

SUMMARY
The co-engagement of fragment crystallizable (Fc) gamma receptors (FcγRs) with the Fc region of recombinant immunoglobulin monoclonal antibodies (mAbs) and its contribution to therapeutic activity has been extensively studied. For example, Fc-FcγR interactions have been shown to be important for mAb-directed effector cell activities, as well as mAb-dependent forward signaling into target cells via receptor clustering. Here we identify a function of mAbs targeting T cell-expressed antigens that involves FcγR co-engagement on antigen-presenting cells (APCs). In the case of mAbs targeting CTLA-4 and TIGIT, the interaction with FcγR on APCs enhanced antigen-specific T cell responses and tumoricidal activity. This mechanism extended to an anti-CD45RB mAb, which led to FcγR-dependent regulatory T cell expansion in mice.
Graphical Abstract
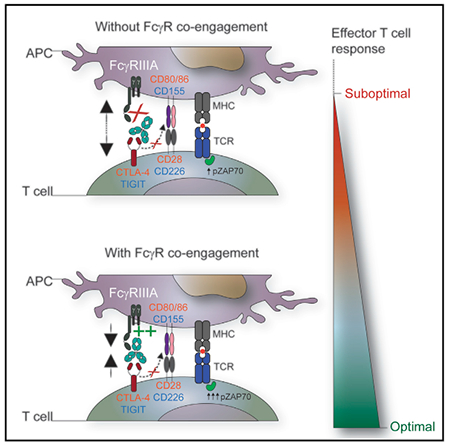
In Brief
Waight et al. report an FcγR-dependent, but independent of Treg depletion, mechanism of action of anti-CTLA-4 antibodies and show that Fc-FcγR co-engagement by anti-CTLA-4 antibodies improves T cell signaling and function. This mechanism also applies to anti-TIGIT and anti-CD45RB antibodies.
INTRODUCTION
Therapeutic immunoglobulin (IgG)-based monoclonal antibodies (mAbs) elicit a range of functional activities, many of which can be fine-tuned by optimizing the interaction of the fragment crystallizable gamma receptor (FcγR) region, with FcγRs expressed on immune and non-immune cell populations (Kim and Ashkenazi, 2013; Offringa and Glennie, 2015; Waight et al., 2017). Two broad subclasses of FcγRs, activating and inhibitory, interact with therapeutic mAbs (Nimmerjahn et al., 2015). The activating subclass of FcγRs signal through an intracellular immunoreceptor tyrosine-based activation motif (ITAM) or via the ITAM-containing common γ chain. A range of effector cell-mediated activities involving activating FcγRs have been described, including mAb-dependent cell-mediated cytotoxicity or phagocytosis (ADCC/P) (Kim and Ashkenazi, 2013; Nimmerjahn and Ravetch, 2008; Stewart et al., 2014). By contrast, the inhibitory receptor, FcγRIIB (CD32B), contains a cytoplasmic immunoreceptor tyrosine-based inhibitory motif (ITIM), which counteracts the function ITAM-containing receptors (Nimmerjahn and Ravetch, 2008; Stewart et al., 2014). FcγRIIB can also facilitate the clustering of agonist mAbs targeting tumor necrosis factor receptor (TNFR) superfamily members, including CD262, CD264, CD40, CD137, and CD28 (Li and Ravetch, 2011; White et al., 2015; Wilson et al., 2011). Recent studies show that attenuation of Fc-FcγR interactions may improve the therapeutic activity of mAbs targeting the PD-1 pathway (Arlauckas et al., 2017; Dahan et al., 2015). Taken together, FcγRs are involved in modulating the activity of a range of therapeutic mAbs. Therefore, an improved understanding of Fc-FcγR crosstalk may be leveraged in the design of more efficacious molecules.
Preclinical studies in mice using mAbs targeting glucocorticoid-induced TNFR-related protein GITR (CD357), OX40 (CD134), and CTLA-4 (CD152) revealed that engagement of activating FcγRs was required for their respective anti-tumor activity (Bulliard et al., 2013, 2014; Kim et al., 2015; Selby et al., 2013; Simpson et al., 2013). A common mechanism was defined as the selective depletion of intratumoral regulatory T (Treg) cells, which was attributed to overexpression of GITR, OX40, and CTLA-4 on Treg cells within the tumor microenvironment. As a central negative regulator of effector T cell function, CTLA-4 is rapidly translocated from intracellular protein stores to the cell surface in response to T cell receptor (TCR) stimulation (Krummel and Allison, 1995). Following engagement with CD80 and CD86 on antigen-presenting cells (APCs), CD28 enhances T cell cytokine and chemokine production, proliferation, and survival (Acuto and Michel, 2003). CTLA-4 has a higher affinity for CD80 and CD86, allowing it to effectively outcompete CD28 for ligand binding, thereby attenuating T cell priming (Krummel and Allison, 1995). In addition to competition for shared CD28 ligands, a range of other cell-intrinsic and -extrinsic functions have been ascribed to the role of CTLA-4 in maintaining immune homeostasis (Walker and Sansom, 2011). For instance, emerging evidence suggests that CTLA-4 promotes T cell motility by antagonizing TCR-induced zeta chain-associated protein 70 (ZAP70) microcluster formation, leading to reduced APC-T cell dwell time (Schneider et al., 2008). To date, three anti-CTLA-4 mAbs have demonstrated single-agent anti-tumor activity in patients, although the contribution of FcγR-associated mechanism(s) to the therapeutic activity of these antibodies remains controversial (Arce Vargas et al., 2018; Gombos et al., 2018; Ribas and Flaherty, 2015; Romano et al., 2015).
In the present study, we investigated the contribution of FcγR co-engagement on APCs for the mechanism of action of antagonistic antibodies targeting CTLA-4 and TIGIT, in the context of existing therapeutic mAbs targeting T cell antigens, as well as in the development of the next generation of therapeutic mAbs through Fc engineering.
RESULTS
Anti-tumor Activity of Anti-CTLA-4 mAb Is Dependent on FcγR Co-engagement Despite Suboptimal Intratumoral Treg Cell Depletion
The anti-tumor activity of mAbs targeting CTLA-4 was initially defined by their ability to block CD80 and CD86 from engaging the MYPPPY motif on CTLA-4, thereby allowing CD28 to access these shared ligands and provide T cell stimulation (Pentcheva-Hoang et al., 2004). However, the co-engagement of FcγRs has since been shown to be required for the anti-tumor activity of CTLA-4 targeted mAbs in a range of preclinical models (Bulliard et al., 2013; Ingram et al., 2018; Selby et al., 2013; Simpson et al., 2013). Consistent with the dependence on FcγR co-engagement, a mouse IgG2a (mIgG2a) anti-CTLA-4 mAb (clone 9D9) provided robust tumor control, compared with a variant (mIgG2a-N297A) with impaired binding to mouse FcγRs (Figures 1A and S1A). FcγR-dependent anti-tumor activity was previously correlated with an acute reduction in Treg cells within the tumor microenvironment (Figures 1B, 1C, S1B, and S1C) (Bulliard et al., 2013; Selby et al., 2013; Simpson et al., 2013). A similar correlation between anti-tumor efficacy and intratumoral Treg cell depletion was also described for mIgG2a versus mIgG2a-N297A mAbs targeting OX40 and GITR (Figures 1B and S1D) (Bulliard et al., 2013, 2014). This observation was refined by showing that the tumoricidal activity of anti-OX40 and anti-GITR mIgG2a mAbs was abrogated in mice deficient for activating FcγRs (I, III, and IV), and was independent of the inhibitory FcγR, FcγRIIB. The overexpression of OX40, GITR, and CTLA-4 by intratumoral Treg cells combined with the abundance of activating FcγR-expressing myeloid and natural killer (NK) cells effector populations supported that Treg cell depletion was by an FcγR-mediated ADCC/P mechanism (Arce Vargas et al., 2018; Bulliard et al., 2013; Ingram et al., 2018; Kim and Ashkenazi, 2013). However, despite a common requirement for activating FcγR co-engagement for the anti-tumor activity mediated by anti-CTLA-4 and anti-GITR mAbs, we observed a quantitative and kinetic difference in the extent of Treg cell depletion mediated between mIgG2a mAbs (Figure 1B). Across three independent experiments, acute (24 hr) Treg cell depletion was between 26.5% ± 5% for an anti-CTLA-4 mIgG2a mAb and 76.6% ± 8% for an anti-GITR-mIgG2a mAb (clone DTA-1) (Figure 1C). Given this quantitative difference in Treg cell depletion, we sought to re-evaluate the strict dependence on activating FcγR binding for the pharmacologic activity of mAbs targeting CTLA-4.
Figure 1. Quantitative Differences in Selective Intratumoral Treg Cell Depletion by mAbs Targeting CTLA-4 and GITR.
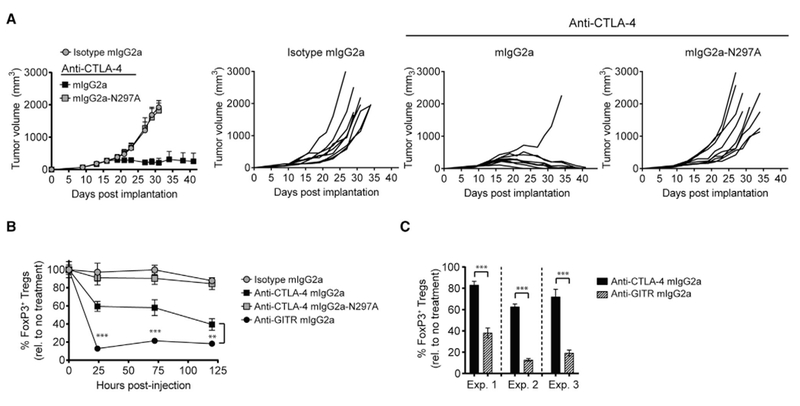
(A) BALB/c mice with established CT26 tumors (50–80 mm3) were treated with a single 100-μg intraperitoneal (i.p.) dose of anti-CTLA-4, or mIgG2a isotype control mAb. Individual tumor growth rates (n = 9 mice/group) are shown on the right.
(B) CT26 tumor-bearing (50–80 mm3) mice were treated with a single 100-μg i.p. dose of anti-CTLA-4, anti-GITR mIgG2a, or mIgG2a isotype control mAb. Intratumoral FoxP3+ Treg cell depletion was evaluated by flow cytometry pre- (t = 0 hr) and post-mAb injection (t = 24, 72, and 120 hr) (n = 4 mice/treatment time point).
(C) Percent (%) reduction in intratumoral FoxP3+ Treg cells 24 hr post-anti-CTLA-4 mIgG2a (Experiment [Exp.]: 1, 17 ± 3; 2, 35 ± 3; 3, 28 ± 7) or GITR mIgG2a ((Exp.): 1,62 ± 4; 2 87 ± 1; 3, 81 ± 3) mAb administration, relative to untreated mice (n = 3–4 animals/group).
Data are representative of three or more experiments. A Student’s t test was used to calculate significance in (B and C). Error bars indicate the SEM. **p < 0.01; ***p < 0.001. See also Figure S1.
FcγR Co-engagement Is Required for Enhanced T Cell Responses In Vivo
Administration of staphylococcal enterotoxin B (SEB peptide) elicits an antigen-specific TCR (Vβ8+) T cell response in mice (Miyahara et al., 2012). This model provided an opportunity to evaluate the role of FcγR co-engagement by an anti-CTLA-4 mAb in non-tumor-bearing animals. SEB-reactive and non-reactive T cell proliferation were evaluated following SEB administration, together with an anti-CTLA-4 mIgG2a, anti-CTLA-4 mIgG2a-N297A, or isotype control mAb (Figures 2A and 2B). In this model, the anti-CTLA-4-mIgG2a mAb produced a superior antigen-specific T cell response, compared with the same mAb devoid of FcγR binding or isotype control mAb (Figure 2A). Increased T cell proliferation mediated by the anti-CTLA-4 mIgG2a mAb corresponded with a subsequent increase in the frequency of Vβ8+ CD4+ and CD8+ T cells in blood and spleen (Figures 2B and S2A). In addition, a greater proportion of −β8+ T cells at the peak of expansion (day 6) produced interleukin-2 (IL-2) in mice that received SEB together with an anti-CTLA-4 mIgG2a mAb (Figures 2C and S2B). The expansion of SEB-specific T cells induced by the anti-CTLA-4 mIgG2a mAb was restricted to the Vβ8+ TCR repertoire of effector T cells, with no significant change in the frequency of naive or memory T cell subsets observed (Figures 2D and S2C). Notably, at an early time point (day 3), both anti-CTLA-4 mIgG2a and anti-CTLA-4 mIgG2a-N297A mAbs increased the Vβ8+ effector T cell expansion relative to isotype control mice. However, only the anti-CTLA-4 mIgG2a mAb promoted durable antigen-specific T cell expansion and IL-2 production. These findings revealed an unexpected dependence on FcγR co-engagement for the CTLA-4 mAb-driven expansion and function of antigen-specific T cells in the absence of a tumor microenvironment.
Figure 2. Antigen-Specific T Cell Responses Enhanced by Anti-CTLA-4 mAb Require Intact FcγR Co-engagement.
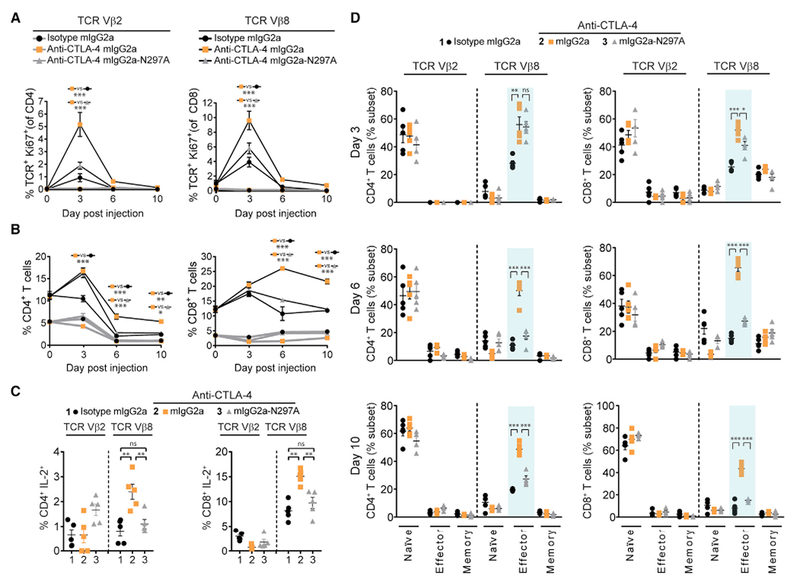
(A and B) Proliferation (Ki67+) of SEB-specific (Vβ8+, black lines) and non-specific (Vβ2+, gray lines) T cells (A) and the percentage of Vβ8+ and Vβ2+ T cells (B) evaluated by flow cytometry in the peripheral blood pre- (day 0) and post-treatment (days 3, 6, and 10) of C57BL/6 mice administered i.p. with 150 μg of SEB together with a 100-μg dose of anti-CTLA-4 mAb or mIgG2a isotype control mAb (n = 5 mice/group). A two-way ANOVA was used to calculate significance.
(C and D) Intracellular flow cytometry for IL-2 produced by Vβ8+ and Vβ2+ T cells on day 6 post-SEB/mAb administration (C) and naive (CD44− CD62L+), effector (CD44+ CD62L+) and memory (CD44+ CD62L+) T cells represented as a percentage of the total Vβ8+ or Vβ2+ T cell fraction (D) in the samples in (A and B). A Student’s t test was used to calculate significance.
Data are representative of three or more experiments. Error bars indicate SEM; ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001. See also Figure S2.
T Cell Responses Mediated by Anti-CTLA-4 mAb Are Independent of Regulatory T Cells
We next sought to understand if Treg cell functions associated with the CTLA-4 pathway might be involved in the superior pharmacologic activity of an anti-CTLA-4 mIgG2a Fc variant (Kong et al., 2014; Li and Rudensky, 2016). We first evaluated changes in the peripheral Treg cell compartment following SEB peptide administration alone, or in combination with an anti-CTLA-4 mIgG2a, anti-CTLA-4 mIgG2a-N297A, or a mIgG2a isotype mAb (Figure 3A). SEB peptide administration promoted a transient increase in the frequency of Treg cells on day 3; however, expansion and subsequent Treg cell contraction was independent of anti-CTLA-4 mAb co-administration. To assess directly if CTLA-4-associated Treg cell function contributed to the activity of anti-CTLA-4 mAbs, Treg cells were pre-depleted using two in vivo strategies. First, anti-CD25 (clone PC61, rat (r)IgG1) or isotype control mAbs were administered 10 days prior to administration of SEB peptide together with anti-CTLA-4 mIgG2a or mIgG2a-N297A mAbs (Figure 3B). As reported previously, anti-CD25 mAb administration selectively reduced FoxP3+ Treg cells by approximately 50% throughout the assay (Figures 3B and S3A) (Arce Vargas et al., 2017). Despite Treg cell depletion, we observed a significant difference in the expansion of antigen-specific Vβ8+ effector T cells in anti-CTLA-4 mIgG2a mAb-injected mice, compared with the SEB peptide plus isotype mAb cohort (Figure 3C). To further discount a role for Treg cells, we utilized a transgenic mouse model that expressed the human diphtheria toxin (DT) receptor under the control of the FoxP3 promoter (FoxP3DTR mice) (Kim et al., 2007). As reported previously, DT administration dramatically reduced (routinely >95%) Treg cells, without changes in other T cell compartments (Figures 3D and S3B–S3D). Anti-CTLA-4 or isotype control mAb co-administration with SEB peptide did not alter DT-mediated Treg cell depletion efficiency in FoxP3DTR mice (Figure 3D). Given the role of Treg cells in maintaining peripheral tolerance, we observed that the systemic depletion of Treg cells in DT-treated FoxP3DTR mice increased the frequency of SEB-reactive T cells in control animals (Sakaguchi et al., 2008). However, consistent with our findings using an anti-CD25 mAb to target Treg cells, the DT-treated FoxP3DTR mice maintained a significant difference in the expansion of total antigen-specific T cells between anti-CTLA-4 mIgG2a and isotype mAb-administered animals (Figures 3E and 3F). Taken together, the dependence on FcγR co-engagement for optimal antigen-specific T cell responses mediated by anti-CTLA-4 mAbs was maintained in the absence of Treg cells.
Figure 3. FcγR Co-engagement by Anti-CTLA-4 mAb Enhances Antigen-Specific T Cell Responses via a Treg Cell-Independent Mechanism.
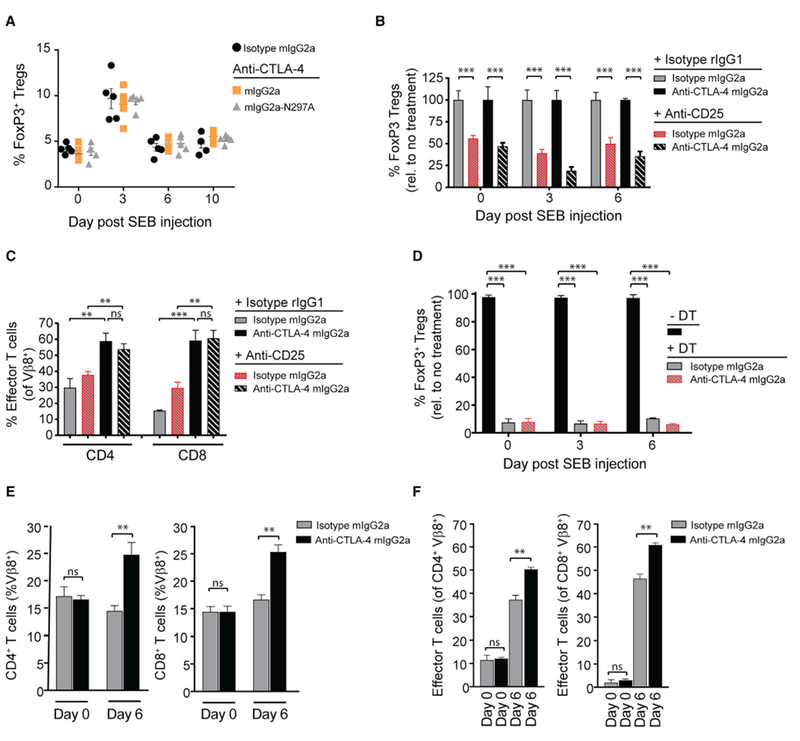
(A) C57BL/6 mice were administered i.p. with 150 μg of SEB together with a 100-μg i.p. dose of anti-CTLA-4, or a mIgG2a isotype control mAb (n = 5 mice/group). FoxP3+ Treg cell frequency was evaluated in the peripheral blood by flow cytometry pre- (day 0) and post-treatment (days 3, 6, and 10).
(B) On day −10, C57BL/6 mice were administered i.p. with a single 250-μg dose of anti-CD25 mAb (clone PC61). On day 0, mice were given a 150-μg i.p. injection of SEB and 100 μg of anti-CTLA-4 mIgG2a mAb (n = 4 mice/group). FoxP3+ Treg cell frequency in the peripheral blood was assessed by flow cytometry pre- (day 0) and post-SEB/mAb injection (days 3 and 6).
(C) Percentage of Vβ8+ effector (CD44+ CD62L−) T cells in anti-CD25 mAb pre-conditioned mice on day 6 post-administration of SEB together with anti-CTLA-4 mIgG2a or mIgG2a isotype control mAb, as measured by flow cytometry.
(D) FoxP3DTR transgenic mice were administered with 100 μg i.p. of diphtheria toxin (DT) on days −2 and −1 to systemically deplete FoxP3+ Treg cells. Frequency of FoxP3+ Treg cells in DT-treated and untreated (normalized to 100%) FoxP3DTR mice pre- (day 0) and post-SEB/mAb injection (days 3 and 6) was assessed in the peripheral blood by flow cytometry.
(E and F) DT-treated mice in (D) were injected i.p. on day 0 with 150 μg of SEB and 100 μg anti-CTLA-4 mIgG2a or isotype control mAb (n = 4 mice/group). Total Vβ8+ T cells (E) and Vβ8+ effector (CD44+ CD62L−) T cells (F) were evaluated in the peripheral blood on day 0 (pre-dose) and day 6 (post-treatment) by flow cytometry.
A Student’s t test was used to calculate significance in (B–F). Data are representative of three or more experiments. Error bars indicate SEM; ns, not significant; **p < 0.01; ***p < 0.001. See also Figure S3.
FcγR Dependence Is Conserved between Mouse and Human Anti-CTLA-4 mAbs
To evaluate the translational implications of our findings, we utilized a human immune cell assay to compare Fc-competent human IgG1 (hIgG1) and Fc-silent hIgG1-N297A mAbs targeting human CTLA-4 (Shields et al., 2001). The anti-CTLA-4 hIgG1 mAb was found to have similar biophysical and functional attributes to another anti-human CTLA-4 mAb, ipilimumab (Figures S4A–S4C). To assess CTLA-4 pathway modulation in the context of FcγR-expressing APCs and T cells, human peripheral blood mononuclear cells (PBMCs) were stimulated with staphylococcal enterotoxin A (SEA peptide) in the presence of an anti-CTLA-4 mAb (Spaulding et al., 2013). As with our observations using a bacterial superantigen (SEB peptide) in mice, the anti-CTLA-4 hIgG1 mAb was shown to be functionally superior to an anti-CTLA-4 hIgG1-N297A mAb in producing T cell cytokine production (IL-2) (Figure 4A). Importantly, the production of IL-2 by T cells in this assay was confirmed by pre-depleting CD3+ cells prior to SEA peptide stimulation (Figure S4D).
Figure 4. Optimal Antigen-Specific T Cell Responses Produced by CTLA-4 mAbs Require FcγRIIIA (Human) or FcγRIV (Mouse) Co-engagement.
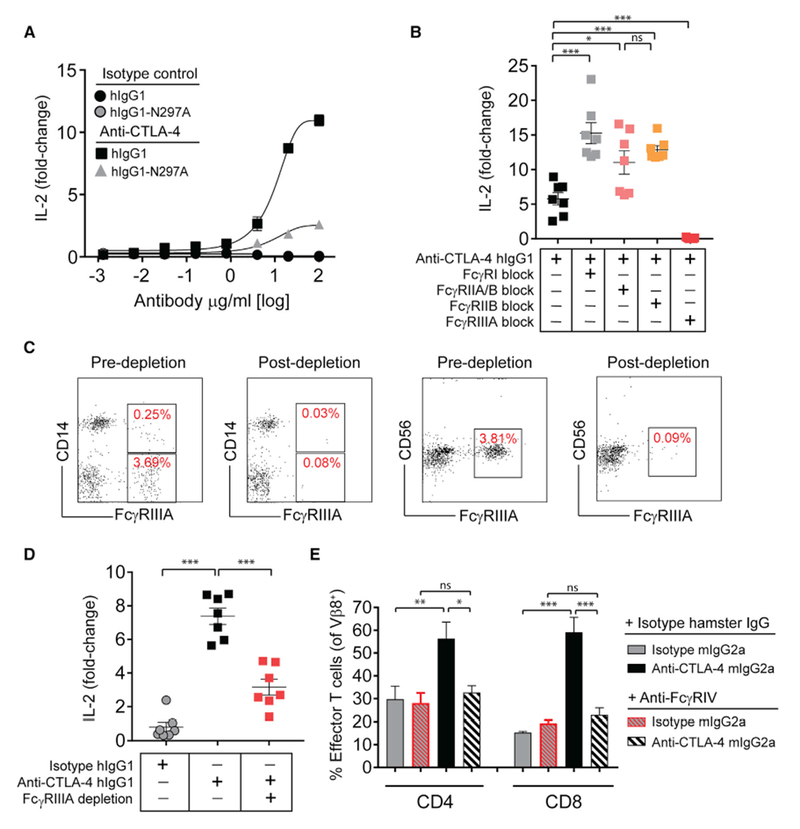
(A) IL-2 production (day 4) by human PBMCs stimulated with 100 ng/mL of SEA peptide together with increasing concentrations of anti-CTLA-4, or isotype control mAbs.
(B) IL-2 production (day 4) by PBMCs following blockade of the indicated FcγRs with FcγR-specific mAbs (10 μg/mL) for 15 min prior to co-incubation with SEA peptide (100 ng/mL) and anti-CTLA-4 hIgG1 mAb (10 μg/mL).
(C) Representative flow cytometry plots of PBMCs depleted of FcγRIIIA+ cells (CD14+ and CD56+ cells were evaluated).
(D) IL-2 production (day 4) by PBMCs stimulated with SEA peptide and anti-CTLA-4 hIgG1 mAb with or without pre-depletion of FcγRIIIA+ cells.
(E) C57BL/6 mice were given i.p. injections of anti-FcγRIV mAb (clone 9E9) or hamster IgG isotype control (200 μg), together with SEB peptide (150 μg) and anti-CTLA-4 mIgG2a or isotype control mAb (100 μg) (n = 4 mice). The frequency of Vβ8+ effector (CD44+ CD62L−) T cells was evaluated on day 6 by flow cytometry.
Fold-change in IL-2 was calculated relative to a no mAb control (A, B, and D). A Student’s t test was used to calculate significance in (B, D, and E). Data are representative of three or more experiments. Error bars indicate SEM; ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001. See also Figure S4.
To explore the relative contribution of particular FcγRs to the function of the anti-CTLA-4 hIgG1 mAb, we utilized a panel of mAb reagents to selectively block individual FcγR-Fc interactions (Figures 4B and S4E) (Veri et al., 2007; Yu et al., 2016). Remarkably, only blockade of the FcγRIIIA-hIgG1 interaction significantly reduced anti-CTLA-4 mAb-induced T cell IL-2 responses. By contrast, blockade of the hIgG1 interaction with mAbs selective for FcγRI, FcγRIIB, or a pan FcγRIIA/B mAb enhanced IL-2 production. Dependence on FcγRIIIA co-engagement was further confirmed using deglycosylated versions of the anti-FcγR blocking mAbs, thereby eliminating the potential for these reagents to co-engage FcγRs via glycans in the Fc region (Figures S4F and S4G). An orthogonal approach to confirm these findings was to pre-deplete the FcγRIIIA-expressing cell fraction prior to SEA peptide stimulation, which included CD56+ NK cells and CD14−/+ cells (Figure 4C). Consistent with mAb-mediated blockade of the FcγRIIIA-hIgG1 interaction, depleting the FcγRIIIA+ cell fraction attenuated IL-2 production (Figure 4D). Notably, we consistently saw a greater reduction in IL-2 production by mAb-mediated FcγRIIIA blockade versus the depletion of FcγRIIIA+ cells. This difference may be attributed to the presence of FcγR blocking mAbs throughout the experiment, versus incomplete depletion of FcγRIIIA+ cells or de novo receptor expression during the assay.
In mice, FcγRIV has been described as a functional ortholog of human FcγRIIIA (Nimmerjahn et al., 2005). Moreover, anti-CTLA-4 mAb-mediated anti-tumor efficacy was found to be severely diminished in mice deficient for FcγRIV (Simpson et al., 2013). Therefore, we sought to evaluate the importance of FcγRIV in the antigen-specific effector T cell response in mice administered SEB peptide together with an anti-CTLA-4-mIgG2a mAb. To test this, we utilized an anti-FcγRIV mAb (clone 9E9, hamster IgG) to selectively block the Fc region of mIgG2a antibodies binding to FcγRIV (Figure S4H). Consistent with our findings implicating human FcγRIIIA, the in vivo blockade of mouse FcγRIV with glycosylated or deglycosylated variants of anti-FcγRIV mAb significantly reduced the expansion of Vβ8+ effector T cells induced by anti-CTLA-4 mIgG2a mAb (Figures 4E, S4I, and S4J). These data therefore support that this FcγR-dependent mechanism showed high concordance between results obtained in either mouse or human assay systems.
FcγRIIIA Co-engagement Is Required for Optimal T Cell Stimulation by Human CTLA-4 Antibodies
It has been shown that FcγR-mediated clustering can enhance the activity of mAbs targeting certain TNFR superfamily members (Li and Ravetch, 2011; Wilson et al., 2011). To exclude that mAb crosslinking might contribute to the enhanced IL-2 response elicited by anti-CTLA-4 mAbs, purified human T cells were stimulated with an anti-CD3 mAb in the presence of either a soluble or crosslinked anti-CTLA-4 hIgG1 mAb (Figure S5A). The anti-CTLA-4 mAb failed to impact TCR-induced IL-2 production in either format, contrasting with an anti-GITR hIgG1 mAb that functioned optimally when crosslinked in the same assay format (Figure S5B). To further explore the dependence on human FcγRIIIA in the T cell response elicited by an anti-CTLA-4 hIgG1 mAb, we took advantage of two previously characterized Fc-engineered hIgG1 scaffolds that alter the affinity for activating or inhibitory FcγRs (Lazar et al., 2006; Smith and Clatworthy, 2010). A hIgG1-S239D/A330L/I332E (hIgG1-DLE) Fc variant had a higher affinity for FcγRIIIA, including both low- (158F/F) and high-affinity (158V/V) allotypes (Figures S5C and S5D) (Lazar et al., 2006). This compared with an anti-CTLA-4 hIgG1 S267E/L328F (hIgG1-SELF) Fc mAb variant with enhanced binding to the inhibitory receptor, FcγRIIB. The anti-CTLA-4 hIgG1-SELF and anti-CTLA-4 hIgG1-DLE mAbs were functionally compared with parental anti-CTLA-4 hIgG1 or anti-CTLA-4 hIgG1-N297A mAbs in the PBMC-SEA peptide assay. Consistent with our findings using FcγR-specific blocking antibodies, the importance of FcγRIIIA co-engagement was confirmed by a >50-fold increase in IL-2 production with the anti-CTLA-4 IgG1-DLE mAb compared with the parental hIgG1 mAb (Figure 5A). Notably, the pharmacologic activity of the anti-CTLA-4 hIgG1-DLE mAb on day 4 was not associated with any change in Treg cell frequency (Figures 5B and 5C). The correlation with FcγRIIIA affinity and T cell IL-2 production was also mirrored by a reduction in IL-10 (Figure 5D). To further exemplify the importance of FcγRIIIA, we generated an afucosylated anti-CTLA-4 hIgG1 mAb (Figure S5E). Afucosylated mAbs are devoid of fucose on the Fc, leading to enhanced FcγRIIIA binding (Shields et al., 2002). Consistent with our findings using the hIgG1-DLE anti-CTLA-4 mAb, an afucosylated variant also showed comparable T cell stimulatory activity in SEA-stimulated PBMC assay (Figure S5E). These findings were contrasted with the anti-CTLA-4 hIgG1-SELF Fc variant with enhanced binding to FcγRIIB, which only weakly induced IL-2 production, in fact to a similar level to that of an anti-CTLA-4 hIgG1-N297A mAb (Figures 5A and S5E).
Figure 5. Improved Functionality of Anti-CTLA-4 mAb with Fc Engineering.
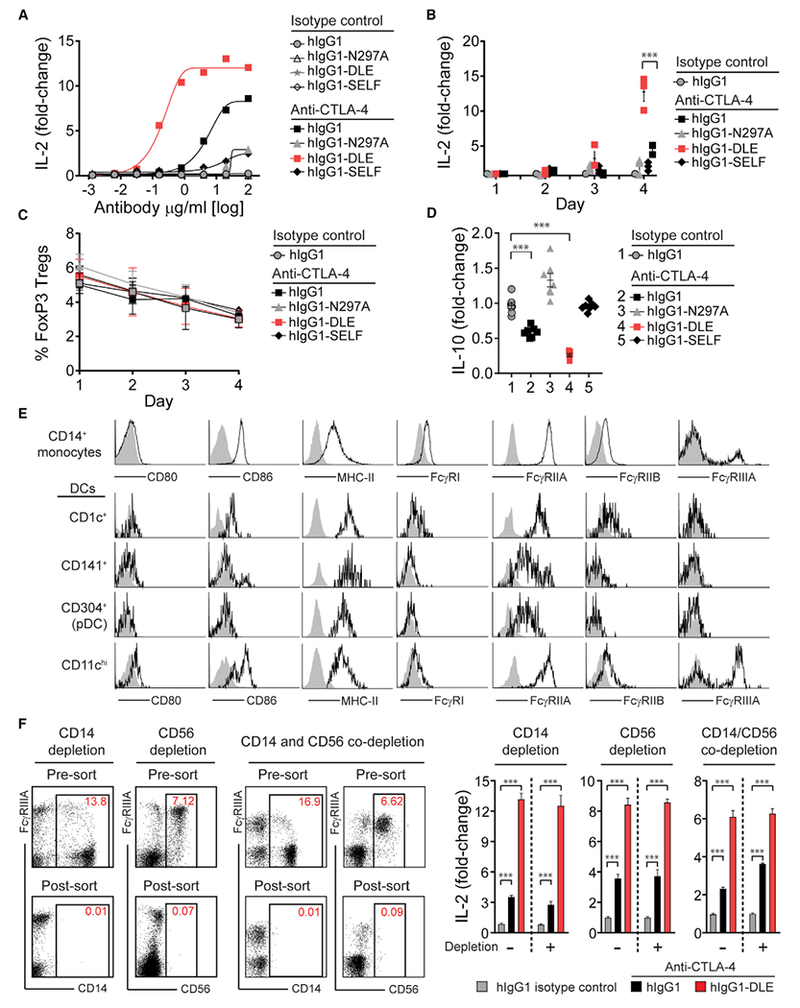
(A) IL-2 production (day 4) by human PBMCs stimulated with 100 ng/mL of SEA peptide together with anti-CTLA-4 mAb variants or corresponding hIgG1 isotype control mAbs.
(B and C) Kinetics of PBMC IL-2 production (B) and FoxP3+ Treg cell frequency (C) following stimulation with SEA peptide and the anti-CTLA-4 hIgG1 mAb variants in (A) or hIgG1 isotype control (10 μg/mL).
(D) IL-10 production by human PBMCs following stimulation with SEA peptide and the anti-CTLA-4 hIgG1 mAb variants in (A) or hIgG1 isotype control (10 μg/mL).
(E) Representative flow cytometry profiles of CD80, CD86, MHC class II, and FcγRs on PBMC-derived CD14+ monocytes or lineage-negative CD14− dendritic cell populations.
(F) Representative flow cytometry plots and associated IL-2 production (day 4) from PBMCs with or without depletion of CD14+, CD56+, or co-depletion of CD14+ and CD56+ cells prior to stimulation with SEA peptide and anti-CTLA-4 mAbs (hIgG1 or hIgG1-DLE) or hIgG 1 isotype control (10 μg/mL).
Fold-change in cytokine production (IL-2 or IL-10) was calculated relative to a no mAb control (A, B, D, and F). Data are representative of three or more experiments. A Student’s t test was used to calculate significance in (B–D and F). Error bars indicate SEM; ***p < 0.001. See also Figure S5.
Superantigens activate T cells by crosslinking major histocompatibility class II (MHC class II) with TCRs (Spaulding et al., 2013). To further define the relevant MHC class II-expressing APCs in PBMC fraction, we evaluated co-expression of FcγRs, CD80 and CD86 (Figure 5E). Monocytes (CD14+) and four dendritic cell (DC) populations were defined and enumerated in PBMCs from healthy donors (Figures S5F and S5G) (Collin et al., 2013; Romano et al., 2015). A subpopulation of MHC class II-expressing CD14+ monocytes expressing FcγRIIIA was present (Figure 5E). However, depletion of CD14+ cells failed to impact the T cell response following stimulation with SEA peptide and either anti-CTLA-4 hIgG1 or anti-CTLA-4 hIgG1-DLE mAbs (Figure 5F). Of the DC populations, only a population of MHC class II-expressing CD11chi DC expressed detectable and homogeneous levels of FcγRIIIA (Figures 5E, S5F, and S5G). The depletion of NK cells, which also express FcγRIIIA, or co-depletion with CD14-expressing cells did not impact the anti-CTLA-4 mAb-mediated IL-2 induction (Figure 5F) (Vivier et al., 2008). Together, these functional and phenotypic data support that an FcγRIIIA+ APC, most likely a specialized DC subset, was required for antigen-specific T cell responses mediated by mAbs targeting CTLA-4.
Fc-FcγR Co-engagement within the Immunological Synapse Modulates Apical TCR Signaling Events
Several models have been proposed to explain the regulation of TCR signaling within the immune synapse, including physical exclusion of the phosphatase CD45 (coined the “kinetic segregation” model) or conformational changes to components of the TCR signaling complex (Chang et al., 2016; Li et al., 2010; Xu et al., 2008). We hypothesized that signaling events modified by the Fc region of the anti-CTLA-4 mAb involved crosstalk between FcγR engaged on the APC and the bound T cell target antigen. To specifically evaluate whether Fc-FcγR co-engagement by anti-CTLA-4 mAb could regulate early TCR signaling events, a human T cell line (Jurkat) was engineered to constitutively express cell surface CTLA-4, together with a luciferase reporter gene under the control of an IL-2 promoter (IL-2-luc) (Figure 6A). In this system, the induction of IL-2 promoter activity was dependent on TCR activation and CD28 co-stimulation, which is repressed by CTLA-4. IL-2-luc T cells were co-cultured with APCs (Raji B cells) that endogenously expressed CD80 and CD86, together with a plasma membrane-expressed anti-CD3 mAb fragment, to trigger TCR activation (Figure 6A). Notably, tethered forms of anti-CD3 antibodies have been previously shown to induce mature immune synapses in T cells (Yokosuka et al., 2005). The APC line used also endogenously expressed a single FcγR, the ITIM-containing FcγRIIB, comparable with primary human CD3− CD20+ B cells (Figures 6B and S6). This system therefore allowed us to ask whether: (1) increasing the affinity of the Fc-FcγR interaction alone could mediate a T cell-intrinsic increase in TCR-induced IL-2 reporter activation, and (2) whether using an APC line expressing the ITIM-containing FcγRIIB would attenuate cell extrinsic factors produced by the APC via the ITAM-containing FcγRIIIA. Confirming the importance of Fc-FcγR co-engagement observed from primary immune cell assays, the Fc-silent anti-CTLA-4 mAb hIgG1-N297A variant weakly induced IL-2 reporter gene activation compared with the parental hIgGI (Figure 6C). Conversely, the hIgG1-SELF Fc-engineered anti-CTLA-4 mAb with improved affinity for FcγRIIB on the APC was 7-fold more potent than the parental hIgGI mAb at inducing IL-2-lucT cell reporter activation (Figures 6C, S5C, and S5D). Taken together, these results support that the activity mAbs targeting CTLA-4 expressed within the immune synapse can be dynamically modulated by altering binding affinity to FcγRs expressed by APCs. Moreover, that FcγR co-engagement on APCs impacted proximal TCR signaling, without evidence for APC conditioning via reverse signaling mediated by activating FcγRs.
Figure 6. Fc-FcγR Co-engagement by Anti-CTLA-4 mAbs Modulates TCR Signaling.
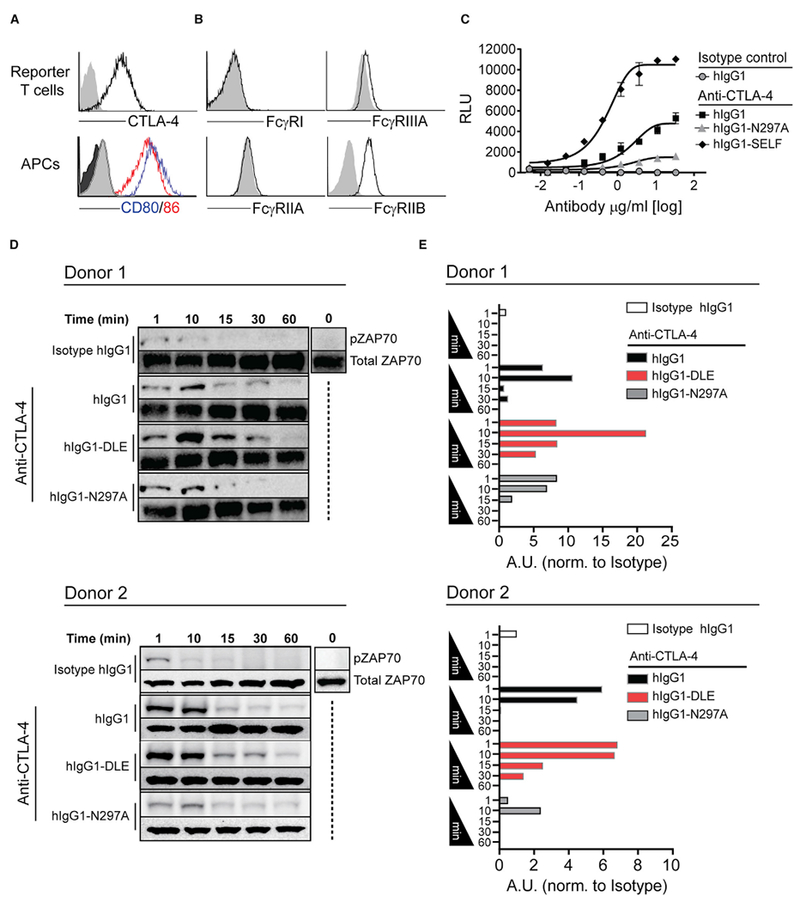
(A) Enforced cell surface expression of CTLA-4 by Jurkat IL-2-luc T cells and endogenous expression of CD80 and CD86 on Raji APCs as compared with isotype control mAbs (gray).
(B) Expression of FcγRs on Raji APCs, as compared with isotype control mAbs (gray).
(C) Jurkat IL-2-luc T cell reporter activation following 8 hr co-culture with Raji APCs in the presence of increasing doses of anti-CTLA-4 mAb variants or hIgG1 isotype control mAb. Error bars indicate SEM.
(D and E) Kinetic immunoblot analysis (D) and associated quantitative densitometry (E) of phosphorylated ZAP70 (Y-493) in human PBMCs following stimulation with 50 ng/mL of SEA peptide and 10 μg/mL of anti-CTLA-4 mAb variants (hIgG1, hIgG1-N297A, or hIgG1-DLE) or hIgG1 isotype control.
See also Figure S6.
To translate this finding back into a primary T cell assay where FcγRIIIA co-engagement on APCs was required, we monitored the kinetics of ZAP70 tyrosine phosphorylation, which is activated upon TCR ligation (Klammt et al., 2015). Unlike TCR activation, CD28 signaling initiated by ligation of CD80 or CD86 does not modulate ZAP70 activation (Michel et al., 2001). However, CTLA-4 has been reported to inhibit TCR-induced ZAP70 cluster formation, thereby providing a potential mechanism for CD28-independent feedback and regulation of TCR-ZAP70 pathway (Schneider et al., 2008). Quantitative immunoblot analysis of SEA-stimulated PBMCs together with a hIgG1 isotype control mAb showed that ZAP70 phosphorylation (pZAP70) transiently increased within 10 min after stimulation, and rapidly diminished with no detectable levels after 15 min (Figures 6D and 6E). A modest increase in ZAP70 phosphorylation was observed with SEA-stimulated PBMCs co-incubated with the Fc-silent hIgG1-N297A anti-CTLA-4 mAb. However, the addition of anti-CTLA-4 hIgG1 or anti-CTLA-4 hIgG1-DLE mAb variants with improved binding to FcγRIIIA prolonged and enhanced ZAP70 activation, with the most pronounced activity observed with the hIgG1-DLE mAb (Figures 6D and 6E). Overall, these findings support a model in which FcγR co-engagement on APCs can be harnessed to modulate TCR signaling by antibodies targeting CTLA-4 on T cells.
The Importance of Fc-FcγR Co-engagement for Improved T Cell Responses Extends to Targets Other than CTLA-4
Analogous to CTLA-4, blockade of the co-inhibitory receptor TIGIT has been shown to augment T cell activity and to support tumor rejection in preclinical animal models (Johnston et al., 2014). Given the similarity of the nature of CTLA-4 and TIGIT signaling networks, we sought to evaluate the importance of FcγR co-engagement for the functional activity of anti-TIGIT mAbs. We first tested the capacity of different Fc variants of a murine cross-reactive TIGIT mAb (clone 10A7) to control tumor growth. While an anti-TIGIT mIgG2a mAb was able to control tumor growth, a TIGIT mAb Fc variant with attenuated FcγR binding (mIgG2a-N297Q) did not demonstrate activity (Figure 7A) (Veri et al., 2007). Strikingly, the anti-tumor efficacy of TIGIT-mIgG2a was observed despite a complete lack of intratumoral Treg cell depletion (Figure 7B). To address if this translated into a human assay system, we generated a hIgG1 and hIgG1-N297A variant of the same human TIGIT cross-reactive mAb (clone 10A7). Analogous to our finding with mAbs targeting CTLA-4, the hIgG1 variant of TIGIT mAb was superior to the Fc-silent hIgG1-N297A mAb variant in the propagation of SEA-reactive T cell responses (Figure 7C). Moreover, consistent with dependence on FcγRIIIA for CTLA-4 targeted antibodies, the IL-2 response elicited by anti-TIGIT mAb (hIgG1) was significantly impaired following blockade of FcγRIIIA using either glycosylated ordegly-cosylated FcγR-specific mAbs (Figures 7D and 7E, respectively).
Figure 7. FcγR Co-engagement Is Important for Enhanced T Cell Responses Elicited by mAbs Targeting Other Immune Receptors.
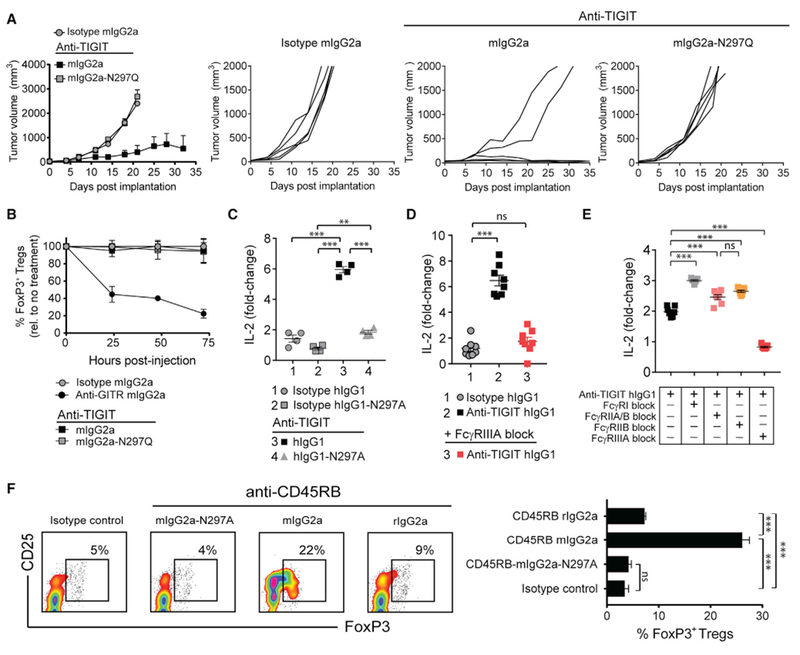
(A) BALB/c mice with established CT26 tumors (50–80 mm3) were administered i.p. twice weekly with 200 μg of anti-TIGIT mAbs or mIgG2a isotype control mAb (n = 5 mice/group). Tumor growth rates for individual mice are shown to the right.
(B) Percent (%) reduction in intratumoral FoxP3+ Treg cells relative to untreated mice 24 hr post-administration of anti-TIGIT mAbs (200 μg, mIgG2a or mIgG2a-N297Q), anti-GITR mIgG2a (100 μg), or mIgG2a isotype control mAb (200 μg) (n = 4 mice/time point).
(C) IL-2 production (day 4) by human PBMCs stimulated with 100 ng/mL of SEA peptide and 10 μg/mL of anti-TIGIT mAbs (hIgG1 or hIgG1-N297A) or corresponding isotype control mAbs.
(D and E) IL-2 production (day 4) by PBMCs stimulated with SEA peptide (100 ng/mL) and anti-TIGIT hIgG1 mAb (10 μg/mL) with or without pre-blockade of the indicated FcγRs with glycosylated (D) or deglycosylated FcγR-specific mAbs (10 μg/mL) (E).
(F) BALB/c mice were administered i.p. on day 0, 1, and 5 with 100 μg of anti-CD45RB mAb variants (rIgG2a, mIgG2a, and mIgG2a-N297A) or mIgG2a isotype control mAb. Spleens were collected on day 10, and FoxP3+ Treg cell frequency was evaluated by flow cytometry. Representative flow cytometry profiles (left), and quantitation (right) (n = 5 mice/group).
Data are representative of three or more experiments. A Student’s t test was used to calculate significance in (C–F). Error bars indicate SEM; ns, not significant; **p < 0.01; ***p < 0.001.
To understand the broader applicability of our mechanistic findings of mAb targeting T cell antigens beyond cancer immunotherapy, we investigated a mAb targeting CD45RB. CD45RB is an isoform of CD45 (ABC) with a reduced extracellular domain and identical cytoplasmic domain, allowing it to mediate phosphatase activity (Gao et al., 1999). Engagement of CD45RB with an anti-CD45RB mAb has been shown to reduce the motility of Treg cells, thereby facilitating a longer interaction or “dwell time” with APCs (Camirand et al., 2014). This culminates in improved TCR signaling, enhanced Treg cell proliferation, and disease amelioration in animal models (Sho et al., 2005). Given this previous finding in the context of our results with antibodies targeting CTLA-4 and TIGIT, we sought to explore if Treg cell expansion could also be modulated in vivo by engineering the Fc region of the CD45RB mAb. Consistent with earlier reports, injection of parental anti-CD45RB mAb (clone MB23G2, rIgG2a) into mice elicited a modest expansion of Treg cells in the peripheral blood (Figure 7F). This expansion was enhanced when the mAb was generated on the mIgG2a Fc backbone, with a higher affinity to FcγRIV (Figure 7F) (Vonderheide and Glennie, 2013). Consistent with a common mechanism with TIGIT and CTLA-4 targeted mAbs, Treg cell expansion was attenuated compared with an anti-CD45RB mIgG2a-N297A Fc variant. In summary, we find that the efficacy of mAbs targeting other receptors on distinct T cell populations can be enhanced by using Fc variants that increase the co-engagement with specific FcγRs on APCs.
DISCUSSION
The activity of mAbs is defined not only by the binding of target antigen with the Fab variable regions of the mAb, but also through co-engagement of the Fc region with FcγRs expressed on immune and non-immune cell populations (Offringa and Glennie, 2015). Recent examples of mechanisms associated with Fc-FcγR crosstalk and the therapeutic activity of immunomodulatory mAbs include engagement of activating FcγR for ADCC/P-mediated depletion of intratumoral Treg cells overexpressing target antigens, or engagement of FcγRs, particularly the inhibitory FcγR (FcγRIIB), which drives mAb-mediated clustering and enhanced receptor forward signaling in target cells (Kim and Ashkenazi, 2013; Wilson et al., 2011). In addition to the potential benefits of engaging FcγRs, other reports have identified that attenuated Fc-FcγR interactions might improve the therapeutic activity of mAbs targeting the co-inhibitory PD-1 pathway (Arlauckas et al., 2017; Dahan et al., 2015).
Antibodies targeting the CTLA-4 and PD-1/PDL-1 pathways are currently the only US Food and Drug Administration-approved immunomodulatory mAbs for the treatment of cancer. Antibody-mediated CTLA-4 pathway blockade alone was originally assumed to be sufficient for tumor-specific T cell immune responses, and the isotype selection was expected to have no impact on the pharmacologic activity of the anti-CTLA-4 mAbs. However, it was recently shown in preclinical models that the anti-tumor activity of anti-CTLA-4 was dependent on engagement with activating FcγRs in mice, particularly FcγRIV (Bulliard et al., 2013; Ingram et al., 2018; Simpson et al., 2013). These studies implicated an ADCC/P mechanism to eliminate CTLA-4-expressing intratumoral Treg cells, with a common mechanism defined for mAbs targeting Treg cell-expressed GITR and OX40 (Bulliard et al., 2013, 2014). Consistent with these preclinical results, melanoma patients exhibiting higher frequencies of FcγRIIIA+ myeloid effector cells in the peripheral blood showed an increased response to ipilimumab treatment, which was attributed to ADCC/P-mediated depletion of Treg cells within the tumor microenvironment (Romano et al., 2015). Recently, others have identified that melanoma patients expressing the high-affinity FcγRIIIA-V158 polymorphism in the context of high tumor mutational burden exhibited improved clinical response rates to ipilimumab (Arce Vargas et al., 2018). This observation was interpreted as further support for the selective depletion of intratumoral Treg cells. However, the contribution of FcγR interactions to treatment efficacy for CTLA-4 antibodies ipilumumab (hIgG1) and tremelimumab (hIgG2) in patients remains controversial. Our own recent pharmacologic assessment of two CTLA-4 antibodies with identical Fab variable regions and distinct Fc regions (hIgG1 versus hIgG2) revealed an unexpected 40-fold difference in the potency of the hIgG1 variant in a T effector cell assay (Gombos et al., 2018). Notably, this result was not attributed to potential differences in ADCC/P activity between these two Fc variants, but rather supported a yet undefined contribution of the Fc region.
Here we have identified a role for the Fc-FcγR interaction contributing to activity of mAbs targeting T cell antigens suggested to function within the immunological synapse. This activity was dependent on activating FcγRs, but notably distinct from previously described mechanisms involving the targeted elimination of Treg cells within tumors. Our findings are supported using a variety of Fc variants and engineering approaches to alter the binding anti-CTLA-4 mAbs to FcγRs, with striking concordance of the pharmacologic activity in mouse (in vitro and in vivo) and human (in vitro) assay systems. In mice, we took advantage of a non-tumor-bearing model to study antigen-specific T cell responses in combination with CTLA-4 mAb blockade in the presence and absence of Treg cells. We found strict dependence on Fc-FcγR engagement for T cell cytokine and proliferative responses in vivo, independent of the presence of Treg cells. In human assays, we established that FcγRIIIA engagement was required for T cell responsiveness to mAb-mediated CTLA-4 pathway blockade, and implicated a population of FcγRIIIA+ dendritic cells that are phenotypically distinct from CD14+ monocytes (Ziegler-Heitbrock et al., 2010). Interestingly, this population phenotypically resembles a similar population of immune cells that were correlated with patient responses to ipilimumab (Romano et al., 2015).
Here we established that Fc variants of anti-CTLA-4 or anti-TIGIT mAbs with enhanced binding to FcγRIIIA provided superior antigen-specific T cell response. These results were supported by strategies to either block the interaction of the Fc region of hIgG1 or deplete FcγRIIIA+ cells. Interestingly, blocking FcγRI, FcγRIIA, or FcγRIIA/IIB binding to the anti-CTLA-4 mAb resulted in increased T cell IL-2 production, which we interpret as providing increased access of the Fc to FcγRIIIA on APCs. In murine models, the dependence on FcγRIV for optimal T effector cell cytokine and proliferative responses was revealed using an anti-FcγRIV mAb to selectively neutralize anti-CTLA-4 mAb co-engagement. One recognized limitation of this reagent is the possibility that it might bind non-specifically to other FcγRs via its Fc domain (Tipton et al., 2015). To mitigate this possibility, we confirmed our in vivo findings using a deglycosylated version of anti-FcγRIV mAb. Importantly, our results with an anti-CTLA-4 mAb were reproduced using an anti-TIGIT mAb, in both human (in vitro) and mouse (in vivo) assays.
The mechanisms leading to phosphorylation of the TCR remain poorly defined (Brownlie and Zamoyska, 2013). Without intrinsic kinase activity, the TCR relies on recruitment of tyrosine kinase Lck. Lck phosphorylates the TCR (via ITAMs) on the CD3 signaling complex. The phosphorylated ITAMs bind a second kinase, ZAP70, which drives downstream signaling cascades (van der Merwe and Dushek, 2011). Some models suggest that MHC-peptide binding promotes conformational changes in the TCR that enable its cytoplasmic ITAM domains to be more accessible to Lck (Xu et al., 2008). Alternative hypotheses include the kinetic segregation model, where TCR phosphorylation is constantly countered by the presence of CD45, with segregation of the protein tyrosine phosphatase favoring Lck-mediated activation of the pathway (Chang et al., 2016; Mustelin et al., 2005; van der Merwe and Dushek, 2011; Li et al., 2017). Here we characterized both CD28-dependent and -independent events involving regulation of the TCR mAbs targeting CTLA-4. In the first assay, IL-2 reporter gene activation was driven by anti-CTLA mAb-mediated blockade of CD80 and CD86 binding to CTLA-4, thereby promoting CD28-CD80/CD86 co-ligation. Notably, the addition of an anti-CTLA-4 mAb with enhanced binding to the FcγR on the APC increased CD28-dependent IL-2 reporter gene activity. In the second assay, PBMCs were stimulated with SEA peptide together with anti-CTLA-4 mAbs, and TCR-induced ZAP70 activation was evaluated. ZAP70 phosphorylation has been described to occur independently of CD28 signaling (Michel et al., 2001). Further, bacterial superantigens are described to engage and directly stimulate CD28, thereby directly providing stimulation to TCR-activated T cells (Levy et al., 2016). This model therefore provided us with an opportunity to assess the impact of anti-CTLA-4 mAbs on the ability of CTLA-4 to antagonize TCR-induced ZAP70 microcluster formation, which has been associated with reduced APC-T cell dwell time (Schneider et al., 2008). Similar to the IL-2 T cell reporter assay (CD28 dependent), only anti-CTLA-4 mAbs that efficiently engaged an FcγRIIIA on peripheral blood DCs led to improved (CD28 independent) ZAP70 phosphorylation. Despite our results favoring the kinetic segregation model, we were unable to find direct evidence for a difference in dwell time or the extent of CD45 exclusion, despite the pronounced effect FcγR co-engagement by anti-CTLA-4 mAbs had on the TCR-CD28 signaling axis.
Our findings highlight the impact of Fc region selection for mAbs targeting CTLA-4 and TIGIT, as well as other T cell targets acting within the immune synapse, including antigens expressed by Treg cells. Moreover, this model may extend to settings of autoimmunity and transplantation, and to other target antigens on cell types that form an immune synapse with APCs, therefore highlighting the need for the selection of an appropriate Fc region for therapeutic antibodies. Currently eight antibodies targeting CTLA-4 are being investigated in clinical trials: ipililumab (human IgG1), tremelimumab (human IgG2), AGEN1884 (IgG1), MK-1308 (IgG information unavailable), BMS-986218 (afucosylated hIgG1), BMS-986249 (hIgG1 probody), CS1002 (hIgG1), and BCD-145 (IgG information unavailable). Similarly, four TIGIT programs have recently entered clinical development: OMP-313M32 (hIgG1), MTIG7192A (hIgG1), MK-7684 (IgG information unavailable), and BMS-986207 (hIgG1). The impact of Fc selection on the therapeutic activity of either CTLA-4 or TIGIT mAb responses remains to be investigated. Taken together, we have discovered a property of Fc-FcγR co-engagement within the immune synapse of T cells and APCs that modulates the activity of therapeutic mAbs targeting T cell-associated antigens. Our findings provide a foundation for a class of next-generation recombinant mAbs that can be optimized to sculpt both pro- and anti-inflammatory T cell immune responses in patients.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse GITR (clone DTA-1) | Fc variants generated in-house | RRID: AB_1107688 |
| Anti-mouse CTLA-4 (clone 9D9) | Patent literature, Fc variants generated in-house | RRID: AB_10949609 |
| Anti-mouse/human TIGIT (clone 10A7) | Patent literature, Fc variants generated in-house | US patent number: 9,499,596 |
| Anti-mouse CD45RB (clone MB23G2) | MB23G2 hybridoma, Fc variants generated in-house | RRID: AB_1107653 |
| Anti-human CTLA-4 (internal clone) | Clone and Fc variants generated in-house | Unpublished patent application |
| Anti-human GITR (clone 6C8, Tolerx) | Patent literature | US patent number: 7,812,135 |
| Anti-mouse FoxP3 (clone FJK-16s) | Thermo Fisher | RRID: AB_469916 |
| Anti-human FOXP3 (clone PCH101) | eBioscience | RRID: AB_1834365 |
| Anti-mouse CD25 (clone PC-61.5.3) | BioXcell | RRID: AB_1107619 |
| Anti-mouse CD25 (clone 3C7) | Biolegend | RRID: AB_2616761 |
| Anti-human CD25 (clone M-A251) | Biolegend | RRID: AB_2561975 |
| Anti-mouse CD45.2 (clone 104) | Biolegend | RRID: AB_313444 |
| Anti-mouse CD4 (clone RM4-5) | Biolegend | RRID: AB_11126142 |
| Anti-mouse CD8 (clone 53-6.7) | Biolegend | RRID: AB_493426 |
| Anti-mouse Vβ2 (clone B20.6) | Biolegend | RRID: AB_1089254 |
| Anti-mouse Vβ8.1, 8.2 (clone KJ16-133.18) | Biolegend | RRID: AB_1186101 |
| Anti-mouse IL-2 (clone JES6-5H4) | Thermo Fisher | RRID: AB_315303 |
| Anti-human IL-2 (clone MQ1-17H12) | Biolegend | RRID: AB_315097 |
| Anti-mouse/human Ki67 (clone B56) | BD Biosciences | RRID: AB_396302 |
| Anti-mouse CD62L (clone MEL-14) | Biolegend | RRID: AB_2563058 |
| Anti-mouse CD44 (clone IM7) | Biolegend | RRID: AB_2564214 |
| Anti-human CD56 (clone HCD56) | Biolegend | RRID: AB_11218798 |
| Anti-human NKp46 (clone 9E2) | Miltenyi Biotec | RRID: AB_2660316 |
| Anti-human CD14 (clone 63D3) | Biolegend | RRID: AB_2687384 |
| Anti-human CD3 (clone OKT3) | Biolegend | RRID: AB_2563352 |
| Anti-human CD3 (clone SP34) | BD Biosciences | RRID: AB_396483 |
| Anti-human CD19 (clone HIB19) | Biolegend | RRID: AB_2562097 |
| Anti-human MHC-II (clone L243) | Biolegend | RRID: AB_2616625 |
| Anti-human IFNγ (clone 4S.B3) | Biolegend | RRID: AB_315237 |
| Anti-human CD11c (clone 3.9) | Biolegend | RRID: AB_389351 |
| Anti-human CD1c (clone L161) | Biolegend | RRID: AB_2629759 |
| Anti-human CD141 (clone M80) | Biolegend | RRID: AB_2572198 |
| Anti-human CD304 (clone 12C2) | Biolegend | RRID: AB_2563872 |
| Anti-human CD4 (clone OKT4) | Biolegend | RRID: AB_11204077 |
| Anti-human CD80 (clone 2D10) | Biolegend | RRID: AB_2076147 |
| Anti-human CD86 (clone IT2.2) | Biolegend | RRID: AB_10899582 |
| Anti-human CD16 (clone 3G8) | Biolegend | RRID: AB_2572006 |
| Anti-mouse CD16.2 (clone 9E9) | Biolegend | RRID: AB_2565302 |
| Anti-human CD32A (clone IV.3) | STEMCELL Technologies | RRID: AB_519584 |
| Anti-human CD32B (clone 2B6), hIgG1-N297A and mIgG2a-N297A variants | Patent literature, Fc variants generated in-house | Patent:US8968730B2 |
| Anti-human CD32A/B (clone 6C4) | Thermo Fisher | RRID: AB_1311188 |
| Anti-human CD64 (clone 10.1) | BD Biosciences | RRID: AB_627153 |
| Anti-human ZAP70 (clone 99F2) | Cell Signaling Technology | RRID: AB_10691455 |
| Anti-human pZAP70 (2704) | Cell Signaling Technology | RRID: AB_2217457 |
| Biological Samples | ||
| Healthy human peripheral blood (unpurified buffy coats and leukopheresis) | Research Blood Components, LLC: http://researchbloodcomponents.com/products.html |
N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Staphylococcal enterotoxin A (SEA) | Toxin Technology, Inc | Cat# AT101red |
| Staphylococcal enterotoxin B (SEB) | Toxin Technology, Inc | Cat# BT202red |
| Recombinant human CTLA-4 (polyhistidine tag) | Sino Biological | Cat# 11159-H08H |
| Recombinant human CTLA-4 (Fc-fusion) | R&D Systems | Cat# 7268-CT |
| Recombinant human CD80 (Fc-fusion) | R&D Systems | Cat# 140-B1 |
| Recombinant human CD86 (Fc-fusion) | R&D Systems | Cat# 141-B2 |
| Diphtheria toxin | Sigma Aldrich | D0564; MDL: MFCD00163490 |
| Critical Commercial Assays | ||
| CTLA-4 Blockade Bioassay | Promega | Cat# JA3001 |
| FOXP3 staining buffer set | eBioscience | Cat# 00-5523-00 |
| Zombie NIR fixable viability kit | Biolegend | Cat# 423106 |
| Experimental Models: Cell Lines | ||
| CT26 colon carcinoma | ATCC | RRID: CVCL_7256 |
| CHO-mouse-FcγRI | CNCM, Institut Pasteur | Described: Mancardi et al., 2008 |
| CHO-mouse-FcγRIIB | CNCM, Institut Pasteur | Described: Mancardi et al., 2008 |
| CHO-mouse-FcγRIII | CNCM, Institut Pasteur | Described: Mancardi et al., 2008 |
| CHO-mouse-FcγRIV | CNCM, Institut Pasteur | Described: Mancardi et al., 2008 |
| CHO-human-FcγRI | CNCM, Institut Pasteur | CNCM I-4383 |
| CHO-human-FcγRIIA-H/H131 | CNCM, Institut Pasteur | CNCM I-4384 |
| CHO-human-FcγRIIA-R/R131 | CNCM, Institut Pasteur | CNCM I-4385 |
| CHO-human-FcγRIIB | CNCM, Institut Pasteur | CNCM I-4386 |
| CHO-human-FcγRIIIA-F/F131 | CNCM, Institut Pasteur | CNCM I-4388 |
| CHO-human-FcγRIIIA-V/V131 | CNCM, Institut Pasteur | CNCM I-4389 |
| Jurkat-FcγRIIA-H/H131 | Promega | Cat# G9991 |
| Jurkat-CTLA-4 (forced extracellular expression) | This paper | Described: Nakaseko et al., 1999 JEM |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | The Jackson Laboratory | Stock# 000664 |
| Mouse: BALB/cJ | The Jackson Laboratory | Stock# 000651 |
| Mouse: B6.129-FoxP3tm3(DTR/GFP)Ayr/J | The Jackson Laboratory | Stock# 016958 |
| Software and Algorithms | ||
| WEHI Weasel software v3.2.1 | WEHI Institute | Information |
| Prism 7 | Graphpad | Information |
| FACSDIVA | BD Biosciences | Information |
| ImageJ | Wayne Rasband, NIH | Information |
CONTACT FOR REAGENT AND RESOURCE SHARING
All information and requests for reagents may be directed to, and will be fulfilled by the Lead Contact, Nicholas Wilson (nicholas.wilson@gilead.com).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Subjects and Samples
Human PBMCs were obtained from healthy, consenting, and anonymized adult volunteers through Research Blood Components, LLC (Boston, MA). All blood collection followed American Association of Blood Banks guidelines and was approved by New England Independent Review Board (NEIRB, 120160613).
Mice and In Vivo Studies
C57BL/6, BALB/c (BALB/cJ), and C57BL/6.129-FoxP3tm3(DTR/GFP)Ayr/J (referred to here as FoxP3DTR) mice were purchased from Jackson Laboratories. For CT26 tumor studies, 5×104 cells were suspended in 100 μl PBS and injected subcutaneously. Following engraftment (approximately 50-80mm3) mice were randomized and treated via intraperitoneal (i.p.) administration of anti-CTLA-4 (mIgG2a and mIgG2a-N297A, single 100 mg dose), anti-TIGIT (mIgG2a and mIgG2a-N297Q, 200 μg twice weekly), anti-GITR (mIgG2a, single 100 μg dose), or an isotype control (mIgG2a, single 100 μg dose). In separate experiments, mice were either measured bi-weekly for tumor growth or sacrificed at 0, 24, 72, or 120 hr post-treatment for tissue collection. To monitor antigen-specific responses in vivo, C57BL/6 mice were administered with a single i.p. injection of 150 μg SEB superantigen (Toxin Technologies) and anti-CTLA-4 (mIgG2a or mIgG2a-N297A, single 100 μg dose) or a mIgG2a isotype control (single 100 μg dose). To test the importance of FcγRIV for the function of anti-CTLA-4, mice were concurrently injected with a single dose of FcγRIV-specific blocking mAb (glycosylated or deglycosylated clone 9E9, 200 μg) or hamster IgG isotype control (200 μg). Blood and/or spleens were collected at 0, 72, 144, or 240 hr post-treatment and analyzed as described (Miyahara et al., 2012). For mAb-mediated depletion of FoxP3+ Treg cells, C57BL/6 mice were treated with a single i.p. injection of anti-CD25 mAb (250 μg, clone PC61), as described (Setiady et al., 2010). Mice were evaluated before anti-CD25 mAb treatment (day −10) and after (days 0,3, and 6) SEB/mAb treatment by flow cytometry to ensure sufficient Treg cell depletion. For transgenic ablation of FoxP3+ Treg cells, FoxP3DTR mice were injected with diphtheria toxin (DT) (100 μg, i.p.) 48 and 24 hr prior to study initiation as described (Kim et al., 2007). As with anti-CD25 mAb treated mice, flow cytometry was conducted to ensure effective Treg cell depletion. For CD45RB-induced expansion of Treg cells, BALB/c mice were treated with different CD45RB mAb Fc variants or isotype control mAb, as previously described (Camirand et al., 2014). Spleens from treated mice were collected on day 10 and evaluated for FoxP3+ Treg cell expansion by flow cytometry. Procedures involving animals were reviewed and approved by IACUC, Agenus, and conform to the relevant regulatory standards.
Cell Lines
The CT26 colon cancer cell line was obtained from American Type Culture Collection (ATCC) and maintained in RPMI 1640 supplemented with 10% heat inactivated FBS. CHO cells expressing murine FcγRs (I, IIB, III, and IV) and human FcγRs (I, IIA-R/R131, IIB, IIIA-F/F158, and IIIA-V/V158) were obtained from the Pasteur Institute (CNCM) (Mancardi et al., 2008) or human (Bruhns et al., 2009). Jurkat cells engineered to express the high affinity allotype of FcγRIIA (H/H131) were obtained from Promega. Jurkat cells with enforced cell surface expression of human CTLA-4 were generated by transduction of ICD-truncated CTLA-4 as described (Nakaseko et al., 1999 JEM). FcγR-expressing cell lines and Jurkat-CTLA-4+ cells were cultured in supplemented RPM11640. All cell lines were kept at 37°C in a 5% CO2 incubator.
METHOD DETAILS
Flow Cytometry
A detailed list of the flow cytometry antibodies used can be found in the Key Resource Table. Cell surface and intracellular (FoxP3 and Ki67) flow cytometry analysis of both mouse and human samples was performed as previously described (Waight et al., 2015). Briefly, cells were stained with the indicated mAbs in flow buffer (PBS-BSA 0.5%, for all cell surface antigens) or permeabilization buffer (eBioscience Foxp3 / Transcription Factor Staining Buffer Set for all intracellular antigens) for a minimum of 45 minutes on ice. For IL-2 cytokine staining, cells were incubated for 5 hr with brefeldin-A to inhibit protein transport prior to staining. All cytometric analysis was conducted on an LSRFortessa instrument (BD Biosciences).
Immunoblot Analysis
Human PBMCs were incubated with SEA peptide and 10μg/mL of the indicated CTLA-4 mAb Fc variants or relevant isotype control antibodies. Cells were then incubated at 37°C for 0 (pre) 1, 5, 10, 30, or 60 minutes. At the end of the incubation, cells were lysed with cold 1× radioimmunoprecipitation assay buffer (RIPA buffer) supplemented with a phosphatase/protease inhibitor cocktail (Cell Signaling Technologies). Following supernatant clarification, protein concentration was quantified using bicinchoninic acid assay (BCA) (Pierce Biotechnology). Cell lysates (20μg/lane) were prepared in Bolt LDS sample buffer and heated for 10 minutes at 70°C before being loaded onto a 4-12% Bolt Bis Tris gels (Novex). Proteins were separated in 1× Bolt MOPS-buffer (ThermoFisher) and then blotted onto a PVDF membrane. Following blockade with 5% bovine serum albumin (BSA, 1 hr), samples were incubated with primary anti-human rabbit ZAP70 (Y-493)/Syk (Y-526) mAb (Cell Signaling Technologies) in blocking buffer overnight at 4°C. Membranes were probed with goat anti-rabbit secondary HRP-conjugate and visualized with SignalFire ECL reagent (Cell Signaling Technology). Images were captured using the Chemidoc imaging system (BioRad). As a control, total ZAP70 protein was evaluated following membrane stripping with Restore™ PLUS Western Blot Stripping Buffer. Densitometric analysis of ZAP70 phosphorylation was performed using ImageJ (Wayne Rasband; National Institute of Mental Health) and normalized to total ZAP70 protein.
Fc-FcγR Binding and FcγR Blockade Studies
For blockade of anti-CTLA-4 Fc:FcγR binding, cells expressing recombinant mouse or human FcγRs were pre-incubated with 10 μg FcγR-specific antibodies for 15-30 minutes at room temperature (see the Key Resource Table for clone information). For human FcγRI blockade 20 μg/ml of FcγRI mAb was used. Following receptor blockade or for experiments without blockade, cells were incubated with serial dilutions anti-human or anti-mouse CTLA-4 mAb Fc variants for 1 hr at 4°C. Binding was detected by flow cytometry using a PE-conjugated anti-human or anti-mouse F(ab’)2 secondary mAb (Jackson Immunoresearch). To ensure accurate Fc:FcγR characterization and prevent potential receptor cross-blockade, FcγR-specific antibodies were deglycosylated using PGNase-F. Briefly, anti-human (clones 3G8, 6C4, and 10.1) and anti-mouse (clone 9E9) FcγR-specific antibodies were deglycosylated with PNGase F (10 ng/10μg of antibody) for 16 hr at 37°C. Residual PNGase F (6×His) was removed with nickel (Ni)-NTA agarose beads. Glycosylation-dependent band shifts of the IgG heavy chain were evaluated by reducing SDS-PAGE (Figures S4F and S4I). The anti-human FcγRIIB-specific mAb (2B6) was produced on a mIgG2a-N297A (aglycosylated) Fc backbone and thus was excluded from PNGase F treatment. For FcγR blockade in human T cell stimulation assays, FcγR blocking antibodies were added to PBMCs for 15 minutes at 37°C, 5% CO2 (at 10 μg/ml) prior to stimulation with SEA peptide (100 ng/ml, Toxin Technology, Inc.) and anti-CTLA-4 or anti-TIGIT mAbs.
T Cell Stimulation Assays
PBMC from healthy donors were plated at 1×105 cells/well in supplemented RPMI-1640 media. Cells were stimulated with SEA peptide (100 ng/ml, Toxin Technology, Inc.) in the presence of titrated or fixed concentrations of indicated antibodies for 4 days at 37°C, 5% CO2. Where indicated, magnetic bead-based sorting (Miltenyi MACS) was used to deplete PBMCs of FcγRIIIA-expressing CD14+ monocytes, CD56+ NK cells, or T cells (CD3+ cell depletion) prior to stimulation. To assess the agonistic potential of anti-CTLA-4 mAb, increasing doses of anti-CTLA-4 mAb or an isotype control (both hIgG1) were cross-linked on a 96-well plate overnight at 4°C. T cells were isolated from human PBMCs by magnetic bead-based sorting (CD4 and CD8; Miltenyi MACS pan T cell isolation kit) and then added to the pre-coated plates (or control uncoated plates with anti-CTLA-4 in solution) in the presence of 5 μg/mL of anti-CD3 mAb for 4 days at 37°C. Cell-free supernatants were harvested at the indicated time-points and stored at −80°C until analysis. IL-2 and IL-10 cytokine levels were quantified using AlpaLISA(Perkin-Elmer). As a positive control for agonistic activity, purified T cells were stimulated with increasing doses of plate-bound or soluble anti-GITR mAb (6C8, hIgG1) for 3 days at 37°C and 5% CO2. Following incubation, intracellular IFNγ levels were assessed by flow cytometry (anti-IFNγ mAb, clone 4S.B3).
TCR Signaling Reporter Assay
The contribution of FcγR co-engagement to TCR signaling was evaluated using the CTLA-4 blockade bioassay (Promega). In this system, the human Jurkat T cell line was engineered to constitutively express cell surface CTLA-4, together with a luciferase reporter gene under the control of an IL-2 promoter (IL-2-luc). Jurkat IL-2-luc T cells were co-cultured anti-CTLA-4 mAbs (hIgG1, hIgG1-N297A, hIgG1-SELF) or a hIgG1 isotype control, together with Raji B cells that endogenously expressed FcγRIIB, CD80, CD86, and a plasma membrane expressed anti-CD3 mAb fragment to trigger TCR activation. Following an 8 hr incubation, IL-2-luc activity was determined using Bio-GloTM Luciferase Assay System (Promega).
Surface Plasmon-Resonance Analysis
Affinities of the utilized anti-human CTLA-4 mAb with human CTLA-4, were determined using a Biacore 2000 instrument (GE Healthcare) equipped with a research-grade CM5 sensor chip. Each mAb was immobilized on the CM5 sensor chip by amine coupling chemistry. The surfaces of flow cells 1, 2, 3 and 4 were activated for 7 minutes with a 1:1 mixture of 0.1 M NHS (N-hydroxysuccinimide) and 0.1 M EDC (3-(N,N-dimethylamino) propyl-N-ethylcarbodiimide) at a flow rate of 5 μl/minute. The ligand, at a concentration of 25 μg/ml, was immobilized on flow cells 2, 3, and 4. Surfaces were blocked with a 7 minute injection of 1M ethanolamine at a pH of 8.0. To collect kinetic binding data, recombinant human CTLA-4 his-tagged (Sino Biological) or Fc-fusion (R&D Systems) was injected over the flow cells at concentrations of 20, 6.7, 2.2, and 0.7 nM at a flow rate of 30 μl/minute and at a temperature of 25°C. The complex was allowed to associate for 90 s and dissociate for >300 s. The surfaces were regenerated with a 30 s injection of 3 M MgCl2. The data were fit to a simple 1:1 interaction model using the global data analysis option available within BiaEvaluation 3.1 software (GE Healthcare).
Blockade of CTLA-4 Binding to CD80 and CD86
To facilitate flow cytometry detection of CTLA-4 ligand binding, recombinant CD80 and CD86 proteins (Fc-fusion, R&D Systems) were conjugated to Alexa Fluor 647 (Invitrogen). Jurkat cells with forced cell surface expression of human CTLA-4 were incubated with serial dilutions of anti-human CTLA-4 mAb or an isotype control (both hIgG1) for 1 hr at 4°C. Cells were washed and subsequently incubated with fluorescently-labelled CD80-Fc or CD86-Fc proteins for 1 hr at 4°C. Following incubation, CD80 and CD86 binding was analyzed by flow cytometry.
QUANTIFICATION AND STATISTICAL ANALYSIS
Flow cytometry data were analyzed with WEHI Weasel software v3.2.1. Statistical analyses were done with Prism 7 (GraphPad Software). p values were calculated using two-way ANOVA or a Student’s t test where indicated (ns, not significant; *, p<0.05; **, p<0.01; ***, p< 0.001).
Supplementary Material
Highlights.
CTLA-4 and TIGIT mAbs required human FcγRIIIA on APCs for optimal T cell responses
FcγRIV contributed to CTLA-4 mAb activity in mice, independent of Treg cells
Fc-FcγR co-engagement by anti-CTLA-4 mAbs modulated both TCR and CD28 signaling
Fc-FcγR co-engagement enhanced Treg cell expansion by an mAb targeting CD45RB
Significance.
Therapeutic mAbs targeting T cell co-inhibitory pathways, such as cytotoxic T lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein-1 (PD-1), have emerged as an important class of cancer therapies. Insights into the function of mAbs with different IgG isotypes have enabled modulation of their biological activity, providing opportunity to enhance their therapeutic effect. We have discovered a property of Fc-FcγR co-engagement within the T cell-APC immune synapse that modulates the activity of mAbs targeting effector and regulatory T cell antigens. Our findings highlight the importance of tailoring the IgG-associated Fc region to each target and provide a foundation for a class of next-generation mAbs that can be optimized to sculpt T cell immune responses in patients.
ACKNOWLEDGMENTS
We thank Dr. Michael Robinson for helpful discussion and his assistance with manuscript finalization.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and can be found with this article online at https://doi.org/10.1016/j.ccell.2018.05.005.
DECLARATION OF INTERESTS
J.D.W., D.C., S.D., R.G., T.H., A.M.G., M.M., L.S., B.M., C.B., A.T., B.A., J.S.B., R.S., D.A.S., and N.S.W. have ownership of equity securities and/or are currently employed by Agenus. This does not alter adherence to Cancer Cell policies on sharing data and materials.
REFERENCES
- Acuto O, and Michel F (2003). CD28-mediated co-stimulation:aquantitative support for TCR signalling. Nat. Rev. Immunol 3, 939–951. [DOI] [PubMed] [Google Scholar]
- Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, Solomon I, Lesko MH, Ruef N, Roddie C, et al. (2018). Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell 33, 649–663 e644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arce Vargas F, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, Miranda Rota E, Dahan R, Georgiou A, Sledzinska A, et al. (2017). Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory T cells and synergizes with PD-1 blockade to eradicate established tumors. Immunity 46, 577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, Miller MA, Carlson JC, Freeman GJ, Anthony RM, et al. (2017). In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci. Transl. Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlie RJ, and Zamoyska R (2013). T cell receptor signalling networks: branched, diversified and bounded. Nat. Rev. Immunol 13, 257–269. [DOI] [PubMed] [Google Scholar]
- Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, and Daeron M (2009). Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 113, 3716–3725. [DOI] [PubMed] [Google Scholar]
- Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, Wilson NS, Dranoff G, and Brogdon JL (2013). Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J. Exp. Med 210, 1685–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulliard Y, Jolicoeur R, Zhang J, Dranoff G, Wilson NS, and Brogdon JL (2014). OX40 engagement depletes intratumoral Tregs via activating FcgammaRs, leading to antitumor efficacy. Immunol. Cell Biol 92, 475–480. [DOI] [PubMed] [Google Scholar]
- Camirand G, Wang Y, Lu Y, Wan YY, Lin Y, Deng S, Guz G, Perkins DL, Finn PW, Farber DL, et al. (2014). CD45 ligation expands Tregs by promoting interactions with DCs. J. Clin. Invest 124, 4603–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang VT, Fernandes RA, Ganzinger KA, Lee SF, Siebold C, McColl J, Jonsson P, Palayret M, Harlos K, Coles CH, et al. (2016). Initiation of T cell signaling by CD45 segregation at ‘close contacts’. Nat. Immunol 17, 574–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin M, McGovern N, and Haniffa M (2013). Human dendritic cell subsets. Immunology 140, 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahan R, Sega E, Engelhardt J, Selby M, Korman AJ, and Ravetch JV (2015). FcgammaRs modulate the anti-tumor activity of antibodies targeting the PD-1/PD-L1 Axis. Cancer Cell 28, 543. [DOI] [PubMed] [Google Scholar]
- Gao Z, Zhong R, Jiang J, Garcia B, Xing JJ, White MJ, and Lazarovits AI (1999). Adoptively transferable tolerance induced by CD45RB monoclonal antibody. J. Am. Soc. Nephrol 10, 374–381. [DOI] [PubMed] [Google Scholar]
- Gombos RB, Gonzalez A, Manrique M, Chand D, Savitsky D, Morin B, Breous-Nystrom E, Dupont C, Ward RA, Mundt C, et al. (2018). Toxicological and pharmacological assessment of AGEN1884, a novel human IgG1 anti-CTLA-4 antibody. PLoS One 13, e0191926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram JR, Blomberg OS, Rashidian M, Ali L, Garforth S, Fedorov E, Fedorov AA, Bonanno JB, Le Gall C, Crowley S, et al. (2018). Anti-CTLA-4 therapy requires an Fc domain for efficacy. Proc. Natl. Acad. Sci. USA 115, 3912–3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, Park S, Javinal V, Chiu H, Irving B, et al. (2014). The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 26, 923–937. [DOI] [PubMed] [Google Scholar]
- Kim JM, and Ashkenazi A (2013). Fcgamma receptors enable anticancer action of proapoptotic and immune-modulatory antibodies. J. Exp. Med 210, 1647–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JM, Rasmussen JP, and Rudensky AY (2007). Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol 8, 191–197. [DOI] [PubMed] [Google Scholar]
- Kim YH, Shin SM, Choi BK, Oh HS, Kim CH, Lee SJ, Kim KH, Lee DG, Park SH, and Kwon BS (2015). Authentic GITR signaling fails to induce tumor regression unless Foxp3+ regulatory T cells are depleted. J. Immunol 105, 4721–4729. [DOI] [PubMed] [Google Scholar]
- Klammt C, Novotna L, Li DT, Wolf M, Blount A, Zhang K, Fitchett JR, and Lillemeier BF (2015). T cell receptor dwell times control the kinase activity of Zap70. Nat. Immunol 16, 961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong KF, Fu G, Zhang Y, Yokosuka T, Casas J, Canonigo-Balancio AJ, Becart S, Kim G, Yates JR 3rd, Kronenberg M, et al. (2014). Protein kinase C-eta controls CTLA-4-mediated regulatory T cell function. Nat. Immunol 15, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummel MF, and Allison JP (1995). CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med 182, 459–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, Chan C, Chung HS, Eivazi A, Yoder SC, et al. (2006). Engineered antibody Fc variants with enhanced effector function. Proc. Natl. Acad. Sci. USA 103, 4005–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy R, Rotfogel Z, Hillman D, Popugailo A, Arad G, Supper E, Osman F, and Kaempfer R (2016). Superantigens hyperinduce inflammatory cytokines by enhancing the B7-2/CD28 costimulatory receptor interaction. Proc. Natl. Acad. Sci. USA 113, E6437–E6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, and Ravetch JV (2011). Inhibitory Fcgamma receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science 333, 1030–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Stagg NJ, Johnston J, Harris MJ, Menzies SA, DiCara D, Clark V, Hristopoulos M, Cook R, Slaga D, et al. (2017). Membrane-proximal epitope facilitates efficient T cell synapse formation by anti-FcRH5/CD3 and is a requirement for myeloma cell killing. Cancer Cell 31, 383–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MO, and Rudensky AY (2016). T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat. Rev. Immunol 16, 220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YC, Chen BM, Wu PC, Cheng TL, Kao LS, Tao MH, Lieber A, and Roffler SR (2010). Cutting edge: mechanical forces acting on T cells immobilized via the TCR complex can trigger TCR signaling. J. Immunol 184, 5959–5963. [DOI] [PubMed] [Google Scholar]
- Mancardi DA, Iannascoli B, Hoos S, England P, Daeron M, and Bruhns P (2008). FcgammaRIV is a mouse IgE receptor that resembles macrophage FcepsilonRI in humans and promotes IgE-induced lung inflammation. J. Clin. Invest 118, 3738–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel F, Attal-Bonnefoy G, Mangino G, Mise-Omata S, and Acuto O (2001). CD28 as a molecular amplifier extending TCR ligation and signaling capabilities. Immunity 15, 935–945. [DOI] [PubMed] [Google Scholar]
- Miyahara Y, Khattar M, Schroder PM, Mierzejewska B, Deng R, Han R, Hancock WW, Chen W, and Stepkowski SM (2012). Anti-TCRbeta mAb induces long-term allograft survival by reducing antigen-reactive T cells and sparing regulatory T cells. Am. J. Transplant 12, 1409–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustelin T, Vang T, and Bottini N (2005). Protein tyrosine phosphatases and the immune response. Nat. Rev. Immunol 5, 43–57. [DOI] [PubMed] [Google Scholar]
- Nakaseko C, Miyatake S, Iida T, Hara S, Abe R, Ohno H, Saito Y, and Saito T (1999). Cytotoxic T lymphocyte antigen 4 (CTLA-4) engagement delivers an inhibitory signal through the membrane-proximal region in the absence of the tyrosine motif in the cytoplasmic tail. J. Exp. Med 190, 765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn F, Bruhns P, Horiuchi K, and Ravetch JV (2005). FcgammaRIV: a novel FcR with distinct IgG subclass specificity. Immunity 23, 41–51. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn F, Gordan S, and Lux A (2015). FcgammaR dependent mechanisms of cytotoxic, agonistic, and neutralizing antibody activities. Trends Immunol 36, 325–336. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn F, and Ravetch JV (2008). Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol 8, 34–47. [DOI] [PubMed] [Google Scholar]
- Offringa R, and Glennie MJ (2015). Development of next-generation immunomodulatory antibodies for cancer therapy through optimization of the IgG framework. Cancer Cell 28, 273–275. [DOI] [PubMed] [Google Scholar]
- Pentcheva-Hoang T, Egen JG, Wojnoonski K, and Allison JP (2004). B7-1 and B7-2 selectively recruit CTLA-4 and CD28 to the immunological synapse. Immunity 21, 401–413. [DOI] [PubMed] [Google Scholar]
- Ribas A, and Flaherty KT (2015). Gauging the long-term benefits of ipilimumab in melanoma. J. Clin. Oncol 33, 1865–1866. [DOI] [PubMed] [Google Scholar]
- Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, Michielin O, Weide B, Romero P, and Speiser DE (2015). Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl. Acad. Sci. USA 112,6140–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Yamaguchi T, Nomura T, and Ono M (2008). Regulatory T cells and immune tolerance. Cell 133, 775–787. [DOI] [PubMed] [Google Scholar]
- Schneider H, Smith X, Liu H, Bismuth G, and Rudd CE (2008). CTLA-4 disrupts ZAP70 microcluster formation with reduced T cell/APC dwell times and calcium mobilization. Eur. J. Immunol 38, 40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, and Korman AJ (2013). Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res 1, 32–42. [DOI] [PubMed] [Google Scholar]
- Setiady YY, Coccia JA, and Park PU (2010). In vivo depletion of CD4+FOXP3+ Treg cells by the PC61 anti-CD25 monoclonal antibody is mediated by FcgammaRIII+ phagocytes. Eur. J. Immunol 40, 780–786. [DOI] [PubMed] [Google Scholar]
- Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, Weikert SH, and Presta LG (2002). Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J. Biol. Chem 277, 26733–26740. [DOI] [PubMed] [Google Scholar]
- Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J,Xie D, Lai J, Stadlen A, Li B, et al. (2001). High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J. Biol. Chem 276, 6591–6604. [DOI] [PubMed] [Google Scholar]
- Sho M, Kishimoto K, Harada H, Livak M, Sanchez-Fueyo A, Yamada A, Zheng XX, Strom TB, Basadonna GP, Sayegh MH, and Rothstein DM (2005). Requirements for induction and maintenance of peripheral tolerance in stringent allograft models. Proc. Natl. Acad. Sci. USA 102, 13230–13235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, et al. (2013). Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med 210, 1695–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KG, and Clatworthy MR (2010). FcgammaRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat. Rev. Immunol 10, 328–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaulding AR, Salgado-Pabon W, Kohler PL, Horswill AR, Leung DY, and Schlievert PM (2013). Staphylococcal and streptococcal superantigen exotoxins. Clin. Microbiol. Rev 26, 422–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart R, Hammond SA, Oberst M, and Wilkinson RW (2014). The role of Fc gamma receptors in the activity of immunomodulatory antibodies for cancer. J. ImmunoTher. Cancer 2, 29. [Google Scholar]
- Tipton TR, Mockridge CI, French RR, Tutt AL, Cragg MS, and Beers SA (2015). Anti-mouse FcgammaRIV antibody 9E9 also blocks FcgammaRIII in vivo. Blood 126, 2643–2645. [DOI] [PubMed] [Google Scholar]
- van der Merwe PA, and Dushek O (2011). Mechanisms for T cell receptor triggering. Nat. Rev. Immunol 11, 47–55. [DOI] [PubMed] [Google Scholar]
- Veri MC, Gorlatov S, Li H, Burke S, Johnson S, Stavenhagen J, Stein KE, Bonvini E, and Koenig S (2007). Monoclonal antibodies capable of discriminating the human inhibitory Fcgamma-receptor IIB (CD32B) from the activating Fcgamma-receptor IIA (CD32A): biochemical, biological and functional characterization. Immunology 121, 392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivier E, Tomasello E, Baratin M, Walzer T, and Ugolini S (2008). Functions of natural killer cells. Nat. Immunol 9, 503–510. [DOI] [PubMed] [Google Scholar]
- Vonderheide RH, and Glennie MJ (2013). Agonistic CD40 antibodies and cancer therapy. Clin. Cancer Res 19, 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waight JD, Gombos RB, and Wilson NS (2017). Harnessing co-stimulatory TNF receptors for cancer immunotherapy: current approaches and future opportunities. Hum. Antibodies 25, 87–109. [DOI] [PubMed] [Google Scholar]
- Waight JD, Takai S, Marelli B, Qin G, Hance KW, Zhang D, Tighe R, Lan Y, Lo KM, Sabzevari H, et al. (2015). Cutting edge: epigenetic regulation of Foxp3 defines a stable population of CD4+ regulatory T cells in tumors from mice and humans. J. Immunol 194, 878–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LS, and Sansom DM (2011). The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat. Rev. Immunol 11, 852–863. [DOI] [PubMed] [Google Scholar]
- White AL, Chan HT, French RR, Willoughby J, Mockridge CI, Roghanian A, Penfold CA, Booth SG, Dodhy A, Polak ME, et al. (2015). Conformation of the human immunoglobulin G2 hinge imparts supera-gonistic properties to immunostimulatory anticancer antibodies. Cancer Cell 27, 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NS, Yang B, Yang A, Loeser S, Marsters S, Lawrence D, Li Y, Pitti R, Totpal K, Yee S, et al. (2011). An Fcgamma receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell 19, 101–113. [DOI] [PubMed] [Google Scholar]
- Xu C, Gagnon E, Call ME, Schnell JR, Schwieters CD, Carman CV, Chou JJ, and Wucherpfennig KW (2008). Regulation of T cell receptor activation by dynamic membrane binding of the CD3epsilon cytoplasmic tyrosine-based motif. Cell 135, 702–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokosuka T, Sakata-Sogawa K, Kobayashi W, Hiroshima M, Hashimoto-Tane A, Tokunaga M, Dustin ML, and Saito T (2005). Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat. Immunol 6, 1253–1262. [DOI] [PubMed] [Google Scholar]
- Yu X, Menard M, Prechl J, Bhakta V, Sheffield WP, and Lazarus AH (2016). Monovalent Fc receptor blockade by an anti-Fcgamma receptor/albumin fusion protein ameliorates murine ITP with abrogated toxicity. Blood 127, 132–138. [DOI] [PubMed] [Google Scholar]
- Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, Leenen PJ, Liu YJ, MacPherson G, Randolph GJ, et al. (2010). Nomenclature of monocytes and dendritic cells in blood. Blood 116, e74–e80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.