

Abstract
The connecting cilium of the rod photoreceptor is a tubular structure that bridges two adjacent cellular compartments, the inner segment, the major site of biosynthesis and energy metabolism, and the outer segment, a highly specialized ciliary structure responsible for phototransduction. The connecting cilium allows for active processes of protein sorting and transport to occur between them. Mutations affecting the cargo, their transporters, and the structural components of the primary cilium and basal body lead to aberrant trafficking and photoreceptor cell death. Understanding the overall design of the cilium, its architectural organization, and the function of varied protein complexes within the structural hierarchy of the cilium requires techniques for visualizing their native three-dimensional structures at high magnification. Here we describe methods for isolating retinas from mice, purifying fragments of rod cells that include much of the inner segment and the rod photoreceptor cilia, vitrifying the cell fragments, and determining their structures by cryo-electron tomography.
Keywords: Cryo-electron tomography, Intraflagellar transport, Primary cilia, Rod outer segment
1. Introduction
The biosynthetic machinery of the rod photoreceptor neuron is sequestered in the inner segment compartment of the cell. Following synthesis and posttranslational modification of key phototransduction proteins in the inner segment, vesicles consisting of these proteins and specific membrane lipids are constructed and transported to the rod outer segment, a modified primary cilium [1, 2]. The rod outer segment contains a highly organized system of membrane disks, each of which contains a high density of rhodopsin, ~25,000 molecules per μm2 [3]. The daily cycle of disk membrane turnover and renewal replaces ~10 % of the outer segment each day [2], which suggests that a minimum of 2,000 rhodopsin molecules per minute [4] are trafficked to the outer segment through the connecting cilium in a process called intraflagellar transport [5, 6]. The transport of different complexes through the connecting cilium, which corresponds roughly to the transition zone of other primary cilia, is a coordinated series of steps that, at minimum, include cargo sorting, cargo docking, active trafficking, and cargo release. Although many proteins such as rhodopsin remain sequestered in the outer segment compartment of the cell, other proteins such as transducin are reportedly able to be reversibly redistributed between the inner and outer segments, via the primary cilium [7]. These complex processes are contained within a structure that is approximately 300 nm in diameter and a little more than 1,000 nm in length [8], dimensions which challenge the limits of most conventional imaging techniques.
Cryo-electron tomography (cryo-ET) is an imaging technique capable of capturing biological structures in their native, hydrated form while preserving their three-dimensional arrangements [9]. Specimens are applied to a copper EM grid, excess liquid is blotted away, and the grid is plunged into liquid ethane (melting temperature at −183 °C) so that it is frozen at a rate exceeding 104 °C/s [10]. This allows specimens up to 10 μm thick (typical specimen thickness ranges from 200 to 500 nm) to be preserved in noncrystalline (vitreous) ice, which prevents disruption of cellular membranes and redistribution of cellular contents, artifacts known to result from crystalline ice formation. Electron tomography, the process of acquiring micrograph images of vitrified biological specimen at incremental angular steps (called a tilt series), is then carried out on an electron microscope with a computer-controlled [11, 12] stage while maintaining the sample at −196 °C (specimen must always be kept below −140 °C, which is the devitrification temperature of water where ice crystals form). Using computational methods, two-dimensional tilt series micrograph projections are aligned and merged to build a three-dimensional density map (tomogram) capable of revealing the original specimen at high resolution (5–50 nm range) [13–15]. The final resolution of the tomogram is directly dependent upon the cumulative electron dose (typically 100 Å/μm2 or less to prevent radiation damage to biological samples) and the incremental angle size between each successive projection micrograph. Physical limitations, caused by increased electron scattering at higher tilt angles due to increasing electron path length through the sample, degrade high angle image quality and practically limit tilt series to ±70° [16]. The limited angular range in electron tomography creates a data void in Fourier space during computational tomogram reconstruction known as the “missing wedge” artifact [13, 17, 18]. The missing wedge of data causes a “stretching” of structural data along the z-axis of the reconstructed tomogram and confounds interpretation of biological structures.
Because of its dimensions and the intricacies of its membrane and microtubule-based structures, the mammalian rod cilium is an excellent subject for cryo-ET [8, 19]. It allows a detailed three-dimensional view of the arrangement of its molecular components under minimally perturbing conditions. We describe here methods for isolating retinas from mice, purifying fragments of the rod containing the outer segments, connecting cilia and adjacent region of the inner segments by isoosmotic density gradient ultracentrifugation, and vitrifying the cell fragments for cryo-ET by plunge freezing in liquid ethane. We also describe image collection and processing procedures for cryo-ET.
2. Materials
2.1. Mouse Retinas
Cell fragments containing rod outer segments (ROS) with attached connecting cilia and portions of inner segment (here referred to as “ROS”) are isolated from maturely developed retinas of 21-day-old mice or older (Fig. 1). Widely available inbred animal strains can be obtained from commercial vendors if they are not housed directly in an institutional vivarium. We recommend preparing ROS from 10 to 12 retinas for easier visualization of purified ROS bands using the current protocol. Animals younger than 21 days or animals containing genetic mutations suspected to alter retinal development or to be subject to retinal degeneration may decrease the yield of ROS in the preparation. It may be necessary to scale the preparation to optimize yield for EM grid preparation.
Fig. 1.
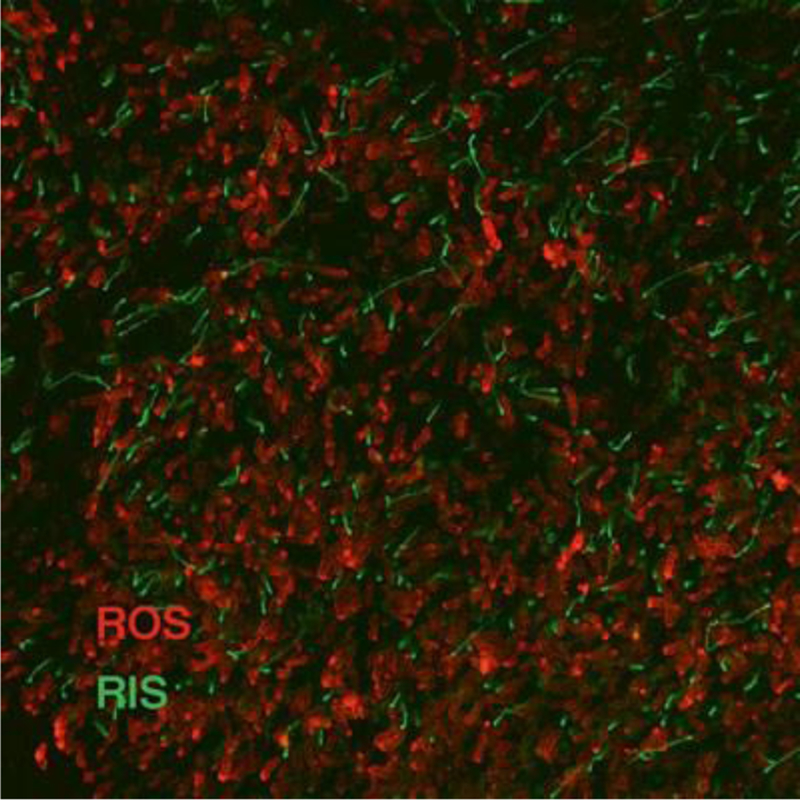
Immunofluorescence of a whole-mount retina. Whole-mount retina was stained with antibodies against rhodopsin (red) and rootletin (green). Rhodopsin staining corresponds to the photoreceptor outer segment, while rootletin staining corresponds to the inner segment
2.2. Buffers and Assay Reagents
Ringer’s Buffer (5×): 50 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), pH 7.4, 650 mM NaCl, 18 mM KCl, 60 mM MgCl2, 6 mM CaCl2, 0.1 mM ethylenediamine-tetraacetic acid (EDTA). Filter through a 0.2 μm pore nitro-cellulose filter and store at 4 °C. Preparing a 100 ml volume is sufficient for at least ten ROS preparations.
Ringer’s Buffer (1×): 10 mM HEPES, pH 7.4, 130 mM NaCl, 3.6mMKCl,12mMMgCl2,1.2mMCaCl2,0.02mMEDTA.Prepare 50 ml by diluting 1:5 from the stock of 5× Ringer’s Buffer.
OptiPrep (Axis-Shield), 60 % (w/v) iodixanol solution, 5,5′-[(2-hydroxy-1–3propanediyl)-bis(acetylamino)]bis[N,N′-bis(2,3-dihydroxypropyl-2,4,6-triiodo-1,3-benzenecarboxamide)] as supplied.
Acetone, EM grade.
BSA-coated gold fiducial markers, 15 nm diameter (Electron Microscopy Sciences). Colloidal gold markers are used to aid in alignment of the micrographs during the tomographic reconstruction (see Note 1).
2.3. Density Gradient Centrifugation Media
40 % OptiPrep (15 ml): 40 % OptiPrep, 1× Ringer’s Buffer. Add 10 ml of 60 % OptiPrep to a 15 ml conical tube or other sealable container. Add 3 ml of 5× Ringer’s Buffer and bring the final volume to 15 ml with water. Mix thoroughly by pipetting or inversion and store at 4 °C on ice.
30 % OptiPrep (4 ml): Add 3 ml of 40 % OptiPrep to 1 ml of 1× Ringer’s Buffer.
25 % OptiPrep (4 ml): Add 2.5 ml of 40 % OptiPrep to 1.5 ml of 1× Ringer’s Buffer.
20 % OptiPrep (4 ml): Add 2 ml of 40 % OptiPrep to 2 ml of 1× Ringer’s Buffer.
15 % OptiPrep (4 ml): Add 1.5 ml of 40 % OptiPrep to 2.5 ml of 1× Ringer’s Buffer.
10 % OptiPrep (4 ml): Add 1 ml of 40 % OptiPrep to 3 ml of 1× Ringer’s Buffer.
8 % OptiPrep (4 ml): Add 0.8 ml of 40 % OptiPrep to 3.2 ml of 1× Ringer’s Buffer.
2.4. Dissection Tools and Apparatus
Stereoscopic dissection microscope with an appropriate light source (see Note 2).
Variable-speed benchtop vortexer (e.g., Vortex-Genie 2).
Wide-bore micropipette tips: for 20, 200, and 1,000 μl volumes (see Note 3).
Dumont #5-style forceps (Fine Science Tools) or equivalent: two pairs required for tissue manipulation.
Standard scissors (Fine Science Tools) or equivalent.
Spring-loaded, Vannas-style microscissors (Fine Science Tools) or equivalent, required for precision cutting of the mouse eye (see Note 4).
Beveled, hypodermic needles, 18 G and 27 G.
Syringes, 1.0 ml, for gradient formation and ROS collection.
Microcentrifuge tubes, 1.5 ml siliconized, for tissue collection and purification.
Polypropylene tissue culture dish, 60 mm or 100 mm diameter, for use as a reservoir during retina dissection.
Ice pack for maintaining temperature during the dissection procedure.
Parafilm.
2.5. Infrared Visualization
Infrared visualization is not required for preparing ROS fragments for cryo-ET, as sample can be easily prepared under room light or dim red light. However, some physiological differences should be expected from photoreceptor cells conditioned to different intensities of light. We provide the additional materials for this optional condition should they be necessary for a more exacting experimental design:
Dark room that has been treated to limit light bleed through (see Note 5).
Infrared LED panel for light-free illumination of the work surface (see Note 6).
Night vision goggles. Binocular, head-mounted goggles allow for unobstructed, hands-free experimentation.
Aluminum foil.
Black permanent marker (see Note 7).
2.6. Ultracentrifuge, Rotor, and Tubes
Beckman TL-100 tabletop ultracentrifuge or equivalent.
TLA-100.3 fixed-angle ultracentrifuge rotor or equivalent.
TLS-55 swinging bucket ultracentrifuge rotor or equivalent.
Microcentrifuge-style 1.5 ml polyallomer tubes (Beckman) with adapters for TLA-100.3.
Thinwall 2.2 ml polyallomer tubes (Beckman) for TLS-55.
Analytical scale for balancing ultracentrifuge samples.
2.7. Cryo-supplies and EM Grids
EM grids: Quantifoil® Holey Carbon Films, 200 mesh, R2/2 (see Note 8).
Empty grid storage boxes (e.g., Electron Microscopy Sciences).
Vacuum dessicator for grid storage.
Insulated forceps for grid manipulation (e.g., Electron Microscopy Sciences).
Glass Petri dishes with lids, 100 mm diameter.
Whatman #1 round filter paper, 90 mm outer diameter.
Whatman #1 filter papers for Vitrobot 60 mm or precut blotting paper (Ted Pella).
Liquid nitrogen.
Ethane gas, flow regulator, and fitted Tygon® tubing.
Dewar transfer flask with lid (Cole-Parmer).
Large cryosample storage freezer (e.g., Taylor-Wharton).
Cryo-grid storage box (blue 4-grid “button” holder) and Teflon® handling wand (Ted Pella).
Glass beakers, 100 ml volume.
Glass microscope slides.
Hair dryer.
Large, long-handle tweezers for handling items submerged in liquid nitrogen.
2.8. Cryo-apparatus
Glow discharge grid cleaning system (Ted Pella).
Vitrobot Mark III or equivalent (FEI).
Vitrobot tweezers assembly (Ted Pella).
Vitrobot cryogen container for plunge freezing.
Cryo-EM microscope setup for tomography with goniometer, and computer control.
Gatan 655 dry pumping station.
Gatan 626 70° cryo-transfer holder or equivalent.
Gatan workstation and cover for 70° cryo-transfer holder.
Fitted plug to seal the support cylinder extension of the workstation.
Clip ring tool for 70° cryo-transfer holder.
Fine tweezers.
Small plastic funnel.
2.9. Software
Gatan DigitalMicrograph.
SerialEM.
IMOD [14] (http://bio3d.colorado.edu/imod/) or equivalent for micrograph alignment and reconstruction.
Amira 3D analysis (FEI).
3. Methods
3.1. Pre-cleaning the EM Grids
All efforts should be taken to minimize particulate contamination of the grids. Process the grids in a dust-free area, and prevent the grids from being exposed to the environment for extended times throughout the process. To limit lipid transfer to the grids, wear clean gloves and only manipulate grids with clean forceps:
Place a 90 mm filter paper inside each of two 100 mm glass Petri dishes, and fill a single dish with 25 ml of acetone. Leave the second dish empty and cover the dish with its lid.
Using forceps, transfer 10–20 new Quantifoil® EM grids to the acetone wash. Lay the grids, carbon side up (see Note 9), on the filter paper. Ensure the grids are completely covered in acetone. Cover the Petri dish and incubate undisturbed for 15 min.
Gently lift a single grid using forceps and remove it from the acetone wash. Without releasing the grid, sequentially immerse the grid in two clean glass beakers, each filled with deionized water to dilute excess acetone.
Place the clean grid, carbon side up, onto the clean 90 mm filter paper inside the second glass Petri dish. Repeat this wash process for each EM grid used. Partially cover the dish with its lid and allow the grids to dry completely for 15–30 min (see Note 10).
Place clean, dry EM grids in a dedicated grid box and store in a dessicator under vacuum.
3.2. Retina Dissection
For dissection of high-quality retinas, carefully immobilize the head of the euthanized animal with one hand against the work surface. Using the thumb and forefinger, stretch the eyelids open while also applying gentle downward pressure against the skull to cause the eye to protrude for easier manipulation (see Note 11).
With the free hand, use dissecting scissors to sever the extraocular muscles and optic nerve (see Note 12).
Collect the eye in a plastic tissue culture dish filled with chilled Ringer’s Buffer. Place the dish on an ice pack under the dissection microscope.
Repeat steps 1–3 to excise the second eye and store it in chilled Ringer’s Buffer.
Using the dissecting microscope, immobilize the eyes with forceps, and use a 27 G needle to puncture the cornea (see Note 13).
Beginning with the corneal puncture site, use spring-loaded scissors to cut a straight line along the cornea to its edge at the ora serrata. Continue cutting around the cornea by following the ora serrata hemisphere. Completely remove the cornea and the lens from the eyecup (see Note 14).
Using two sets of forceps, carefully tease the neural retina away from the pigmented epithelium. Complete this process around the entire circumference of the eyecup until the retina remains attached to the eyecup by only the optic nerve. Cut the optic nerve to completely release the retina from the eyecup.
Transfer retinas to a 1.5 ml siliconized tube filled with 300 μl 8 % OptiPrep in Ringer’s Buffer and store on ice.
Repeat the dissection until the necessary number of retinas, generally 10–12, has been collected.
3.3. Crude Rod Outer Segment Preparation
Vortex the retinas for 1 min on the lowest vortexing setting to dissociate rod outer segments from the retina (see Note 15).
Centrifuge the mixture at 200 × g for 1 min to separate the intact retinas from the detached rod outer segments.
Using a wide-bore 1,000 μl pipette tip, transfer 250 μl of supernatant to a fresh 1.5 ml siliconized tube.
Add 250 μl of 8 % OptiPrep buffer to the retinas.
Repeat the isolation procedure (steps 1–4) four times to collect ~1,250 μl of rod outer segments. All supernatant aliquots should be collected in a single 1.5 ml siliconized tube and stored on ice.
Prepare the 10–30 % OptiPrep gradient.
3.4. 10–30 % OptiPrep Discontinuous Gradient Preparation
The following procedure is for preparing two balanced 1,500 μl discontinuous OptiPrep gradients. While the gradient solutions should be prepared before the procedure begins, the gradient should be made immediately before the centrifugation step to prevent unwanted mixing of the discrete gradient layers:
Mix the pre-prepared OptiPrep gradient solutions to ensure homogeneity.
Add 300 μl of 30 % OptiPrep to each of two 2.2 ml (11 × 35 mm) polyallomer tubes.
Overlay the preceding layer with 300 μl of 25 % OptiPrep using a syringe fitted with an 18 G needle.
Overlay each new layer with 300 μl of the next linear dilution of OptiPrep to create five layers (300 μl per layer) from 10 to 30 % of total OptiPrep concentration.
The layer interfaces should be clearly visible with no mixing of the layers.
Mark the outside of the 2.2 ml polyallomer tubes with black permanent marker to indicate the layer interface locations and to aid future assessment of ROS band quality.
Weigh the tubes on an analytical scale to ensure the gradients are balanced prior to addition of rod outer segments.
3.5. ROS Ultracentrifugation
Gently mix the crudely isolated ROS by inversion.
Use a 1.0 ml syringe capped with an 18 G needle to slowly collect 625 μl of crude ROS preparation. To reduce pressure-induced shearing of the ROS during collection, we recommend an approximate flow rate of 600 μl/min (see Note 16).
Slowly overlay the 625 μl of ROS on top of the 10 % OptiPrep gradient layer with an approximate flow rate of 600 μl/min.
Repeat steps 1–3 to generate the second gradient.
Weigh the tubes to ensure proper weight balance prior to ultracentrifugation. Any balance offset should be corrected with cold 8 % OptiPrep solution to maintain identical centers of gravity between the two tubes.
To obtain a higher separation resolution of the ROS, it is recommended to create a continuous gradient to reduce the density barrier effect that occurs at layer interfaces. Cover the 2.2 ml polyallomer tube openings with Parafilm. Apply finger pressure to firmly seal the openings, and quickly invert the tubes 45° to disturb the gradient layer interfaces.
Centrifuge the ROS gradients for 60 min at 24,700 × g at 4 °C using a TLS-55 swinging bucket rotor. A slow acceleration should be used, and no brake should be applied to the rotor during deceleration (i.e., for the Optima ultracentrifuge: acceleration = 9, deceleration = 9).
Unbleached, intact ROS is visible as a single, bright red band that equilibrates between the 10 and 15 % OptiPrep concentration layers (see Note 17). Black, pigmented epithelial cells and granules will pellet at the bottom of the gradient. Using a 1 ml syringe fitted with an 18 G needle, puncture the sidewall of the 2.2 ml polyallomer just below the meniscus of the ROS band and angle the bevel upward. Collect the ROS band from below the meniscus while being careful to not disrupt the band (see Note 18).
Deposit the ROS into 1.5 ml polyallomer tubes, and dilute the concentration of OptiPrep by adding 1× Ringer’s Buffer up to 1.5 ml. Mix the tubes by inversion, and pellet the ROS by centrifugation for 30 min at 26,500 × g at 4 °C using a TLA 100.3 rotor. Remove all supernatant, as excess OptiPrep can adversely affect the imaging quality of vitrified specimens, and place ROS pellets on ice (see Note 19).
3.6. Vitrification of Purified ROS
The following procedures involve diluting the purified ROS to an appropriate concentration, adding an appropriate concentration of electron-dense gold markers (both variables are determined empirically) (see Note 20), and preparing vitrified samples that meet a number of criteria for optimal tomographic imaging. As a general caution, vitrified EM grids are exceptionally sensitive to ice crystallization resulting from condensation of atmospheric water throughout the stages of sample preparation, storage, and imaging. As such, it is recommended to invest time to optimize the freezing conditions by screening individual vitrified grids on the microscope prior to batch freezing of grids from a single ROS prep. A key parameter in successful imaging is ice thickness. Ice becomes opaque to the electron beam as the ice thickness approaches 1,000 nm. However, if the ice layer is not at least as thick as the specimen (300 nm in the case of cilia), the sample will tend to flatten, distorting some features of the structure [20]. The thickness is best assessed using the microscope:
Resuspend a single ROS pellet in 20 μl of 1× Ringer’s Buffer with gentle finger tapping to aid in disrupting the solid pellet (see Note 21).
Add 8 μl of 15 nm BSA-gold fiducial marker to the resuspended ROS (see Note 22).
Glow discharging the pre-cleaned, carbon-coated Quantifoil® grids renders them hydrophilic and aids in uniform distribution of the ROS across the grid surface. Prepare 4–8 precleaned grids by placing them on an uncoated glass microscope slide, carbon side up (see Note 23). Glow discharge the grids under vacuum for 10 s.
Condensation of the secondary cryogen using the Vitrobot cryogen holder. Cool the cryogen holder and its inner brass cup using liquid nitrogen. As the temperature of the holder equilibrates, the rate of nitrogen evaporation will decrease. Fill the holder’s outer annulus with liquid nitrogen, leaving the inner ring and brass cup free of nitrogen. Condense the ethane by slowly filling the inner brass cup with a 1,000 μl pipette tip attached to the tubing. After a small quantity of ethane condenses, it will be possible to hear the ethane gently bubbling. Keep the pipette tip submerged under the surface of the liquid ethane to prevent the tip from clogging.
Carefully select a single grid using a dedicated pair of Vitrobot forceps, and engage the sliding tweezers lock to secure the grid. Place the forceps on the Vitrobot shaft, ensuring the carbon-coated side of the grid is facing the sample-loading port of the Vitrobot.
Move the grid-tweezer assembly into the enclosed blotting chamber. Allow the relative humidity of the chamber to reach 100 % to maintain sample hydration. Place the cryogen reservoir in the fitted Vitrobot holder, and ensure the surface of the ethane has not frozen (see Note 24).
Gently tap the ROS sample to mix and apply 2.5 μl of sample to the grid. The ROS sample should spread evenly across the surface of a properly glow-discharged grid. After adding the sample, wait 10 s and immediately blot the grid 1× for 2 s and plunge the grid into liquid ethane (see Note 25).
Allow the white, evaporated gas to clear from the work area. Refill the outer annulus with nitrogen, ensuring the grid holder is covered in liquid nitrogen.
Remove the tweezers from the Vitrobot shaft, being careful to not lift the grid from the ethane bath or to bend the grid against the wall of the brass cup (see Note 26).
Quickly transfer the grid from the liquid ethane into the nitrogen contained in the outer annulus. Transfer the grid into the blue 4-grid “button” holder and release the grid from the tweezers (see Note 27).
Warm tweezers using a hair dryer and remove condensation with an absorbent laboratory wipe.
Repeat steps 5–11 until the grid holder is filled or the desired number of grids has been prepared.
Secure the 4-grid holder with the Teflon® rod, and transfer the samples to a transfer dewar.
Once the optimal freezing conditions have been verified on the cryo-electron microscope, batch-frozen grids can be stored long term in a large cryosample storage freezer under liquid nitrogen.
3.7. Cryo-specimen Holder Preparation and Sample Grid Loading
Before a sample can be loaded into the cryo-electron microscope for imaging, it is necessary to precool the microscope by filling the refrigeration tank with nitrogen. The microscope should be initialized to bring it up to the proper acceleration voltage (typically 200 or 300 kV) and beam operating current. Since this process is specific to each microscope model, follow the manufacturer’s specifications to ensure the microscope is functioning properly before proceeding. The procedures below apply to a JEM2100 (JEOL) microscope with a Gatan cryo-stage:
Seal the workstation ports by inserting the fitted plug into the support cylinder that extends from the workstation.
Precool the workstation by filling its dewar with liquid nitrogen, and cover the station with the clear cover. Several iterations of this are necessary until the workstation temperature equilibrates and nitrogen bubbles stably rather than evaporates. Fill the workstation with sufficient nitrogen to fill the small reservoir.
Using precooled large tweezers, transfer the combined Teflon® rod and blue cryo-grid holder from a nitrogen dewar to the workstation. Insert the Teflon® rod through the loading port of the clear workstation cover and submerge in liquid nitrogen. Place the blue cryo-grid holder in the notched reservoir of the workstation, and carefully detach the Teflon® rod without disturbing the grids held in the blue holder. Submerge the Teflon® rod back into liquid nitrogen and set aside. Ensure the blue grid holder is covered with nitrogen.
Carefully remove the plug from the workstation, and insert the cryo-transfer holder into the workstation through the support cylinder (see Note 28).
Fill the specimen holder dewar with liquid nitrogen (see Note 29). If using a digital thermometer, fit the plug into the port on the side of the transfer holder dewar. Refill the dewar as needed until the temperature equilibrates (see Note 30).
Precool the special clip ring tool (specific to 70° holder) in the workstation reservoir (see Note 31).
Open the specimen holder shutter and place the tool over the clip ring (located on the tip of the transfer holder) and turn the knob to engage the clip ring and lock it to the tool. Lift the tool to remove the clip ring from the holder. Keep the tool submerged in nitrogen within the workstation reservoir.
Using precooled fine tweezers, transfer a prepared grid from the blue grid holder to the transfer holder. Ensure the grid is centered in the beveled slot to prevent bending the grid with the clip ring.
Place the clip ring tool over the grid, and press the clip ring firmly to snap it into the transfer holder. Turn the tool knob to release the clip ring and lock the grid in place. Close the specimen holder shutter to shield the grid.
Prepare the microscope for specimen transfer by initiating any vacuum sequences in the airlock. Remove the room temperature specimen holder from the goniometer.
Remove the cryo-specimen holder from the workstation, and carefully insert the tip into the goniometer airlock (see Note 32). Wait for the vacuum sequences to finish before inserting the holder fully into the microscope vacuum column (see Note 33).
Rotate the holder to the upright position as it is fully inserted into the goniometer, and fill the dewar to the top with liquid nitrogen (see Note 34).
3.8. Digital Image Acquisition of Vitrified Rod Outer Segments
After inserting the cryo-specimen holder into the microscope, it is necessary to carry out standard procedures to turn on the beam filament and find the electron beam. Gun alignment should be conducted by inserting and centering the condenser lens aperture, correcting for gun tilt, correcting for condenser lens astigmatism, and aligning the condenser lens system. Following these steps, the objective lens aperture is then centered and corrected for astigmatism. The microscope should be set up in transmission electron microscopy (TEM) mode with Spot size = 1 and Alpha = 3 for a low magnification power of 4,000× and a high magnification range between 12,000× and 30,000× (see Notes 35 and 36). Carry out these procedures as specified by the manufacturer’s instructions. SerialEM, the software used to collect micrographs and track incremental tilt shifts during tomography, is a user-friendly package, but it is suggested that users familiarize themselves with the program (http://bio3d.colorado.edu/SerialEM/) [21]:
Initialize DigitalMicrograph for acquiring and analyzing single micrographs in TEM mode.
After the sample is inserted into the microscope, it is useful to take 4,000× low-magnification images of the grid to map the ice thickness gradient across the grid and to identify unique features for correlating the location of images taken at higher magnification. Begin by taking an image of the center of the grid followed by images along the x- and y-axes of the grid, moving outward toward the edge of the grid (see Note 37).
Using the low-magnification map images, identify grid squares of interest where the carbon mesh remains intact and has acceptable sample contrast (see Note 38).
Set up the low-dose search mode to screen the grid while preventing excess electron damage to the cells on the grid (see Note 39).
In low-magnification, low-dose search mode, screen regions of the grid for closer inspection. Often, intact outer segment regions, with the cilium still attached, can be seen under low magnification as individual, oval, or cylindrical dark spots within a grid square (Fig. 2). These cells are more visually distinctive than a generally blurry and diffuse region, which is often correlated with disrupted cellular membranes and broken cilia.
When a sample has been identified for tilt series acquisition, initialize SerialEM. Remove the sample from the field of view by moving to an adjacent grid square to prevent specimen damage during software setup (see Note 40).
Using SerialEM, take a trial micrograph image to examine the contrast and brightness of the empty grid square. Set the desired magnification and brightness under which the sample micrographs will be acquired.
Collect a gain reference measurement consistent with the imaging parameters (magnification and spot size) of the current microscope settings (see Note 41).
Begin “eucentric – rough” from the task menu.
Re-center the region of interest (ROI) to be imaged. Run “eucentric – fine” from the task menu.
Test the maximum tilt range for tomography under “Low Dose” mode using the “walkup and anchor” feature under the task menu (see Note 42).
Set the electron beam intensity to give a cumulative dose of 50–100 electrons per Å2 using the “Set Intensity” command in the “Tasks” menu (see Note 43).
Take a final trial image and center the ROI on the desired cellular features.
Verify that the exposure settings for the focus, trial, and record camera modes provide images without drift.
Under the “Tilt Series” menu, select “Startup/Setup Tilt Series” to bring up the “Tilt Series Control Setup” window for setting the tomography parameters. Specify the tilt angle range based upon the “walkup” test. Set a basic increment of 2° per micrograph (see Note 44). Set the defocus target within a range from 7 to 12 μm (see Note 45).
Begin automated tilt series by selecting “Go” on the “Tilt Series Control Setup” graphical user interface (see Note 46). Figure 3a shows single micrograph corresponding to 0o tilt angle of a ROS taken during a tilt series.
Once the tilt series is complete, end the program from the “Tilt Series” menu and close the file.
Fig. 2.

Vitrified ROS specimens. (a) Vitrified ROS specimens on holey carbon EM grid at 50× magnification. (b) Magnification of single EM grid square (white box in a) at 250× magnification. Dark, elongated shapes are individual ROS
Fig. 3.
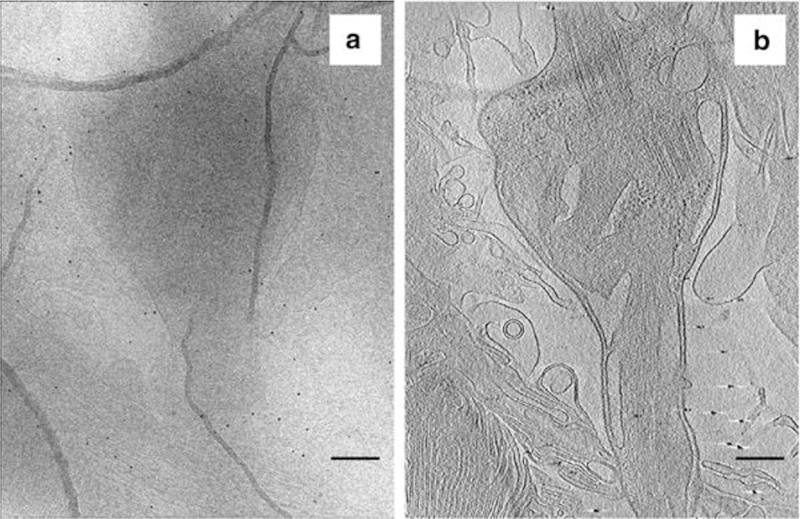
Single micrograph of a ROS taken during a tilt series. (a) Image corresponds to the 0° tilt angle. (b) Projection (6 nm thick) from a tomogram (same cell as in a) of the inner segment, containing the base of the connecting cilium and attached structures. This projection reveals the contrast gained from tomographic reconstruction of the tilt series micrographs. Scale bars = 300 nm
3.9. Alignment, Reconstruction, and Segmentation of Rod Photoreceptor Tomograms
The process of generating three-dimensional tomograms from tilted specimens requires two closely related steps. The first step, called alignment, is required to compensate for small specimen movements that occurred within the field of view due to goniometer inaccuracies and limited accuracy of the tracking procedure. The alignment process is used to assign all projections to a common coordinate system, assign an angle of the tilt axis to all projections, and account for small variations or specimen rotations that may have occurred during imaging. The second step, called reconstruction, results in an x–y–z coordinate system to which volume elements (voxels) are mapped [22]. Tomograms can be generated with the IMOD suite (http://bio3d.colorado.edu/imod/), and it is advisable for the user to view the IMOD Tomography Guide (http://bio3d.colorado.edu/imod/doc/tomoguide.html#TOP) and the ETomo Tutorial for IMOD (http://bio3d.colorado.edu/imod/doc/etomoTutorial.html#TOP) to learn more about the functions and operation of these tools [14, 15].
Following tomogram reconstruction, manual segmentation can be carried out using commercial software packages, such as Amira, to aid in visualization of biological structures. The surface contours of structural features can be seen in the grayscale tomograms and are traced by hand through the tomogram in slice-by-slice fashion to create iso-dense surfaces within the tomogram. These surfaces can be assigned different colors to highlight the structures, which allow their surfaces to be easily visualized in context within the tomogram. Manual, hand-drawn segmentation is a subjective, time-consuming process that is prone to interpretation errors by the investigator. For this reason, it is essential to compare the artistic renderings alongside the original grayscale tomogram images to which they correspond and to provide an opportunity for objective comparison to the scientific community in the published literature:
Initialize ETomo to begin the tilt series alignment. Select “Build Tomogram” to begin a new dataset.
Select the dataset to be used for single-axis tomograms. Select “Scan Header” to retrieve pixel size and image rotation information from the dataset (see Note 47). Manually enter the fiducial diameter (15 nm unless a different size is used). Press “Create Coms Scripts” to begin processing the images.
Press the “Course Alignment” button to create a coarsely pre-aligned stack from the tilt micrographs.
To begin creating a fiducial marker-based alignment, select “Make seed and track” where fiducial markers can be chosen manually by the user or automatically through the software. Between 15 and 25, fiducial markers should be selected to align the micrographs (see Note 48). Follow the interface prompts to generate a seed model and automatically select fiducial markers.
After the software has finished its routine, the preselected markers are indicated by green and magenta indicator points. At this point it is essential to manually verify that the majority of the preselected fiducials are correctly tracked throughout the entire tilt series. Any fiducials that were incompletely tracked at high tilt angles should be manually selected to fill in the gaps the software was unable to complete.
Once the fiducial reference map has been created by the software, press the “Compute Alignment” button under the “Fine Alignment” section of the graphical user interface to create a high-quality alignment of the data.
Before the final reconstruction process can occur, it is necessary to limit the dimensions of the aligned stack to focus on the specimen volume and exclude extracellular, non-specimen volume (see Note 49). Follow the on-screen prompts to create the boundary models which will limit the reconstruction to the true sample thickness.
To complete the tomogram reconstruction, press the “Create Final Alignment” button followed by “Generate Tomogram.” Figure 3b shows projection from a tomogram of the inner segment.
4. Notes
Electron-dense gold particles provide high-contrast markers for aligning the micrographs acquired at various tilt angles during the reconstruction of tomograms. Gold particles coated with bovine serum albumin (BSA) are more uniformly distributed than non-coated particles. We find that a particle diameter of 15 nm is suitable at magnifications of 10,000× to 20,000×, while a diameter of 10 nm is suitable from 30,000× to 40,000× magnification. This interdependence on gold particle size and magnification power results from the computational limitations of tomogram reconstruction where larger fiducials cover a greater number of pixels, which creates difficulty in accurately determining the center of the fiducial for tracking purposes, potentially introducing alignment errors. Tomograms constructed from higher-magnification micrographs will require adjustment of the BSA-fiducial concentration, because increasing the magnification reduces the field of view and restricts the number of fiducial gold particles that can be used in the tomogram reconstruction process.
For this type of procedure, a fiber-optic episcopic illumination source provides a maneuverable overhead light source with the greatest control of illumination direction throughout the dissection.
Wide-bore pipette tips can be purchased or created by cutting the tips using scissors or a razor blade. Any solutions containing rod outer segments should be transferred using wide-bore tips to reduce cellular shearing.
The angled cutting edge of the spring-loaded scissors minimizes the need to adjust hand position during dissection and can improve user efficiency and comfort during the procedure.
The dark room can be treated for light leak by using black fabric hangings to cover the entrance. By sitting in the dark room with the lights off for 20–30 min to allow for eyes to adjust to the dark, it becomes possible to detect sources of light from cracks or seams around the door frame. These openings can be sealed using black photographic masking tape. All electrical equipment in the room should be treated to cover indicator lights or LED displays that emit light.
If absolute darkness is not necessary for rod outer segment preparation, low light conditions can be used instead for dark adaptation of mouse retinas. Dim light conditions can be created with low-intensity red lighting via overhead bulbs in the dark room. Additionally, standard light bulbs can be covered with red gel filters to reduce light activation of rhodopsin protein. Night vision goggles are not essential under dim light conditions but can still enhance the user’s ability to visualize the work surface and improve sample preparation.
Black permanent marker is visible under infrared illumination, while other colors are invisible or difficult to see.
EM grids with 200 mesh (compared with 400 mesh) provide grids that are large enough such that the grid partition bars are less likely to interfere with the tilt series by obstructing the field of view. Our choice to use the R2/2 spaced grids is based upon the relative balance between the ratio of carbon to hole area and the overall size of the grid hole. We found that membranous ROS more often associated with the carbon film than covered the grid holes. In general, an intact cell was likely to have regions that overlapped several contiguous holes, which provides ample opportunity to image the high-contrast specimen over the holes.
Copper EM grids are carbon coated on a single side. The uncoated side has a bright orange, metallic reflective appearance, while the carbon-coated side has a dull, brown appearance.
By placing the Petri dish in a chemical fume hood, the air flow can increase grid drying. Ensure the grids are shielded from the direct air current by the dish lid to prevent dust deposits on the clean grids.
The most rapid isolation of retinas can be obtained with an altered procedure. While the eye is held with forceps, a singleedge razor blade can be touched against the cornea, perpendicular to the circumference of the eye. By holding the blade angle constant and firmly pushing the blade down toward the work surface, tangent to the cornea (i.e., not using the blade to directly slice the eye), the cornea will rupture and expel the lens onto the side of the blade. Using the forceps which hold the eye, gently squeeze the tissue and firmly slide the forceps toward the cornea. This movement will force the retina out of the puncture site for collection into a 1.5 ml siliconized tube filled with 8 % OptiPrep. The rapid procedure bypasses steps 2–7 of the standard method. In our experience, this rapid procedure provides high-quality rod outer segments and cilia suitable for tomography, but at the expense of a lower yield (typically 30–40 % less tissue) and a higher proportion of broken cells and cilia than the detailed method outlined in Subheading 3.2. While this method may be advantageous for some studies, it is not advised for retinal degeneration studies as isolation artifacts can obscure genuine structural phenotypes.
Once the eye is removed from the orbit, it is not necessary to remove all remnants of ocular muscle from the sclera. Attached tissue remnants provide a convenient anchor that can be grasped with forceps to aid in the dissection procedure.
In our experience, isolating the retina under a buffered solution allows for easier manipulation of the eye and a more careful separation of the tissues when compared with “dry” dissecting the eye.
As the cornea cut site is enlarged, it will be easier to grip the edges of the cornea and sclera with forceps and rotate the eye for more efficient dissection.
Because different variations on the Vortex-Genie shakers exist, the lowest “vortex” setting is at roughly the 10 o’clock position on the rotary dial or roughly 1/3 of the entire rotary span of movement. Higher vortexing speeds will provide larger numbers of ROS fragments and give the initial impression of being more efficient; however, ROS fragments isolated under higher speeds are less likely to separate from the retina with the cilium attached. At the preferred speed, roughly 50–60 % of ROS remain attached to the cilium, but at higher speeds this number can drop to 25 % or below.
If using a 1 ml syringe without a Luer lock fitting, the tip of the syringe barrel can be inserted directly into the OptiPrep solution to collect rod outer segments. Because the barrel tip diameter is larger than that of the 18 G needle, this will further reduce shearing of the tissue. If this procedure is carried out under infrared or dim light conditions, it is helpful to premeasure a 625 μl volume of 8 % OptiPrep and mark this plunger location by wrapping labeling tape around the syringe barrel. In this way, the marking tape will provide a visual and tactile cue that is easier to identify in the dark.
As rhodopsin is bleached by light, the rod outer segment band will change from dark red to faint orange in color and will become more difficult to visualize if an insufficient number of cells has been collected. For ROS preps where dark-adapted retinas are not used, it can still be advantageous to minimize ROS bleaching (as unbleached ROS is easier to see) by covering ROS samples in aluminum foil and by limiting direct exposure to light throughout the procedure. This additional precaution can aid in ROS band visibility.
The needle cannula can be moved slowly from side to side while collecting the ROS band to increase yield around the perimeter of the tube wall, but quick movements will cause mixing of the gradient and diffuse the ROS band. Ideally, each ROS band should be collected in 600 μl or less of volume, since larger collection volumes will carry more OptiPrep into the final purification step and cannot be effectively diluted with 1× Ringer’s Buffer (see Subheading 3.5, step 9).
If too much OptiPrep is collected along with the ROS band and is not sufficiently diluted prior to pelleting, the ROS pellet will not reach the bottom of the 1.5 ml polyallomer tube and will float on a dense OptiPrep layer that forms at the bottom of the tube. If this occurs, it will be necessary to collect as much of the OptiPrep-containing supernatant, without disturbing the ROS layer, and discard. Again fill the tubes with 1× Ringer’s Buffer to further dilute the remaining OptiPrep and invert the tubes to mix. Ensure the tubes are correctly balanced, and centrifuge 30 min at 26,500 × g at 4 °C using a TLA 100.3 rotor to correctly pellet the ROS. Remove all traces of OptiPrep and discard the supernatant.
The optimum concentration of ROS is such that there are ~1–2 cells per grid square to yield high-probability imaging sessions. This optimum ROS concentration can be attained through a combination of (a) manipulating the final ROS pellet resuspension volume, (b) adjusting the volume of sample applied to the grid, (c) adjusting the wait time before grid blotting, and (d) adjusting the blotting time. The desired concentration of gold fiducial markers is such that 15–20 gold markers are present in any field of view where a tilt series will be collected. Software designed to reconstruct the tomogram requires a lower limit of ten easily visible particles evenly distributed across the field of view. A smaller field of view (i.e., higher magnification) will generally require a higher gold fiducial concentration to provide the necessary parameters to avoid alignment errors.
If the pellet is resistant to solubilization, improved resuspension can be achieved by interspersing 2–5 min incubations on ice, which seems to aid by letting the pellet disaggregate. Mechanical trituration of the pellet with a wide-bore 20 μl micropipette tip can be helpful. If a wide-bore tip is used, extreme caution should be used to prevent “smearing” the ROS pellet and destroying a large proportion of the ROS and cilia contained in the sample.
If a volume larger than 20 μl is used to resuspend the ROS pellet, it will be necessary to concentrate the BSA-colloidal gold. To concentrate the fiducial marker, add 8.0 μl of BSA-gold to a microcentrifuge tube, and centrifuge 2 min at 12,000 × g at 4 °C in a tabletop microcentrifuge. Discard the supernatant and resuspend the BSA-gold (visible as a red pellet) in an appropriate volume before adding to the ROS sample.
Larger numbers of grids can be simultaneously glow discharged if desired, but grids not used within 1 h should be discarded as they readily attract debris from the environment. Practically, it is easier to glow discharge smaller batches of grids, since the process is quickly completed and can be interleaved easily with other steps under Subheading 3.6.
If enough time has elapsed since the ethane has been condensed, the surface will freeze over, which results in grid bending when it collides with the frozen surface (ethane gas condenses at −160 °C and solidifies at −188 °C). Frozen ethane can be thawed by bubbling more gaseous ethane against the frozen surface. As the surface thaws, the attached pipette tip can be stirred in the ethane to further break up the ice.
Ice thickness on each grid will vary depending on the blotting length and inconsistencies in the filter paper surface, which warps as the filter absorbs liquid from successive samples and from the humidified Vitrobot chamber. Some variation of the blotting protocol may be appropriate, but setting the Vitrobot software for a single blot with a 2 s duration is a reliable starting point for 2.5 μl of volume with ROS.
As the grid is lifted out of the liquid ethane, it has the likelihood of condensing atmospheric water that is visible in the cryo-electron microscope as dense, ice crystals. By keeping the grid submerged in the ethane, ice crystallization can be minimized.
When lifting the grid out of the liquid ethane, keep the grid as close to the liquid surface as possible. As the ethane and nitrogen evaporate, they create a gas barrier at the liquid-gas interface that excludes atmospheric moisture. By moving the grid through this gas layer, the grid can be transferred to the outer annulus and immersed in liquid nitrogen while limiting ice crystallization on the grid.
Take care not to damage the metal tip of the holder upon transfer to the workstation.
Using a small funnel fitted into the transfer holder, dewar will aid with nitrogen filling.
This process takes approximately 5–10 min.
Working through the ports of the workstation cover will limit frost formation from atmospheric water.
Many airlocks are designed such that the transfer holder must be tilted prior to insertion. The holder is then rotated to its upright position after the vacuum pump sequences are complete.
The aspects of this process are specific to the make and model of microscope used. Refer to the manufacturer’s specifications before proceeding.
At this point, it may take from 20 to 45 min for the transfer holder temperature to equilibrate with the microscope. This thermal equilibration is essential to quality imaging since it reduces vibrations that lead to blurred images and reduced image quality.
This setting will vary according to the desired magnification power, brightness level, image contrast, resolution required, and specifics of the electron microscope. In TEM mode, the indicated electron beam spot-size diameter and aperture angle (Alpha) are consistent with imaging at 20,000× magnification while minimizing the electron dose that damages biological samples.
Magnification is chosen based upon the region of the cell to be imaged as well as the level of information desired from the data. A magnification of 12,000× is useful for imaging large regions of cells, including both the outer segment and the connecting cilium, which corresponds to the transition zone of other primary cilia. A magnification of 12,000× can provide gross structural anatomy (membranes, basal bodies, and microtubules are visible) and is useful for quickly evaluating the quality of a ROS preparation or grid freezing conditions, but it is unlikely to provide more detailed information for an in-depth research investigation. A magnification of 20,000× provides a balance between field of view size and high-resolution information. At 20,000×, the entire connecting cilium can easily be contained within the field of view. Alternatively, large regions of the inner segment (containing the rootlet) or the outer segment can be viewed. At 30,000× magnification, data collection is almost exclusively limited to the connecting cilium or the cell periphery of the outer segment or inner segment. Two problems complicate magnifications of 30,000× or greater: (a) the number of gold fiducial markers is often insufficient to accurately align the micrographs during the tomographic reconstruction steps since the markers are exclusively located outside of the cell membrane; and (b) while the magnification theoretically provides higherresolution data, the resolution comes at the price of greatly reduced image contrast. Reduced micrograph contrast coupled with low numbers of fiducial gold greatly reduces the ability to detect biological structures away from the periphery of the cell. From our experience, a 40,000× magnification is nearly impossible to use with our experimental setup since the micrograph contrast is low enough that the automated tracking feature of SerialEM no longer functions reliably.
These map images can be kept active on the desktop for easy access to help track movement across grid squares once the imaging process begins. Correlating these images with the microscope specimen location window will help orient the user through the experimental process.
At low magnification, the ice thickness gradient can be seen as a uniform level of contrast that gradually changes from dark (low contrast) at one edge of the grid to light (high contrast) at the opposite grid edge. An initial inspection of the low-magnification grid map will indicate regions of the grid where ice crystals may have formed on the grid. Heavy ice contamination should be avoided as it creates dense objects capable of interfering with digital image acquisition during the sample tilt process. Qualitatively, ice thickness is greatest in regions of the grid where contrast is lowest, whereas highest contrast indicates regions where ice is thin. While regions with more contrast are easier to visualize and tend to draw the user’s interest for ease of data collection, our experience has been that more cellular deformation occurs in these areas. More cilia and cell membranes are ruptured in these regions (likely resulting from the blotting procedure described under Subheading 3.6) and, more importantly, outer segments and cilia are more compressed and less circular in diameter. These two structural confounds make comparisons difficult between wild-type structures and aberrant structures where deformation may be caused by genetic mutation. While lower-contrast (darker) regions are more difficult for identifying rod cells, we have found the data collected from these regions to be more rewarding since these regions consistently provided the most intact and uniformly shaped structures across multiple grids under different freezing conditions.
Compared with plastic-embedded biological specimens for TEM (which can sustain electron doses of 10,000 e−/Å2), cryo-specimen is more sensitive to electron dose. Therefore, the goal for imaging cryo-specimens is to maintain the cumulative electron dose at 100 e−/Å2 or less (this includes all ancillary images taken during setup and eucentricity, as well as images taken during the tilt series). Dose tolerance varies depending on the specimen, but electron damage can be detected as “bubbling” that occurs within the specimen. Rod outer segments seem fairly resilient to damage and can often be imaged at doses above 100 e−/Å2, though we avoid this practice by convention. In contrast, rod cilia are best imaged below the upper threshold since bubbling becomes clearly visible along the length of the basal body and axoneme microtubules.
At this point, the stage movement and brightness settings can be controlled through SerialEM. To prevent excess dose delivery to the cell, the “Focus and Trial/Tracking” images taken during the automated tracking cycles are displaced along the tilt axis and do not overlap the ROI captured in the “Record” images.
The gain reference is necessary for the software to correctly calculate the electron dose and distribute it across the different micrographs to be taken as the tilt angle is modified. It is useful to collect a new gain reference before each tilt series is initiated since sample parameters change across different regions of the grid.
At this point, it is beneficial to test the maximum tilt range under which the sample can be imaged under the “Low Dose” mode since not every sample can be imaged under the full −70° to 70° range (tilt angle walk test). The “anchor” function collects an image at a specified tilt angle that is saved by SerialEM and the image is used as a reference when the tilt series reaches that angle to enhance the tracking accuracy and image alignment.
The maximum electron dose for rod outer segments and rod cilia should be kept at 100 e−/Å2 or less. As a general starting point, doses closer to this maximum should be reserved for cells embedded in thicker ice, while lower doses are better for thinner specimen to prevent damage.
A finer tilt increment (1.0° or 1.5°) can be chosen, but the cumulative electron dose is delivered over a larger number of micrographs, each of lower contrast due to reduced image dose. We have not noticed a significant difference in data quality by using a finer tilt increment.
Image contrast for unstained, frozen-hydrated (vitrified) specimens predominantly arises from interference between scattered and unscattered electrons interacting with the specimen, a phenomenon called phase contrast. Contrast is characterized by a low signal-to-noise ratio due to the similar electrostatic potential (electron density) between the biological specimen and the surrounding vitreous ice. For this reason, image contrast is low when specimens are in focus, making cellular details and structures difficult to see. To improve phase contrast, specimens are often imaged below focus (defocused), but this contrast advantage comes with a drawback. The defocus phase contrast is influenced by the contrast transfer function (CTF), which depends on the microscope acceleration voltage and the spherical aberration of the lens. The effect of the CTF, as defocus values increase, is the loss of fine, high-frequency details within the image. This compromise between image contrast and resolution means that selecting a defocus value dramatically affects the level of detail that can be extracted from the data. For experimentalists looking to maintain the highest resolution possible, defocus values of −2 to −6 μm are often appropriate. However, if the goal is to identify larger macromolecular structures, larger defocus values may be necessary (−7 to −12 μm).
This process is automated through SerialEM, but it is advisable for the user to be present to observe the entire tilt processes as challenges do often arise. Low-contrast micrographs are prone to cause tracking difficulties, especially at higher tilt angles where specimen thickness increases dramatically, and can cause unwanted stage shifts that remove the cell from view.
Pixel size is dependent upon the specifics of the microscope, the size of the camera charge-coupled device (CCD), and the magnification. Our experimental setup, which imaged on a 4k × 4k CCD at 15,000× to 30,000× magnification, routinely provided pixel sizes ranging from 1.7 to 3 nm/pixel.
While allowing the software to automatically choose fiducial markers can simplify many tomogram alignments, it is essential that all fiducials be verified by the user across all micrographs in the tilt series. Markers that are visible in the zero-degree tilt image, where contrast is highest, are often difficult or impossible to see at higher tilt angles. For this reason, it is preferred to begin the fine alignment procedure with a large number of fiducials (20 or more) when using the automated fiducial selection tool.
By limiting the reconstruction to the dimensions of the desired cellular region and the contents within the cell (thus excluding the majority of the volume in the original field of view that is extracellular to the specimen of interest), the reconstruction time can be greatly shortened by reducing the computational load of the computer as it computes a smaller three-dimensional tomogram. With some very large specimen, it is necessary to computationally bin the data during the reconstruction process to reduce the CPU computation load and prevent operating system crashes.
References
- 1.Wang J, Deretic D (2014) Molecular complexes that direct rhodopsin transport to primary cilia. Prog Retin Eye Res 38:1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Young RW (1967) The renewal of photoreceptor cell outer segments. J Cell Biol 33:61–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wensel TG (2012) Molecular biology of vision. In: Brady ST, Albers RW, Price D, Siegel JG (eds) Basic neurochemistry: principles of molecular, cellular, and medical neurobiology, 8th edn. Elsevier, London, pp 889–903 [Google Scholar]
- 4.Besharse JC, Horst CJ (1990) The photoreceptor connecting cilium. A model for the transition zone. In: Bloodgood RA (ed) Ciliary and flagellar membranes Springer, New York, pp 389–417 [Google Scholar]
- 5.Kozminski KG, Johnson KA, Forscher P et al. (1993) A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc Natl Acad Sci U S A 90:5519–5523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenbaum JL, Cole DG, Diener DR (1999) Intraflagellar transport: the eyes have it. J Cell Biol 144:385–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nair KS, Hanson SM, Mendez A et al. (2005) Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions. Neuron 46:555–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilliam JC, Chang JT, Sandoval IM et al. (2012) Three-dimensional architecture of the rod sensory cilium and its disruption in retinal neurodegeneration. Cell 151:1029–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yahav T, Maimon T, Grossman E et al. (2011) Cryo-electron tomography: gaining insight into cellular processes by structural approaches. Curr Opin Struct Biol 21:670–677 [DOI] [PubMed] [Google Scholar]
- 10.Dubochet J, Adrian M, Chang JJ et al. (1988) Cryo-electron microscopy of vitrified specimens. Q Rev Biophys 21:129–228 [DOI] [PubMed] [Google Scholar]
- 11.Dierksen K, Typke D, Hegerl R et al. (1992) Towards automatic electron tomography. Ultramicroscopy 40:71–87 [Google Scholar]
- 12.Dierksen K, Typke D, Hegerl R, Baumeister W (1993) Towards automatic electron tomography. II. Implementation of autofocus and lowdose procedures. Ultramicroscopy 49:109–120 [Google Scholar]
- 13.Frank J (1992) Electron tomography: three-dimensional imaging with the transmission electron microscope Plenum, New York [Google Scholar]
- 14.Kremer JR, Mastronarde DN, McIntosh JR (1996) Computer visualization of three-dimensional image data using IMOD. J Struct Biol 116:71–76 [DOI] [PubMed] [Google Scholar]
- 15.Mastronarde DN (1997) Dual-axis tomography: an approach with alignment methods that preserve resolution. J Struct Biol 120:343–352 [DOI] [PubMed] [Google Scholar]
- 16.Grimm R, Singh H, Rachel R et al. (1998) Electron tomography of ice-embedded prokaryotic cells. Biophys J 74:1031–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grimm R, Typke D, Barmann M et al. (1996) Determination of the inelastic mean free path in ice by examination of tilted vesicles and automated most probable loss imaging. Ultramicroscopy 63:169–179 [DOI] [PubMed] [Google Scholar]
- 18.Leis A, Rockel B, Andrees L et al. (2009) Visualizing cells at the nanoscale. Trends Biochem Sci 34:60–70 [DOI] [PubMed] [Google Scholar]
- 19.Nickell S, Park PS, Baumeister W et al. (2007) Three-dimensional architecture of murine rod outer segments determined by cryoelectron tomography. J Cell Biol 177:917–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nicastro D (2009) Cryo-electron microscope tomography to study axonemal organization. Methods Cell Biol 91:1–39 [DOI] [PubMed] [Google Scholar]
- 21.Mastronarde DN (2005) Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152:36–51 [DOI] [PubMed] [Google Scholar]
- 22.Sandberg K, Mastronarde DN, Beylkin G (2003) A fast reconstruction algorithm for electron microscope tomography. J Struct Biol 144:61–72 [DOI] [PubMed] [Google Scholar]