

Abstract
Influenza neuraminidase is responsible for the escape of new viral particles from the infected cell surface. Several neuraminidase inhibitors are used clinically to treat patients or stockpiled for emergencies. However, the increasing development of viral resistance against approved inhibitors has underscored the need for the development of new antivirals effective against resistant influenza strains. A facile, sensitive, and inexpensive screening method would help achieve this goal. Recently, we described a multiwell plate-based DNA-linked inhibitor antibody assay (DIANA). This highly sensitive method can quantify femtomolar concentrations of enzymes. DIANA also has been applied to high-throughput enzyme inhibitor screening, allowing the evaluation of inhibition constants from a single inhibitor concentration. Here, we report the design, synthesis, and structural characterization of a tamiphosphor derivative linked to a reporter DNA oligonucleotide for the development of a DIANA-type assay to screen potential influenza neuraminidase inhibitors. The neuraminidase is first captured by an immobilized antibody, and the test compound competes for binding to the enzyme with the oligo-linked detection probe, which is then quantified by qPCR. We validated this novel assay by comparing it with the standard fluorometric assay and demonstrated its usefulness for sensitive neuraminidase detection as well as high-throughput screening of potential new neuraminidase inhibitors.
Keywords: assay, crystallography, DIANA, influenza neuraminidase
Introduction
Influenza virus causes severe respiratory infections associated with significant morbidity and mortality. Worldwide, annual influenza epidemics result in three to five million cases of severe acute viral infection and roughly 290 000–650 000 deaths [1]. Several influenza pandemics have occurred in the last century, each caused by a new strain of the virus in humans. Two types of influenza antivirals, which target different steps in the viral lifecycle, have been approved by the Food and Drug Administration (FDA). The first type blocks the viral ion channel formed by the M2 protein, preventing the virus from infecting the cell. However, use of M2 inhibitors leads to rapid development of drug resistance, and these drugs are no longer routinely used. The second type inhibits neuraminidase (NA).
NA is an influenza virus membrane glycoprotein that is essential for release of new virions from an infected cell. NA inhibitors are the first line treatment for patients in need of anti-influenza drug therapy. Nevertheless, the increasing development of viral resistance against approved NA inhibitors has underscored the need for new anti-influenza drugs active against resistant viral strains and different NA subtypes. While several methods for NA inhibitor characterization based on enzymatic activity measurements have been described, most of them are time-consuming, sensitive to interference, and expensive [2–5].
Recently, our group developed a multiwell plate-based DNA-linked inhibitor antibody assay (DIANA) as a simple and reliable technique for enzyme detection and screening of small-molecule inhibitors [6]. First, the target enzyme is captured by an antibody immobilized on the well. The enzyme then binds a detection probe consisting of a DNA oligonucleotide covalently linked to a known competitive inhibitor. The bound probe is measured by quantitative PCR (qPCR, Figure 1A). DIANA showed a several-order-of-magnitude higher sensitivity toward a model enzyme when compared with sandwich ELISA (enzyme-linked immunosorbent assay) and enabled the detection of target enzymes in complex biological matrices. DIANA also has been used to screen inhibitors of two clinically relevant enzymes: glutamate carboxypeptidase II and carbonic anhydrase IX [6]. The assay enables inhibition constants (Ki) to be calculated directly from single-well measurements.
Figure 1. Schematic representation of evaluation of neuraminidase inhibitors using DIANA (A) and preparation of a detection probe (B).

(A) The detection probe binds to the active site of neuraminidase captured on the well by an immobilized anti-NA antibody. The amount of bound detection probe is determined by qPCR. The inhibition potency of the tested compound can be determined from the difference in the amount of bound probe after incubation of neuraminidase in the presence and absence of the compound. (B) Preparation of a covalent conjugate consisting of a reporter DNA oligonucleotide and a neuraminidase inhibitor (compound 1) used as a DIANA detection probe. Only one regioisomer of SPAAC product is shown.
Here, we propose DIANA as a new reliable and rapid method for screening influenza NA inhibitors. We report preparation of an active-site-directed detection probe and structurally characterize the interactions of the inhibitor incorporated in the probe with recombinant NA. Additionally, we compare inhibition constants for various compounds tested by DIANA with results from the standard fluorometric assay.
Experimental
Cloning, expression, and purification of recombinant NA2009wt
DNA encoding the neuraminidase ectodomain (residues 82–469) from the A/California/07/2009 (H1N1) influenza virus was prepared by GenScript USA, Inc. (Genbank Source Sequence CY121682). DNA was inserted into the pMT/BiP/V5-HisA vector (Invitrogen) with an N-terminal tag containing two Strep-tags, a FLAG-tag and a thrombin cleavage site. This construct was used to transfect Drosophila Schneider S2 cells (Invitrogen), and large-scale expression was performed as previously described [7]. Recombinant neuraminidase expressed into the cell culture medium was subsequently purified using one-step purification on Strep-Tactin agarose resin (IBA GmbH) [8,9]. First, Strep-Tactin resin was equilibrated in buffer W (100 mM Tris–HCl, pH 8.0, 150 mM NaCl), and medium with added BioLock biotin blocking solution (IBA GmbH) was applied. The matrix with bound tagged protein was thoroughly washed with buffer W, and elution was performed with 10 mM desthiobiotin in buffer W. The resin was regenerated with buffer W containing 1 mM 2-(4-hydroxyphenylazo)benzoic acid (Sigma–Aldrich) and stored at 4°C for later use. The purification process was monitored by SDS–PAGE and Western blot using murine monoclonal anti-FLAG M2-peroxidase antibody clone M2 (Sigma–Aldrich). The N-terminal tag was removed by cleavage with thrombin protease immobilized on agarose beads (Sigma–Aldrich).
Synthesis of inhibitors
The compounds presented here were either prepared by the same procedures described recently [10] (compounds 2–7) or as outlined in Figures 2 and 3. For preparation of ω-hexylazido tamiphosphor 1, we used our previously described approach to a key Barton ester based on a modified version of Gunasekera's procedure (Figure 2). Briefly, the phosphonate salt of ethyl oseltamivir carboxylate was free-based with aqueous bicarbonate and Boc-protected to provide 12. The ethyl ester moiety was hydrolyzed, and acid 13 was treated with HOTT [N,N,N′,N′-tetramethyl-S-(1-oxido-2-pyridyl)thiuronium hexafluorophosphate] reagent to yield the key Barton thioester. Upon irradiation with a flood lamp in a solution of bromotrichloromethane, bromide 14 was obtained. Palladium-catalyzed Hirao coupling of 14 with dimethyl phosphite afforded dimethyl phosphonate 15 in very good yield. The dimethyl ester was subjected to selective mono-O-demethylation with sodium hydroxide, yielding intermediate 16. Surprisingly, alkylation of 16 at 60°C furnished alkylated product 17, which lacks the methyl ester moiety. Apparently, under these conditions, the desired alkylation was followed with demethylation mediated by halide ions [11]. This serendipitous finding led to a shortcut in the synthesis of ω-hexylazido tamiphosphor that resulted not only in omission of the problematic second O-demethylation by thiophenolate but also improvement of overall yield. The last step of the reaction sequence was Boc deprotection with trifluoroacetic acid (TFA). Compound 2 was prepared by the same procedure and was isolated as a mixture of diastereomeric monoalkyl esters because the phosphorus atom is an additional stereogenic center. As expected, we were not able to observe separation of these diastereomers on analytical HPLC (high-performance liquid chromatography), and thus, the material was used as such.
Figure 2. Synthesis of ω-azidohexyl tamiphosphor derivative 1.

The following reagents and conditions were used: (a) NaHCO3/H2O; (b) Boc2O, Et3N, 98%; (c) 0.5 M NaOH, 1,4-dioxane, 95%; (d) S-(1-oxido-2-pyridyl)-N,N,N′,N′-tetramethylthiuronium hexafluorophosphate, Et3N, DMAP, THF; (e) bromotrichloromethane, DCM, hv, 78%; (f) dimethyl phosphite, tetrakis(triphenylphosphine)palladium, Et3N, toluene, 86%; (g) 1-azido-6-bromohexane, NaI, DIPEA, DMF, 60°C, 56%; (h) trifluoroacetic acid.
Figure 3. Synthesis of oseltamivir derivatives.

The following reagents and conditions were used: (a) ethyl 2-bromopropionate, NaHCO3; (b) 0.5 M NaOH, 1,4-dioxane; (c) 4-nitrobenzoyl chloride, Et3N; (d) (i) NaHCO3 and (ii) dimethyl maleate; (e) 2-[2-(2-methoxyethoxy)ethoxy]acetic acid, TBTU, Et3N, DMF.
Oseltamivir derivatives with an alkyl moiety at the C-5 amino functionality were also prepared from oseltamivir phosphonate. In this case, the starting compound was treated with ethyl 2-bromopropionate to yield a mixture of diastereomers 18 and 19. After separation followed by saponification and Boc deprotection with TFA (Figure 3), diastereomers 8 and 9 were produced in 38% and 34% yield, respectively. To determine the absolute configuration of the introduced polar moiety, we performed crystallization attempts in different solvent systems. However, our attempts to obtain a monocrystal of 19 suitable for X-ray analysis failed, and we decided to introduce 4-nitrobenzoyl moiety to the structure of 19 to facilitate crystallization.
Diastereomer 19 was treated with 4-nitrobenzoyl chloride in the presence of triethylamine. Crystallization of the resulting derivative 20 eventually yielded a suitable monocrystal, the X-ray structure of which is shown in Supplementary Figure S1. This structural information allowed us to assign an (R) configuration to the newly formed stereocenter in 20. We surmise that the stereogenic center in 9 has the same configuration since it is not altered by N-acylation.
Alkylation of the same starting material with dimethyl maleate produced a diastereomeric mixture of triester 21 in good yield (Figure 3). Cleavage of alkylesters followed by preparative HPLC resulted in 10, which was used as a mixture of diastereomers. Amide 11 has an intentionally impaired basic functionality at C-5. All potent NA inhibitors possess either basic amino or guanidino functionalities at C-5, as their interaction with three acidic residues of NA contributes significantly to strong inhibitor-NA binding. Therefore, the binding affinity of 11, which has a non-basic moiety at C-5, should be greatly diminished. This compound was prepared to demonstrate the viability of DIANA assay on a broader range of Ki values. An acyl was introduced by standard amide coupling mediated with TBTU (N,N,N′,N′-tetramethyl-O-(benzotriazol-1-yl)uronium tetrafluoroborate), yielding ethyl ester 22, which was subsequently hydrolyzed to give sialylmimetic 11 in 65% overall yield.
Preparation of DIANA detection probe
The detection probe was prepared by copper-free click-chemistry [12]. The probe consists of an oligonucleotide of sequence 5′-CCT GCC AGT TGA GCA TTT TTA TCT GCC ACC TTC TCC ACC AGA CAA AAG CTG GAA A-3′ with the terminal 3′-phosphate moiety modified by a 6-amino-2-(hydroxymethyl)hexyl group (Generi-Biotech, OPC purification). The modified oligonucleotide was reacted with NHS-DBCO (dibenzocyclooctyne-N-hydroxysuccinimidyl ester, Sigma–Aldrich) in a 1 : 50 ratio. The product (designated as DNA_DBCO) was conjugated with an NA inhibitor containing an azido group (1). The conjugation was performed at a 1 : 10 ratio of DNA_DBCO conjugate to 1. The DNA conjugates were purified from the unconjugated small molecule by ultrafiltration with a 10 kDa cutoff and then analyzed by LC–MS (liquid chromatography-mass spectrometry).
Determination of inhibition constants (Ki) by the fluorometric assay
Enzyme inhibition constants (Ki) were determined in 0.1 M MES, pH 6.15, 150 mM NaCl, and 10 mM CaCl2 at 37°C by the fluorometric assay using 2′-(4-methylumbelliferyl)-α-d-N-acetylneuraminic acid (4-MUNANA, Sigma–Aldrich) as a substrate [2]. Substrate cleavage was monitored with an Infinite M1000 fluorescence reader (TECAN) using an excitation wavelength of 355 nm and an emission wavelength of 450 nm.
Each 40 µl reaction contained 17 nM (34 ng) NA2009wt and 500 µM 4-MUNANA. The reactions were performed for 20 min at 37°C in black fluorescence 96-well plates and terminated by the addition of 40 µl of 1 M sodium carbonate. Inhibition constants were determined by measuring the reduction in fluorescence of the product 4-MU in the presence of different inhibitor concentrations. The data were analyzed using the equation for competitive inhibition according to Williams and Morrison or Dixon analysis when the Ki value was expected to be above 100 nM [13,14].
General DIANA protocol
The protocol described as ‘general assay conditions’ by Navrátil et al. [6] was used with the following modifications. A sheep antibody against NA of influenza A virus H1N1 (cat. no. AF4858, R&D systems) was used as the capture antibody and was immobilized onto the plate by applying 5 µl antibody solution [10 ng/µl in TBS (Tris-buffered saline): 20 mM Tris–HCl, 150 mM NaCl] to the bottom of the wells. Then, the immobilized antibody was blocked with casein blocker and incubated overnight, followed by washing using a microplate washer (405™ Microplate Washer LS, BioTek). Next, 2 ng of NA in 5 µl Q1 buffer [20 mM Tris–HCl, 150 mM NaCl, 0.1% (w/v) Tween 20, and 5 mM CaCl2] was applied to the bottom of the wells and incubated for 2 h, followed by another wash in the microplate washer. Afterward, 5 µl of detection probe at a concentration of 200 pM diluted in Q2 buffer [20 mM Tris–HCl, 150 mM NaCl, 0.1% (w/v) Tween 20, 5 mM CaCl2, and 0.0055% (w/v) casein] mixed in a 9 : 1 ratio with inhibitor dissolved in 100% DMSO was applied to the bottom of the wells (final concentration of 10% DMSO) and incubated for 1 h. The plate was again washed in the microplate washer to remove unbound probe. Finally, the amount of bound probe was determined by qPCR as described recently [6].
Determination of inhibition constants by DIANA
Inhibitors were tested at concentrations ranging from 10 nM to 10 µM for tight-binding inhibitors and 316 nM to 316 µM for weak inhibitors. The model for the determination of Ki in the presence of detection probe with serial dilutions of inhibitor was described recently [6]. Ki was determined as follows: the ΔCq values for each inhibitor were obtained as the difference in Cq between the well(s) incubated with the inhibitor and the mean of wells without the inhibitor (typically 12 wells per experiment). The Ki values of the inhibitors were computed from their ΔCq values and the concentration of inhibitor according to the formula: , where the Itot is the total inhibitor concentration, Ptot is the total concentration of the probe (200 pM), and Kd is the dissociation constant of the probe determined by incubating serial dilutions of the probe with a constant amount of enzyme (Kd = 3.9 nM). The final Ki value for each inhibitor was calculated as the average of Ki values determined from each inhibitor concentration and corresponding Cq.
Protein crystallography
Enzyme–inhibitor complexes for crystallization were prepared by mixing NA2009wt in 5 mM Tris–HCl, pH 8.0, with 1 and 3. Mixtures were concentrated by ultrafiltration to a final concentration of 8 mg/ml (3-fold molar excess of inhibitor in the mixture). Crystals were grown using the hanging-drop vapor diffusion method at 19°C. Drops consisted of 1 µl NA–inhibitor complex and 1 µl reservoir solution. The reservoir solutions for NA2009wt-1 and NA2009wt-3 were 100 mM HEPES, pH 7.5, 10% PEG (polyethylene glycol) 8000 and 100 mM HEPES, pH 7.0, 8% PEG 8000, respectively. All crystals were transferred into a cryoprotectant consisting of reservoir solution supplemented with 20% (v/v) ethylene glycol and flash-cooled in liquid nitrogen.
Data collection and structure determination
Diffraction data were collected at 100 K on BL14.1 at the BESSY II electron storage ring operated by the Helmholtz-Zentrum Berlin MX14.1 of BESSY, Berlin, Germany [15]. The dataset was processed using XDSAPP [16]. The crystal parameters and data collection statistics are listed in Table 1. The structure was determined by molecular replacement with the program Molrep [17] using the crystal structure of NA2009wt complexed with tamiphosphor [10]. Model refinement was performed using the program REFMAC 5.7.0032 [18] from the CCP4 (Collaborative Computational Project Number 4) package [19] in combination with manual adjustments in Coot software [20]. Compounds were modeled after complete refinement of the protein chains and solvent model. The Molprobity server [21] was used to evaluate the final model quality. The final refinement statistics are summarized in Table 1. The structures were analyzed using the programs lsqkab (superpose) [22], baverage, and contact from the CCP4 package [19].
Table 1. Crystal data and diffraction data collection and refinement statistics.
The data in parentheses refer to the highest-resolution shell.
| NA2009wt/compound 3 | NA2009wt/compound 1 | |
|---|---|---|
| PDB code | 6G01 | 6G02 |
| Data collection statistics | ||
| Space group | C2221 | P4 |
| Cell parameters (Å; °) | 118.88, 137.64, 118.81; 90, 90, 90 | 114.89, 114.89, 63.93; 90, 90, 90 |
| Number of molecules in AU | 2 | 2 |
| Wavelength (Å) | 0.9184 | 0.9184 |
| Resolution (Å) | 44.89–1.61 (1.71–1.61) | 42.72–1.86 (1.97–1.86) |
| Number of unique reflections | 121 339 (19 127) | 70 090 (11 223) |
| Redundancy | 3.12 (2.79) | 3.75 (3.58) |
| Completeness (%) | 98.2 (96.4) | 99.8 (99.6) |
| Rmeas1 | 0.095 (0.645) | 0.136 (0.738) |
| Average I/σ(I) | 10.29 (1.88) | 9.24 (1.86) |
| CC1/2 (%) | 99.7 (75.8) | 99.4 (61.4) |
| Wilson B (Å2) | 22.3 | 24.1 |
| Refinement statistics | ||
| Resolution range (Å) | 44.89–1.61 (1.656–1.614) | 42.71–1.84 (1.891–1.84) |
| No. of reflections in working set | 119 520 (8443) | 70 147 (5127) |
| No. of reflections in test set | 1821 (129) | 1799 (131) |
| R value (%)2 | 17.8 (31.7) | 15.3 (27.9) |
| Rfree value (%)3 | 20.5 (30.3) | 19.7 (28.9) |
| RMSD bond length (Å) | 0.014 | 0.015 |
| RMSD angle (°) | 1.637 | 1.670 |
| Number of atoms in AU | 7153 | 6995 |
| Number of protein atoms in AU | 6056 | 6006 |
| Number of water molecules in AU | 825 | 809 |
| Mean B-value protein/waters/compounds (Å2) | 17.7/28.7/14.8 | 20.0/31.0/22.6 |
| Ramachandran plot statistics4 | ||
| Residues in favored regions (%) | 96.4 | 96.1 |
| Residues in allowed regions (%) | 3.6 | 3.9 |
Rmeas = Σhkl(n/(n − 1))1/2ΣiIin(hkl) − 〈I(hkl)〉|/ΣhklΣi·Ii(hkl), where the Ii(hkl) is an individual intensity of the ith observation of reflection hkl and 〈I(hkl)〉 is the average intensity of reflection hkl with summation over all data.
R-value = ||Fo| − |Fc||/|Fo|, where Fo and Fc are the observed and calculated structure factors, respectively.
Rfree is equivalent to R-value, but is calculated for 5% of the reflections chosen at random and omitted from the refinement process [38].
As determined by Molprobity [39].
Results
Design of the detection probe
The FDA-approved drug oseltamivir carboxylate is the most widely used NA inhibitor, and we aimed to prepare our detection probe by linking this compound or a derivative to a DNA oligonucleotide. First, we sought to determine an appropriate way to link the oligonucleotide with the inhibitor without compromising inhibitor binding to NA. The main features of oseltamivir carboxylate that mimics the sialic acid substrate of influenza NA are as follows: (i) a negatively charged carboxylate at C-1 interacting with three arginine residues (also known as the arginine triad conserved in sialidases), (ii) a C-3 pentyloxy moiety accommodated in the hydrophobic pocket, and (iii) the C-4 acetamide and basic amino group at carbon C-5 that interact with an aspartic acid and two glutamic acids. According to the crystal structure of NA in complex with oseltamivir (PDB: 3TI6) [23], the C-1 carboxylate group is a suitable site for the attachment of a linker. However, a negative charge at C-1 is indispensable for inhibitor tight binding, as demonstrated on a series of oseltamivir derivatives substituted at the carboxylate moiety and a series of phospha-congeners (tamiphosphor derivatives) [24–26]. As attachment of a linker to the carboxylate moiety via an ester bond would lead to loss of the negative charge, we used an oseltamivir derivative with a negatively charged C-1 phosphonate group, which maintains its negative charge after linker.
In 2009, Streicher and coworkers demonstrated that replacement of the C-1 carboxylate with a monoalkyl phosphonate moiety does not diminish inhibitory activity [27,28]. This so-called tamiphosphor binds NA with similar potency as oseltamivir carboxylate, as we recently confirmed by protein microcalorimetry [10]. Moreover, the same group in 2011 demonstrated immobilization/conjugation of the ω-azidohexyl ester of tamiphosphor by CuAAC (copper(I)-catalyzed azide alkyne cycloaddition) click chemistry. Subsequently, conjugates of this modified tamiphosphor with biotin and fluorescein were designed for fluorometric detection and quantification of influenza viruses. Both conjugates displayed selective, high-affinity binding to influenza NA and showed promise for the development of various diagnostic tools for biological research [29,30]. Together, these findings indicate that monoalkylated tamiphosphor derivatives are suitable sialylmimetics. Moreover, the phosphonate functionality is a well-suited chemical handle for further modification to develop tethered and very potent NA inhibitors.
The construction of DIANA detection probes requires a linker of appropriate length equipped with bioorthogonal functional groups. In this particular case, ligation of the sialylmimetic inhibitor to the oligonucleotide was performed by strain-promoted alkyne-azide cycloaddition (SPAAC). Tamiphosphor was modified with a clickable ω-azidoalkyl moiety (yielding 1) suitable for conjugation to a DNA oligonucleotide equipped with a dibenzocyclooctyne moiety, and the probe was prepared by click-chemistry (Figure 1B) [12]. The quality and quantity of the probe were monitored by LC–MS.
To determine the effect of linker attachment to tamiphosphor on enzyme binding, we determined the inhibition potencies of 1, oseltamivir carboxylate (4), and tamiphosphor (2) using a standard kinetic assay. All three compounds bound NA with comparable affinity (Ki values of 24, 24, and 26 nM, respectively), supporting use of a tamiphosphor–oligonucleotide conjugate as a DIANA detection probe (Figure 1B).
X-ray structures of neuraminidase in complex with tamiphosphor derivatives
We recently reported the structure of tamiphosphor in complex with NA at 1.8 Å resolution [10]. The structure revealed that the O3 atom of the phosphonate functionality is oriented out of the active site and does not engage in direct interactions with protein residues [10]. This observation led us to prepare a modified inhibitor for the DIANA detection probe by substituting one hydroxyl in tamiphosphor with a large linker connected to an oligonucleotide.
To analyze the binding of tamiphosphor derivatives to NA, we solved X-ray crystal structures of complexes of 1 (ω-azidoalkyl ester) and 3 (methyl ester) with NA (strain NA2009wt) at high resolution. These structures revealed that the compounds bind into the active site with a pose very similar to that of tamiphosphor [10] (Figure 4A,B). The RMSD (root-mean-square deviation) values for superposition of tamiphosphor with the corresponding atoms in 1 and 3 were 0.22 Å. The methyl ester of 3 was modeled into well-defined electron density map in a position pointing out of the active site (Figure 4A). Most of the atoms of 1 could be traced in the electron density map, with the exception of the three terminal nitrogen atoms of the tamiphosphor extension. The azide group does not interact with the protein and is fully exposed to the solvent, and thus, its electron density map was of lower quality. Of the linker atoms, only C24 appears to interact with NA. This atom is within van der Waals distance (3.7–4.0 Å) of residues Pro431 and Ile149 (Figure 4C). This interaction may explain the slight improvement in Ki value observed for 1 (24 nM) compared with 3 (39 nM) (Table 2). The RMSD value for superposition of the corresponding atoms of 1 and 3 was 0.15 Å.
Figure 4. X-ray analysis of ligand interactions.
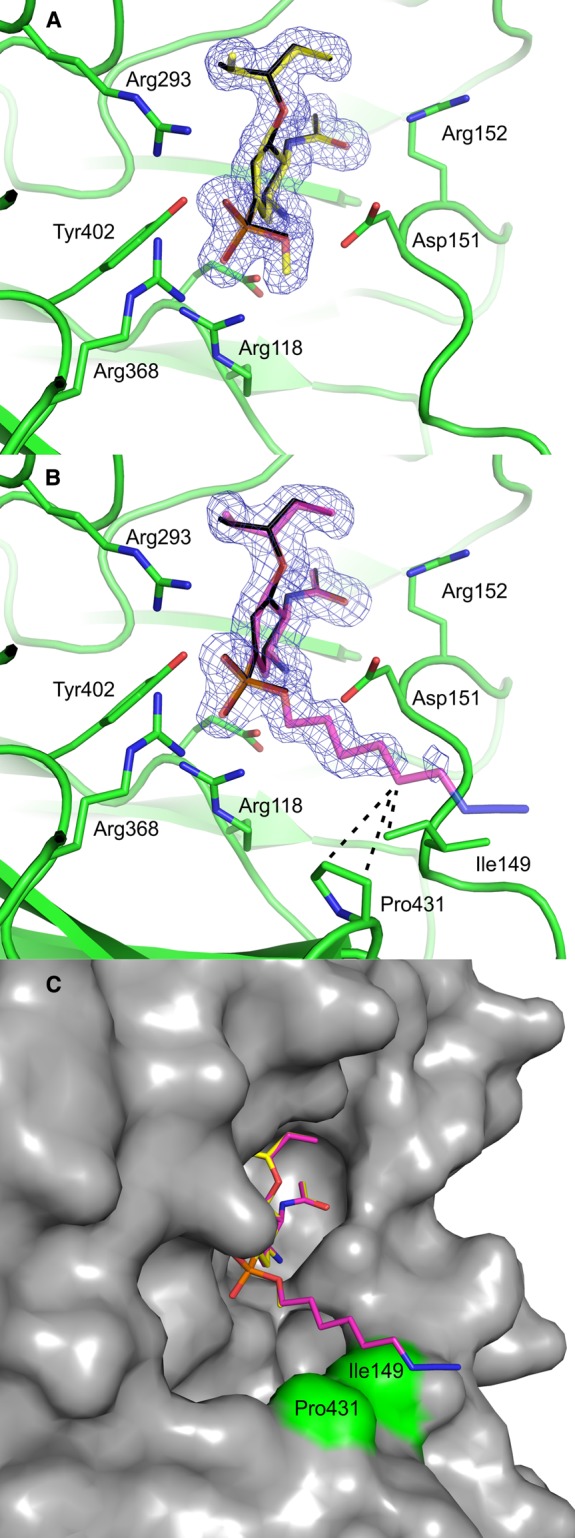
Crystal structure of NA2009wt in complex with 3 (A) and 1 (B). Compounds are represented as sticks with carbon atoms colored yellow (3) or maroon (1), oxygen atoms red, nitrogen atoms blue, and phosphorus atoms orange. The tamiphosphor molecule from the previously reported structure [10] is overlaid and colored black. The protein is shown in green with residues forming polar interactions highlighted as sticks. The 2Fo − Fc electron density maps are contoured at 1.0 σ. In (B), van der Waals interactions are represented by dashed lines. (C) Overlay of 1 and 3. The protein is represented by its solvent accessible surface (gray), and Pro431 and Ile149 are highlighted in green.
Table 2. Inhibition constants determined by the standard kinetic assay and DIANA.
| Compound | Chemical formula | Ki (kinetic assay) (pH 6.15) (nM) | Ki (DIANA) (pH 7.4) (nM) | Fold Ki |
|---|---|---|---|---|
| 1 | 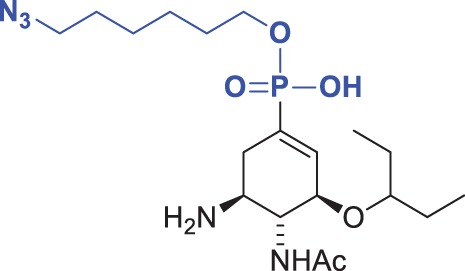 |
24 ± 5 | 33 ± 8 | 1.4 |
| 2 | 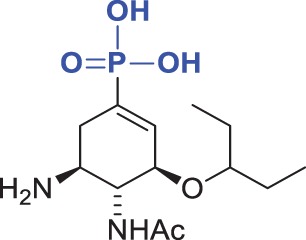 |
26 ± 4 | 52 ± 9 | 2.0 |
| 3 | 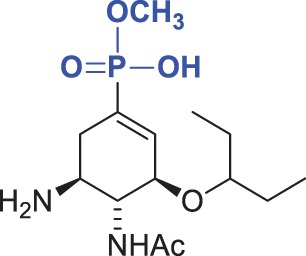 |
39 ± 1 | 100 ± 20 | 2.6 |
| 4 | 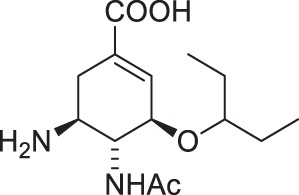 |
24 ± 4 | 31 ± 6 | 1.3 |
| 5 | 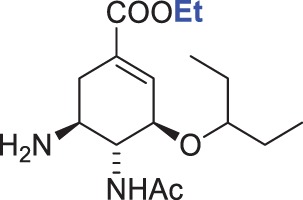 |
2100 ± 200 | 2000 ± 500 | 1.0 |
| 6 | 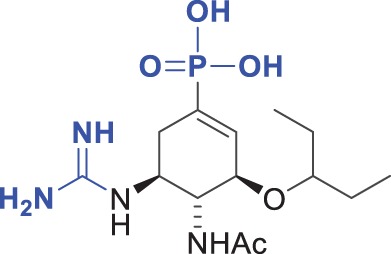 |
140 ± 30 | 570 ± 180 | 4.1 |
| 7 | 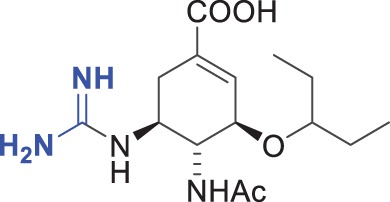 |
46 ± 12 | 570 ± 200 | 12 |
| 8 | 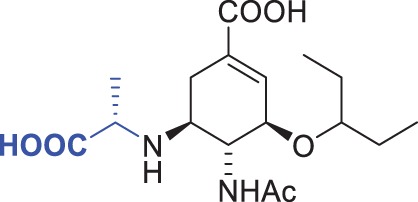 |
15 ± 2 | 170 ± 60 | 11 |
| 9 | 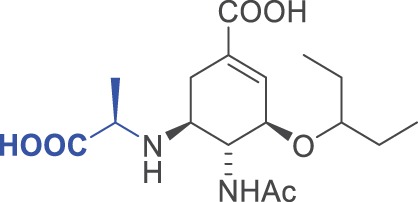 |
250 ± 40 | 960 ± 260 | 3.8 |
| 10 | 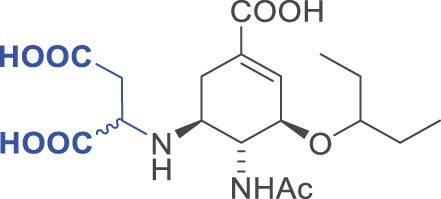 |
13 000 ± 1000 | 16 000 ± 5000 | 1.3 |
| 11 | 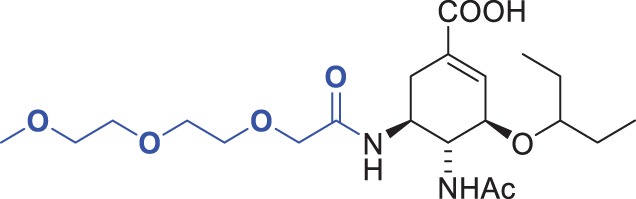 |
43 000 ± 11 000 | 210 000 ± 110 000 | 5 |
The chemical formulae of the inhibitors are also shown. The ‘fold Ki’ parameter represents the ratio Ki (DIANA, pH 7.4)/Ki (kinetic assay, pH 6.15). Substitutions of the moieties in oseltamivir carboxylate (4) are highlighted in blue. The Ki values for both assays were calculated from two independent experiments.
Development of DIANA for neuraminidase inhibitor screening
We captured recombinant NA with an immobilized polyclonal anti-NA antibody and detected it with the DNA probe based on 1 (Figure 1). Using this set-up, potential inhibitors can be screened by incubating the test compounds with the captured enzyme in the presence of the probe. The amount of bound probe is quantifiable by qPCR, and the inhibitory potency of the compounds can be calculated from the difference in qPCR cycle number between wells incubated with and without the test compound (ΔCq) [6].
We found that it is important to include Ca2+ in the assay buffer, as Ca2+ is essential for NA activity and stabilization of the active site [31,32]. Because small-molecule libraries are typically dissolved in DMSO, we also tested the influence of including up to 10% DMSO in the assay and found no significant effect.
Using serial dilutions of NA and the probe, we determined optimal working concentrations of 2 ng of NA and 200 pM probe per well. These concentrations led to a signal-to-background ratio of ∼7 qPCR cycles (over two orders of magnitude) and a Z′-score of ∼0.83 (average value calculated from several experiments with known inhibitors). According to Zhang et al. [33], a Z′-score between 0.5 and 1.0 is indicative of an excellent assay for high-throughput screening.
We next assessed whether we could use this novel assay to determine the Ki value from a single inhibitor concentration. We determined inhibition constants from single-well measurements, as described in Experimental section, with serial dilutions of tamiphosphor. The resulting Ki values were constant within the range of the assay (i.e. over tamiphosphor concentrations spanning 43 nM to 12 µM; see Figure 5A). The average calculated Ki was 52 ± 9 nM, in good agreement with the value of 26 ± 4 nM obtained from standard enzyme kinetics. We conclude that DIANA is suitable for accurate Ki determinations from single-well measurements.
Figure 5. Determination of inhibition constants from a single inhibitor concentration by DIANA (A) and comparison with kinetic measurement (B).

(A) Serially diluted tamiphosphor (TMF, x-axis) was tested using DIANA, and Ki values were calculated from each data point (y-axis). The dashed horizontal line shows the average Ki calculated as the mean of Ki values determined from concentrations of tamiphosphor in the 43 nM to 12 µM range. Error bars indicate the standard error of mean from two independent experiments. Both axes are shown as log-scales. (B) Plot of Ki values of 11 neuraminidase inhibitors determined either by kinetic measurement (fluorometric assay; x-axis) or by DIANA (y-axis). Error bars indicate the standard error of mean from two independent experiments. Both axes are shown as log-scales. The diagonal solid line represents a 1 : 1 ratio of values from both methods. The dashed line is the linear regression of the Ki values determined by both methods (Ki values determined by DIANA were on average 4-fold higher than those determined by the kinetic assay).
Comparison of DIANA with the standard enzymatic assay
To compare the ability of DIANA and a standard enzymatic assay to determine inhibition constants of novel compounds, we prepared a series of 11 modified oseltamivir and tamiphosphor derivatives with various inhibition potencies. Compounds 2–4, known inhibitors of NA with nanomolar Ki values, served as standards for validation of the assay for tight-binding inhibitors. Compounds 5, an ethyl ester prodrug of oseltamivir phosphate, and 10–11 represent inhibitors with several-order-of-magnitude lower inhibition potency and were chosen to demonstrate the ability of the assay to identify and characterize weakly binding compounds. Guanidylated compounds 6 and 7 also have been described previously, with contradictory data on their inhibitory activity compared with oseltamivir [10,34,35]. In addition to these known inhibitors, we prepared and tested three new compounds that feature stereogenic centers adjacent to the former C-5 amino functionality. Compounds 8 and 9 are diastereomers, allowing us to validate the assay on two very structurally similar inhibitors.
We first used an established fluorometric assay to determine the inhibition constants of all compounds (see Table 2). This analysis was performed at pH 6.15, which is optimal for NA2009wt activity and is necessary to achieve a sufficient signal-to-background ratio, using the fluorescent substrate 4-MUNANA [2,36]. The Ki values of 1–11 ranged from 20 nM to 40 µM. As expected, 1–4 exhibited the highest potency. Oseltamivir ethylester (5) was a roughly 100-fold inhibitor than oseltamivir carboxylate (4), which corroborates the necessity of a negatively charged moiety in the C-1 position. Interestingly, substitution of the basic amine moiety with the basic and bulkier guanidium group led to a several-fold decrease in potency (6 and 7). On the other hand, the effect of modifying the C-5 amino group with a propanoate moiety was dependent on the configuration of the stereogenic center: the S-configuration in 8 led to inhibitory potency comparable to that of oseltamivir carboxylate, whereas the R-configuration in 9 led to an ∼10-fold decrease in potency. These results also indicate that an acidic substituent is tolerated at the C-5 position. Finally, 10 and 11 showed the lowest inhibitory activity of all compounds, with binding constants in the micromolar range.
We next evaluated the inhibition constants of all 11 compounds using DIANA. Because there was no requirement for acidic pH, this assay was performed at physiological pH (pH 7.4). We tested serial dilutions of each inhibitor (seven concentrations spanning a three-order-of-magnitude range) in duplicate in two independent experiments and calculated Ki values as the average Ki determined from each well. However, only wells with Cq-values at least two cycles lower than the background Cq and at least one cycle higher than Cq in wells incubated without the inhibitor were considered for Ki determination (note that cycle number is indirectly proportional to the logarithm of the probe concentration, i.e. the higher the probe concentration, the lower the cycle number). These cutoff values were applied to avoid potential errors in Ki values caused by considering Cq values near the limits of the assay window. Indeed, we found that the Ki values for individual compounds were constant over the concentrations tested (Table 2).
Discussion
We previously described DIANA as an ultrasensitive tool with a large dynamic range for the determination of enzyme inhibition constants [6]. Here, we adapted the protocol for the evaluation of NA inhibitors. In our setup, NA was captured by an immobilized antibody, and tested compounds competed with an active site-directed detection probe, which was then quantified by qPCR (Figure 1A). The detection probe was prepared by linking a tamiphosphor derivative 1 to a reporter DNA oligonucleotide (Figure 1B). The crystal structure of complex of ω-azidoalkylester of tamiphosphor (compound 1) with NA2009wt revealed that modification of the phosphate moiety of tamiphosphor does not affect its interactions with active site residues. The esterification results in very minor interactions with residues at the edge of the active site of the enzyme, and the terminal azide group is fully exposed to solvent and thus available for further modifications. We thus concluded that modification of the azide group with a DNA oligonucleotide in the DIANA probe will not significantly affect binding of 1 to the NA2009wt active site.
We then characterized and validated the assay setup using oseltamivir carboxylate as a reference inhibitor. We synthesized a series of novel oseltamivir derivatives and showed that DIANA faithfully reproduced the structure–activity relationship of the series determined by the standard kinetic assay (Figure 5B). The absolute Ki values obtained by DIANA are, however, on average roughly 4-fold higher than those obtained by standard kinetics (ranging from equivalent to 12-fold higher). The most pronounced differences were observed for inhibitors containing substituents targeting 150-loop identified as very dynamic part of NA sensitive to protonation [37]. These inconsistencies may thus result from the different protonation of the enzyme under different conditions used for individual methods, such as use of different pH and buffers. The advantage of DIANA assay is that it uses the experimental conditions that are similar to the physiological ones while the kinetic assay has to be performed in artificial conditions needed for efficient cleavage of the substrate.
To analyze whether our method is suitable for the testing of inhibitors of known resistant mutants of NA, we determined Ki values of the compound 1 with the wild-type NA and resistant variants containing oseltamivir major resistance mutations H275Y and H275Y/I223V. Ki values for these mutants were 1700- and 11 100-fold higher than for the wt NA, respectively (data not shown). Owing to these unfavorable binding characteristics of compound 1, the DNA probe would not be suitable for sensitive DIANA testing with resistant NA mutants.
In conclusion, our results demonstrate that DIANA is useful for determining inhibition constants of wt NA inhibitors and consumes only a small amount of enzyme (the kinetic assay consumes an amount more than 10-fold higher of NA per measurement). Moreover, the multi-well plate format, the possibility to determine inhibition potency from a single-point measurement, and the possibility to use up to 10% DMSO make this assay a promising choice for screening small-molecule libraries for new influenza NA inhibitors.
Database Depositions
Atomic co-ordinates and structure factors have been deposited in the PDB database under accession codes 6G01 and 6G02.
Acknowledgements
The authors thank Jana Starková and Iva Flaisigová for technical assistance and Hillary Hoffman for proofreading of the manuscript. Diffraction data were collected on BL14.1 at the BESSY II electron storage ring operated by the Helmholtz-Zentrum Berlin.
Abbreviations
- 4-MUNANA
2′-(4-methylumbelliferyl)-α-d-N-acetylneuraminic acid
- AU
asymmetric unit
- CCP4
Collaborative Computational Project Number 4
- DCM
dichloromethane
- DIANA
DNA-linked inhibitor antibody assay
- DIPEA
N,N-diisopropylethylamine
- DMAP
4-dimethylaminopyridine
- DMSO
dimethyl sulfoxide
- FDA
Food and Drug Administration
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HPLC
high-performance liquid chromatography
- Kd
dissociation constant
- Ki
inhibition constant
- LC–MS
liquid chromatography-mass spectrometry
- NA
neuraminidase
- PDB
Protein Data Bank
- PEG
polyethylene glycol
- qPCR
quantitative polymerase chain reaction
- RMSD
root-mean-square deviation
- SPAAC
strain-promoted alkyne-azide cycloaddition
- TBTU
N,N,N′,N′-tetramethyl-O-(benzotriazol-1-yl)uronium tetrafluoroborate
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- TMF
tamiphosphor
- Tris
tris(hydroxymethyl)aminomethane
Author Contribution
M.K. conceived idea, designed and performed experiments, analyzed results, wrote the paper, and communicated. V.N. designed experiments and wrote the paper. K.R. performed experiments and analyzed data. J.P. analyzed and validated the data. C.B.A. designed and performed experiments. P.P. analyzed and validated the data. J.Z. designed and performed experiments. A.M. and P.Š. designed experiments. J.H. and I.C. performed experiments. P.Ř. analyzed data, visualized results, and wrote the paper. J.K. supervised the studies and revised the paper.
Funding
This work was supported by the project InterBioMed LO1302 from the Ministry of Education of the Czech Republic.
Competing Interests
The Authors declare that there are no competing interests associated with the manuscript.
Supplementary Material
References
- 1.Iuliano A.D., Roguski K.M., Chang H.H., Muscatello D.J., Palekar R., Tempia S. et al. (2018) Estimates of global seasonal influenza-associated respiratory mortality: a modelling study. Lancet 391, 1285–1300 10.1016/S0140-6736(17)33293-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Potier M., Mameli L., Bélisle M., Dallaire L. and Melançon S.B. (1979) Fluorometric assay of neuraminidase with a sodium (4-methylumbelliferyl-α-d-N-acetylneuraminate) substrate. Anal. Biochem. 94, 287–296 10.1016/0003-2697(79)90362-2 [DOI] [PubMed] [Google Scholar]
- 3.Li A.F., Wang W.H., Xu W.F. and Gong J.Z. (2009) A microplate-based screening assay for neuraminidase inhibitors. Drug Discov. Ther. 3, 260–265 PMID: [PubMed] [Google Scholar]
- 4.Marjuki H., Mishin V.P., Sleeman K., Okomo-Adhiambo M., Sheu T.G., Guo L. et al. (2013) Bioluminescence-based neuraminidase inhibition assay for monitoring influenza virus drug susceptibility in clinical specimens. Antimicrob. Agents Chemother. 57, 5209–5215 10.1128/AAC.01086-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okomo-Adhiambo M., Sheu T.G. and Gubareva L.V. (2013) Assays for monitoring susceptibility of influenza viruses to neuraminidase inhibitors. Influenza Other Respir. Viruses 7 (Suppl 1), 44–49 10.1111/irv.12051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Navrátil V., Schimer J., Tykvart J., Knedlík T., Vik V., Majer P. et al. (2017) DNA-linked inhibitor antibody assay (DIANA) for sensitive and selective enzyme detection and inhibitor screening. Nucleic Acids Res. 45, e10 10.1093/nar/gkw853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bařinka C., Rinnová M., Šácha P., Rojas C., Majer P., Slusher B.S. et al. (2002) Substrate specificity, inhibition and enzymological analysis of recombinant human glutamate carboxypeptidase II. J. Neurochem. 80, 477–487 10.1046/j.0022-3042.2001.00715.x [DOI] [PubMed] [Google Scholar]
- 8.Schmidt T.G. and Skerra A. (2007) The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2, 1528–1535 10.1038/nprot.2007.209 [DOI] [PubMed] [Google Scholar]
- 9.Schmidt T.G.M., Batz L., Bonet L., Carl U., Holzapfel G., Kiem K. et al. (2013) Development of the Twin-Strep-tag (R) and its application for purification of recombinant proteins from cell culture supernatants. Protein Expr. Purif. 92, 54–61 10.1016/j.pep.2013.08.021 [DOI] [PubMed] [Google Scholar]
- 10.Albiñana C.B., Machara A., Řezáčová P., Pachl P., Konvalinka J. and Kožíšek M. (2016) Kinetic, thermodynamic and structural analysis of tamiphosphor binding to neuraminidase of H1N1 (2009) pandemic influenza. Eur. J. Med. Chem. 121, 100–109 10.1016/j.ejmech.2016.05.016 [DOI] [PubMed] [Google Scholar]
- 11.Boutevin B., Hervaud Y., Jeanmaire T., Boulahna A. and Elasri M. (2001) Monodealkylation des esters phosphoniques synthese de monosels et de monoacides phosphoniques. Phosphorus Sulfur Silicon Relat. Elem. 174, 1–14 10.1080/10426500108040229 [DOI] [Google Scholar]
- 12.Baskin J.M., Prescher J.A., Laughlin S.T., Agard N.J., Chang P.V., Miller I.A. et al. (2007) Copper-free click chemistry for dynamic in vivo imaging. Proc. Natl Acad. Sci. U.S.A. 104, 16793–16797 10.1073/pnas.0707090104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams J.W. and Morrison J.F. (1979) The kinetics of reversible tight-binding inhibition. Methods Enzymol. 63, 437–467 10.1016/0076-6879(79)63019-7 [DOI] [PubMed] [Google Scholar]
- 14.Dixon M. (1953) The determination of enzyme inhibitor constants. Biochem. J. 55, 170–171 10.1042/bj0550170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mueller U., Förster R., Hellmig M., Huschmann F.U., Kastner A., Malecki P. et al. (2015) The macromolecular crystallography beamlines at BESSY II of the Helmholtz-Zentrum Berlin: current status and perspectives. Eur. Phys. J. Plus 130, 141 10.1140/epjp/i2015-15141-2 [DOI] [Google Scholar]
- 16.Krug M., Weiss M.S., Heinemann U. and Mueller U. (2012) XDSAPP: a graphical user interface for the convenient processing of diffraction data using XDS. J. Appl. Crystallogr. 45, 568–572 10.1107/S0021889812011715 [DOI] [Google Scholar]
- 17.Vagin A. and Teplyakov A. (2000) An approach to multi-copy search in molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 56 (Pt 12), 1622–1624 10.1107/S0907444900013780 [DOI] [PubMed] [Google Scholar]
- 18.Murshudov G.N., Vagin A.A. and Dodson E.J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53 (Pt 3), 240–255 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 19.Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50 (Pt 5), 760–763 10.1107/S0907444994003112 [DOI] [PubMed] [Google Scholar]
- 20.Emsley P. and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60 (Pt 12 Pt 1), 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 21.Lovell S.C., Davis I.W., Arendall W.B., de Bakker P.I., Word J.M., Prisant M.G. et al. (2003) Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins 50, 437–450 10.1002/prot.10286 [DOI] [PubMed] [Google Scholar]
- 22.Krissinel E. and Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 10.1107/S0907444904026460 [DOI] [PubMed] [Google Scholar]
- 23.Vavricka C.J., Li Q., Wu Y., Qi J., Wang M., Liu Y. et al. (2011) Structural and functional analysis of laninamivir and its octanoate prodrug reveals group specific mechanisms for influenza NA inhibition. PLoS Pathog. 7, e1002249 10.1371/journal.ppat.1002249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen C.L., Lin T.C., Wang S.Y., Shie J.J., Tsai K.C., Cheng Y.S.E. et al. (2014) Tamiphosphor monoesters as effective anti-influenza agents. Eur. J. Med. Chem. 81, 106–118 10.1016/j.ejmech.2014.04.082 [DOI] [PubMed] [Google Scholar]
- 25.Lew W., Chen X.W. and Kim C.U. (2000) Discovery and development of GS 4104 (oseltamivir): an orally active influenza neuraminidase inhibitor. Curr. Med. Chem. 7, 663–672 10.2174/0929867003374886 [DOI] [PubMed] [Google Scholar]
- 26.Shie J.J., Fang J.M., Wang S.Y., Tsai K.C., Cheng Y.S.E., Yang A.S. et al. (2007) Synthesis of Tamiflu and its phosphonate congeners possessing potent anti-influenza activity. J. Am. Chem. Soc. 129, 11892–11893 10.1021/ja073992i [DOI] [PubMed] [Google Scholar]
- 27.Carbain B., Martin S.R., Collins P.J., Hitchcock P.B. and Streicher H. (2009) Galactose-conjugates of the oseltamivir pharmacophore-new tools for the characterization of influenza virus neuraminidases. Org. Biomol. Chem. 7, 2570–2575 10.1039/b903394g [DOI] [PubMed] [Google Scholar]
- 28.Carbain B., Collins P.J., Callum L., Martin S.R., Hay A.J., McCauley J. et al. (2009) Efficient synthesis of highly active phospha-isosteres of the influenza neuraminidase inhibitor oseltamivir. ChemMedChem 4, 335–337 10.1002/cmdc.200800379 [DOI] [PubMed] [Google Scholar]
- 29.Stanley M., Martin S.R., Birge M., Carbain B. and Streicher H. (2011) Biotin-, fluorescein- and ‘clickable’ conjugates of phospha-oseltamivir as probes for the influenza virus which utilize selective binding to the neuraminidase. Org. Biomol. Chem. 9, 5625–5629 10.1039/c1ob05384a [DOI] [PubMed] [Google Scholar]
- 30.Streicher H., Martin S.R., Coombs P.J., McCauley J., Neill-Hall D. and Stanley M. (2014) A phospha-oseltamivir-biotin conjugate as a strong and selective adhesive for the influenza virus. Bioorg. Med. Chem. Lett. 24, 1805–1807 10.1016/j.bmcl.2014.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chong A.K., Pegg M.S. and von Itzstein M. (1991) Influenza virus sialidase: effect of calcium on steady-state kinetic parameters. Biochim. Biophys. Acta 1077, 65–71 10.1016/0167-4838(91)90526-6 [DOI] [PubMed] [Google Scholar]
- 32.Burmeister W.P., Cusack S. and Ruigrok R.W. (1994) Calcium is needed for the thermostability of influenza B virus neuraminidase. J. Gen. Virol. 75 (Pt 2), 381–388 10.1099/0022-1317-75-2-381 [DOI] [PubMed] [Google Scholar]
- 33.Zhang J.H., Chung T.D.Y. and Oldenburg K.R. (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen 4, 67–73 10.1177/108705719900400206 [DOI] [PubMed] [Google Scholar]
- 34.Hilfinger J. and Shen W. (2011) Neuraminidase inhibitors. Google Patents, International Publication Number WO 2011/123856 Al https://patentimages.storage.googleapis.com/a7/f6/bd/96edbe41cf1edc/WO2011123856A1.pdf
- 35.Mooney C.A., Johnson S.A., ‘t Hart P., van Ufford L.Q., de Haan C.A.M., Moret E.E. et al. (2014) Oseltamivir analogues bearing N-substituted guanidines as potent neuraminidase inhibitors. J. Med. Chem. 57, 3154–3160 10.1021/jm401977j [DOI] [PubMed] [Google Scholar]
- 36.Xu R., Zhu X., McBride R., Nycholat C.M., Yu W., Paulson J.C. et al. (2012) Functional balance of the hemagglutinin and neuraminidase activities accompanies the emergence of the 2009 H1N1 influenza pandemic. J. Virol. 86, 9221–9232 10.1128/JVI.00697-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu Y., Qin G., Gao F., Liu Y., Vavricka C.J., Qi J. et al. (2013) Induced opening of influenza virus neuraminidase N2 150-loop suggests an important role in inhibitor binding. Sci. Rep. 3, 1551 10.1038/srep01551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brünger A.T. (1992) Free R-value: a novel statistical quantity for assessing the accuracy of crystal-structures. Nature 355, 472–475 10.1038/355472a0 [DOI] [PubMed] [Google Scholar]
- 39.Chen V.B., Arendall W.B., Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J. et al. (2010) Molprobity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.