental footprint of cattle production (Hegarty et al., 2007). Residual feed intake (RFI) is 1 measurement of feed efficiency and is the difference between actual and predicted feed intake (Koch et al., 1963; Berry and Crowley, 2013). However, the expense of measuring RFI limits its use in breeding programs (Pryce et al., 2015). Given the complexity of the trait, it has proven difficult to find the causal genes by genome-wide association (GWA) studies (Bolormaa et al., 2013). Nevertheless, GEBV of RFI have already been calculated using reference populations of dairy and beef cattle with low to moderate accuracy (Khansefid et al., 2014; Pryce et al., 2015). To find the genes influencing RFI, the association between variation in gene expression and RFI can be studied as an alternative method to GWAS because the expression of genes may influence traits (Albert and Kruglyak, 2015). However, the correlation between gene expression and RFI may be different in different tissues and between growing and lactating cattle.
The aim of this study was to find the genes, biological processes, and pathways that are associated with RFI variation in growing and lactating cattle using RNA sequencing (RNA-Seq). Residual feed intake in beef cows is correlated with RFI in growing heifers (Jeyaruban et al., 2009; Khansefid et al., 2014), so we expected some of the same biological processes to be involved. However, a second aim was to compare the correlation between gene expression in lactating cows and growing cattle. Therefore, we analyzed gene expression data from beef and dairy cattle separately and then compared the results.
MATERIAL AND METHODS
Animals and RFI Records
The beef cattle used in our study were Angus bulls from lines of cattle divergently selected for low and high RFI on the basis of their individual RFI values at the Agricultural Research Centre in Trangie, NSW, Australia (Arthur et al., 2001). Since the second generation of divergent selection, the animals in the low-RFI line consumed significantly (P < 0.05) less feed than those selected for high RFI (Arthur and Herd, 2008). Liver samples were taken from 37 bulls (including 11 paternal half-sibling groups) from a single cohort after approximately 3 generations of selection. Semitendinosus muscle samples were taken from 43 bulls from a different cohort that had been selected for RFI for approximately 4 generations (consisting of 8 paternal half-sibling groups).
The 19 Holstein cows used in this study were selected using their phenotypes as growing heifers from a feed efficiency trial at the Agriculture Victoria Rutherglen Research Station (n = 843, average RFI = 0, and SD = 0.19; Williams et al., 2011). The extreme animals for RFI (top and bottom 10% using phenotype data) were used in this study. These calves were then evaluated in first lactation to determine if they were still divergent for RFI in lactation (Macdonald et al., 2014). Blood and liver samples were taken from the 19 lactating Holsteins (including 3 paternal half-sibling groups) at 38 d in milk (±10 d). The RFI of the lactating cows was calculated (Macdonald et al., 2014), and the RFI phenotypes for Holstein cattle in this study refer to cows in lactation. In this study, we used the GEBV for RFI in lactating Holstein and GEBV for RFI during growth in Angus cattle reported by Pryce et al. (2015) and Khansefid et al. (2014), respectively. The GEBV of the Holsteins come from a multitrait model in which the 2 traits were RFI as heifers and RFI as cows, but it is the GEBV for RFI as cows that is used here. A summary of animal RFI records is shown in Table 1.
Table 1.
Residual feed intake (RFI) phenotypes and GEBV mean and SD in each group
| Breed (No.) | Phenotype | GEBV | |||
|---|---|---|---|---|---|
| Tissue | Mean | SD | Mean | SD | |
| Angus (43) | Muscle | 0.22 | 1.03 | 0.16 | 0.58 |
| High-RFI line (22) | 0.44 | 1.15 | 0.39 | 0.60 | |
| Low-RFI line (21) | −0.01 | 0.85 | -0.08 | 0.45 | |
| Angus (37) | Liver | 0.06 | 0.98 | 0.22 | 0.64 |
| High-RFI line (23) | 0.47 | 0.88 | 0.59 | 0.42 | |
| Low-RFI line (14) | −0.61 | 0.76 | −0.39 | 0.44 | |
| Holstein (19) | Liver and blood | 0.05 | 1.27 | −0.01 | 0.20 |
| High-RFI (9) | 0.09 | 1.09 | 0.16 | 0.11 | |
| Low-RFI (10) | 0.02 | 1.47 | −0.17 | 0.12 | |
Sampling and RNA-Seq Data
Liver and Muscle Samples from Angus Bulls
Liver tissue from Angus animals was sampled according to the procedure described by Chen et al. (2011) under University of New England Animal Ethics Committee application number AEC 06/123. (We used a subset of the animals used by Chen et al., 2011.) Semitendinosus muscle samples were taken from growing bulls (about 9 mo of age) by biopsy and then transferred into 2 mL RNAlater solution (Ambion, Applied Biosystems, Austin, TX) under Orange Animal Ethics Committee application number ORA09/015. Total RNA was extracted from liver and muscle tissue by TRI Reagent (Ambion) and Qiagen RNeasy MinElute kit (Qiagen, Hilden, Germany) using a modified protocol. In brief, approximately 30 mg of liver tissue (100 mg of muscle tissue) were finely minced and then mixed with 500 μL TRIzol Reagent (Life Technologies, Carlsbad, CA) and immediately homogenized for approximately 45 to 50 s and then incubated at room temperature for 5 min. The resulting lysate was mixed with 100 μL 1-bromo-3-chloropropane (BCP) incubated at room temperature for 10 min, followed by centrifugation at 10,000 × g at 4°C for 15 min. The top aqueous layer was transferred to a new microfuge tube and mixed with an equal volume of 75% ethanol. The resulting lysate was then loaded onto the Qiagen RNeasy MinElute column, and RNA was purified as per protocol with on-column DNA digestion. The extracted RNA quality was assessed using an Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA); only those with RNA integrity number (RIN) greater than 7 were used. Ten micrograms of the extracted RNA from the sampled tissues were enriched with a Dynabeads mRNA Purification Kit (Invitrogen, Carlsbad, CA). The mRNA molecules were randomly fragmented by heating in the presence of Mg2+. First-strand cDNA synthesis was performed using random primers and SuperScriptII reverse transcriptase (Invitrogen) following the manufacturer's recommendation. The second-strand cDNA was synthesized with DNA Polymerase I and RNase H (Invitrogen), and ends were polished by addition of T4 DNA Polymerase and Klenow DNA Polymerase. Twenty-four unique bar-coded adapters were developed, adenylated with Klenow Fragment (3′→5′ exo), and ligated to the ends of the double-stranded cDNA fragments by using T4 DNA ligase (New England Biolabs Ltd., Ipswich, MA). The libraries were enriched by PCR with Phusion High-Fidelity DNA Polymeras (New England Biolabs Ltd.), purified with Agencourt AMPure XP (Beckman Coulter, Brea, CA), and selected for a target size of 200 bp. The bar-coded libraries were pooled and run on a single lane of a HiSeq 2000 (Illumina Inc., San Diego, CA) in a 101-cycle paired-end run.
Liver and Blood Samples from Holstein Cows
Blood sampling and liver tissue biopsy from Holstein cows, RNA extraction, and sequencing were described by Chamberlain et al. (2015; Department of Economic Development, Jobs, Transport and Resources, Animal Ethics application number 2011-23). In brief, 10 mL of whole blood were collected by venipuncture of the coccygeal vein using a BD Vacutainer (BD Vacutainer Systems, Franklin Lakes, NJ). The white blood cells were separated by centrifugation at 2,000 g for 15 min at 4°C, then stored in 1.2 mL RNAlater RNA Stabilization Solution (Ambion, Applied Biosystems). Two to three grams of liver tissue were collected by biopsy and then were frozen in liquid nitrogen. The RNA was extracted from white blood cells using a RiboPure Blood Kit (Ambion, Applied Biosystems) and from liver samples using a RiboPure Kit (Ambion, Applied Biosystems) according to the manufacturer's instructions. All samples had RIN greater than 8.0. Sequencing libraries were prepared using the TruSeq RNA Sample Preparation Kit v2 Set A (Illumina Inc.) and selected for a size of 200 bp. All libraries were uniquely bar-coded, pooled, and sequenced on a HiSeq 2000 (Illumina Inc.) in a 105-cycle paired-end run.
Reference Genome
The bovine genome assembly UMD3.1 was customized for each animal to improve the mapability of the reads by reducing the mismatches in aligning RNA-Seq reads to the reference genome (Chamberlain et al., 2015). The Holsteins were genotyped with an Illumina HD Bovine SNP chip (Pryce et al., 2012). The 37 Angus bulls with liver samples were genotyped (Bolormaa et al., 2014) with the low-density 50K genotypes (Illumina BovineSNP50K chip), and then HD genotypes were imputed using BEAGLE (Browning and Browning, 2009). The 43 Angus bulls with muscle samples had whole genome sequence data (WGS) with average 6.7-fold coverage (Daetwyler et al., 2014). FImpute was used to impute WGS from HD genotypes and phase all WGS genotypes (Sargolzaei et al., 2014). Parental haplotypes for each animal were then used to make a maternal and a paternal reference genome as described by Chamberlain et al. (2015).
Quality Control and Alignment of Reads
The quality of raw RNA-Seq reads was checked using FastQC (Andrews, 2010). Reads were trimmed of adaptor sequence and bases with Phred+33 score < 15, where 3 consecutive bases had a Phred+33 score less than 15; the rest of the sequence was removed. Reads with average Phred+33 score < 20 or read length < 50 after trimming were removed, and only the paired reads were used in alignment (Ross et al., 2012). Reads were aligned to maternal and paternal customized reference genomes for each animal using TopHat2 (Kim et al., 2013) using Ensembl GFF (General Feature Format) release 75 for bovine genome assembly UMD3.1 and allowing 2 mismatches per read. The mismatches can be due to different nucleotides in the reference genome and the RNA-Seq read (real mismatches) or sequencing errors in RNA-Seq data. So the number of “real mismatches” can be reduced using the actual genome for each animal as the reference genome. The mean and SD for the distribution of inner distances between mate pairs in each animal were estimated using Burrows-Wheeler Aligner (BWA) software (Li and Durbin, 2009).
Gene Expression Measurement
The expression abundance of each gene was estimated by counting the number of reads mapped to each gene in the reference using HTSeq (Anders et al., 2015). The RNA-Seq libraries were not prepared with a strand-specific protocol, so we used the HTSeq option of stranded=no. As 2 reference genomes (maternal and paternal) were used for aligning reads in each animal, 2 counts for each gene were generated, which were expected to be similar. Where the reads mapped to 1 reference genome slightly better, we used the higher counts as a measure for expression of the gene. Gene read counts for the animals in each group were first normalized with a weighted trimmed mean using edgeR (Robinson and Oshlack, 2010). Then, the normalized counts were log transformed to make normal distributions for gene read counts.
Gene Expression Associated with RFI
To find the differentially expressed genes related to feed efficiency, we tested the association between the normalized gene read counts and RFI variation. The regression of each gene count on RFI phenotypic records and GEBV were calculated separately in each RNA-Seq data set using ASReml (Gilmour et al., 2009) to find genes significantly associated with RFI phenotypes (GSAphenotype) and GEBV (GSAGEBV), respectively (P < 0.05). The sign of the regression coefficient can be used to determine whether a genes was upregulated (+) or downregulated (−) in animals with higher RFI. The false discovery rate (FDR) in each RNA-Seq data set was calculated as (Bolormaa et al., 2013)
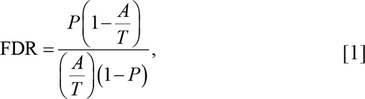 |
where P is the P-value of the test (i.e., 0.05), A is the number of GSAphenotype or GSAGEB at P, and T is the total number of genes tested.
Differentially Expressed Gene Annotation
The genes whose expression was significantly correlated with RFI phenotype or GEBV (i.e., GSAphenotype and GSAGEBV) in each of the 4 RNA-Seq analyses were used to investigate the biological themes associated with RFI variation. The lists of differentially expressed genes in each data set were analyzed with DAVID online software (Huang et al., 2009) separately to test if any functional annotation or biological pathways were overrepresented among the genes. The gene ontology (GO) at the biological process level and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of GSAphenotype and GSAGEBV were investigated to find biological mechanisms (P < 0.05 and fold enrichment > 2) and pathways (Fisher Exact Statistics for gene-enrichment analysis, EASE scores < 0.1) associated with RFI. In each tissue, the genes expressed in that tissue were used as background in biological process and KEGG pathway analyses. As the blood had more than 3,000 differentially expressed genes, the 3,000 most significant genes associated with RFI phenotypes were used because of a limitation for the number of genes in DAVID.
RESULTS
Animals and RFI Records
The correlation between RFI records and GEBV (rPhenotypes,GEBV) was high in Angus bulls (0.83 ± 0.09 for bulls with muscle samples and 0.86 ± 0.09 for bulls with liver samples) but low in Holstein cows (0.27 ± 0.23), which was not significantly different from zero (P = 0.26). The low rPhenotypes,GEBV in dairy cattle was probably due to the limited number of cows used to calculate GEBV for RFI in lactating cows and the fact that in calculation of GEBV, data from the animals as heifers were used by Pryce et al. (2015). The high rPhenotypes,GEBV in beef cattle was because the beef animals were from divergent RFI lines, the RFI phenotype was used in the calculation of GEBV, and a comparatively large number of animals (n = 820) were used to calculate GEBV for RFI.
RNA-Seq Data
On average, about 75% of the raw RNA-Seq reads for the Angus muscle and liver libraries passed Quality control (QC) filters (paired reads). For the Holstein blood and liver libraries, on average, about 80% of the reads passed QC filters (paired reads). The details of RNA-Seq data in addition to RFI phenotype and GEBV for each individual are provided in Supplementary File 1. A brief summary of the alignment of reads and their concordant pair rate is shown in Table 2. The concordant pair alignment rate indicates the percentage of reads that both forward and reverse sequence map to the reference genome correctly with proper distance. The approximate distance or inferred external insert size in each RNA-Seq data set was estimated by BWA (Li and Durbin, 2009); on average, it was roughly 175 (±50) in liver and blood samples of Holstein cows and 185 (±60) in liver and 200 (±60) in muscle samples of Angus bulls.
Table 2.
The mean (and SD) of raw, quality control (QC) passed paired sequences and concordant of paired alignment rate of RNA sequencing reads in each data set
| Breed | Tissue | Raw reads | Reads passing QC (Paired) | Paired aligned | Concordance |
|---|---|---|---|---|---|
| Angus | Muscle | 17,906,533 (4,372,921) | 6,366,128 (1,866,319) | 5,796,348 (1,710,722) | 90% (2%) |
| Angus | Liver | 13,616,027 (7,751,116) | 5,437,947 (3,065,337) | 4,478,339 (2,739,338) | 77 (8%) |
| Holstein | Liver | 34,964,230 (7,205,821) | 14,764,421 (3,385,481) | 13,701,770 (3,052,165) | 92% (1%) |
| Holstein | Blood | 40,042,855 (6,307,797) | 16,246,344 (2,841,461) | 14,710,129 (2,543,539) | 89 (1%) |
Analysis of Gene Expression
Eight separate analyses were performed for each of 4 sets of tissue samples (Angus liver, Angus muscle, Holstein liver, and Holstein blood) for 2 measures of RFI (phenotype and GEBV). The number of genes expressed in more than 25% of animals in each group and also the number of genes associated with RFI phenotypes and GEBV (P < 0.05) are shown in Table 3, and details of individual genes are available in Supplementary File 2. Among the 16,039 genes expressed in at least 1 tissue, 6,143 GSAphenotype and 2,343 GSAGEBV were found (P < 0.05). However, in most data sets the FDR was high. Therefore, we focused on genes whose expression was correlated with RFI in more than 1 data set. The expression of 1,850 genes was significantly associated with RFI in more than 1 RNA-Seq data set. The overlap between GSAphenotype and GSAGEBV within tissue samples in Angus bulls was much higher (475 for muscle and 460 for liver samples) than in Holstein cows (23 for liver and 6 for blood samples), which is mainly due to higher correlation between RFI phenotypes and GEBV in Angus bulls and probably higher accuracy of GEBV for RFI in growing animals. The overlap between GSAphenotype and GSAGEBV in different tissues is shown in Fig. 1 A and 1B, respectively. Comparing the GSA (phenotypes or GEBV) in different tissues showed the direction of the correlation between gene expression and RFI records was not necessarily the same across different tissues. Table 4 shows the number of genes whose expression was correlated with RFI in more than 1 analysis, and these genes are listed in Supplementary File 3. For instance, the expression of 363 genes was correlated with RFI phenotype in both Holstein blood and Angus muscle, but for 259 (363-104) genes the direction of the correlation reversed.
Table 3.
Total number of genes expressed in each data set in more than 25% of animals and the number of genes significantly associated with residual feed intake (RFI) phenotypes (GSAphenotype) and GEBV (GSAGEBV; P < 0.05) that were downregulated (–) or up-regulated (+) in animals with higher RFI
| No. of genes expressed | GSAphenotype | GSAGEBV | ||||||
|---|---|---|---|---|---|---|---|---|
| Breed | Tissue | All (FDR1) | – | + | All (FDR1) | – | + | |
| Angus | Muscle | 12,278 | 922 (0.65) | 468 | 454 | 791 (0.76) | 381 | 410 |
| Angus | Liver | 12,233 | 768 (0.79) | 295 | 473 | 1028 (0.57) | 438 | 590 |
| Holstein | Liver | 14,374 | 473 (1.55) | 233 | 240 | 526 (1.39) | 285 | 241 |
| Holstein | Blood | 14,176 | 4817 (0.10) | 2561 | 2256 | 137 (5.39) | 43 | 94 |
False discovery rate.
Figure 1.

The overlap between genes significantly associated (P < 0.05) with (A) residual feed intake phenotypes and (B) GEBV in different tissues sampled from growing Angus bulls (muscle and liver tissues) and lactating Holstein cows (liver tissue and white blood cells).
Table 4.
The number of genes whose expression was correlated with residual feed intake in more than 1 analysis1
| Angus muscle | Angus liver | Holstein liver | Holstein blood | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Data set | RFI | Phenotype | GEBV | Phenotype | GEBV | Phenotype | GEBV | Phenotype | GEBV |
| Angus muscle | Phenotype | — | 475 | 58 | 78 | 30 | 27 | 363 | 5 |
| GEBV | 475 | — | 53 | 66 | 24 | 24 | 301 | 7 | |
| Angus liver | Phenotype | 35 | 29 | — | 460 | 35 | 22 | 254 | 3 |
| GEBV | 47 | 40 | 460 | — | 39 | 36 | 413 | 4 | |
| Holstein liver | Phenotype | 13 | 12 | 19 | 16 | — | 23 | 140 | 3 |
| GEBV | 15 | 12 | 11 | 12 | 23 | — | 137 | 6 | |
| Holstein blood | Phenotype | 104 | 81 | 85 | 54 | 120 | 110 | — | 6 |
| GEBV | 2 | 5 | 1 | 1 | 2 | 4 | 6 | — | |
On the upper triangle elements are the total number of genes, and on the lower triangle elements are those with effects in the same direction.
Enriched Biological Processes and KEGG Pathways
In 8 analyses performed separately for each tissue-breed combination, in total, the GSAphenotype were enriched for 158 biological processes (EASE score < 0.1) and overrepresented with 13 KEGG pathways (P < 0.05 and fold enrichment > 2). The GSAGEBV were enriched in 78 biological processes and overrepresented in 13 KEGG pathways.
There were 39 biological processes that were enriched in more than 1 of the 8 analyses and 29 that were enriched repeatedly in different RNA-Seq data sets (Table 5). The list of the genes associated with all biological processes is available in Supplementary File 4. There were also 3 KEGG pathways that were overrepresented in more than 1 analysis. All of the overrepresented KEGG pathways are shown in Table 6.
Table 5.
The gene ontology (GO) terms of biological processes enriched in genes significantly associated with residual feed intake phenotypes (GSAphenotype) and GEBV (GSAGEBV) repeatedly in different analyses
| GO term | GO definition | Analysis (No. of genes) | Common genes1 |
|---|---|---|---|
| GO:0006412 | Translation | Angus liver, GEBV (32); Angus muscle, GEBV (27); Angus muscle, phenotype (38); Holstein blood, phenotype (142) | 58 |
| GO:0019941 | Modification-dependent protein catabolic process | Angus liver, GEBV (24); Angus muscle, GEBV (19); Angus muscle, phenotype (22); Holstein blood, phenotype (81) | 32 |
| GO:0043632 | Modification-dependent macromolecule catabolic process | Angus liver, GEBV (24); Angus muscle, GEBV (19); Angus muscle, phenotype (22); Holstein blood, phenotype (81) | 32 |
| GO:0006508 | Proteolysis | Angus liver, GEBV (42); Angus muscle, GEBV (41); Angus muscle, phenotype (38) | 32 |
| GO:0030163 | Protein catabolic process | Angus liver, GEBV (30); Angus muscle, GEBV (23); Holstein blood, phenotype (96) | 25 |
| GO:0044257 | Cellular protein catabolic process | Angus liver, GEBV (28); Angus muscle, GEBV (21); Holstein blood, phenotype (88) | 22 |
| GO:0055114 | Oxidation reduction | Angus liver, GEBV (54); Angus liver, phenotype (36); Holstein liver, phenotype (20) | 29 |
| GO:0006325 | Chromatin organization | Angus muscle, phenotype (15); Holstein blood, phenotype (59) | 8 |
| GO:0006396 | RNA processing | Angus muscle, phenotype (26); Holstein blood, phenotype (113) | 10 |
| GO:0007049 | Cell cycle | Angus liver, phenotype (19); Holstein blood, phenotype (92) | 7 |
| GO:0007166 | Cell surface receptor linked signal transduction | Angus muscle, GEBV (29); Holstein blood, GEBV (7) | 1 |
| GO:0009057 | Macromolecule catabolic process | Angus liver, GEBV (35); Holstein blood, phenotype (115) | 14 |
| GO:0009063 | Cellular AA catabolic process | Angus muscle, GEBV (5); Holstein liver, phenotype (5) | 2 |
| GO:0016071 | mRNA metabolic process | Angus muscle, phenotype (16); Holstein blood, phenotype (71) | 6 |
| GO:0016568 | Chromatin modification | Angus muscle, phenotype (11); Holstein blood, phenotype (42) | 7 |
| GO:0032313 | Regulation of Rab GTPase activity | Angus liver, phenotype (6); Holstein blood, phenotype (16) | 3 |
| GO:0032318 | Regulation of Ras GTPase activity | Angus liver, phenotype (7); Holstein blood, phenotype (23) | 4 |
| GO:0032483 | Regulation of Rab protein signal transduction | Angus liver, phenotype (6); Holstein blood, phenotype (16) | 3 |
| GO:0034621 | Cellular macromolecular complex subunit organization | Holstein blood, phenotype (60); Holstein liver, GEBV (10) | 3 |
| GO:0044265 | Cellular macromolecule catabolic process | Angus liver, GEBV (32); Angus muscle, phenotype (26) | 4 |
| GO:0045449 | Regulation of transcription | Angus muscle, phenotype (67); Holstein blood, phenotype (319) | 29 |
| GO:0045597 | Positive regulation of cell differentiation | Angus liver, phenotype (9); Angus muscle, phenotype (9) | 1 |
| GO:0046578 | Regulation of Ras protein signal transduction | Angus liver, phenotype (13); Holstein liver, GEBV (11) | 1 |
| GO:0050870 | Positive regulation of T cell activation | Angus muscle, phenotype (6); Holstein blood, phenotype (22) | 3 |
| GO:0051056 | Regulation of small GTPase mediated signal transduction | Angus liver, phenotype (14); Holstein liver, GEBV (12) | 1 |
| GO:0051094 | Positive regulation of developmental process | Angus liver, phenotype (10); Angus muscle, phenotype (11) | 1 |
| GO:0051276 | Chromosome organization | Angus muscle, phenotype (19); Holstein blood, phenotype (76) | 10 |
| GO:0051603 | Proteolysis involved in cellular protein catabolic process | Angus liver, GEBV (28); Holstein blood, phenotype (88) | 12 |
| GO:0070647 | Protein modification by small protein conjugation or removal | Angus liver, GEBV (11); Holstein blood, phenotype (33) | 6 |
Number of genes that were significantly associated with residual feed intake in more than 1 data set.
Table 6.
The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways overrepresented with genes significantly associated with residual feed intake phenotypes (GSAphenotype) and GEBV (GSAGEBV)
| GO term | GO definition | Data sets (No. of genes) | Common genes1 |
|---|---|---|---|
| bta03010 | Ribosome | Angus muscle, GEBV (16) | 25 |
| Angus muscle, phenotype (25) | |||
| Holstein blood, phenotype (75) | |||
| bta00280 | Valine, leucine, and isoleucine degradation | Angus muscle, GEBV (7) | 7 |
| Holstein liver, phenotype (12) | |||
| bta03050 | Proteasome | Angus liver, GEBV (10) | 7 |
| Holstein liver, GEBV (7) | |||
| bta00062 | Fatty acid elongation in mitochondria | Holstein liver, phenotype (3) | — |
| bta00071 | Fatty acid metabolism | Holstein liver, phenotype (7) | — |
| bta00100 | Steroid biosynthesis | Angus muscle, phenotype (6) | — |
| bta00120 | Primary bile acid biosynthesis | Angus liver, GEBV (5) | — |
| bta00190 | Oxidative phosphorylation | Angus liver, GEBV (21) | — |
| bta00310 | Lysine degradation | Angus muscle, phenotype (7) | — |
| bta00330 | Arginine and proline metabolism | Angus liver, phenotype (8) | — |
| bta00650 | Butanoate metabolism | Holstein liver, phenotype (4) | — |
| bta00830 | Retinol metabolism | Angus liver, phenotype (7) | — |
| bta00980 | Metabolism of xenobiotics by cytochrome P450 | Angus liver, phenotype (8) | — |
| bta00982 | Drug metabolism | Angus liver, phenotype (9) | — |
| bta04060 | Cytokine-cytokine receptor interaction | Angus muscle, phenotype (12) | — |
| bta04360 | Axon guidance | Holstein liver, GEBV (8) | — |
| bta04520 | Adherens junction | Angus muscle, GEBV (9) | — |
| bta04670 | Leukocyte transendothelial migration | Holstein liver, GEBV (10) | — |
| bta04810 | Regulation of actin cytoskeleton | Holstein liver, GEBV (12) | — |
| bta04914 | Progesterone-mediated oocyte maturation | Holstein liver, GEBV (7) | — |
| bta05012 | Parkinson's disease | Angus liver, GEBV (24) | — |
| bta05016 | Huntington's disease | Angus liver, GEBV (29) | — |
Number of genes that were significantly associated residual feed intake in more than 1 data set.
The descriptions of biological processes and KEGG pathways sometimes use different words for similar processes. For instance, the biological process “translation” overlaps with the KEGG pathway “ribosome.” Taken together, the biological processes and KEGG pathways implicate many different processes in RFI, which was not unexpected. The major processes involved might be summarized as follows: cell signaling regulating transcription; translation; energy metabolism, including oxidation-reduction; cell cycle; division and apoptosis; and catabolism, especially proteolysis. The details of the enriched KEGG pathways in each tissue are available in Supplementary File 5.
DISCUSSION
Variation in RFI is influenced by many biological factors (Moore et al., 2009). Among the biological mechanisms associated with RFI are feeding pattern, digestibility, activity, protein turnover and tissue metabolism, stress, thermoregulation, heat increment, and body composition (Richardson and Herd, 2004; Herd and Arthur, 2009). Therefore, finding genes with polymorphisms affecting RFI is difficult and made more so by the cost of RFI measurement, which limits the sample size.
Complex traits, such as RFI, can be associated with variation in gene expression between individuals (Albert and Kruglyak, 2015). The number of mRNA copies of a gene, indicating the expression of the gene in each individual for a specific tissue, can be measured by RNA-Seq (Morin et al., 2008). Hence, an alternative method for finding genes associated with RFI might be to study the expression level of the genes in efficient and inefficient animals. However, a correlation between the level of expression of a gene and RFI does not mean that polymorphisms in that gene influence RFI. The expression of a gene may be correlated with RFI but not a cause of variation in RFI. Despite this reservation, finding genes whose expression is correlated with RFI should help us to understand the physiological pathways controlling RFI and, ultimately, to find genes and polymorphisms that cause variation in RFI (Chen et al., 2012).
In our study, in individual data sets, the power to find genes whose expression is correlated with RFI is low (as shown by the high FDR), partly because of the small number of animals per data set and limited depth of RNA-Seq. However, by considering pathways and genes that were significant in more than 1 data set some consistent patterns emerged. For each of the 4 sets of tissue samples, we considered the correlation between expression and both RFI phenotype and GEBV. Since the phenotypes of these animals were used in calculating the GEBV, they are not independent measures. In the Angus bull data set, the phenotype and GEBV are highly correlated (r = 0.88), but in the Holstein cow data set they are not (r = 0.21). This difference is because we had a smaller data set for dairy cattle and the RFI phenotype in the Holstein cows was recorded during lactation (when the tissue samples were taken) but the GEBV was for lactating cows but calculated with a multitrait model using their RFI records during growth (as heifers) and lactation (Pryce et al., 2015). Consequently, the phenotypes were more highly correlated with gene expression than the GEBV were because the phenotypes were measured at the same time as the tissue samples were taken for gene expression measurements. Even among the Angus cattle, there were more genes whose expression was correlated with phenotype than with GEBV. This result may indicate that the correlations with phenotype could be environmental correlations not genetic. Even if the correlations are genetic, it does not necessarily mean that the expression of a gene causes differences in RFI. For instance, the coordinated up- and downregulation of genes for ribosomal proteins may not cause differences in RFI but may be the result of signaling mechanisms within the cell that are controlling multiple pathways, leading to changes in RFI and to expression of ribosomal genes.
When we consider individual genes, the number whose expression is significantly correlated with RFI in multiple data sets is small. However, within each data set there are GO terms and KEGG pathways that are enriched, and there are some biological processes that are enriched in multiple data sets. In the processes related to translation and protein catabolism or proteolysis there are many genes whose expression is correlated with RFI across more than 1 data set. The genes associated with translation include many genes for ribosomal proteins, and the genes associated with proteolysis include many ubiquitin ligases such as Itchy E3 Ubiquitin Protein Ligase Homolog ITCH. It is not surprising that mechanisms that increase translation of mRNA into protein and that catabolize proteins are associated with RFI.
However, a closer look at the results reveals a surprising finding: for genes, represented by the GO terms translation, energy generation, oxidation-reduction, and proteolysis, the direction of the correlation in Holsteins is often the opposite of that in Angus. The level of expression of individual genes within the biological pathways may be positively or negatively correlated with RFI. A negative correlation means that increased expression is associated with lower-RFI phenotypes or GEBV, which we interpret as greater phenotypic and genetic feed efficiency. For instance, the RFI phenotype was correlated negatively (P < 0.05) with expression of 25 genes for ribosomal proteins, which are part of the biological process “translation,” in Angus muscle samples. This negative correlation suggests that increased translation of mRNA into protein in muscle is associated with more efficient growth. However, in Holstein blood samples, RFI was positively correlated with expression of 67 ribosomal protein genes. This positive correlation suggests that high efficiency in a milking cow is associated with reduced translation in white blood cells and perhaps other tissues, so that resources are spared for the mammary gland. This reversal in the direction of effects between Angus and Holstein is also seen in some other pathways, for example, proteolysis and oxidation-reduction.
The GEBV for RFI in Angus were negatively correlated with expression of 11 ATP synthase genes and for genes that are part of the biological process oxidation-reduction, such as NADH dehydrogenases. Most of these genes function in the mitochondria and suggest that high efficiency of growth is associated with high levels of energy generation in the liver and with protection against the oxygen stress associated with energy generation. Some of the same genes were highlighted in the KEGG pathway “Parkinson's disease.” This pathway includes not only genes involved in mitochondrial function but also Phosphatase and Tensin Induced Putative Kinase 1 (PINK1), Parkinson Protein 2 (PARK2), Parkinson Protein 7 (PARK7), and Leucine-Rich Repeat Kinase 2 (LRRK2). Again, where the expression of these genes was significantly correlated with RFI phenotype in Holstein cows, the sign of the correlation was opposite to that in Angus.
The GEBV for RFI were positively correlated with expression of many genes from the biological process proteolysis in Angus muscle and liver tissue. For instance, this pathway includes genes for ubiquitin-specific peptidases, ubiquitin ligases (e.g., ITCH), and peptidases, suggesting that high levels of proteolysis are associated with inefficient growth. However, in Holstein blood expression of ubiquitin-specific proteases was negatively correlated with RFI phenotype. It is possible that proteolysis in organs other than the mammary gland provides substrates for milk synthesis and hence improves efficiency of milk production.
A broader view of all genes associated with RFI variation in Table 4 supports this tendency for a reversal of sign in the correlations with RFI between Angus and Holstein data sets. When comparing the 2 Angus data sets or the 2 Holstein data sets, the proportion of genes with an effect in the same direction is slightly above 50%, indicating that these results are not random. However, when an Angus data set is compared to a Holstein data set, less than 50% of genes have correlations with RFI in the same direction.
The pathways that were associated with variation in RFI in multiple data sets show some consistency. In growing Angus bulls, there were significant pathways associated with low RFI that corresponded to high metabolic activity (transcription, translation, energy generation) but low catabolism in the tissue experiencing the most growth (i.e., muscle) and, to a lesser extent, in the liver, which supports that growth. However, in lactating cows, the high efficiency of milk production (i.e., low RFI) may be associated with low metabolic activity in the rest of the body. The results support the hypothesis, based on previous research, that high levels of proteolysis contribute to high maintenance requirements and hence high RFI (Richardson and Herd, 2004). The biological processes that are most consistently enriched in the genes correlated with RFI in more than 1 of our 4 data sets can be summarized as signaling pathways controlling transcription, translation, proteolysis, and oxidation-reduction. Other published studies in pigs and cattle correlating RFI with gene expression support this conclusion.
Chen et al. (2011) used liver samples from most of the same Angus bulls as reported here but measured gene expression using a microarray assay. They found 161 genes differentially expressed between bulls selected for high and low RFI. In our Angus liver samples the association between 111 of these 161 DE genes and RFI phenotypes and genotypes was calculated. We found that 29 of the 111 genes were significantly correlated with the RFI phenotype, of which 26 genes displayed the same direction of correlation as reported by Chen et al. (2011), and 31 genes were significantly correlated with GEBV, of which 26 correlations were in the same direction. Of these, there were 23 genes whose expression was associated with both RFI phenotypes and genotypes. However, in the Angus muscle samples, where 97 of the same 161 DE genes were tested, only 6 were significantly correlated with RFI phenotype, and 8 were correlated with GEBV. The expression of 5 genes was associated with both RFI phenotypes and GEBV. The failure to fully replicate the results of Chen et al. (2011) in Angus liver samples in our study is due to small differences in the animals used but mainly to differences between microarray and RNA-Seq technologies. The low overlap between genes we found in our Angus muscle RNA-Seq analyses and the reported genes by Chen et al. (2011) from liver could be due to the use of different animals and the difference between muscle and liver.
Liu et al. (2016) reported 1,972 differentially expressed genes (adjusted P-values for multiple testing or q < 0.15) between the low- and high-RFI postweaning gilts using RNA-Seq of blood samples. Although the digestive systems of pigs and cattle are quite different, we investigated the association between gene expression and RFI in 1,536 DE genes identified by Liu et al. (2016). There were 587 GSAphenotype that we also found in Holstein blood, and 528 had an effect in the same direction as reported by Liu et al. (2016). Common pathways among these 587 genes include signaling controlling gene expression (e.g., Interleukin 2 Receptor, Gamma; IL2RG), translation (e.g., RPL38), and proteolysis (e.g., Ubiquitin Specific Peptidase 1; USP1). Many of the same pathways are also enriched in the genes shared by Liu et al. (2016) and our Angus liver and muscle and Holstein liver data sets (e.g., USP1). Among them are regulators of transcription that affect many genes (e.g., Ring Finger Protein 1; RING1, B Lymphoma Mo-MLV Insertion Region 1 Polycomb Ring Finger Oncogene; BMI1, and SERTA domain containing 1; SERTAD1).
Tizioto et al. (2015) found 112 genes whose expression in liver samples from Nellore steers (Bos indicus) was correlated with RFI. The highest overlap between their reported DE genes and our findings was in our liver samples from Angus bulls, which was 8 GSAGEBV and 8 GSAphenotype. Tizioto et al. (2016) identified 73 differentially expressed genes in longissimus thoracis muscle of Nellore steers genetically divergent for RFI. In this case, the highest overlap between their DE genes and our results was in our muscle samples from Angus bulls, which was 7 GSAGEBV and 6 GSAphenotype. There were also 2 common enriched biological processes, cell death and regulation of transcription, for GSAphenotype in Angus muscle and 4 enriched biological processes, transcription, regulation of transcription–DNA-dependent, regulation of transcription, and regulation of RNA metabolic process, for GSAphenotype in Holstein blood samples. Comparing the reported KEGG pathways, we also found metabolism of xenobiotics by cytochrome P450 and butanoate metabolism pathways were enriched by DE genes in Angus and Holstein liver, respectively.
Salleh et al. (2017) reported 70 and 19 DE genes in Holstein and Jersey liver samples with low and high RFI. Although there was limited overlap between their and our DE genes, 3 out of 5 KEGG pathways reported enriched by DE genes were also identified in our study. These pathways include leukocyte transendothelial migration, which both studies found in Holstein liver samples, and retinol metabolism and metabolism of xenobiotics by cytochrome P450, which Salleh et al. (2017) found in Jersey liver and we identified in Angus liver samples. Kong et al. (2016) found 122 DE genes by transcriptome analysis of rumen epithelium of beef cattle with different RFI. The low overlap between their results and ours was expected because the tissues we studied were different. However, 3 out of 6 reported KEGG pathways were rediscovered in our studies in Angus liver (proteasome and oxidative phosphorylation) and Holstein liver (regulation of actin cytoskeleton). Weber et al. (2016) studied gene expression of 5 tissues (adipose, duodenum, liver, muscle, and pituitary gland) in progenies of 2 sires with low and high RFI. We found 199 of their 633 DE genes in our study. The highest overlap was observed between GSAphenotype in Holstein blood and their DE genes in different tissues and in pathways associated with fat deposition and metabolisms.
Thus, although there is little consistency in the genes whose expression is correlated with RFI, especially when expression is measured in different tissues of cattle in different physiological states, there are some biological processes and pathways found by different studies. The list of DE genes found by Chen et al. (2011), Tizioto et al. (2015, 2016), Kong et al. (2016), Liu et al. (2016), Weber et al. (2016), and Salleh et al. (2017) that were also discovered in our study is available in Supplementary File 6.
The reversal between Angus and Holstein data sets of direction of the correlation between RFI and gene expression that we found is also supported by the other published studies. The genes found by Liu et al. (2016) in the blood of gilts have the same direction of effect as the same genes in our Holstein blood for 528 out of 587 genes. However, less than 50% of the genes from Liu et al. (2016) that we also found in Angus liver and muscle are in the same direction (38 out 94 for muscle and 36 out of 76 for liver). Even more dramatic is that among the 45 genes correlated with RFI in both our Holstein blood and the rumen epithelium of Kong et al. (2016), 44 reverse direction. This finding suggests that the high efficiency of muscle growth (low RFI) is associated with high rates of transcription, translation, and general metabolism but low proteolysis in growing tissues (e.g., muscle) and tissues supporting this growth (e.g., liver and rumen). However, low rates of transcription, translation, and general metabolism in other tissue (e.g., blood) may be correlated with efficiency of milk yield (our data) or growth in pigs (Liu et al., 2016).
The KEGG pathway results also indicated that some of the functions of the genes we found were consistent with previous reports. For example, Chen et al. (2011) found 7 pathways associated with RFI, and of these we found 2 pathways. The pathway related to drug metabolism was exactly the same, and we found it in the same tissue, that is, Angus live tissue. For the other pathway, cellular growth and proliferation, we found some closely related pathways such as ribosome (Angus muscle and Holstein blood); proteasome (Angus and Holstein liver); valine, leucine, and isoleucine degradation (Angus muscle and Holstein liver); arginine and proline metabolism (Angus liver); steroid biosynthesis (Angus muscle); and fatty acid and butanoate metabolism (Holstein liver). Also, in our gene ontology analysis we found pathways related to those reported by Chen et al. (2011) such as translation, proteolysis, protein and macromolecule catabolic processes in muscle and liver, oxidation-reduction, hydrogen transport, oxidative phosphorylation and ATP metabolic processes in liver, cell proliferation and death, and regulation of dephosphorylation and homeostatic processes (such as metal ion, cation and cellular chemicals) in the muscle of growing bulls and eicosanoid, unsaturated fatty acid, and lipoprotein metabolic processes in liver of lactating cows.
Conclusions
The genes whose expression were associated with RI, did not extensively overlap in different tissues, but the same biological processes and pathways were often involved. The biological processes influencing RFI in different tissues are diverse: We identified 39 biological processes that were enriched in more than 1 tissue and 3 KEGG pathways influencing RFI in multiple tissues. Many genes correlated with RFI in more than 1 tissue and in published studies are involved in proteolysis, energy metabolism, regulation of transcription, translation, the cell cycle, and apoptosis. However, the direction of the correlation between RFI and gene expression often reverses between lactating cows and growing bulls and between tissues.
LITERATURE CITED
- Albert F. W., Kruglyak L. 2015. The role of regulatory variation in complex traits and disease. Nat. Rev. Genet. 16:197–212. doi: 10.1038/nrg3891 [DOI] [PubMed] [Google Scholar]
- Anders S., Pyl P. T., Huber W. 2015. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. doi: 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. 2010. FastQC: A quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc. Accessed 20 November, 2015.
- Arthur P. F., Archer J. A., Herd R. M., Melville G. J. 2001. Response to selection for net feed intake in beef cattle. Proc. Assoc. Adv. Anim. Breed. Genet. 14:135–138. [Google Scholar]
- Arthur P. F., Herd R. M. 2008. Residual feed intake in beef cattle. Rev. Bras. Zootec. 37:269–279. doi: 10.1590/S1516-35982008001300031 [DOI] [Google Scholar]
- Beever D. E., Doyle P. T. 2007. Feed conversion efficiency as a key determinant of dairy herd performance: A review. Aust. J. Exp. Agric. 47:645–657. doi: 10.1071/EA06048 [DOI] [Google Scholar]
- Berry D. P., Crowley J. J. 2013. Cell Biology Symposium: Genetics of feed efficiency in dairy and beef cattle. J. Anim. Sci. 91:1594–1613. doi: 10.2527/jas.2012-5862 [DOI] [PubMed] [Google Scholar]
- Bolormaa S., Pryce J. E., Kemper K., Savin K., Hayes B. J., Barendse W., Zhang Y., Reich C. M., Mason B. A., Bunch R. J., Harrison B. E., Reverter A., Herd R. M., Tier B., Graser H. U., Goddard M. E. 2013. Accuracy of prediction of genomic breeding values for residual feed intake and carcass and meat quality traits in Bos taurus, Bos indicus, and composite beef cattle. J. Anim. Sci. 91:3088–3104. doi: 10.2527/jas.2012-5827 [DOI] [PubMed] [Google Scholar]
- Bolormaa S., Pryce J. E., Reverter A., Zhang Y., Barendse W., Kemper K., Tier B., Savin K., Hayes B. J., Goddard M. E. 2014. A multi-trait, meta-analysis for detecting pleiotropic polymorphisms for stature, fatness and reproduction in beef cattle. PLoS Genet. 10:e1004198. doi: 10.1371/journal.pgen.1004198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning B. L., Browning S. R. 2009. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am. J. Hum. Genet. 84:210–223. doi: 10.1016/j.ajhg.2009.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain A. J., Vander Jagt C. J., Hayes B. J., Khansefid M., Marett L. C., Millen C. A., Nguyen T. T., Goddard M. E. 2015. Extensive variation between tissues in allele specific expression in an outbred mammal. BMC Genom. 16:993. doi: 10.1186/s12864-015-2174-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Arthur P. F., Herd R. M., Quinn K., Barchia I. M. 2012. Using genes differentially expressed in bulls to classify steers divergently selected for high and low residual feed intake. Anim. Prod. Sci. 52:608–612. [Google Scholar]
- Chen Y., Gondro C., Quinn K., Herd R. M., Parnell P. F., Vanselow B. 2011. Global gene expression profiling reveals genes expressed differentially in cattle with high and low residual feed intake. Anim. Genet. 42:475–490. doi: 10.1111/j.1365-2052.2011.02182.x [DOI] [PubMed] [Google Scholar]
- Daetwyler H. D., Capitan A., Pausch H., Stothard P., van Binsbergen R., Brøndum R. F., Liao X., Djari A., Rodriguez S. C., Grohs C., Esquerré D., Bouchez O., Rossignol M. N., Klopp C., Rocha D., Fritz S., Eggen A., Bowman P. J., Coote D., Chamberlain A. J., Anderson C., VanTassell C. P., Hulsegge I., Goddard M. E., Guldbrandtsen B., Lund M. S., Veerkamp R. F., Boichard D. A., Fries R., Hayes B. J. 2014. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat. Genet. 46:858–865. doi: 10.1038/ng.3034 [DOI] [PubMed] [Google Scholar]
- Gilmour A. R., Gogel B. J., Cullis B. R., Thompson R., Butler D. 2009. ASReml user guide release 3.0. VSN Int. Ltd., Hemel Hempstead, UK. [Google Scholar]
- Hegarty R. S., Goopy J. P., Herd R. M., McCorkell B. 2007. Cattle selected for lower residual feed intake have reduced daily methane production. J. Anim. Sci. 85:1479–1486. doi: 10.2527/jas.2006-236 [DOI] [PubMed] [Google Scholar]
- Herd R. M., Arthur P. F. 2009. Physiological basis for residual feed intake. J. Anim. Sci. 87:E64–E71. doi: 10.2527/jas.2008-1345 [DOI] [PubMed] [Google Scholar]
- Huang W., Sherman B. T., Lempicki R. A. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4:44–57. doi: 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- Jeyaruban M. G., Johnston D. J., Graser H. U. 2009. Genetic association of net feed intake measured at two stages with insulin-like growth factor-I, growth and ultrasound scanned traits in Angus cattle. Proc. Assoc. Adv. Anim. Breed. Genet. 18:584–587. [Google Scholar]
- Khansefid M., Pryce J. E., Bolormaa S., Miller S. P., Wang Z., Li C., Goddard M. E. 2014. Estimation of genomic breeding values for residual feed intake in a multibreed cattle population. J. Anim. Sci. 92:3270–3283. doi: 10.2527/jas.2014-7375 [DOI] [PubMed] [Google Scholar]
- Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S. L. 2013. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14:R36. doi: 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch R. M., Swiger L. A., Chambers D., Gregory K. E. 1963. Efficiency of feed use in beef cattle. J. Anim. Sci. 22:486–494. [Google Scholar]
- Kong R. S., Liang G., Chen Y., Stothard P., Guan L. 2016. Transcriptome profiling of the rumen epithelium of beef cattle differing in residual feed intake. BMC Genom. 17:592. doi: 10.1186/s12864-016-2935-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 25:1754–1760. doi: 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H., Nguyen Y. T., Nettleton D., Dekkers J. C., Tuggle C. K. 2016. Post-weaning blood transcriptomic differences between Yorkshire pigs divergently selected for residual feed intake. BMC Genom. 17:73. doi: 10.1186/s12864-016-2395-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald K. A., Pryce J. E., Spelman R. J., Davis S. R., Wales W. J., Waghorn G. C., Williams Y. J., Marett L. C., Hayes B. J. 2014. Holstein-Friesian calves selected for divergence in residual feed intake during growth exhibited significant but reduced residual feed intake divergence in their first lactation. J. Dairy Sci. 97:1427–1435. doi: 10.3168/jds.2013-7227 [DOI] [PubMed] [Google Scholar]
- Moore S. S., Mujibi F. D., Sherman E. L. 2009. Molecular basis for residual feed intake in beef cattle. J. Anim. Sci. 87:E41–E47. doi: 10.2527/jas.2008-1418 [DOI] [PubMed] [Google Scholar]
- Morin R., Bainbridge M., Fejes A., Hirst M., Krzywinski M., Pugh T., McDonald H., Varhol R., Jones S., Marra M. 2008. Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing. Biotechniques 45:81–94. doi: 10.2144/000112900 [DOI] [PubMed] [Google Scholar]
- Pryce J. E., Arias J., Bowman P. J., Davis S. R., Macdonald K. A., Waghorn G. C., Wales W. J., Williams Y. J., Spelman R. J., Hayes B. J. 2012. Accuracy of genomic predictions of residual feed intake and 250-day body weight in growing heifers using 625,000 single nucleotide polymorphism markers. J. Dairy Sci. 95:2108–2119. doi: 10.3168/jds.2011-4628 [DOI] [PubMed] [Google Scholar]
- Pryce J. E., Gonzalez-Recio O., Nieuwhof G., Wales W. J., Coffey M. P., Hayes B. J., Goddard M. E. 2015. Hot topic: Definition and implementation of a breeding value for feed efficiency in dairy cows. J. Dairy Sci. 98:7340–7350. doi: 10.3168/jds.2015-9621 [DOI] [PubMed] [Google Scholar]
- Richardson E. C., Herd R. M. 2004. Biological basis for variation in residual feed intake in beef cattle. 2. Synthesis of results following divergent selection. Aust. J. Exp. Agric. 44:431–440. doi: 10.1071/EA02221 [DOI] [Google Scholar]
- Robinson M. D., Oshlack A. 2010. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11:R25. doi: 10.1186/gb-2010-11-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross E. M., Moate P. J., Bath C. R., Davidson S. E., Sawbridge T. I., Guthridge K. M., Cocks B. G., Hayes B. J. 2012. High throughput whole rumen metagenome profiling using untargeted massively parallel sequencing. BMC Genet. 13:53. doi: 10.1186/1471-2156-13-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salleh M. S., Mazzoni G., Höglund J. K., Olijhoek D. W., Lund P., Løvendahl P., Kadarmideen H. N. 2017. RNA-Seq transcriptomics and pathway analyses reveal potential regulatory genes and molecular mechanisms in high- and low-residual feed intake in Nordic dairy cattle. BMC Genom. 18(1):258. doi: 10.1186/s12864-017-3622-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargolzaei M., Chesnais J. P., Schenkel F. S. 2014. A new approach for efficient genotype imputation using information from relatives. BMC Genom. 15:478. doi: 10.1186/1471-2164-15-478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tizioto P. C., Coutinho L. L., Decker J. E., Schnabel R. D., Rosa K. O., Oliveira P. S., Souza M. M., Mourão G. B., Tullio R. R., Chaves A. S., Lanna D. P., Zerlotini-Neto A., Mudadu M. A., Taylor J. F., Regitano L. C. 2015. Global liver gene expression differences in Nelore steers with divergent residual feed intake phenotypes. BMC Genom. 16:242. doi: 10.1186/s12864-015-1464-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tizioto P. C., Coutinho L. L., Oliveira P. S., Cesar A. S., Diniz W. J., Lima A. O., Rocha M. I., Decker J. E., Schnabel R. D., Mourão G. B., Tullio R. R., Zerlotini A., Taylor J. F., Regitano L. C. 2016. Gene expression differences in longissimus muscle of Nelore steers genetically divergent for residual feed intake. Sci. Rep. 6:39493. doi: 10.1038/srep39493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber K. L., Welly B. T., Van Eenennaam A. L., Young A. E., Porto-Neto L. R., Reverter A., Rincon G. 2016. Identification of gene networks for residual feed intake in Angus cattle using genomic prediction and RNA-seq. PLoS One 11(3):e0152274. doi: 10.1371/journal.pone.0152274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams Y. J., Pryce J. E., Grainger C., Wales W. J., Linden N., Porker M., Hayes B. J. 2011. Variation in residual feed intake in Holstein-Friesian dairy heifers in southern Australia. J. Dairy Sci. 94:4715–4725. doi: 10.3168/jds.2010-4015 [DOI] [PubMed] [Google Scholar]