

Abstract
Immune responses to pathogens are complex and not well understood in many diseases, and this is especially true for infections by persistent pathogens. One mechanism that allows for long-term control of infection while also preventing an over-zealous inflammatory response from causing extensive tissue damage is for the immune system to balance pro- and anti-inflammatory cells and signals. This balance is dynamic and the immune system responds to cues from both host and pathogen, maintaining a steady state across multiple scales through continuous feedback. Identifying the signals, cells, cytokines, and other immune response factors that mediate this balance over time has been difficult using traditional research strategies. Computational modeling studies based on data from traditional systems can identify how this balance contributes to immunity. Here we provide evidence from both experimental and mathematical/computational studies to support the concept of a dynamic balance operating during persistent and other infection scenarios. We focus mainly on tuberculosis, currently the leading cause of death due to infectious disease in the world, and also provide evidence for other infections. A better understanding of the dynamically balanced immune response can help shape treatment strategies that utilize both drugs and host-directed therapies.
Keywords: cytokines infectious disease, granuloma, immune response, mathematical modelling, mycobacterium tuberculosis
1 |. INTRODUCTION
The immune system of an organism is charged with protecting the host against pathogenic microbes, but this response must be appropriate and measured. It is increasingly clear that inappropriately calibrated responses are pathologic, where insufficient responses may enable an infection to spread quickly and overly aggressive responses may clear the pathogen but result in severe incidental tissue damage. A successful host immune response is generally the result of both pro- and anti-inflammatory elements that are carefully tuned with the goal of clearing the pathogen and limiting host damage. Our focus here is on persistent infections, where we often think of the immune response as being “balanced” between pro- and anti-inflammatory responses that produce steady-state outcomes that are acceptable to both host and pathogen–enabling both to survive long-term. The balance is dynamic as it requires (and responds to) continuous feedback from both pathogen and host and might be achieved in multiple ways: between pro- and anti-inflammatory cytokines and cells at the molecular, organ, and whole-host levels; between isolating and engaging the pathogen; and over space and time. This balance is not necessarily quantitative (eg, equal concentrations of pro- and anti-inflammatory cytokines), but it is a qualitative harmonization in downstream activation and inhibition. Our primary example here will be tuberculosis (TB), but these principles also apply to other persistent diseases such as Helicobacter pylori infection and Hepatitis C (HCV) and to acute scenarios like sepsis. In this review, we provide evidence that a dynamic balance between pro- and anti-inflammatory cells and cytokines generated following infection results in an optimized trade-off between pathogen clearance and increased pathology. We demonstrate that this balance occurs not only spatiotemporally but also at both local and whole-host scales.
Figure 1 illustrates conceptual inflammatory response profiles over time (Panel A) and their corresponding outcome trajectories following infection (Panel B). If the inflammatory response is too weak or too slow, the pathogen will replicate freely and the host may succumb to infection (outcome i). If the inflammatory response is too strong, the pathogen may be eliminated, but at the price of excessive inflammatory damage to host tissue (outcome ii). In some cases, the immune system may mount a large inflammatory response, but the pathogen is too robust to be properly contained (outcome iii). The other 2 trajectories are examples where a pathogen is either contained or eliminated. In one case, the immune system does its job properly, mounting an appropriate inflammatory response that eliminates a pathogen while simultaneously decreasing inflammation via an effective and appropriate anti-inflammatory response (outcome iv). The other scenario is persistent infection, where the immune system and pathogen reach a stalemate, or steady state. The immune system produces a level of mild inflammation, and total pathogen burden eventually reaches a level where it does not significantly increase or decrease over time (outcome v). The trajectory of a given infection arises from the complex dynamics of the interaction between host and pathogen.
FIGURE 1.
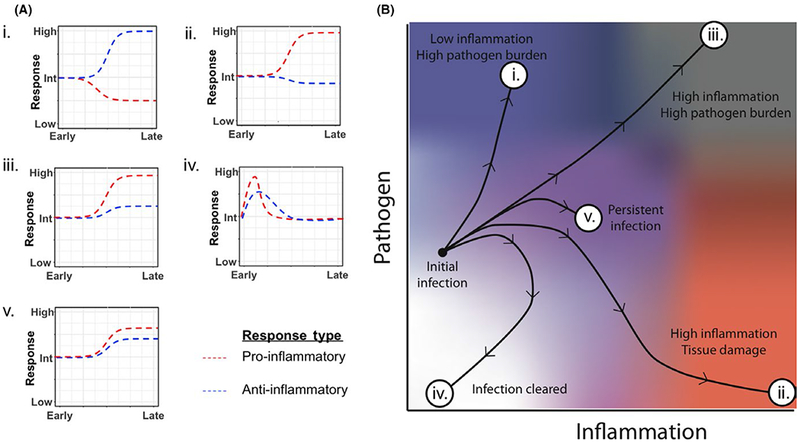
Dynamics of pro- and anti-inflammatory immune responses steer disease progression along various trajectories. Schematic of relative pro- and anti-inflammatory responses to a pathogen over time corresponding to various host outcomes (A). Schematic of disease trajectories corresponding to qualitative ratios of pro- and anti-inflammatory responses (B). The outcomes are (i) high pathogen burden (blue); (ii) severe tissue damage (red); (iii) high pathogen burden along with a large amount of tissue damage (gray); (iv) a cleared infection and return to base levels of inflammation (white); (v) a dynamically balanced pro- and anti-inflammatory response to control pathogen growth and limit host pathology (purple)
2 |. PRO- AND ANTI-INFLAMMATORY MEDIATORS RELEVANT TO PERSISTENT DISEASES INCLUDE BOTH CELLS AND MOLECULES
The role of cells trafficking between sites of infection and the draining lymph nodes (LN) is crucial in immune dynamics. Lymphocytes work individually and coordinate with each other to perform their immune functions. Specific immune cells, such as macrophages and some CD4+ T cells, secrete pro-inflammatory signals (IFN-γ, TNF, etc.) and perform inflammatory functions. Cytotoxic CD8+ T cells and activated macrophages kill pathogens and infected host cells either directly or by induction of cell apoptosis.1–3 In contrast, other cell types, such as regulatory T cells, serve to suppress or shut down the immune response via cell-cell contact and cytokine secretion.
Pro-inflammatory cytokines, including chemokines, are signaling molecules that are secreted from immune cells, such as T cells and macrophages, and other cells that promote inflammation and immunity. Some of the most studied pro-inflammatory cytokines include interleukin-1 (IL-1), IL-12, tumor necrosis factor alpha (TNF), interferon gamma (IFN-γ), and granulocyte-macrophage colony stimulating factor (G-MCSF). (For a more comprehensive list see4,5). Pro-inflammatory cytokines are predominantly produced by and involved in the upregulation of immune responses, including activation of macrophages, induction of apoptosis, and recruitment of additional immune cells. In contrast, anti-inflammatory cytokines secreted from immune cells, such as regulatory T cells and some macrophages, serve to suppress inflammation and immunity. The list of anti-inflammatory cytokines is also large, but the primary ones that we focus on here include IL-4, IL-10, and transforming growth factor beta (TGF-β)4,5
In order to achieve pathogen control without inducing excessive inflammation and tissue damage, an appropriate and dynamic balance between pro- and anti-inflammatory mediators must exist and evolve over time. Here, we review both experimental and mathematical/computational studies that support the concept of an immune balance during persistent infections. Experimental studies are critical to uncovering the identities of pro- and anti-inflammatory players in the immune response and identifying the consequences of disrupting balance. Mathematical and computational studies can be used to generate understanding, test hypotheses, fill in gaps, and make predictions. The ultimate goal is to harness knowledge and isolate factors influencing this dynamic balance to provide strategies for better patient outcomes. For example, analysis of models can identify immunomodulatory factors that, combined with antibiotics, enable patients to achieve pathogen clearance. We demonstrate that the type of balance required for controlling a pathogen and limiting host damage differs among different diseases and over spatiotemporal scales.
3 |. MYCOBACTERIUM TUBERCULOSIS INFECTION
TB is a deadly infectious disease caused by the bacterium M. tuberculosis (Mtb). Figure 2 outlines the multi-scale and multi-organ interactions occurring during infection. Briefly, infection occurs after Mtb inhalation into the lungs, where it is taken up by resident antigen-presenting cells (APCs), such as macrophages and DCs that initiate granuloma formation. Granulomas are organized cellular structures comprised primarily of lymphocytes, neutrophils, and macrophages, with or without centralized caseous necrosis and are the pathologic hallmark of TB.6–8 Humans experience either latent tuberculosis infection (LTBI)–a sub-clinical but long-term, persistent Mtb infection (about 90% of cases), or active primary disease (about 10% of cases).7,9,10 Protection against TB can be defined as host responses that prevent development of active disease, or in other words, long-term controlled infection (clinically latent).11 A human with LTBI likely retains relatively small numbers of Mtb within granulomas in lungs or LN granulomas, does not have any symptoms of disease, and is not contagious, but has a 10% lifetime risk of reactivation to active TB.12 Compromising the immune system by pharmacologic manipulation of TNF or HIV infection substantially increases the risk of reactivation, emphasizing that immunity maintains control over clinically latent infection.13,14
FIGURE 2.
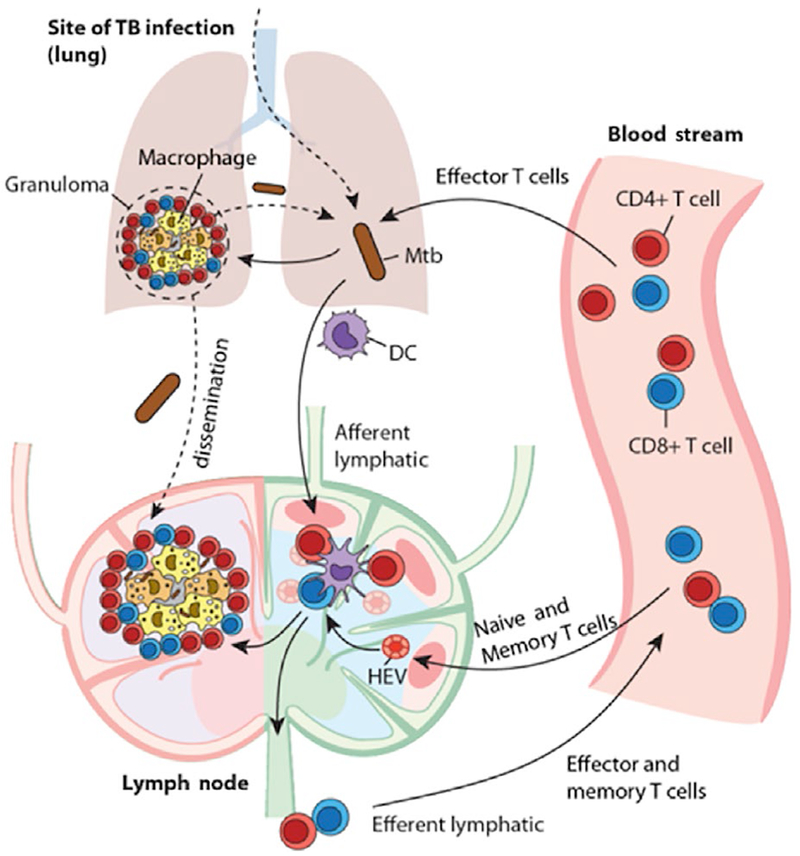
Multi-organ events following infection with Mycobacterium tuberculosis. Mtb is inhaled into lungs and engulfed by macrophages where intracellular replication occurs. Additionally, DCs take up Mtb and traffic to lung-draining LNs via afferent lymphatics, where they prime T cells that have been recruited from high endothelial venules (HEV). These primed T cells migrate to lungs via efferent lymphatics and participate in granuloma formation and function by activating macrophages, secreting cytokines, and participating in adaptive immune responses
Granulomas are organized immune structures, and while granuloma microenvironments promote control of infection, they also provide an intra-host niche for bacterial survival. Studies on Mtb-infected humans and non-human primates (NHPs) demonstrate that an individual may contain a spectrum of granuloma types (eg, solid cellular, caseous-necrotic, and disseminating), each with different bacterial loads.15–19 The mechanisms driving protection and pathology at the individual granuloma level are poorly understood but are likely to be important for determining whether Mtb infection progresses to active or latent disease at the organism scale.6,17,20 Furthermore, identifying these mechanisms may lead to advances in treatment and host-directed therapies that shift the balance of protection and pathology to the host’s favor.
4 |. EXPERIMENTAL SUPPORT FOR A BALANCED RESPONSE IN TB
There is substantial evidence that both pro- and anti-inflammatory elements are present in the immune response to Mtb infection, and that both play essential roles.21 A pro-inflammatory TH1 response driven by CD4+ T cells producing IL-2, IFN-γ, and TNF is essential for controlling Mtb infection.22–27 CD4+ and CD8+ T cells produce cytokines that activate other immune cells, including activation of macrophages that can kill Mtb,1,28 and cytotoxic CD8+ T cells that can kill Mtb-infected macrophages.29–31 Most Mtb-infected people, including people with active TB, have Mtb-specific TH1 responses suggesting TH1 responses are necessary, but not sufficient, factors in infection control. T cells are necessary to control Mtb infection, as mice or NHP without CD4+ or CD8+ T cells progress rapidly to active TB.28,31–35 HIV or SIV co-infection, which depletes CD4+ T cells, greatly increases risk of active TB in patients and macaques.13,36,37
Anti-inflammatory responses are important for preventing excessive lung pathology during Mtb infection.38–43 Macrophages influence inflammation in TB44 and can be divided into subsets based on activation status (M1 or classically activated macrophages–CAM–and M2 or alternatively activated macrophages–AAM).45–49 Anti-inflammatory cytokines produced in granulomas, such as IL-27, IL-10, and TGF-β, can act on T cells. Other cells that participate in anti-inflammatory responses to Mtb include TH17 cells,50–53 regulatory T cells (Tregs, CD4+Foxp3+ T cells),54–56 and neutrophils.57,58 There are limited data on the role of these cell subsets in Mtb infection. In mice, TH17 cells are important in optimizing vaccination against TB59–62 by enhancing induction or recruitment of TH1 cells to lungs. Tregs modulate immune responses by inhibiting proliferation and cytokine production by T cells, through production of IL-10 or TGF-β, or direct contact. In mice, Tregs inhibit control of Mtb by downregulating T-cell responses.63,64 One recent study in NHPs supports that balanced pro- and anti-inflammatory responses are associated with some sterilized granulomas; meaning that the ability of granulomas to clear bacteria on a per granuloma basis is based on the presence of both pro- and anti-inflammatory mediators, which correlates with good outcomes.42
Some T cells (at least at some point in their lifecycle) have been identified as producing multiple cytokines simultaneously. The presence of these so-called multifunctional T cells (MFTs) has been shown to correlate with a protective response in some diseases, while there is evidence that MFTs can act to increase pathology and inflammation in others.65–72 The role of MFTs in TB remains poorly characterized. There is conflicting evidence as to whether the presence of MFTs confers protection, with most human TB studies focusing on T cells derived from the periphery where the frequency of MFTs is often, although not always, associated with active disease rather than protection.73,74 A study by Qiu et al75 found that the percentage of MFTs in TB is significantly higher in active TB cases than in those with clinically latent TB or compared to healthy controls. Conversely, it has also been reported that active TB patients have increased numbers of single positive, TNF-secreting T cells and less MFT cells than those with clinically latent TB.76 Elevated numbers of MFTs have been observed following TB vaccination.77 At the granuloma level, a study recently showed that the majority of T cells in NHP TB infection secrete a single cytokine, but that there is a subset of T cells that secrete up to 5 pro-inflammatory cytokines concurrently. In addition, a small subset of cells secreting both pro- and anti-inflammatory cytokines, namely IL-17 and IL-10, were also observed, suggesting that a dynamic balance can be executed even at a single-cell level.42 While there are still limited data available to determine what role MFTs play in controlling infection and limiting pathology in TB and other diseases, the fact that MFTs are able to secrete either or both pro- and anti-inflammatory cytokines throughout disease establishes them as players in achieving and maintaining balance.
5 |. MATHEMATICAL AND COMPUTATIONAL MODELING STUDIES AT THE WHOLE LUNG LEVEL SUPPORT A BALANCE OF PRO- AND ANTI-INFLAMMATORY FACTORS IN TB
Models of the immune mediators have furthered our understanding of immune responses and also of viral infection (see this special issue and also cf.78–82), and such models have proved particularly insightful for TB.83 Model development can be specific to molecular, organ, host, or population scales, or can involve combining different-scale models (eg, using a multi-scale approach); multi-scale models can be particularly valuable to further understanding of the processes active in disease development as they account for molecular and cellular dynamics operating over time as well as physiological scales, ranging from molecular to organ to the entire host.83
Early modeling studies of pro- and anti-inflammatory factors in TB were focused on the whole lung as data available were derived from studies on whole lungs of infected animals. Wigginton and Kirschner84 highlighted the importance of maintaining a balance of concentrations of pro- and anti-inflammatory cytokines across time within the lung during TB infection. The data used to calibrate the model were derived from what was then the only known measurement from humans in vivo, broncho-alveolar lavage fluid, as blood data do not provide local infection information. Ordinary differential equations (ODEs) track whole lung dynamics of cells, cytokines, and bacteria. More specifically, the model tracks the temporal dynamics of macrophages, divided into resting, activated, and infected states; T lymphocytes, including individual populations of activated CD4+ naive T cells and the type 1 helper and type 2 helper they differentiate into (CD8+ T cells were not directly included, but their cytotoxic effects were accounted for indirectly); as well as concentrations of IFN-γ, and IL-4, IL-10, and IL-12. To assess the impact of levels of pro- and anti-inflammatory cytokines, the authors vary sources of cytokines as well as production and decay rates. In addition, virtual deletion and depletion experiments were simulated. In a framework with an increased anti-inflammatory or decreased pro-inflammatory response, the system moves to active infection. In a framework with an increased pro-inflammatory or decreased anti-inflammatory response, the system increases the ability to control infection but also increases damage to tissue (Figure 1). Results from this study suggest that a dynamic balance must be struck between pro- and anti-inflammatory cytokines in order to suppress bacterial infection in the most efficient way possible and thereby minimize tissue damage. In addition, while mouse studies suggested that there was no phenotype to an IL-10 knockout mouse,85 this model predicted that IL-10 plays a key role in stabilizing immune response dynamics. This supports the idea that both pro- and anti-inflammatory signals trade-off dynamically during infection to produce an optimized, controlled response that limits bacterial load without inducing significant damage.
6 |. MACROPHAGE POLARIZATION AND ITS ROLE IN BALANCE
This idea of balance is explored further in next-generation models that track additional relevant immune cell types (including DCs, cytotoxic T cells, and macrophages of specific phenotype), and investigate multi-organ dynamics of cell trafficking, antigen presentation, and cell priming occurring between the lung and lung-draining LNs.86–92 One goal of these studies is to focus on the role of cytokines on the interplay between the spectrum of phagocytic populations (macrophages and DCs) and inflammation. The idea is to identify control mechanisms for immune protection by dissecting the effects of pro-inflammatory (eg, TNF) and anti-inflammatory (eg, IL-10) cytokines in lung and LN environments. It is not clear how this balance translates into macrophage polarization and their plasticity, or how the relative contribution of these macrophages, broadly classified as M1 (or classically activated macrophages-CAM) and M2 (or alternatively activated macrophages-AAM), evolves over time and leads to an effective immune response, at the scale of a granuloma.
The mathematical representation used in these studies is a multi-compartmental model including separate systems of ODEs representing lungs84 and is linked to ODE models of LN and blood. The model is calibrated to datasets from murine samples over multiple time points.86,87,90 Based on a study by Day et al93, the model describes 4 different macrophage subpopulations, both in lung and LN: uncommitted/resting/resident (M0), infected (M1), classically activated (M1, also denoted MA), and alternatively activated (M2).89–91 A pool of macrophages that reside in the blood (as monocytes) and cells are continuously recruited to the lung based on danger signals and inflammatory chemokine/cytokine gradients.94 Macrophage polarization is defined as a function of IFN-γ (pro), TNF (pro), IL-10 (anti), and bacteria (both) that collectively drive macrophage differentiation into either an M1 or M2 phenotype.89
The model defines the concept of switching time93 as the time needed to switch from an AAM-dominated (M2) to a CAM-dominated (M1) lung environment during Mtb infection. Studies found that switching time is negatively affected by higher IL-10 saturation thresholds: more IL-10 is needed to skew macrophage populations to an AAM phenotype. The biological relevance of increasing a switching time (on the order of days) is that a delay in CAM presence and pro-inflammatory cues in lung may be responsible for Mtb gaining an initial foothold. The concept of a switching time also supports the hypothesis of a dynamic balance between pro- and anti-inflammatory cues that can shape infection progression and ultimately affect the outcome.89
7 |. COMPUTATIONAL MODEL OF GRANULOMA FORMATION AND FUNCTION
As described above, Mtb infection largely plays out at the level of individual granulomas. Some granulomas may sterilize (clear all bacteria), but clinically latent TB is the result of 1 or more granulomas which contain, but do not eliminate, the pathogen.18 Granulomas evolve largely independently from one another,95 and thus it is likely that a balance of pro- and anti-inflammatory factors are responsible for the outcome of infection at an individual granuloma level. The structure of a granuloma–a characteristic cuff of lymphocytes surrounding an inner core of macrophages, in humans and NHPs–likely plays an important role in infection progression and in treatment. Granulomas are difficult to study, and generally can be examined in detail only at necropsy in animal studies. However, recently obtained PET/CT imaging data from individual NHP granulomas can report granuloma size and metabolic activity at multiple time points prior to necropsy.95,96
Use of computational models of single granulomas can generate insights into how the structures form and how a balance of pro- and anti-inflammatory factors may play out in the evolution of the infection at the single granuloma level. Importantly, these models account for changes in cellular and molecular concentrations in both space and time. This versatility allows for exploration of the role of dynamic balance of pro- and anti-inflammatory mediators in infection outcomes at both single granuloma and collective scales.
GranSim is a hybrid computational model that integrates agent-based, ODE, and PDE models. It captures the formation and behavior of a granuloma is calibrated to biological data and has been continuously curated since 2004.97–101 Segovia-Juarez et al created the first iteration of GranSim as an agent-based model (ABM) that describes cellular behavior, including recruitment, changes of state (activation, infection, etc.), and movement during granuloma formation. Subsequent iterations contain macrophages (M1 and M2) and T cells (CD4+, CD8+, and Tregs) as agents that can have multiple states and phenotypes (eg, infected, activated, etc.),89 and bacteria represented as either continuous functions or as agents in the extra- or intracellular environment.102 Probabilistic interactions between immune cells and bacterial populations are described by a well-defined set of rules that are continually updated with new biological datasets.
The diffusion of relevant chemokines, cytokines, and other soluble ligands is determined by solving partial differential equations. GranSim can capture receptor-ligand binding and intracellular signaling events of cytokines, such as TNF or IL-10, either with ODEs that are solved within each agent or a linear model.97,98,101,103 The precise model used is determined by the questions being asked. Using an approach termed “tunable resolution”, model resolution can toggle between fine-grained (more detailed) and coarse-grained (less detailed) as needed, based on the biological events described.104
In GranSim, information is continually exchanged across scales in a computationally efficient manner, incorporating molecular and cellular events explicitly with tissue-scale behavior (granuloma formation) as an emergent feature of the model. Rules and typical parameters for GranSim are available online at http://malthus.micro.med.umich.edu/GranSim. Simulations are representative of a 4 mm2 or 16 mm2 cross-section of lung tissue (Figure 3A). Each simulation follows events over several 100 days, building over time to track thousands of individual cells (agents). One strength of this approach is that generated in silico data can be tracked in both space and time. Marino et al88 have generated an in silico library of tens of thousands of virtual granulomas that can be used as a virtual repository for repeated study. The model can also track temporal and spatial gradients of molecular concentrations on the grid (Figure 3B). These virtual granulomas can be validated experimentally by comparing with spatial images generated using immunohistochemistry and imaging derived from both human and NHP granulomas (Figure 3C). GranSim is able to recapitulate these spatial dynamics, and also temporal dynamics, such as fitting to NHP data on colony-forming units (CFU) (Figure 3D).
FIGURE 3.
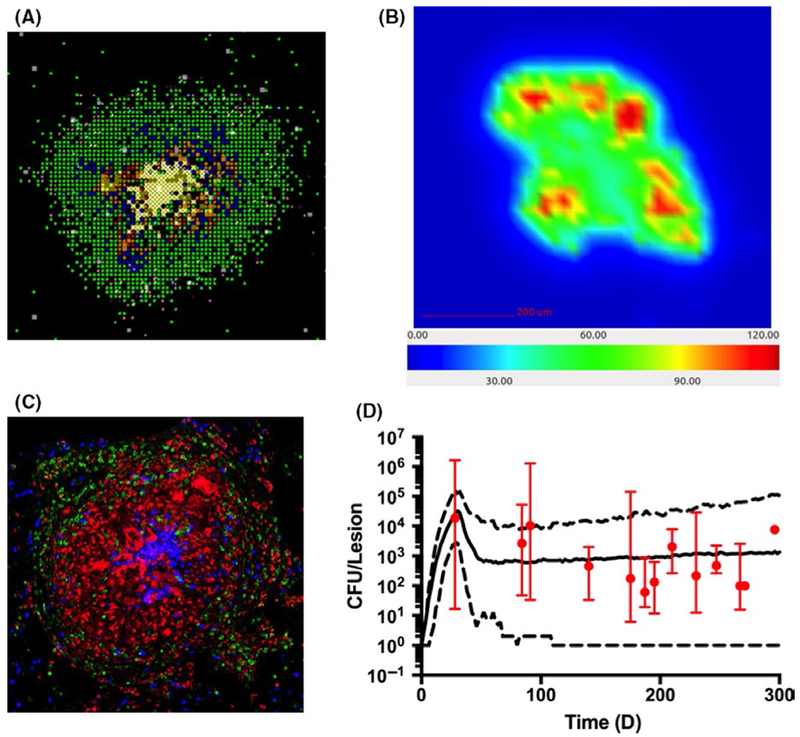
Computational and experimental visualizations of various granulomas. A zoomed in 2-D GranSim snapshot (A), which shows macrophage substates (green-resting, blue-activated, orange-infected, red-chronically infected), T-cell phenotypes (pink IFN-producing, purple-cytotoxic, light blue-Tregs), extracellular bacteria (yellow), necrosis (white). (B) is a GranSim snapshot of a different granuloma that shows the concentration gradient of TNF (units on colorbar are molecules of TNF per grid compartment). (C) is a macaque granuloma image stained for macrophages (Red CD68), T cells (Green CD3), and neutrophils (Blue calprotectin). (D) is a time plot of CFU/lesion NHP data (red, median with error bars indicating min/max) along with GranSim simulations with model predictions (median–solid line, min/max–dashed lines). Simulations plotted as in 103 with a newer generation of GranSim and data derived from 32 NHP in previously published works 6,17,95,212,213
8 |. COORDINATED ROLES FOR IL-10, TNF, AND TGF-β IN SPATIOTEMPORAL DYNAMICS OF GRANULOMA FORMATION AND FUNCTION
GranSim simulations predict that a balance between pro- and anti-inflammatory factors is necessary to achieve, at the granuloma level, control over Mtb and the immune response (Figure 4). Granulomas that favor anti-inflammatory responses fail to control bacterial growth, whereas granulomas that favor pro-inflammatory responses may sterilize a granuloma, but at the price of increased necrosis and caseum. GranSim has been used to explore the individual roles of pro- and anti-inflammatory cytokines TGF-β, TNF, and IL-10 and then evaluate their roles in combination to provide evidence that a balanced response to these inflammatory cytokines is necessary for granuloma formation and maintenance.97,98,105–107
FIGURE 4.
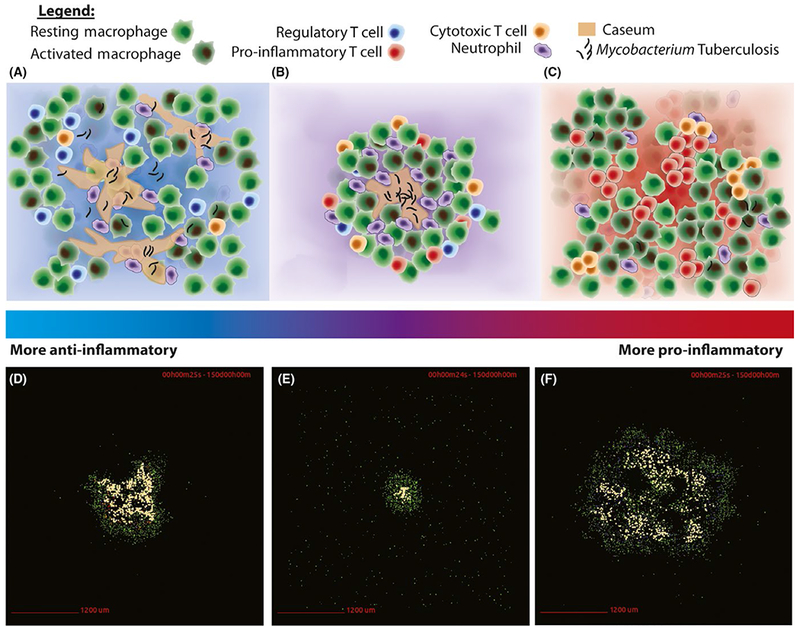
Balanced immune responses lead to contained Mtb granulomas. The top row (A, B, C) displays conceptual images of granuloma formation across a spectrum of immune responses. During a primarily anti-inflammatory response (A), a granuloma shows a lack of proper formation, and large amounts of caseum develop as the host struggles to contain the pathogen. A balanced response (B) displays proper granuloma formation and containment of the pathogen. A primarily pro-inflammatory response (C) clears the pathogen, but at the cost of widespread cellular activation, death, and inflammation. The bottom row (D, E, F) displays GranSim simulation snapshots of these scenarios at day 150 post infection. A containment simulation from baseline parameter ranges is demonstrated in (E), whereas a simulation from a TNF-depletion parameter set is shown in (D), and a simulation from an IL-10 knockout parameter set that also exhibits more pro-inflammatory behavior (F). (D), (E), and (F) correspond to the conceptual immune responses displayed in (A), (B), and (C), respectively
Within a granuloma, the major producers of the pleiotropic cytokine TNF are macrophages, effector T cells, and DCs. TNF has also been thought to play a role in the activation of macrophages–stimulating further cytokine or chemokine production as well as initiating bacterial killing, and inducing macrophage apoptosis.44,99,108–112 However, without the ability to measure an in vivo gradient of TNF or to generally measure TNF activities in vivo, the spatiotemporal dynamics of TNF are unable to be analyzed experimentally. Therefore, Fallahi-Sichani et al98,106 explored the role of TNF using GranSim, by creating a multi-scale molecular to tissue model. The model predicts that there is an interplay between molecular events occurring at the second to minute timescales and the long-term events of immune response to Mtb. In particular, TNF receptor 1 internalization kinetics modulate a diverse set of outcomes. At low receptor internalization rates, the granuloma sterilizes, but at the cost of excessive inflammation. At high internalization rates, the granuloma cannot control growth of Mtb. Finally, at internalization rates that match experimental values, many in silico granulomas are able to contain Mtb growth without excessive inflammation. Studies have shown that internalization of TNF acts as a regulator of both paracrine and autocrine responses to TNF, influencing activation and apoptosis in macrophages.98,106
The models also predict that there exists a TNF gradient within a single granuloma, with highest concentrations near the center, and that this gradient determines the spatial range of TNF action within the granuloma.98 The gradient is controlled by the TNF receptor internalization dynamics described above. An attempt to detect such a TNF gradient is shown in Figure 5C, using immunofluorescence staining on lung cryo-sections from the well-established PPD bead model of granuloma formation in mice (protocol in106,113–116) Free (unbound) TNF receptor concentrations increase toward the periphery of the granuloma, suggesting a decrease in TNF concentration. As the simulations predict (Figure 5B), a contained granuloma includes a narrow range of TNF activities: as one travels to the periphery of the granuloma, there is less TNF available to bind to TNF receptors.
FIGURE 5.
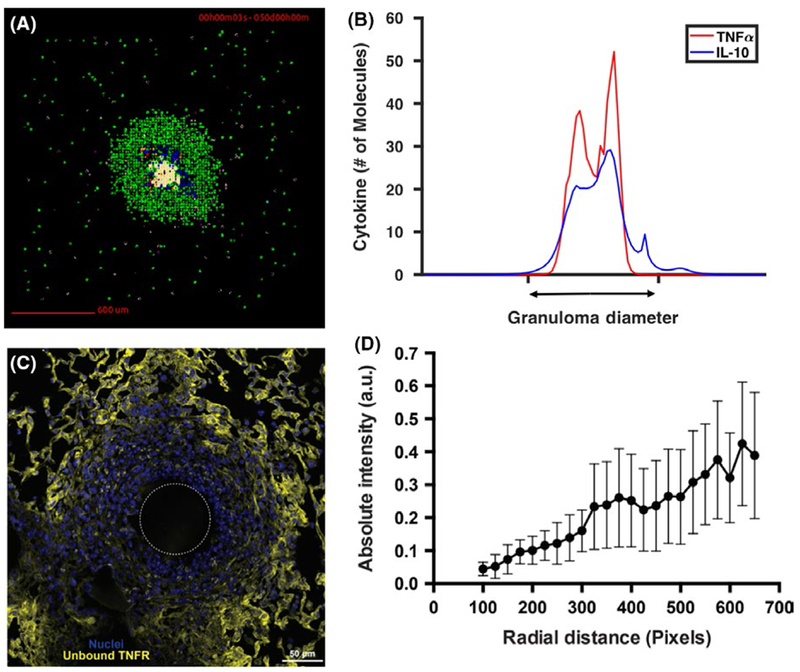
Cytokine distribution across in silico and bead model granulomas. (A) GranSim snapshot of a representative containment granuloma scenario at 50 days post infection. The TNF (red) and IL-10 (blue) gradients of this representative granuloma are shown in (B). In GranSim granulomas, the gradient of TNF is contained within the granuloma (see diameter of granuloma on x-axis), and the gradient of IL-10 spreads along the diameter of the granuloma. This particular set of gradients is representative of containment granulomas and is consistent across time. If the spatial range of TNF is too large, bystander cellular death increases despite little increase in bacterial killing. If TNF spatial distribution is too narrow, simulations show increased bacterial load. Similarly, if IL-10 has a spatial distribution that is too wide, IL-10 decreases bystander cell death, but causes increased bacterial loads in the granuloma. Yet, too narrow of an IL-10 distribution yields too powerful of a pro-inflammatory response. (C) The well-established PPD bead model of granuloma formation in mice (protocol in 106,113–116) can determine whether there is a gradient of soluble TNF within a single granuloma and detect soluble TNF gradients in ex vivo granuloma tissue. Immunofluorescence techniques identify the spatial organization of cells in granulomas, and then this known organization is combined with flow cytometry data,106 a simple receptor-ligand model, and fluorescent microscopy of unbound TNF receptors to reconstruct the relative soluble TNF gradient. A molecular probe from biotinylation of recombinant TNF was constructed to target unbound TNF receptors.214–218 In the representative PPD bead granuloma shown, stains are for free TNFR (yellow) using the biotinylated TNF probe and cell nuclei (blue) using DAPI. The dashed white circle indicates approximate PPD bead location. Entire image was used. (D) Intensity of biotinylated TNF in image from (C) as a function of radial distance from the center of the PPD bead, binned into 25 pixel lengths. Unbound TNFR is roughly inversely proportional to gradient of soluble TNF (derived from the simple receptor-ligand model). Bars indicate standard deviation 219
Simulations with GranSim can also elucidate the role of IL-10 within the granuloma at both molecular and cellular scales. Decreased levels of IL-10 lead to a greater likelihood of sterilization, but at the cost of more caseation and cellular death at earlier time points in infection. Bacterial persistence can be achieved by increasing levels of IL-10 produced by infected macrophages. These increased levels of IL-10 allow for a limited immune response during early time points of infection and prevent lesion sterilization across time.97,103 The role of IL-10 at early time points supports the importance of an appropriate cytokine response across time in controlling granuloma outcomes.
The dynamics of TGF-β, another anti-inflammatory cytokine, can also be examined at both molecular and cellular levels within GranSim.107,117–119 These studies present an in silico TGF-β knockout and predict an increase in bactericidal activity of cytotoxic T cells, thereby decreasing granuloma CFU across time. Furthermore, the presence of TGF-β regulates cytotoxic T cell effector function at the individual cell level. The individual actions of TGF-β on unique cell types within a granuloma indicate that its inhibitory role is less straightforward when evaluated within the context of other cytokine actions.
The computational studies reviewed above evaluate the individual impacts of TNF, IL-10, and TGF-β. To evaluate the impact of sets of pro- and anti-inflammatory cytokines working in concert, Cilfone et al and Warsinske et al performed further sets of in silico experiments. They found 2 distinct results. First, Cilfone et al defined a metric of granuloma outcomes, the host-pathogen index, which includes factors such as bacterial load, number of activated macrophages, and the inverse of the amount of resting macrophage apoptosis; lower scores indicate a better outcome for the host. Simulations show that an optimal result occurs when the ratio of concentrations, [TNF]/[IL-10], was in a middle range: lower values yielded high bacterial loads and higher values lead to excessive inflammation.97 In other words, the required balance of pro- and anti-inflammatory factors can be determined via simulation.
Additionally, simulations show that both anti-inflammatory mediators TGF-β and IL-10 represent distinct regulatory mechanisms within granulomas as each distinctly inhibit effector T cell and macrophage functions, respectively.107 Altogether, the action of each cytokine represents a unique function, but granulomas that effectively contain Mtb require that cytokines perform in harmony with other cytokines in a dynamic and balanced way.
Importantly, in silico experiments also emphasize the importance of cytokine spatial dynamics. Figure 5B shows the IL-10 and TNF gradients in a representative contained granuloma at day 50; these gradients lead to a granuloma that contains bacteria without the cost of high inflammation and bystander cellular death. Immunohistochemical analysis of necrotic NHP granulomas further supports a key role for anti-inflammatory cytokines.107
Simulations predict that proper control of Mtb requires a multi-scale balance of pro- and anti-inflammatory factors both spatially and temporally. This was further supported in later studies with experimental collaborators.39,42,49 While studies have explored the dynamic balance for the case of TNF, IL-10, and TGF-β,97,98,105–107 this concept of balance likely extends beyond these cytokines to include other pro- and anti-inflammatory molecules and cell types.
9 |. NEUTROPHILS CONTRIBUTE TO BALANCING PRO- AND ANTI-INFLAMMATORY FACTORS IN TB
Although the role of neutrophils in the granuloma response to Mtb infection remains largely unknown, neutrophils are thought to dually influence the protective host immune response and the ability to balance inflammatory responses. During Mtb infection, neutrophils secrete both pro- and anti-inflammatory cytokines. TNF, CCL2, CCL3, CCL4, CCL19, CCL20, CXCL2, CXCL9/MIG, S100A8, S100A9, all secreted by neutrophils, recruit monocytes and T lymphocytes to the infection site.49,120,121 Notably, the majority of the cytokines and chemokines that neutrophils secrete in turn function as neutrophil activators or attractants, resulting in a positive feedback loop.121 This self-accumulation can become detrimental if left unchecked, contributing to immune hyper-reactivity121; namely, although neutrophils do not secrete large quantities of pro-inflammatory cytokines at the single-cell level, the accumulated amount that a local population of neutrophils secretes can become harmful to the host. That neutrophil death occurs through the necrosis pathway during Mtb, rather than by the anti-inflammatory efferocytosis, and that neutrophil secretion of the anti-inflammatory cytokines IL-10, IL-4, and IL-13 is reduced, both imply that there is a positive-feedforward neutrophilic, inflammatory response during Mtb.121–123 Through disrupting pro-inflammatory balance checkpoints in this way, infections like Mtb are thought to experience increased pathology.
Easily activated, short-lived, and unable to be depleted in cynomolgus macaque models,6,49,124,125 neutrophils pose unique experimental challenges to wet lab study. The inherent difficulties of experimental work highlight the need for computational modeling, as computational models of the immune response to Mtb infection enable testing of neutrophil-centric hypotheses that are difficult, impossible, or too costly to test experimentally. The value of computational studies in gaining a better understanding of whether Mtb disrupts the ability of neutrophils to modulate their secretion of pro- and anti-inflammatory cytokines thus cannot be overstated. Bru and Cardona126 similarly motivate the importance of using a systems biology approach to study early granuloma formation, and they present a computational model that builds on the techniques presented in Kirschner et al.100 This model is noteworthy in the context of this review as it both includes neutrophils–although allows them limited abilities, eg, only macrophages are able to secrete chemokines–and emphasizes the importance of temporal chemokine distribution and concentrations in the progression of Mtb pathology.126 It also suggests a link between persistent, spatially dependent chemokine secretion and effective granuloma formation, and exemplifies how mathematical models can help us to better understand early inflammatory responses. While further work is needed both experimentally and computationally to elaborate the role of neutrophils in Mtb infection at the granuloma scale, it is clear that their role in maintaining a dynamic balance will be key.
10 |. MODULATING THE DYNAMIC INFLAMMATORY BALANCE FOR TB THERAPY
Standard TB therapy consists of 6 months or longer of multiple antibiotics. There are many factors that cause patients to fail to adhere and complete this lengthy treatment.127 Shortening TB therapy could help increase the number of patients that complete treatment, as well as increase efficacy, but it can be difficult to predict regimen efficacy in humans. Rationalizing new therapy approaches requires an understanding of exposure levels of therapeutic agents within the granuloma, as well as consideration of how therapies can modulate the inflammatory balance that occurs within a granuloma to control TB.
We have argued that the organization and formation of the granuloma require a dynamic balance of pro- and anti-inflammatory responses. Successful granulomas (ie, those that control bacterial levels or sterilize them completely) can help contain infection and prevent Mtb from infecting healthy lung tissue.8 Although this may be considered a positive alternative to active TB, there is strong evidence that granulomas also act as a physical and physiological barrier to antibiotic diffusion.128,129 Imaging techniques, such as matrix-assisted laser desorption/ionization coupled with mass spectrometry (MALDI-MS) allow for semi-quantitative analysis of antibiotic distribution in granuloma tissue samples and can show that many antibiotics fail to evenly distribute throughout granuloma lesions and often accumulate outside of caseous regions.130 This may explain the lack of clinical success of some antibiotics that show promise in preclinical trials.
There is a close relationship between granuloma structure and antibiotic distribution. Because granuloma formation and structure both depend on the dynamic balance of pro- and anti-inflammatory signals, generating models to predict antibiotic efficacy for Mtb requires modeling both immune system phenomena and pharmacokinetics. To this end, Pienaar et al131 added pharmacokinetic and pharmacodynamic (PK and PD) models into GranSim to study how different antibiotics distribute within granulomas and contribute to sterilization. The PK model includes a compartmental, ODE plasma PK model that captures absorption into plasma following an oral dose, exchange with peripheral tissue, and metabolic clearance of the antibiotic. The agent-based simulation references the plasma PK model to deliver antibiotic onto the spatial grid through vascular sources based on local concentration gradients. Solving for spatial antibiotic concentration involves modeling diffusion through the grid, partitioning into macrophages, and metabolic degradation of the antibiotics. The pharmacodynamic model incorporates antibiotic-induced bacterial death by determining antibiotic killing rate constants or probabilities based on Hill curve kinetics.
To incorporate granuloma heterogeneity while simulating antibiotic therapy, parameters that control host immune responses can be varied. Many of these parameters modulate the balance of the pro- and anti-inflammatory cytokine responses, as well as capture sensitivity of immune cells to these signals, such as thresholds for immune cell activation and cellular recruitment. The combined model reveals how the balanced inflammatory response and granuloma formation affect antibiotic distribution and efficacy. Figure 6 shows isoniazid concentration 2 hours following an oral dose in 3 examples of simulated granulomas: one more anti-inflammatory (Figure 4A), one more pro-inflammatory (Figure 4C), and one that is balanced (Figure 4B). Within each simulated granuloma, exposure to drugs is much lower than exposure outside, indicating that isoniazid is prevented from penetrating granulomas that develop under inflammatory conditions across the pro- to anti-inflammatory spectrum. Although isoniazid penetration is limited regardless of the inflammatory balance of a granuloma, an additional feature for why granulomas have low antibiotic exposure is the presence of caseum, which is an avascular region at the center of granulomas due to the build-up of dead cell debris. Analysis of host immune features such as sensitivity to TNF-induced apoptosis, macrophage activation, and amount of caseation each influence treatment outcome.131 In particular, increased caseum formation as a result of increased sensitivity to a pro-inflammatory response negatively impacts antibiotic penetration and treatment outcome.
FIGURE 6.
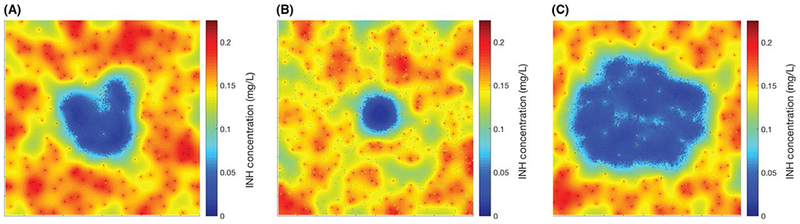
Simulated antibiotic distributions in granulomas using GranSim. Heat maps of isoniazid (INH) concentration for the 3 representative granulomas shown in Figure 4: one that is highly anti-inflammatory (A), one that is contained (B), and one that is highly pro-inflammatory (C). Each type of granuloma has lower concentrations of antibiotic compared to the surrounding tissue. The heat maps show INH concentration in granulomas after 150 days of infection and 2 hours after a single, oral dose of 5 mg/kg. PK/PD modeled as in131,220
In an effort to characterize how caseum affects antibiotic distribution, there have been a number of studies that image the distribution of antibiotics in granulomas and quantify antibiotic molecular binding to caseum.130,132,133 Pienaar et al134 have utilized MALDI-MS images and experimentally determined temporal granuloma antibiotic concentration measurements to compare efficacy of tuberculosis therapy with 3 different fluoroquinolones: moxifloxacin, levofloxacin, and gatifloxacin. The 3 fluoroquinolones display different distribution patterns in granulomas that GranSim can recapitulate. These simulations show moxifloxacin has a slight advantage in sterilizing granulomas over the other 2 fluoroquinolones. This is likely due to higher intracellular accumulation of moxifloxacin, allowing it to better target intracellular bacterial populations and eliminate the population of infected macrophages. This reduction in infected macrophages causes treatment with moxifloxacin to reduce the predicted number of activated macrophages and TNF/IL10 ratios in granulomas more than treatment with gatifloxacin and levofloxacin. Because infection, immune response, and treatment are modeled together, this provides a unique opportunity to study how antibiotics assist the immune system in sterilizing granulomas, as well as how they indirectly modulate the balance of immune responses over the course of therapy.
Because active TB can be considered an immune response that is out of balance–too little pro-inflammatory mediators, or too many anti-inflammatory mediators, another approach to treatment is to modify the levels of 1 or both of the pro- and anti-inflammatory responses to achieve latency. The same idea could also be considered for clinically latent TB, though the idea would be to tip the distribution slightly toward pro-inflammatory responses and thus eliminate Mtb without inducing additional inflammation. These approaches, generally termed host-directed therapies, might be considered separately or in combination with antibiotic treatment.135 One theory for how combining antibiotics and host-directed therapy is beneficial is that these therapies can modulate the inflammatory balance and reduce recruitment of immune cells to a granuloma, resulting in smaller lesions and thus better penetration of antibiotics into lesion. Another theory is that many bacteria in granulomas are non-replicating and less susceptible to antibiotics. Host-directed therapy and reduced immune pressure may encourage bacterial replication, making them more susceptible to certain antibiotics.
Host-directed therapies include TNF inhibitors such as Etanercept and PDE4 inhibitors. There is evidence that TNF is required for protection during Mtb infection and TNF inhibitors can contribute to reactivation of latent TB.136–138 Simulations at the cellular scale by Marino et al indicate low availability of TNF present in granulomas due to anti-TNF therapies increases likelihood of reactivation.14 Additional simulations by Fallahi-Sichani et al105 incorporated molecular scale binding kinetics of anti-TNF molecules, and show that anti-TNF molecules with increased permeability and binding to membrane-bound TNF limit the ability to control bacterial growth in granulomas. However, there have been cases where TNF inhibition or reduced production had some beneficial effects in conjunction with antibiotics in human patients co-infected with HIV139,140 Additionally, murine and rabbit TB models offer evidence that a PDE4 inhibitor given with antibiotics leads to decreased lung involvement and lower bacterial loads compared to antibiotics alone.141–143
Because the inflammatory balance that is required to form and stabilize a granuloma is a complex process, it is difficult to predict how modulating this balance through the use of host-directed therapies will affect the granuloma and treatment outcomes. These potential outcomes include the percent of non-sterile granulomas over the course of treatment, which indicates how well treatment provides sterilizing activity. Other outcomes, such as average CFU and granuloma size over the course of treatment, show how modulating the inflammatory response affects bacterial growth and the ability of antibiotics to penetrate the granuloma. GranSim simulations exhibit immunomodulatory effects by either depleting the amount of TNF or IL-10 present in the simulation, or by modeling the PK of anti-TNF drugs, such as Etanercept, that can bind and neutralize TNF molecules.14,105 Simulating these host-directed therapies along with the delivery of antibiotics to fully formed in silico granulomas shows a reduction in granuloma size compared to antibiotics alone (Figure 7). However, the host-directed therapies with antibiotics tend to have longer sterilization times and greater treatment failure rates compared to just antibiotics, suggesting that host-directed therapies may not be beneficial in treating human granulomas. The median CFU is higher in granulomas treated with host-directed therapies and antibiotics compared to antibiotics alone after about 10 to 20 days of treatment. By comparison, host-directed therapies start to reduce granuloma sizes between 30 and 50 days. Because host-directed therapies reduce the amount of TNF present, they inhibit macrophage activation and ability to control Mtb growth. The timescale over which the host-directed therapies produce potential benefits such as reducing granuloma size for better antibiotic penetration is longer than the timescale over which disrupting the balance of cytokines leads to granulomas with more bacteria. Thus, these simulations predict that modulating this inflammatory balance through the use of host-directed therapies in conjunction with antibiotics to reduce cellular recruitment and granuloma size may not provide any benefits over antibiotics alone.
FIGURE 7.
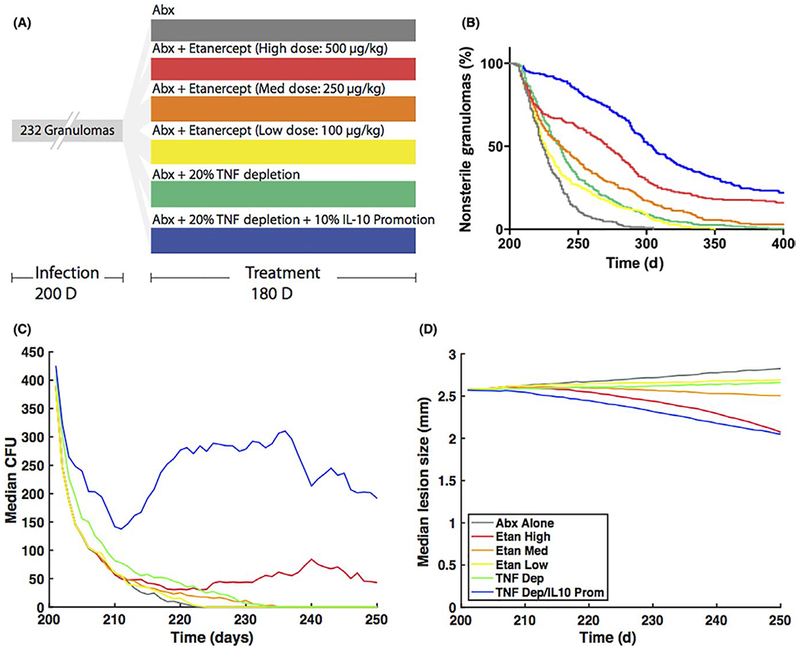
Effects of host-directed therapies on treatment outcome. (A) An in silico biorepository of 232 granulomas was created using GranSim and an infection period of 200 days. Starting at day 200, different treatments were simulated for 180 days: antibiotics alone (gray), antibiotics with varying doses of etanercept (500 μg/kg in red, 250 μg/kg in orange, 100 μg/kg in yellow), antibiotics with a 20% TNF-depletion (green), and antibiotics with 20% TNF-depletion and 10% IL-10 promotion (blue). Percent depletions and promotions were simulated by reducing or increasing the amount of cytokine secreted from immune cells compared to a baseline simulation. (B) The percent of granulomas that contain bacteria over the course of treatment shows that antibiotics alone sterilize more granulomas and sterilize faster than any of the host-directed therapies. (C) Host-directed therapies tend to have higher median CFU compared to antibiotics alone from 10-20 days post-treatment and later. (D) Simulations involving host-directed therapies result in granulomas that gradually decrease in size, compared to those involving only antibiotics (only medians shown)
11 |. THE CONCEPT OF BALANCE IN OTHER DISEASES
The importance of a balanced immune response to promote pathogen clearance and maintain host protection is evident in a wide range of infections. To demonstrate the way in which balance has been highlighted more generally, we considered a range of other diseases including Leishmania (granulomatous), H. Pylori (persistent) infection, sepsis (acute), and HCV (viral).
11.1 |. Leishmania
Another persistent infection that likely requires balance to achieve control is Leishmania. Leishmania is a parasite that causes disease manifestation in the skin or internal organs, including the spleen and bone marrow.144 Macrophages are the primary host cells for Leishmania; however, it is possible for the parasite to inhabit neutrophils, fibroblasts, and DCs.145
During progressive infection, cytokines play different roles in containment of Leishmania infection.146 In general, parasite growth is dependent on IFN-γ, as well as other inflammatory cytokines. IL-12, TNF, IFN-γ are usually associated with parasite clearance,147,148 while IL-4, IL-10, IL-13, and TGF-β are often associated with a dampened immune response and persistence of the parasite.149 As infection advances, granulomas begin to form.150 It was assumed that these were non-necrotizing; however, a recent study found almost 20% of cutaneous leishmaniasis contained caseated granulomas,151 highlighting the need to again understand the dynamic balance of pro- and anti-inflammatory immune responses to this infection.
One of the first models to look at cytokine-related activities in Leishmania infection152 focused on a TH1/TH2 paradigm, using these as proxies for inflammatory cytokines (TH1) and anti-inflammatory cytokines (TH2). The model predicted that stable solutions existed in 2 situations: (i) TH1 response overtakes TH2, resulting in a persistent, active infection; (ii) A balance between TH1/TH2 is reached, resulting in an asymptomatic infection. It was also observed that TH1 was required to be dominant to remove the parasite and that Leishmania exploits the TH2 response to its benefit.
In recent Leishmania models, cytokines have been modeled more explicitly. Siewe et al153 used an ODE model to explore immune response to L. donovani using pro- and anti-inflammatory (M1 and M2) macrophages, DCs, T cells, parasites, and concentrations of cytokines. Through the observation that IFN-γ greatly reduced Leishmania infection, the authors perform simulations for treatment with sodium antimony gluconate (SAG), which enhances T-cell proliferation and response. When looking at the efficacy of treatment, they find that a fixed total amount of drug, given intermittently, was not as effective as continuous treatment. During continuous treatment, an optimal window of time to start SAG was seen 3-10 days post infection, to reduce parasite burden: if started too early, then the immune system likely has insufficient time to be alerted; starting too late allows the parasite load to grow quickly, which diminishes drug effects. Thus, not only should timing be considered in treatment but it should also be considered part of the dynamic balance of the immune system in response to the pathogen.
Some groups have taken interest in specific cytokines and their production sources in Leishmania infection. Albergante et al154 used a Petri net model to look at how source of IL-10 impacts L. donovani parasite burden in granulomas. Inhibiting CD4+ IFN+ Th1 and NK cells from producing IL-10 results in no reduction in peak parasite burden, but does lead to delayed clearance of the parasites. Inhibiting myeloid cells from IL-10 production results in a reduced peak burden and faster clearance of the parasites, but increases the amount of pro-inflammatory cytokines, and therefore, host damage. This further supports the idea that cell secretion of pro- and anti-inflammatory mediators helps create a trade-off between reducing tissue damage and simultaneously controlling pathogen levels.
Investigating how the balance among cytokines affects the host in Leishmania infection in the longer term has only recently been explored.155 Using a cellular automata/lattice-gas model, Ribeiro et al. incorporates TH cells, cytokines, and adenosine inhibition of T cells to study host lesion size for several species of Leishmania. In simulations, they find that using sufficient levels of Adenosine to inhibit T cells in the range of 0%-20% had little effect on the maximum lesion size. When inhibition of T cells increased from 20% to 40%, an increase is seen in host lesion size. Higher inhibition of T cells results in decreased host lesion size. The group also finds that a T-cell inhibition range of 0-15% represents the peak of a lesion that could eventually heal, while anything above 20% disrupts the balance to such an extent that lesions would not heal and would continue to grow. The model implicates that both cells and cytokines play a key role in balance. Strong inhibition from 40% to 100% results in reduced lesions, but with high parasite burden. Weak T-cell inhibition results in better containment of parasites, but more host damage. This appeared worst with intermediate inhibition from 20% to 40%, where there is enough of a host response to cause damage, but the parasite is never fully contained, which results in growing lesion size. Further work should extend this idea to look at the balancing of specific cytokines in Leishmania infection to predict if a dynamically optimized trade-off can reduce host damage during immune response while clearing the parasite.
11.2 |. H. pylori
Helicobacter pylori is a bacterial pathogen that colonizes the intestinal tract of humans. It is estimated that up to 50% of the global population is infected with the bacterium, and as high as 70% in Africa.156 In general, people infected with H. pylori are asymptomatic and there is some evidence that this bacterium confers a benefit to its host. It has been suggested that H. pylori can protect against Mtb infection,157 as well as other diseases including gastroesophageal reflux disease,158 inflammatory bowel disease (IBD),159 and allergic conditions such as asthma.160 However, these findings are often linked to the so-called hygiene hypothesis,161 and H. pylori is more commonly implicated in causing conditions such as persistent gastric inflammation, peptic ulcers, and gastric cancer.162–164
Helicobacter pylori colonizes the stomach, where there is very little microbial competition,165 and deploys a range of adaptive mechanisms to make clearance by the immune system difficult, despite all infected individuals mounting an immune response to the bacteria. In the asymptomatic cases, this immune response, while detectable, is not detrimental to health, and the bacteria and host reach a symbiotic equilibrium.166 The balance between pro- and anti-inflammatory cytokines in the gut following infection with H. pylori has been suggested to be important in determining the severity of symptoms.167 Infection with H. pylori in humans stimulates the infiltration of lymphocytes and monocytes along with significantly increased expression of IL-1, IL-8, and IL-6 in the gut by 2 weeks post infection,166 and persistent infection is associated with increased expression of additional cytokines, including IFN-γ, TNF, and IL-10.168 The T-cell response to H. pylori is primarily driven by Th1, Th17, and Treg cells.
Kirschner and Blaser previously developed models describing the steady-state dynamics of H. pylori colonization depending on nutrient availability, involving an auto-regulatory network in which inflammation leads to nutrient release.169,170 Through these models, they demonstrate that downregulation of the host immune response can be beneficial to the host in the presence of microbial evasion strategies against the immune response, as is the case with H. pylori in humans.170 In this case, the appropriate immune response requires a balance to be reached consisting of an inflammatory response that is necessary for microbe control, and negative feedback to ensure the response is not overly harmful to the host.171 In particular, it has been shown that downregulation of regulatory T-cell function is associated with the development of life threatening peptic ulcers in humans infected with H. pylori.172 This result has been echoed in mice where IL-10 has been demonstrated to be essential for preventing H. pylori induced gastritis in Rag2−/− mice. It has also been shown that downregulated inflammatory responses are associated with reduced gastritis in children.173 The role of pro- and anti-inflammatory cytokines in H. pylori is thoroughly reviewed in.174
Other models have focused on the role of CD4+ T cells in H. pylori infection using a system of ODEs to determine how IL-21, a pro-inflammatory T cell-derived cytokine, maintains a persistent pro-inflammatory T-cell immune response driving chronic gastritis during H. pylori infection.175 The authors also extended this model to an ABM that was used to demonstrate that there is a crucial contribution of Th1 and Th17 effector responses as mediators of histopathological changes in the gastric mucosa during chronic stages of infection.176 These results have also been verified in mice and demonstrate that a balance of different inflammatory mediators is required to achieve an appropriate response in this disease.
11.3 |. Sepsis
Sepsis is an acute systemic inflammatory response to an infection and is the major cause of death among critically ill patients in the developed world. The most common causes of sepsis are Staphylococcus aureus and Escherichia coli, although there are many other bacteria capable of causing the condition.177 Sepsis is characterized by a systemic pro-inflammatory response178 and occurs due to an imbalance of pro- and anti-inflammatory cytokines following dysregulation of an initial, appropriate host response to infection. In contrast to the other infections discussed herein, a sepsis response primarily involves the innate immune pathway that still utilizes both pro- and anti-inflammatory mediators179,180 and occurs on a rapid timescale, and thus is not a persistent infection. The fast timing of immune dysregulation in sepsis makes clinical and experimental studies on disease development difficult to perform, thus in silico studies are important to provide insight into the early immune response in sepsis.
Several immune models have been developed to study sepsis and focus on the role of cytokines in the immune response (reviewed in181). Yang et al182 showed that the outcome of sepsis progression is improved when IL-10 is increased early in infection. Conversely, An et al183 calibrated their previously published Innate Immune Response ABM (IIRABM) to achieve sepsis dynamics and proposed several schemes for intervention that suggested that TNF inhibition almost always contributed to resolution, and inhibition of anti-inflammatory cytokines GCSF and IL-10 also improved outcome. These conflicting results could be a result of the time frame under study as the mathematical model by183 only considered early inflammatory events (up to 7 days), whereas work by Yang et al182 considered a longer time period of 41 days; however, taken together they highlight the important role of immune balance in controlling infection.
The importance of restoring balance for recovery from sepsis has also been highlighted by a number of in vivo and in vitro studies.184–188 It has been suggested that inflammatory and anti-inflammatory responses involving both the innate and adaptive immune systems are each equally important and represent potential targets for immune therapy to improve sepsis outcomes.184–188 Luan et al189 observed that in a murine model of sepsis, inhibiting TGF-β could improve host outcome by reducing immunosuppression. It has also been shown that the important mechanisms in pathology alter over the course of infection, with pro-inflammatory cells and cytokines playing a major role in pathogenesis early in infection, but high levels of immunosuppression by anti-inflammatory cells and cytokines responsible for worsening outcomes if the initial inflammatory phase is survived.190
11.4 |. Hepatitis C
The role of balance in immunity has also been highlighted in the field of virology. Here we present a case study exploring the immune response to hepatitis C virus (HCV) infection. HCV infection is chronic, and it is estimated that up to 170 million individuals are infected worldwide.191 Due to shared transmission routes, co-infection with HIV is common: approximately 16% of HIV positive individuals in the United States and Europe are also infected with HCV.192
HCV infects the hepatocytes in the liver where it continually replicates, establishing an intrahepatic infection.193 The innate immune response to HCV involves the production of interferons and cytokines, and the recruitment of natural killer (NK) cells.194 This phase of the immune response is thought to be critical in determining the outcome of an infection; however, high numbers of NK cells are also associated with tissue injury.195, 196 The subsequent adaptive response requires myeloid cells such as DCs, and macrophages, as well as T and B cells.
The main inflammatory mediator of HCV infection is IFN-γ, produced primarily by NK and NK T cells. IFN-γ stimulates the production of TNF by Kupffer cells (KCs).197 The inflammatory response is essential for viral clearance; however, in hosts where clearance is not achieved, this response is damaging. Regulatory T cells are important in controlling the balance between host damage and viral control and are induced following galectin-9 production by KCs.198 Alternatively, Tregs have also been associated with inducing chronic infection199 and the virus itself can use this pathway to promote immune tolerance.200 Thorough reviews of the immune response to HCV response are available for more details.201,202
Much of the modeling work on HCV infection focuses on viral kinetics and uses target-cell-limited models to determine the effect that the immune response may have on controlling the virus in the presence of HIV co-infection and does not specially examine the role of pro- and anti-inflammatory cells and mediators in HCV infection.203,204 There are still open questions related to the concept of balance in HCV infection and the role of pro- and anti-inflammatory cells in infection outcomes that lead to a chronic, yet stable latent infection. Clinical studies that seek to identify significant differences in cytokine profiles between those that do and do not clear or control virus have had limited success in identifying influential cytokines.205 Modeling and simulation could be a powerful tool in understanding the role that pro- and anti-inflammatory mediators play in controlling HCV infection and answering some of these questions. Based on similarities with other persistent infections discussed herein, we hypothesize that a similar trade-off exists for HCV as well.
12 |. DISCUSSION
The concept of a dynamically achieved, spatially dependent steady state is ubiquitous in biology. Nature has numerous ways to self-regulate and respond to novel or pathologic perturbations, whether localized or systemic; natural systems, such as predator-prey systems or regulation of gene expression, often rely on dynamic adjustments over varying timescales to attain and maintain this sort of balance. It is not surprising, then, that the immune system operates on a similar level of complexity. During infection, the immune system mounts inflammatory responses to neutralize or kill pathogens, but this response can also damage host cells, leading to a trade-off between clearing the pathogen and limiting damage to host tissues. In this review, we present examples where the immune response seeks an optimized dynamic balance with a pathogen that fails to be eliminated, and we explore the role of pro- and anti-inflammatory signals that participate in this process.
The Goldilocks principle (named from the children’s story) refers to responses that are “just right” (ie, not too strong and not too weak).5,206,207 This is a paradigm that is repeated in many instances (from intracellular signaling to porridge) and is closely related to the concept of balance. Recent authors suggested that the Goldilocks principle implies that “…immunity must be choreographed so that invaders are purged quickly, inflammation tempered, and homeostasis preserved,” which is consistent with our hypotheses.206 It should also be noted that balanced immune responses are not split evenly between pro- and anti-inflammatory mediators, but instead are dynamic and vary with respect to each other over the course of infection.
Recent work similarly highlights the importance of balanced pro- and anti-inflammatory responses in combating Mtb infection and extends this concept to spatial and temporal dimensions. These studies also indicate that a better understanding of this aspect of immunity could improve vaccine development and drug treatment.5,208,209 Cytokines embody both good and bad roles in TB, because they can have context-dependent protective and pathologic consequences.5 Monin and Khader208 similarly suggested that chemokine responses can be both good and bad, and that protection in TB is a trade-off between immune control and bacterial replication mediated by appropriately calibrated responses. Our review offers a unique and detailed look at the role of pro- and anti-inflammatory balance by consolidating insights from experimental studies and mathematical models that incorporate the complex, multi-scale interactions involved in host-pathogen dynamics.
We have considered specific cells and cytokines to be either pro- or anti-inflammatory based on functions that are widely accepted in both in vivo and in vitro studies. It is worth noting that our strict classifications of cells and cytokines into either pro- or anti-inflammatory categories may be too simplistic. There are many examples showing that cytokine amount, cell or target cell nature, activating signal cascade, timing, sequence of cytokine action, and even the experimental study system can all greatly influence cytokine properties.210,211 This is demonstrated by plasticity within some T-cell subsets, whereby cells can switch from pro- or anti-inflammatory profiles, or in the case of multifunctional T cells, simultaneously express cytokines with different properties.65–72
In addition to its dynamic nature, inflammatory balance is something that is maintained over various time and length scales. To mount an appropriate immune response during an infection, balance is achieved at the molecular scale by signaling appropriately and inducing proliferative responses within immune cells, which in turn influence tissue-scale behaviors that influence organ-level responses and host-level outcomes. These processes are occurring over time scales ranging from seconds to years and spatial scales ranging from sub-micron to meters. While the relationship between these scales are difficult to study in a traditional wet lab setting, mathematical and computational modeling can be used to integrate data from these multi-scale interactions and outcomes. Modeling these integrated responses over space and time allows the fullest picture of the dynamics of balance, and enables us to make predictions that can have the greatest impact on disease outcomes. In addition, computational modeling is a vital tool to isolate and understand factors from the larger integrated model that regulate such complex biological systems.
While we now better understand how a balance of pro- and anti-inflammatory mediators may be required for control infection, we have not yet fully utilized that concept for therapeutic development. Experimentally driven mathematical modeling will allow us to identify how antibiotics or host-directed therapies can be used to modulate inflammatory balance and promote sterilizing immunity against pathogens or tilt host-pathogen interactions in favor of the host.
ACKNOWLEDGEMENTS
This research was supported by: National Institutes of Health grant numbers R01AI123093, R01HL110811, 1U01HL131072. Any simulations also use resources of the National Energy Research Scientific Computing Center, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. ACI-1053575 and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number MCB140228. We thank Amy Oberlin for her work on the effects of host-directed therapies on treatment outcome in Figure 7. We thank Dr. JoAnne Flynn for almost 20 years of collaborative science and also supplying the NHP data shown in Figure 3. We also thank all of the students and trainees whose fantastic work reviewed herein makes these studies possible.
Funding information
NIH National Institute of Allergy and Infectious Diseases, Grant/Award Number: R01AI123093; NIH National Heart, Lung, and Blood Institute, Grant/Award Number: 1U01HL131072 and R01HL110811; NSF U.S. Department of Energy; National Energy Research Scientific Computing Center; XSEDE
Footnotes
This article is part of a series of reviews covering Modeling Viral Infection and Immunity appearing in Volume 285 of Immunological Reviews.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- 1.Jasenosky LD, Scriba TJ, Hanekom WA, Goldfeld AE. T cells and adaptive immunity to Mycobacterium tuberculosis in humans. Immunol Rev. 2015;264:74–87. [DOI] [PubMed] [Google Scholar]
- 2.Nussing S, Sant S, Koutsakos M, Subbarao K, Nguyen THO, Kedzierska K. Innate and adaptive T cells in influenza disease. Front Med. 2018;12:34–47. [DOI] [PubMed] [Google Scholar]
- 3.Zuniga EI, Macal M, Lewis GM, Harker JA. Innate and adaptive immune regulation during chronic viral infections. Annu Rev Virol. 2015;2:573–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murphy K, Weaver C. Immunobiology, 9th edn. New York, NY: Current Biology Ltd./Garland Publishing Inc.; 2016. [Google Scholar]
- 5.Domingo-Gonzalez R, Prince O, Cooper A, Khader SA. Cytokines and chemokines in Mycobacterium tuberculosis infection. Microbiol Spectr. 2016;4:TBTB2-0018-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin PL, Rodgers M, Smith L, et al. Quantitative comparison of active and latent tuberculosis in the cynomolgus macaque model. Infect Immun. 2009;77:4631–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin PL, Flynn JL. Understanding latent tuberculosis: a moving target. J Immunol. 2010;185:15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramakrishnan L Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol. 2012;12:352–366. [DOI] [PubMed] [Google Scholar]
- 9.Herrera V, Perry S, Parsonnet J, Banaei N. Clinical application and limitations of interferon-gamma release assays for the diagnosis of latent tuberculosis infection. Clin Infect Dis. 2011;52:1031–1037. [DOI] [PubMed] [Google Scholar]
- 10.Al-Orainey IO. Diagnosis of latent tuberculosis: Can we do better? Ann Thor Med. 2009;4:5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flynn JL, Chan J. Tuberculosis: latency and reactivation. Infect Immun. 2001;69:4195–4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zumla A, Raviglione M, Hafner R, von Reyn CF. Tuberculosis. N Engl J Med. 2013;368:745–755. [DOI] [PubMed] [Google Scholar]
- 13.Diedrich CR, Flynn JL. HIV-1/Mycobacterium tuberculosis coinfection immunology: how does HIV-1 exacerbate tuberculosis? Infect Immun. 2011;79:1407–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marino S, Sud D, Plessner H, et al. Differences in reactivation of tuberculosis induced from anti-TNF treatments are based on bioavailability in granulomatous tissue. PLoS Comput Biol. 2007;3:1909–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barry CE 3rd, Boshoff HI, Dartois V, et al. The spectrum of latent tuberculosis: Rethinking the biology and intervention strategies. Nat Rev Microbiol. 2009;7:845–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flynn JL, Klein E. Pulmonary tuberculosis in monkeys In: Leong J, Dick T, eds. A Color Atlas of Comparative Pulmonary Tuberculosis Histopathology. Boca Raton, FL: CRC Press, Taylor & Francis; 2011:83–106. [Google Scholar]
- 17.Lin PL, Ford CB, Coleman MT, et al. Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat Med. 2014;20:75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin CJ, Cadena AM, Leung VW, et al. Digitally barcoding Mycobacterium tuberculosis reveals in vivo infection dynamics in the macaque model of tuberculosis. MBio. 2017;8:e00312–e00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cadena AM, Fortune SM, Flynn JL. Heterogeneity in tuberculosis. Nat Rev Immunol. 2017;17:691–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cadena AM, Flynn JL, Fortune SM. The importance of first impressions: Early events in Mycobacterium tuberculosis infection influence outcome. MBio. 2016;7:e00342–00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lyadova I Inflammation and immunopathogenesis of tuberculosis progression, understanding tuberculosis – Analyzing the origin of Mycobacterium tuberculosis pathogenicity. InTech; 2012. [Google Scholar]
- 22.Leem AY, Song JH, Lee EH, et al. Changes in cytokine responses to TB antigens ESAT-6, CFP-10 and TB 7.7 and inflammatory markers in peripheral blood during therapy. Sci Rep. 2018;8:1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Badell E, Nicolle F, Clark S, et al. Protection against tuberculosis induced by oral prime with Mycobacterium bovis BCG and intranasal subunit boost based on the vaccine candidate Ag85B-ESAT-6 does not correlate with circulating IFN-γ gamma producing T-cells. Vaccine. 2009;27:28–37. [DOI] [PubMed] [Google Scholar]
- 24.Clark S, Cross ML, Smith A, et al. Assessment of different formulations of oral Mycobacterium bovis Bacille Calmette-Guerin (BCG) vaccine in rodent models for immunogenicity and protection against aerosol challenge with M. bovis. Vaccine. 2008;26:5791–5797. [DOI] [PubMed] [Google Scholar]
- 25.Dey B, Jain R, Khera A, et al. Boosting with a DNA vaccine expressing ESAT-6 (DNAE6) obliterates the protection imparted by recombinant BCG (rBCGE6) against aerosol Mycobacterium tuberculosis infection in guinea pigs. Vaccine. 2009;28:63–70. [DOI] [PubMed] [Google Scholar]
- 26.Freches D, Romano M, Korf H, et al. Increased pulmonary tumor necrosis factor alpha, interleukin-6 (IL-6), and IL-17A responses compensate for decreased gamma interferon production in anti-IL-12 autovaccine-treated, Mycobacterium bovis BCG-vaccinated mice. Clin Vaccine Immunol. 2011;18:95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawn SD, Wilkinson RJ, Lipman MC, Wood R. Immune reconstitution and “unmasking” of tuberculosis during antiretroviral therapy. Am J Respir Crit Care Med. 2008;177:680–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Green AM, Difazio R, Flynn JL. IFN-γ from CD4 T cells is essential for host survival and enhances CD8 T cell function during Mycobacterium tuberculosis infection. J Immunol. 2013;190:270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Einarsdottir T, Lockhart E, Flynn JL. Cytotoxicity and secretion of gamma interferon are carried out by distinct CD8 T cells during Mycobacterium tuberculosis infection. Infect Immun. 2009;77:4621–4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lazarevic V, Flynn J. CD8+ T cells in tuberculosis. Am J Respir Crit Care Med. 2002;166:1116–1121. [DOI] [PubMed] [Google Scholar]
- 31.Chen CY, Huang D, Wang RC, et al. A critical role for CD8 T cells in a nonhuman primate model of tuberculosis. PLoS Pathog. 2009;5:e1000392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saunders BM, Frank AA, Orme IM, Cooper AM. CD4 is required for the development of a protective granulomatous response to pulmonary tuberculosis. Cell Immunol. 2002;216:65–72. [DOI] [PubMed] [Google Scholar]
- 33.Lin PL, Rutledge T, Green AM, et al. CD4 T cell depletion exacerbates acute Mycobacterium tuberculosis while reactivation of latent infection is dependent on severity of tissue depletion in cynomolgus macaques. AIDS Res Hum Retroviruses. 2012;28:1693–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scanga CA, Mohan VP, Yu K, et al. Depletion of CD4(+) T cells causes reactivation of murine persistent tuberculosis despite continued expression of interferon gamma and nitric oxide synthase 2. J Exp Med. 2000;192:347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yao S, Huang D, Chen CY, Halliday L, Wang RC, Chen ZW. CD4+ T cells contain early extrapulmonary tuberculosis (TB) dissemination and rapid TB progression and sustain multieffector functions of CD8+ T and CD3-lymphocytes: Mechanisms of CD4+ T cell immunity. J Immunol. 2014;192:2120–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bates M, Marais BJ, Zumla A. Tuberculosis comorbidity with communicable and noncommunicable diseases. Cold Spring Harb Perspect Med. 2015;5:a017889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diedrich CR, Mattila JT, Klein E, et al. Reactivation of latent tuberculosis in cynomolgus macaques infected with SIV is associated with early peripheral T cell depletion and not virus load. PLoS ONE. 2010;5:e9611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barber DL, Mayer-Barber KD, Feng CG, Sharpe AH, Sher A. CD4 T cells promote rather than control tuberculosis in the absence of PD-1-mediated inhibition. J Immunol. 2011;186:1598–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Green AM, Mattila JT, Bigbee CL, Bongers KS, Lin PL, Flynn JL. CD4(+) regulatory T cells in a cynomolgus macaque model of Mycobacterium tuberculosis infection. J Infect Dis. 2010;202:533–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lazar-Molnar E, Chen B, Sweeney KA, et al. Programmed death-1 (PD-1)-deficient mice are extraordinarily sensitive to tuberculosis. Proc Natl Acad Sci USA. 2010;107:13402–13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Windish HP, Lin PL, Mattila JT, et al. Aberrant TGF-beta signaling reduces T regulatory cells in ICAM-1-deficient mice, increasing the inflammatory response to Mycobacterium tuberculosis. J Leukoc Biol. 2009;86:713–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gideon HP, Phuah J, Myers AJ, et al. Variability in tuberculosis granuloma T cell responses exists, but a balance of pro- and anti-inflammatory cytokines is associated with sterilization. PLoS Pathog. 2015;11:e1004603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Orme IM, Robinson RT, Cooper AM. The balance between protective and pathogenic immune responses in the TB-infected lung. Nat Immunol. 2015;16:57–63. [DOI] [PubMed] [Google Scholar]
- 44.Flynn JL, Chan J, Lin PL. Macrophages and control of granulomatous inflammation in tuberculosis. Mucosal Immunol. 2011;4:271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gordon S, Martinez FO. Alternative activation of macrophages: Mechanism and functions. Immunity. 2010;32:593–604. [DOI] [PubMed] [Google Scholar]
- 46.Chan J, Tanaka K, Carroll D, Flynn J, Bloom BR. Effects of nitric oxide synthase inhibitors on murine infection with Mycobacterium tuberculosis. Infect Immun. 1995;63:736–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flynn JL, Scanga CA, Tanaka KE, Chan J. Effects of aminoguanidine on latent murine tuberculosis. J Immunol. 1998;160:1796–1803. [PubMed] [Google Scholar]
- 48.MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci USA. 1997;94:5243–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mattila JT, Ojo OO, Kepka-Lenhart D, et al. Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. J Immunol. 2013;191:773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Cassan SC, Pathan AA, Sander CR, et al. Investigating the induction of vaccine-induced Th17 and regulatory T cells in healthy, Mycobacterium bovis BCG-immunized adults vaccinated with a new tuberculosis vaccine, MVA85A. Clin Vaccine Immunol. 2010;17:1066–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garcia Jacobo RE, Serrano CJ, Enciso Moreno JA, et al. Analysis of Th1, Th17 and regulatory T cells in tuberculosis case contacts. Cell Immunol. 2014;289:167–173. [DOI] [PubMed] [Google Scholar]
- 52.Torrado E, Cooper AM. IL-17 and Th17 cells in tuberculosis. Cytokine Growth Factor Rev. 2010;21:455–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ye ZJ, Zhou Q, Du RH, Li X, Huang B, Shi HZ. Imbalance of Th17 cells and regulatory T cells in tuberculous pleural effusion. Clin Vaccine Immunol. 2011;18:1608–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.He XY, Xiao L, Chen HB, et al. T regulatory cells and Th1/Th2 cytokines in peripheral blood from tuberculosis patients. Eur J Clin Microbiol Infect Dis. 2010;29:643–650. [DOI] [PubMed] [Google Scholar]
- 55.Hougardy JM, Place S, Hildebrand M, et al. Regulatory T cells depress immune responses to protective antigens in active tuberculosis. Am J Respir Crit Care Med. 2007;176:409–416. [DOI] [PubMed] [Google Scholar]
- 56.Lim HJ, Park JS, Cho YJ, et al. CD4(+)FoxP3(+) T regulatory cells in drug-susceptible and multidrug-resistant tuberculosis. Tuberculosis. 2013;93:523–528. [DOI] [PubMed] [Google Scholar]
- 57.Lowe DM, Redford PS, Wilkinson RJ, O’Garra A, Martineau AR. Neutrophils in tuberculosis: Friend or foe? Trends Immunol. 2012;33:14–25. [DOI] [PubMed] [Google Scholar]
- 58.Martineau AR, Newton SM, Wilkinson KA, et al. Neutrophil-mediated innate immune resistance to mycobacteria. J Clin Invest. 2007;117:1988–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Christensen D, Mortensen R, Rosenkrands I, Dietrich J, Andersen P. Vaccine-induced Th17 cells are established as resident memory cells in the lung and promote local IgA responses. Mucosal Immunol. 2017;10:260–270. [DOI] [PubMed] [Google Scholar]
- 60.Khader SA, Bell GK, Pearl JE, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8:369–377. [DOI] [PubMed] [Google Scholar]
- 61.Khader SA, Cooper AM. IL-23 and IL-17 in tuberculosis. Cytokine. 2008;41:79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Khader SA, Gopal R. IL-17 in protective immunity to intracellular pathogens. Virulence. 2010;1:423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Scott-Browne JP, Shafiani S, Tucker-Heard G, et al. Expansion and function of Foxp3-expressing T regulatory cells during tuberculosis. J Exp Med. 2007;204:2159–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shafiani S, Tucker-Heard G, Kariyone A, Takatsu K, Urdahl KB. Pathogen-specific regulatory T cells delay the arrival of effector T cells in the lung during early tuberculosis. J Exp Med. 2010;207:1409–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Darrah PA, Patel DT, De Luca PM, et al. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat Med. 2007;13:843–850. [DOI] [PubMed] [Google Scholar]
- 66.Macedo AB, Sanchez-Arcila JC, Schubach AO, et al. Multifunctional CD4(+) T cells in patients with American cutaneous leishmaniasis. Clin Exp Immunol. 2012;167:505–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Califano D, Sweeney KJ, Le H, et al. Diverting T helper cell trafficking through increased plasticity attenuates autoimmune encephalomyelitis. J Clin Invest. 2014;124:174–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Carbo A, Hontecillas R, Kronsteiner B, et al. Systems modeling of molecular mechanisms controlling cytokine-driven CD4+ T cell differentiation and phenotype plasticity. PLoS Comput Biol. 2013;9:e1003027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Voo KS, Wang YH, Santori FR, et al. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc Natl Acad Sci USA. 2009;106:4793–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hovhannisyan Z, Treatman J, Littman DR, Mayer L. Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology. 2011;140:957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morrison PJ, Bending D, Fouser LA, et al. Th17-cell plasticity in Helicobacter hepaticus-induced intestinal inflammation. Mucosal Immunol. 2013;6:1143–1156. [DOI] [PubMed] [Google Scholar]
- 72.Annunziato F, Cosmi L, Santarlasci V, et al. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wilkinson KA, Wilkinson RJ. Polyfunctional T cells in human tuberculosis. Eur J Immunol. 2010;40:2139–2142. [DOI] [PubMed] [Google Scholar]
- 74.Mattila JT, Diedrich CR, Lin PL, Phuah J, Flynn JL. Simian immunodeficiency virus-induced changes in T cell cytokine responses in cynomolgus macaques with latent Mycobacterium tuberculosis infection are associated with timing of reactivation. J Immunol. 2011;186:3527–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Qiu Z, Zhang M, Zhu Y, et al. Multifunctional CD4 T cell responses in patients with active tuberculosis. Sci Rep. 2012;2:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Harari A, Rozot V, Bellutti Enders F, et al. Dominant TNF-alpha+ Mycobacterium tuberculosis-specific CD4+ T cell responses discriminate between latent infection and active disease. Nat Med. 2011;17:372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Beveridge NE, Price DA, Casazza JP, et al. Immunisation with BCG and recombinant MVA85A induces long-lasting, polyfunctional Mycobacterium tuberculosis-specific CD4+ memory T lymphocyte populations. Eur J Immunol. 2007;37:3089–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Burke MA, Morel BF, Oriss TB, Bray J, McCarthy SA, Morel PA. Modeling the proliferative response of T cells to IL-2 and IL-4. Cell Immunol. 1997;178:42–52. [DOI] [PubMed] [Google Scholar]
- 79.Segel LA, Bar-Or RL. On the role of feedback in promoting conflicting goals of the adaptive immune system. J Immunol. 1999;163:1342–1349. [PubMed] [Google Scholar]
- 80.Yates A, Callard R, Stark J. Combining cytokine signalling with T-bet and GATA-3 regulation in Th1 and Th2 differentiation: A model for cellular decision-making. J Theor Biol. 2004;231:181–196. [DOI] [PubMed] [Google Scholar]
- 81.Fishman MA, Perelson AS. Th1/Th2 cross regulation. J Theor Biol. 1994;170:25–56. [DOI] [PubMed] [Google Scholar]
- 82.Perelson AS. Modelling viral and immune system dynamics. Nat Rev Immunol. 2002;2:28–36. [DOI] [PubMed] [Google Scholar]
- 83.Kirschner D, Pienaar E, Marino S, Linderman JJ. A review of computational and mathematical modeling contributions to our understanding of Mycobacterium tuberculosis within-host infection and treatment. Curr Opin Syst Biol. 2017;3:170–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wigginton JE, Kirschner D. A model to predict cell-mediated immune regulatory mechanisms during human infection with Mycobacterium tuberculosis. J Immunol. 2001;166:1951–1967. [DOI] [PubMed] [Google Scholar]
- 85.Murray PJ, Young RA. Increased antimycobacterial immunity in interleukin-10-deficient mice. Infect Immun. 1999;67:3087–3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Marino S, Kirschner DE. The human immune response to Mycobacterium tuberculosis in lung and lymph node. J Theor Biol. 2004;227:463–486. [DOI] [PubMed] [Google Scholar]
- 87.Marino S, Pawar S, Fuller CL, Reinhart TA, Flynn JL, Kirschner DE. Dendritic cell trafficking and antigen presentation in the human immune response to Mycobacterium tuberculosis. J Immunol. 2004;173:494–506. [DOI] [PubMed] [Google Scholar]
- 88.Marino S, Gideon HP, Gong C, et al. Computational and empirical studies predict Mycobacterium tuberculosis-specific T cells as a biomarker for infection outcome. PLoS Comput Biol. 2016;12:e1004804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Marino S, Cilfone NA, Mattila JT, Linderman JJ, Flynn JL, Kirschner DE. Macrophage polarization drives granuloma outcome during Mycobacterium tuberculosis infection. Infect Immun. 2015;83:324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marino S, Myers A, Flynn JL, Kirschner DE. TNF and IL-10 are major factors in modulation of the phagocytic cell environment in lung and lymph node in tuberculosis: A next-generation two-compartmental model. J Theor Biol. 2010;265:586–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Myers AJ, Marino S, Kirschner DE, Flynn JL. Inoculation dose of Mycobacterium tuberculosis does not influence priming of T cell responses in lymph nodes. J Immunol. 2013;190:4707–4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Marino S, El-Kebir M, Kirschner D. A hybrid multi-compartment model of granuloma formation and T cell priming in Tuberculosis. J Theor Biol. 2011;280:50–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Day J, Friedman A, Schlesinger LS. Modeling the immune rheostat of macrophages in the lung in response to infection. Proc Natl Acad Sci USA. 2009;106:11246–11251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Matzinger P The danger model: A renewed sense of self. Science. 2002;296:301–305. [DOI] [PubMed] [Google Scholar]
- 95.Lin PL, Coleman T, Carney JP, et al. Radiologic responses in cynomolgous macaques for assessing tuberculosis chemotherapy regimens. Antimicrob Agents Chemother. 2013;57:4237–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Coleman MT, Chen RY, Lee M, et al. PET/CT imaging reveals a therapeutic response to oxazolidinones in macaques and humans with tuberculosis. Sci Transl Med. 2014;6:265–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cilfone NA, Perry CR, Kirschner DE, Linderman JJ. Multi-scale modeling predicts a balance of tumor necrosis factor-alpha and interleukin-10 controls the granuloma environment during Mycobacterium tuberculosis infection. PLoS ONE. 2013;8:e68680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fallahi-Sichani M, El-Kebir M, Marino S, Kirschner DE, Linderman JJ. Multiscale computational modeling reveals a critical role for TNF-alpha receptor 1 dynamics in tuberculosis granuloma formation. J Immunol. 2011;186:3472–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ray JC, Flynn JL, Kirschner DE. Synergy between individual TNF-dependent functions determines granuloma performance for controlling Mycobacterium tuberculosis infection. J Immunol. 2009;182:3706–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Segovia-Juarez JL, Ganguli S, Kirschner D. Identifying control mechanisms of granuloma formation during M. tuberculosis infection using an agent-based model. J Theor Biol. 2004;231:357–376. [DOI] [PubMed] [Google Scholar]
- 101.Fallahi-Sichani M, Kirschner DE, Linderman JJ. NF-kappaB signaling dynamics play a key role in infection control in tuberculosis. Front Physiol. 2012;3:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pienaar E, Matern WM, Linderman JJ, Bader JS, Kirschner DE. Multiscale model of Mycobacterium tuberculosis infection maps metabolite and gene perturbations to granuloma sterilization predictions. Infect Immun. 2016;84:1650–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cilfone NA, Ford CB, Marino S, et al. Computational modeling predicts IL-10 control of lesion sterilization by balancing early host immunity-mediated antimicrobial responses with caseation during Mycobacterium tuberculosis infection. J Immunol. 2015;194:664–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kirschner DE, Hunt CA, Marino S, Fallahi-Sichani M, Linderman JJ. Tuneable resolution as a systems biology approach for multi-scale, multi-compartment computational models. Wiley Interdiscip Rev Syst Biol Med. 2014;6:289–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fallahi-Sichani M, Flynn JL, Linderman JJ, Kirschner DE. Differential risk of tuberculosis reactivation among anti-TNF therapies is due to drug binding kinetics and permeability. J Immunol. 2012;188:3169–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fallahi-Sichani M, Schaller MA, Kirschner DE, Kunkel SL, Linderman JJ. Identification of key processes that control tumor necrosis factor availability in a tuberculosis granuloma. PLoS Comput Biol. 2010;6:e1000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Warsinske HC, Pienaar E, Linderman JJ, Mattila JT, Kirschner DE. Deletion of TGF-beta1 increases bacterial clearance by cytotoxic T cells in a tuberculosis granuloma model. Front Immunol. 2017;8:1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fuller CL, Flynn JL, Reinhart TA. In situ study of abundant expression of proinflammatory chemokines and cytokines in pulmonary granulomas that develop in cynomolgus macaques experimentally infected with Mycobacterium tuberculosis. Infect Immun. 2003;71:7023–7034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Clay H, Volkman HE, Ramakrishnan L. Tumor necrosis factor signaling mediates resistance to mycobacteria by inhibiting bacterial growth and macrophage death. Immunity. 2008;29:283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Algood HM, Lin PL, Flynn JL. Tumor necrosis factor and chemokine interactions in the formation and maintenance of granulomas in tuberculosis. Clin Infect Dis. 2005;41(Suppl 3):S189–S193. [DOI] [PubMed] [Google Scholar]
- 111.Algood HM, Lin PL, Yankura D, Jones A, Chan J, Flynn JL. TNF influences chemokine expression of macrophages in vitro and that of CD11b+ cells in vivo during Mycobacterium tuberculosis infection. J Immunol. 2004;172:6846–6857. [DOI] [PubMed] [Google Scholar]
- 112.Lee J, Remold HG, Ieong MH, Kornfeld H. Macrophage apoptosis in response to high intracellular burden of Mycobacterium tuberculosis is mediated by a novel caspase-independent pathway. J Immunol. 2006;176:4267–4274. [DOI] [PubMed] [Google Scholar]
- 113.Ito T, Schaller M, Hogaboam CM, et al. TLR9 regulates the mycobacteria-elicited pulmonary granulomatous immune response in mice through DC-derived Notch ligand delta-like 4. J Clin Invest. 2009;119:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chensue SW, Warmington K, Ruth JH, Lukacs N, Kunkel SL. Mycobacterial and schistosomal antigen-elicited granuloma formation in IFN-γ and IL-4 knockout mice: Analysis of local and regional cytokine and chemokine networks. J Immunol. 1997;159:3565–3573. [PubMed] [Google Scholar]
- 115.Chensue SW, Warmington K, Ruth J, Lincoln P, Kuo MC, Kunkel SL. Cytokine responses during mycobacterial and schistosomal antigen-induced pulmonary granuloma formation. Production of Th1 and Th2 cytokines and relative contribution of tumor necrosis factor. Am J Pathol. 1994;145:1105–1113. [PMC free article] [PubMed] [Google Scholar]
- 116.Chensue SW, Ruth JH, Warmington K, Lincoln P, Kunkel SL. In vivo regulation of macrophage IL-12 production during type 1 and type 2 cytokine-mediated granuloma formation. J Immunol. 1995;155:3546–3551. [PubMed] [Google Scholar]
- 117.Warsinske HC, Ashley SL, Linderman JJ, Moore BB, Kirschner DE. Identifying mechanisms of homeostatic signaling in fibroblast differentiation. Bull Math Biol. 2015;77:1556–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Warsinske HC, Wheaton AK, Kim KK, Linderman JJ, Moore BB, Kirschner DE. Computational modeling predicts simultaneous targeting of fibroblasts and epithelial cells is necessary for treatment of pulmonary fibrosis. Front Pharmacol. 2016;7:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Warsinske HC, DiFazio RM, Linderman JJ, Flynn JL, Kirschner DE. Identifying mechanisms driving formation of granuloma-associated fibrosis during Mycobacterium tuberculosis infection. J Theor Biol. 2017;429:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mayadas TN, Cullere X, Lowell CA. The multifaceted functions of neutrophils. Annu Rev Pathol. 2014;9:181–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lyadova IV. Neutrophils in tuberculosis: Heterogeneity shapes the way? Mediators Inflamm. 2017;2017:8619307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.McCracken JM, Allen LA. Regulation of human neutrophil apoptosis and lifespan in health and disease. J Cell Death. 2014;7:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Witko-Sarsat V, Rieu P, Descamps-Latscha B, Lesavre P, Halbwachs-Mecarelli L. Neutrophils: Molecules, functions and pathophysiological aspects. Lab Invest. 2000;80:617–653. [DOI] [PubMed] [Google Scholar]
- 124.Lin PL, Pawar S, Myers A, et al. Early events in Mycobacterium tuberculosis infection in cynomolgus macaques. Infect Immun. 2006;74:3790–3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mattila JT, Maiello P, Sun T, Via LE, Flynn JL. Granzyme B-expressing neutrophils correlate with bacterial load in granulomas from Mycobacterium tuberculosis-infected cynomolgus macaques. Cell Microbiol. 2015;17:1085–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bru A, Cardona PJ. Mathematical modeling of tuberculosis bacillary counts and cellular populations in the organs of infected mice. PLoS ONE. 2010;5:e12985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bieberly J, Ali J. Treatment adherence of the latently infected tuberculosis population (post-Katrina) at Wetmore TB Clinic, New Orleans, USA. Int J Tuberc Lung Dis. 2008;12:1134–1138. [PubMed] [Google Scholar]
- 128.Dartois V The path of anti-tuberculosis drugs: From blood to lesions to mycobacterial cells. Nat Rev Microbiol. 2014;12:159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kjellsson MC, Via LE, Goh A, et al. Pharmacokinetic evaluation of the penetration of antituberculosis agents in rabbit pulmonary lesions. Antimicrob Agents Chemother. 2012;56:446–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sarathy JP, Zuccotto F, Hsinpin H, et al. Prediction of drug penetration in tuberculosis lesions. ACS Infect Dis. 2016;2:552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pienaar E, Cilfone NA, Lin PL, et al. A computational tool integrating host immunity with antibiotic dynamics to study tuberculosis treatment. J Theor Biol. 2015;367:166–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Prideaux B, Dartois V, Staab D, et al. High-sensitivity MALDI-MRM-MS imaging of moxifloxacin distribution in tuberculosis-infected rabbit lungs and granulomatous lesions. Anal Chem. 2011;83:2112–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zimmerman M, Lestner J, Prideaux B, et al. Ethambutol partitioning in tuberculous pulmonary lesions explains its clinical efficacy. Antimicrob Agents Chemother. 2017;61:e00924–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pienaar E, Sarathy J, Prideaux B, et al. Comparing efficacies of moxifloxacin, levofloxacin and gatifloxacin in tuberculosis granulomas using a multi-scale systems pharmacology approach. PLoS Comput Biol. 2017;13:e1005650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wallis RS, Hafner R. Advancing host-directed therapy for tuberculosis. Nat Rev Immunol. 2015;15:255–263. [DOI] [PubMed] [Google Scholar]
- 136.Flynn JL, Goldstein MM, Chan J, et al. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity. 1995;2:561–572. [DOI] [PubMed] [Google Scholar]
- 137.Wallis RS, Broder M, Wong J, Lee A, Hoq L. Reactivation of latent granulomatous infections by infliximab. Clin Infect Dis. 2005;41(Suppl 3):S194–S198. [DOI] [PubMed] [Google Scholar]
- 138.Lin PL, Myers A, Smith L, et al. Tumor necrosis factor neutralization results in disseminated disease in acute and latent Mycobacterium tuberculosis infection with normal granuloma structure in a cynomolgus macaque model. Arthritis Rheum. 2010;62:340–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wallis RS, Kyambadde P, Johnson JL, et al. A study of the safety, immunology, virology, and microbiology of adjunctive etanercept in HIV-1-associated tuberculosis. AIDS. 2004;18:257–264. [DOI] [PubMed] [Google Scholar]
- 140.Mayanja-Kizza H, Jones-Lopez E, Okwera A, et al. Immunoadjuvant prednisolone therapy for HIV-associated tuberculosis: A phase 2 clinical trial in Uganda. J Infect Dis. 2005;191:856–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Subbian S, Tsenova L, Holloway J, et al. Adjunctive phosphodiesterase-4 inhibitor therapy improves antibiotic response to pulmonary tuberculosis in a rabbit model. EBioMedicine. 2016;4:104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Subbian S, Tsenova L, O’Brien P, et al. Phosphodiesterase-4 inhibition alters gene expression and improves isoniazid-mediated clearance of Mycobacterium tuberculosis in rabbit lungs. PLoS Pathog. 2011;7:e1002262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Koo MS, Manca C, Yang G, et al. Phosphodiesterase 4 inhibition reduces innate immunity and improves isoniazid clearance of Mycobacterium tuberculosis in the lungs of infected mice. PLoS ONE. 2011;6:e17091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kaye P, Scott P. Leishmaniasis: Complexity at the host-pathogen interface. Nat Rev Microbiol. 2011;9:604–615. [DOI] [PubMed] [Google Scholar]
- 145.Contreras I, Estrada JA, Guak H, et al. Impact of Leishmania Mexicana infection on dendritic cell signaling and functions. PLoS Negl Trop Dis. 2014;8:e3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Maioli TU, Takane E, Arantes RM, Fietto JL, Afonso LC. Immune response induced by New World Leishmania species in C57BL/6 mice. Parasitol Res. 2004;94:207–212. [DOI] [PubMed] [Google Scholar]
- 147.Mattner F, Magram J, Ferrante J, et al. Genetically resistant mice lacking interleukin-12 are susceptible to infection with Leishmania major and mount a polarized Th2 cell response. Eur J Immunol. 1996;26:1553–1559. [DOI] [PubMed] [Google Scholar]
- 148.Swihart K, Fruth U, Messmer N, et al. Mice from a genetically resistant background lacking the interferon gamma receptor are susceptible to infection with Leishmania major but mount a polarized T helper cell 1-type CD4+ T cell response. J Exp Med. 1995;181:961–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kopf M, Brombacher F, Kohler G, et al. IL-4-deficient Balb/c mice resist infection with Leishmania major. J Exp Med. 1996;184:1127–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kaye PM, Beattie L. Lessons from other diseases: Granulomatous inflammation in leishmaniasis. Semin Immunopathol. 2016;38:249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Aoun J, Habib R, Charaffeddine K, Taraif S, Loya A, Khalifeh I. Caseating granulomas in cutaneous leishmaniasis. PLoS Negl Trop Dis. 2014;8:e3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.De Almeida MC, Moreira HN. A mathematical model of immune response in cutaneous leishmaniasis. J Biol Syst. 2007;15:313–354. [Google Scholar]
- 153.Siewe N, Yakubu AA, Satoskar AR, Friedman A. Immune response to infection by Leishmania: A mathematical model. Math Biosci. 2016;276:28–43. [DOI] [PubMed] [Google Scholar]
- 154.Albergante L, Timmis J, Beattie L, Kaye PM. A Petri net model of granulomatous inflammation: Implications for IL-10 mediated control of Leishmania donovani infection. PLoS Comput Biol. 2013;9:e1003334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ribeiro HAL, Maioli TU, de Freitas LM, Tieri P, Castiglione F. Modeling immune response to leishmania species indicates adenosine as an important inhibitor of Th-cell activation. Front Cell Infect Microbiol. 2017;7:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Brown LM. Helicobacter pylori: Epidemiology and routes of transmission. Epidemiol Rev. 2000;22:283–297. [DOI] [PubMed] [Google Scholar]
- 157.Perry S, de Jong BC, Solnick JV, et al. Infection with Helicobacter pylori is associated with protection against tuberculosis. PLoS ONE. 2010;5:e8804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Falk GW. The possible role of Helicobacter pylori in GERD. Semin Gastrointest Dis. 2001;12:186–195. [PubMed] [Google Scholar]
- 159.Lord AR, Simms LA, Hanigan K, Sullivan R, Hobson P, Radford-Smith GL. Protective effects of Helicobacter pylori for IBD are related to the cagA-positive strain. Gut. 2018;67:393–394. [DOI] [PubMed] [Google Scholar]
- 160.Blaser MJ, Chen Y, Reibman J. Does Helicobacter pylori protect against asthma and allergy? Gut. 2008;57:561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Matsushima K, Nagai S. Unraveling the mystery of the hygiene hypothesis through Helicobacter pylori infection. J Clin Invest. 2012;122:801–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wroblewski LE, Peek RM Jr, Wilson KT. Helicobacter pylori and gastric cancer: Factors that modulate disease risk. Clin Microbiol Rev. 2010;23:713–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ferreira AC, Isomoto H, Moriyama M, Fujioka T, Machado JC, Yamaoka Y. Helicobacter and gastric malignancies. Helicobacter. 2008;13(Suppl 1):28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. 2006;19:449–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Dooley CP, Cohen H, Fitzgibbons PL, et al. Prevalence of Helicobacter pylori infection and histologic gastritis in asymptomatic persons. N Engl J Med. 1989;321:1562–1566. [DOI] [PubMed] [Google Scholar]
- 166.Lindholm C, Quiding-Jarbrink M, Lonroth H, Hamlet A, Svennerholm AM. Local cytokine response in Helicobacter pylori-infected subjects. Infect Immun. 1998;66:5964–5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bodger K, Crabtree JE. Helicobacter pylori and gastric inflammation. Br Med Bull. 1998;54:139–150. [DOI] [PubMed] [Google Scholar]
- 168.Kivrak Salim D, Sahin M, Koksoy S, Adanir H, Suleymanlar I. Local immune response in Helicobacter pylori infection. Medicine (Baltimore). 2016;95:e3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kirschner DE, Blaser MJ. The dynamics of Helicobacter pylori infection of the human stomach. J Theor Biol. 1995;176:281–290. [DOI] [PubMed] [Google Scholar]
- 170.Blaser MJ, Kirschner D. Dynamics of Helicobacter pylori colonization in relation to the host response. Proc Natl Acad Sci USA. 1999;96:8359–8364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Parsonnet J, Friedman GD, Orentreich N, Vogelman H. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut. 1997;40:297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bagheri N, Shirzad H, Elahi S, et al. Downregulated regulatory T cell function is associated with increased peptic ulcer in Helicobacter pylori-infection. Microb Pathog. 2017;110:165–175. [DOI] [PubMed] [Google Scholar]
- 173.Kern KB, Nelson JR, Norman SA, Milander MM, Hilwig RW. Oxygenation and ventilation during cardiopulmonary resuscitation utilizing continuous oxygen delivery via a modified pharyngeal-tracheal lumened airway. Chest. 1992;101:522–529. [DOI] [PubMed] [Google Scholar]
- 174.Larussa T, Leone I, Suraci E, Imeneo M, Luzza F. Helicobacter pylori and T helper cells: Mechanisms of immune escape and tolerance. J Immunol Res. 2015;2015:981328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Chen MG. Progress and problems in schistosomiasis control in China. Trop Med Parasitol. 1989;40:174–176. [PubMed] [Google Scholar]
- 176.Carbo A, Bassaganya-Riera J, Pedragosa M, et al. Predictive computational modeling of the mucosal immune responses during Helicobacter pylori infection. PLoS ONE. 2013;8:e73365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sriskandan S, Altmann DM. The immunology of sepsis. J Pathol. 2008;214:211–223. [DOI] [PubMed] [Google Scholar]
- 178.Bone RC, Balk RA, Cerra FB, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101:1644–1655. [DOI] [PubMed] [Google Scholar]
- 179.Cohen J The immunopathogenesis of sepsis. Nature. 2002;420: 885–891. [DOI] [PubMed] [Google Scholar]
- 180.Schulte W, Bernhagen J, Bucala R. Cytokines in sepsis: Potent immunoregulators and potential therapeutic targets–an updated view. Mediators Inflamm. 2013;2013:165974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Vodovotz Y, Billiar TR. In silico modeling: Methods and applications to trauma and sepsis. Crit Care Med. 2013;41:2008–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Yang H, Lee EK, Wu CJ, Shi Z, Ben-Arieh D, Simpson SQ. Mathematical modeling of innate immunity responses of sepsis: Modeling and computational studies In: Yang H, Lee EK, eds. Healthcare Analytics: From Data to Knowledge to Healthcare Improvement. 1st ed. Hoboken, NJ: John Wiley & Sons, Inc; 2016:221–259. [Google Scholar]
- 183.An G In silico experiments of existing and hypothetical cytokine-directed clinical trials using agent-based modeling. Crit Care Med. 2004;32:2050–2060. [DOI] [PubMed] [Google Scholar]
- 184.Delano MJ, Ward PA. The immune system’s role in sepsis progression, resolution, and long-term outcome. Immunol Rev. 2016;274:330–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: From cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13:862–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Delano MJ, Moldawer LL. Magic bullets and surrogate biomarkers circa 2009. Crit Care Med. 2009;37:1796–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Bosmann M, Ward PA. The inflammatory response in sepsis. Trends Immunol. 2013;34:129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Hutchins NA, Unsinger J, Hotchkiss RS, Ayala A. The new normal: Immunomodulatory agents against sepsis immune suppression. Trends Mol Med. 2014;20:224–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Luan YY, Dong N, Xie M, Xiao XZ, Yao YM. The significance and regulatory mechanisms of innate immune cells in the development of sepsis. J Interferon Cytokine Res. 2014;34:2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Rimmele T, Payen D, Cantaluppi V, et al. Immune cell phenotype and function in sepsis. Shock. 2016;45:282–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: Considerations for healthcare providers in the United States. Clin Infect Dis. 2012;55(Suppl 1):S10–S15. [DOI] [PubMed] [Google Scholar]
- 192.Kim AY, Onofrey S, Church DR. An epidemiologic update on hepatitis C infection in persons living with or at risk of HIV infection. J Infect Dis. 2013;207(Suppl 1):S1–S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Boltjes A, Movita D, Boonstra A, Woltman AM. The role of Kupffer cells in hepatitis B and hepatitis C virus infections. J Hepatol. 2014;61:660–671. [DOI] [PubMed] [Google Scholar]
- 194.Saha B, Szabo G. Innate immune cell networking in hepatitis C virus infection. J Leukoc Biol. 2014;96:757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Yoon JC, Yang CM, Song Y, Lee JM. Natural killer cells in hepatitis C: Current progress. World J Gastroenterol. 2016;22:1449–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Tian Z, Chen Y, Gao B. Natural killer cells in liver disease. Hepatology. 2013;57:1654–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Nguyen-Lefebvre AT, Horuzsko A. Kupffer cell metabolism and function. J Enzymol Metabol. 2015;1:101. [PMC free article] [PubMed] [Google Scholar]
- 198.Mengshol JA, Golden-Mason L, Arikawa T, et al. A crucial role for Kupffer cell-derived galectin-9 in regulation of T cell immunity in hepatitis C infection. PLoS ONE. 2010;5:e9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hartling HJ, Gaardbo JC, Ronit A, et al. CD4(+) and CD8(+) regulatory T cells (Tregs) are elevated and display an active phenotype in patients with chronic HCV mono-infection and HIV/HCV co-infection. Scand J Immunol. 2012;76:294–305. [DOI] [PubMed] [Google Scholar]
- 200.Barjon C, Dahlqvist G, Calmus Y, Conti F. Role of regulatory T-cells during hepatitis C infection: From the acute phase to post-transplantation recurrence. Digest Liver Dis. 2015;47:913–917. [DOI] [PubMed] [Google Scholar]
- 201.Terilli RR, Cox AL. Immunity and hepatitis C: A review. Curr HIV/AIDS Rep. 2013;10:51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Gremion C, Cerny A. Hepatitis C virus and the immune system: A concise review. Rev Med Virol. 2005;15:235–268. [DOI] [PubMed] [Google Scholar]
- 203.Burg D, Rong L, Neumann AU, Dahari H. Mathematical modeling of viral kinetics under immune control during primary HIV-1 infection. J Theor Biol. 2009;259:751–759. [DOI] [PubMed] [Google Scholar]
- 204.Obaid A, Ahmad J, Naz A, et al. Modeling and analysis of innate immune responses induced by the host cells against hepatitis C virus infection. Integr Biol (Camb). 2015;7:544–559. [DOI] [PubMed] [Google Scholar]
- 205.Osburn WO, Levine JS, Chattergoon MA, Thomas DL, Cox AL. Anti-inflammatory cytokines, pro-fibrogenic chemokines and persistence of acute HCV infection. J Viral Hepatitis. 2013;20:404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Wiesner DL, Klein BS. The balance between immunity and inflammation. Science. 2017;357:973–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Martin SJ. Oncogene-induced autophagy and the Goldilocks principle. Autophagy. 2011;7:922–923. [DOI] [PubMed] [Google Scholar]
- 208.Monin L, Khader SA. Chemokines in tuberculosis: The good, the bad and the ugly. Semin Immunol. 2014;26:552–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Slight SR, Khader SA. Chemokines shape the immune responses to tuberculosis. Cytokine Growth Factor Rev. 2013;24:105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Eizenberg-Magar I, Rimer J, Zaretsky I, Lara-Astiaso D, Reich-Zeliger S, Friedman N. Diverse continuum of CD4(+) T-cell states is determined by hierarchical additive integration of cytokine signals. Proc Natl Acad Sci USA. 2017;114:E6447–E6456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Fang M, Xie H, Dougan SK, Ploegh H, van Oudenaarden A. Stochastic cytokine expression induces mixed T helper cell States. PLoS Biol. 2013;11:e1001618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Lin PL, Dartois V, Johnston PJ, et al. Metronidazole prevents reactivation of latent Mycobacterium tuberculosis infection in macaques. Proc Natl Acad Sci USA. 2012;109:14188–14193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Via LE, Weiner DM, Schimel D, et al. Differential virulence and disease progression following Mycobacterium tuberculosis complex infection of the common marmoset (Callithrix jacchus). Infect Immun. 2013;81:2909–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Stanton P, Richards S, Reeves J, et al. Epidermal growth factor receptor expression by human squamous cell carcinomas of the head and neck, cell lines and xenografts. Br J Cancer. 1994;70:427–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Corti A, Gasparri A, Sacchi A, et al. Tumor targeting with biotinylated tumor necrosis factor alpha: Structure-activity relationships and mechanism of action on avidin pretargeted tumor cells. Cancer Res. 1998;58:3866–3872. [PubMed] [Google Scholar]
- 216.Zimmermann VS, Bondanza A, Monno A, Rovere-Querini P, Corti A, Manfredi AA. TNF-alpha coupled to membrane of apoptotic cells favors the cross-priming to melanoma antigens. J Immunol. 2004;172:2643–2650. [DOI] [PubMed] [Google Scholar]
- 217.Desnoyers L, Simonette RA, Vandlen RL, Fendly BM. Novel non-isotopic method for the localization of receptors in tissue sections. J Histochem Cytochem. 2001;49:1509–1518. [DOI] [PubMed] [Google Scholar]
- 218.Wiley HS, Cunningham DD. The endocytotic rate constant. A cellular parameter for quantitating receptor-mediated endocytosis. J Biol Chem. 1982;257:4222–4229. [PubMed] [Google Scholar]
- 219.Cilfone NA. Understanding and Treating Mycobacterium tuberculosis Infection: A Multi-Scale Modeling Approach: Chemical Engineering. Ann Arbor, MI: University of Michigan; 2014. [Google Scholar]
- 220.Pienaar E, Dartois V, Linderman JJ, Kirschner DE. In silico evaluation and exploration of antibiotic tuberculosis treatment regimens. BMC Syst Biol. 2015;9:79. [DOI] [PMC free article] [PubMed] [Google Scholar]