

The FDA has granted regular approval to vemurafenib for the treatment of adult patients with Erdheim‐Chester Disease (ECD) with BRAFV600 mutation. This article describes the FDA review of the evidence and the clinical implications for this rare patient population.
Keywords: Non‐Langerhans cell histocytosis, Vemurafenib, Zelboraf, Histiocytosis, BRAFV600 mutations
Abstract
On November 6, 2017, the U.S. Food and Drug Administration (FDA) granted regular approval to vemurafenib for the treatment of adult patients with Erdheim‐Chester disease (ECD) with BRAFV600 mutation. ECD is a type of histiocytosis, a rare disorder characterized by an abnormal accumulation and behavior of cells of the mononuclear phagocytic system, which includes antigen‐processing cells, dendritic cells, monocytes, or macrophages. Recently published data confirm a frequency of 54% of BRAFV600E mutations in patients with ECD.
Approval was based on a cohort of 22 patients who received 960 mg of vemurafenib twice daily within the VE Basket Trial (MO28072), a single‐arm, multicenter, multiple cohort study. Patients in the ECD cohort had histologically confirmed ECD with BRAFV600 mutations that were refractory to standard therapy. The ECD cohort achieved an overall response rate of 54.5% (95% confidence interval: 32.2–75.6), with a complete response rate of 4.5%. With a median duration of follow‐up of 26.6 months, the median duration of response has not been reached. The most frequently reported adverse reactions (>50%) in the ECD cohort were arthralgia, rash maculo‐papular, alopecia, fatigue, electrocardiogram QT interval prolonged, and skin papilloma. The median treatment duration for ECD patients in this study was 14.2 months. This article describes the FDA review of the vemurafenib efficacy supplement for patients with ECD with BRAFV600 mutations.
Implications for Practice.
Vemurafenib, an oral monotherapy targeting a mutation in BRAF, is the first U.S. Food and Drug Administration approval for the treatment of Erdheim‐Chester disease (ECD). ECD is an extremely rare hematopoietic neoplasm that represents clonal proliferation of myeloid progenitor cells. ECD may involve bone and one or more organ systems, primarily affecting adults in their 5th and 7th decades of life, with a slight male predominance. This approval provides an effective and reasonably safe therapy for patients with a serious and life‐threatening condition for which no approved therapy exists.
摘要
2017 年 11 月 6 日,美国食品和药品管理局 (FDA) 对用于治疗患有 BRAFV600 突变的 Erdheim‐Chester 病 (ECD) 成人患者的维罗非尼授予常规批准。ECD 是一种组织细胞增生症,这种罕见病以包括抗原提呈细胞、树突细胞、单核细胞或巨噬细胞的单核巨噬细胞系统的细胞异常积聚和行为为特征。最近发布的数据证实,ECD 患者的 BRAFV600E 突变发生率为 54%。
此项批准以一个 22 名患者的队列为依据,这些患者在 VE 篮型试验 (MO28072)(一个单组、多中心、多队列研究)中接受维罗非尼960 mg、每天给药 2 次的治疗。ECD 队列中的患者患有已在组织学上证实的 ECD 和 BRAFV600 突变,并在标准疗法中耐药。ECD 队列取得了 54.5% 的总缓解率(95% 置信区间:32.2–75.6),完全缓解率为 4.5%。随访的中位持续时间为 26.6 个月,未达到缓解的中位持续时间。在 ECD 队列中,最常报告的不良反应 (>50%) 为关节痛、斑状丘疹、脱发、疲劳、心电图 QT 间期延长以及皮肤乳头状瘤。在本次研究中,ECD 患者的中位治疗持续时间为 14.2 个月。本文描述了 FDA 对患有 BRAFV600 突变的 ECD 患者的维罗非尼疗效补充的评审
Introduction
Erdheim‐Chester disease (ECD) is a slow‐growing blood cancer that originates in the bone marrow. ECD causes an increased production of histiocytes, a type of white blood cell. Excess histiocytes can result in tumors infiltrating many organs and tissues throughout the body, including the heart, lungs, brain, and others. ECD is estimated to affect 600–700 patients worldwide. Approximately 54% of patients with ECD have the BRAFV600 mutation [1].
This rare, non‐Langerhans histiocytosis was described by Jakob Erdheim and William Chester in 1930 [2]. Tissue biopsy, preferably of an osteosclerotic bone lesion, is needed to confirm the diagnosis with CD68+/CD1a‐/S‐100‐/Langerin‐ foamy histocytes. This differentiates ECD from Langerhans cell histiocytosis (LCH), in which the Langerhans cells are positive for CD1a, S‐100 protein, and Langerin. The disease primarily affects the bone but can impact any organ system. Approximately 20% of patients present with symptoms, most commonly bone pain, neurological and constitutional symptoms, and diabetes insipidus [3], [4], [5].
Laboratory workup, including complete blood count and chemistries, are usually nondescript for this diagnosis unless urine electrolytes highlight evidence of diabetes insipidus. There is no universally accepted staging, prognostic, or scoring system for ECD. Staging evaluations include magnetic resonance imaging (MRI) of the brain, computed tomography (CT) scan or MRI of the heart and aorta, CT scan of the chest, abdomen, and pelvis, and 18F‐fluoro‐2‐glucose‐positron emission tomography/computed tomography (FDG‐PET/CT), echocardiogram (if heart involvement is suspected), and MRI of the spine (if spine involvement is suspected). The most specific imaging findings would be symmetrical diaphyseal and metaphyseal osteosclerosis of long bone on plain radiographs, increased radiotracer uptake in the proximal and distal ends of tibia and proximal ends of femurs by bone scan or FDG‐PET/CT, respectively, and infiltration of perinephric fat and circumferential soft‐tissue sheathing of the aorta on CT. It has been suggested that FDG‐PET/CT along with C‐reactive protein elevation may predict disease activity. Therefore, the diagnosis of ECD relies heavily on the established radiological and histological criteria, because the clinical picture is variable [5].
Although previously reported in only 600–700 patients, the diagnosis of ECD has dramatically increased in the last 10 years due to increasing recognition of the disease. ECD primarily affects adults between their 5th and 7th decades of life, with a slight male predominance noted. The etiology of the disease is unknown, and there is no evidence that ECD is an inheritable genetic disorder. ECD, and the related histiocytic disorder Langerhans cell histiocytosis, are hematopoietic neoplasms that represent clonal proliferation of myeloid progenitor cells. This was demonstrated by finding the BRAFV600E mutation in subsets of dendritic cells, mature monocytes, committed myeloid progenitors, and CD34+ cells of affected ECD and LCH patients. Somatic mutations in components of the mitogen‐activated protein kinase (MAPK) signaling pathway are present in most patients with ECD. BRAFV600E mutations have been found in approximately half of ECD cases, and the mutation of this serine‐threonine kinase enhances cell proliferation and survival by activating the RAS‐RAF‐MEK‐MAPK signaling pathway [6]. ECD can involve bone, central nervous system, the retroperitoneum, skin, lungs, heart, and endocrine glands. Patients with ECD can be heavily symptomatic and debilitated, but the symptoms are varied between patients due to the heterogeneity of organ involvement between patients. The 1‐year and 5‐year survival rates for patients with ECD have been reported at 96% and 68%, respectively [7], [8].
Vemurafenib is a kinase inhibitor that works by blocking enzymes in a cell growth‐promoting pathway. The U.S. Food and Drug Administration (FDA) first approved vemurafenib in 2011 for the treatment of patients with unresectable or metastatic melanoma with BRAFV600E mutation as detected by an FDA‐approved test. Vemurafenib is not recommended for use in patients with wild‐type melanoma [9]. Vemurafenib was first approved in 2011 for the treatment of patients with unresectable or metastatic melanoma with BRAFV600E mutation as detected by a U.S. Food and Drug Administration (FDA)‐approved test (Table 1). Vemurafenib is not recommended for use in patients with wild‐type melanoma [9].
Table 1. Background information: vemurafenib.
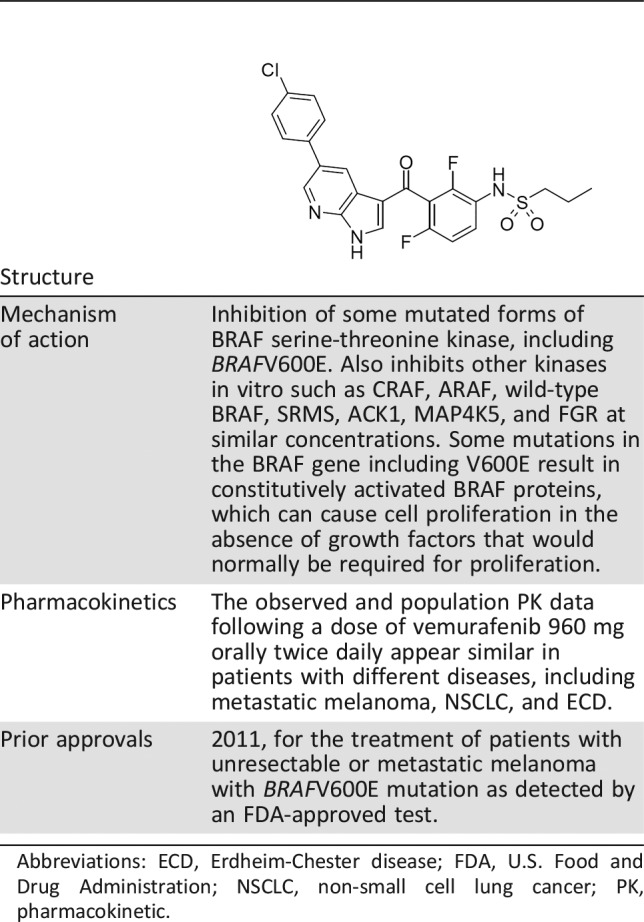
Abbreviations: ECD, Erdheim‐Chester disease; FDA, U.S. Food and Drug Administration; NSCLC, non‐small cell lung cancer; PK, pharmacokinetic.
The FDA granted orphan drug designation to vemurafenib for the treatment of ECD in August 2016. Breakthrough therapy designation was granted to vemurafenib for the treatment of patients with ECD with BRAFV600 mutation in April 2017.
At the time of this review, there were no FDA‐approved products for the treatment of ECD. The treatment of ECD has included off‐label use of pegylated and nonpegylated interferons (IFN‐α), anakinra, cladribine, imatinib, infliximab, tocilizumab, and a combination of sirolimus and prednisone. In 2013, a report [7] was published describing rapid clinical and tumor responses for three patients with BRAFV600E mutation ECD who received vemurafenib. The current approval was granted on November 6, 2017, 1 month prior to the goal date. Herein, we describe the FDA review of the evidence submitted for this application and the clinical implications for the rare ECD population.
Clinical Trial Design
M028072 (ClinicalTrials.gov identifier NCT01524978) was an open‐label, multicenter, multicohort, multinational, nonrandomized basket trial exploring the efficacy and safety of vemurafenib monotherapy in a diverse population of patients with cancers known to harbor BRAFV600 mutations (excluding melanoma and papillary thyroid cancer). Cohort 7A included 22 patients with ECD in addition to 4 patients with LCH. Eligible patients were adults with histologically confirmed cancers (excluding melanoma and papillary thyroid cancer) who harbored BRAFV600 mutation refractory to standard therapy or for which standard or curative therapy did not exist, and who had measurable disease per RECIST 1.1. Patients were selected for enrollment based upon BRAFV600 mutation analysis assays as routinely performed at each participating site according to local procedure. However, sites could submit a tumor sample for retrospective confirmation of the BRAF mutation using the Roche CDx cobas 4800 BRAFV600 test or other standard methodology by a central laboratory.
Patients must have had adequate hematologic function (defined as absolute neutrophil count ≥1.5 × 109/L and platelet count ≥100 × 109/L). Patients with concurrent ECD and LCH, as well as patients with ECD and/or LCH and active or untreated central nervous system involvement, were eligible.
Patients received vemurafenib 960 mg orally twice daily with or without food until the development of progressive disease, unacceptable toxicity, withdrawal of consent, protocol violation endangering the patient's safety, death, reasons deemed critical by the treating physician, or study termination. Patients with ECD could discontinue vemurafenib treatment after 1 year, if the Investigator considered it to be in the best interests of the patient. Patients were eligible to resume treatment if they became symptomatic or if they had radiographic evidence of progressive disease.
Prior to the closure of the trial, patients who had completed the protocol‐mandated minimum 12‐month safety follow‐up and who continued to benefit from vemurafenib therapy were offered the opportunity to receive continued vemurafenib treatment via enrollment in the GO28399 extension trial. Patients in Study GO28399 trial were followed for survival for a minimum period of 12 months after the last patient has been enrolled or until all patients had died, withdrawn consent, or were lost to follow‐up, whichever occurred first.
The primary endpoint of the final analysis for Cohort 7a (patients with ECD) was the overall response rate (ORR), which was defined as the proportion of patients who had a complete response (CR) or partial response (PR) confirmed on two occasions at least 4 weeks apart, as assessed by the Investigator using RECIST 1.1. Secondary endpoints included progression‐free survival (PFS) and time to tumor progression, best overall response, clinical benefit rate, time to response, duration of response (DoR), and overall survival (OS).
Efficacy assessments included baseline tumor assessment by CT/MRI of the chest, abdomen, and pelvis as well as clinically relevant tumor assessment to define baseline extent of disease (i.e., brain MRI, cardiac MRI/echocardiogram, bone scan, 18F‐FDG‐PET). Tumor assessments were conducted every 8 weeks after the start of vemurafenib. There were no formal statistical hypotheses for this cohort.
Pharmacokinetic (PK) samples were collected before the dose on multiple occasions, including Cycle 1 Day 15 (C1D15), C2D1, C3D1, and C4D1 in a single patient with ECD. PK data were available for 21 additional patients enrolled in the basket trial. A population analysis was completed.
Results
Trial M028072 (VE Basket) initiated on April 11, 2012, and completed on October 27, 2016. The data cutoff for the application submission was January 12, 2017. The trial enrolled 208 patients into seven different cohorts. Twenty‐two patients with ECD with BRAFV600 mutation enrolled in Cohort 7a. Enrolled ECD patients had a median age of 58.5 years (34–77 years), had a slight male predominance (55%), and were predominantly white (96%). Eastern Cooperative Oncology Group (ECOG) scores at baseline were mostly 1 (54%), with 23% of patients having a score of 2, 18% having a score of zero, and 5% “not applicable.” Fifteen patients (68%) had at least one prior systemic therapy for ECD. At the time of data cutoff, all patients were no longer receiving study treatment and had discontinued from the study.
Efficacy
The overall response rate (ORR) was 54.5% (95% confidence interval: 32.2–75.6). One patient had a CR, and eleven had PRs (Table 2). The remaining nine patients had stable disease, and one patient's outcome was not measurable. The median DoR was not estimable with a median duration of follow‐up of 26.6 months (3–44.3 months). The median time to response was 11 months (3.7–14.6 months). The median time to treatment discontinuation was 14.2 months (1.6–44.2 months). The median PFS and OS were not estimable.
Table 2. Efficacy results for vemurafenib in BRAFV600‐mutation positive Erdheim‐Chester disease.
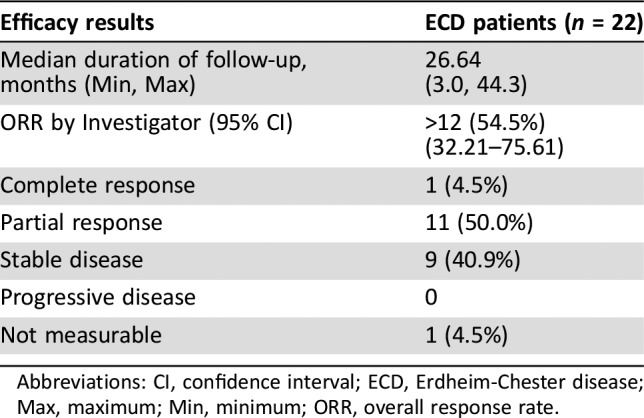
Abbreviations: CI, confidence interval; ECD, Erdheim‐Chester disease; Max, maximum; Min, minimum; ORR, overall response rate.
Efficacy narratives, abstracted from medical records, for all ECD patients were reviewed. The narratives provided statements describing changes in disease symptomatology, physical function, and quality of life while receiving vemurafenib. Fifteeen were documented by the clinician to have had an improvement in disease‐related symptoms or physical function after starting treatment with vemurafenib. Examples include improvements in symptoms (i.e., dysarthria, ataxia, headache, visual disturbances, fatigue, bone pain, lymphedema, pain, skin lesions, night sweats, urinary frequency, arthralgias, dyspnea) and physical function (i.e., resumption of activities of daily living, wheelchair‐bound patient ambulating independently, returning to work, navigating stairs without assistance, and ECOG 2 to 0). The efficacy narratives provided supportive evidence that patients with ECD treated with vemurafenib experienced an improvement in the way they felt and functioned.
Safety
The safety review of vemurafenib in patients with ECD was based on 22 patients with BRAFV600‐mutation ECD (enrolled in trial MO28072 and including longer‐term data from 8 patients who rolled over to the extension study [GO28399]). Due to the small sample size and rarity of the condition, supportive safety data from 3,378 non‐ECD patients who had been treated at the same vemurafenib dose and schedule (including non‐ECD patients from trial MO28072 and patients with metastatic melanoma from trial MO25515) were also included.
The study population was monitored for deaths, serious adverse events, adverse events (graded per National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0), and laboratory test abnormalities. Scheduled clinical visits including safety assessments occurred at baseline; on Cycle 1 Days 1, 15, 29; at the start of every subsequent 28‐day cycle; and 1 month after the final dose.
The most commonly reported adverse reactions (incidence >50%) in 22 patients with BRAFV600 mutation‐positive ECD treated with vemurafenib were arthralgia, rash maculo‐papular, alopecia, fatigue, electrocardiogram QT interval prolonged, and skin papilloma. The most common (≥10%) grade 3–4 adverse reactions were squamous cell carcinoma of the skin, hypertension, rash maculo‐papular, and arthralgia. Nonfatal serious adverse events occurred in 73% of patients with ECD. There were no fatal adverse events (grade 5) reported among the ECD patients within 100 days of the final dose.
All patients with ECD had at least one dose reduction (DR) from the starting dose of 960 mg orally twice daily and one dose interruption (DI) due to an adverse reaction. The most frequent adverse reaction leading to DR or DI were maculopapular rash, fatigue, arthralgia, palmar‐plantar erythrodysaesthesia, and increased lipase. The duration of DI ranged from 1 to 29 days. The dose was reduced to 720 mg orally twice daily in 8 patients, and ultimately to 480 mg orally twice daily in 14 patients. The median duration of treatment was longer following dose reduction to 480 mg (236 days; 21–924 days) compared with 720 mg (77 days; 4–1,325 days). The efficacy was maintained in these patients based on the ORR. These events were similar to those leading to dose modifications in previous trials in non‐small cell lung cancer and metastatic melanoma; however, more patients with ECD needed DR and DI due to adverse reactions. Due to DR and DI in the ECD population, the relative dose intensity in these patients was lower (62%) than that in the non‐ECD patients from Trial MO28072 (80%) and that in the patients with metastatic melanoma from Trial MO25515 (90%).
Discussion
It was agreed in advance of the application submission that the FDA would not require a companion diagnostic for vemurafenib for the BRAFV600‐mutation assay with this application.
The efficacy of vemurafenib was based on response rate as assessed by the Investigator using RECIST 1.1 response criteria. In addition to the overall response rate among the patients with ECD, supportive evidence of symptomatic and physical function improvement was reported in 68% of the 22 patients after starting treatment with vemurafenib. These results were derived from “efficacy narratives” created from excerpts from the patient medical records. The documented evidence of clinician‐reported functional and symptomatic improvement while on vemurafenib does represent a favorable benefit to risk ratio to support regular approval of vemurafenib for patients with ECD. The functional and disease symptom improvements are very likely to be due to vemurafenib treatment, and not a function of disease waxing and waning. Many of these symptomatic responses were rapid and not always identified concurrent with radiographic (RECIST) responses.
This is a common problem in obtaining drug approvals for rare diseases. Typically, the natural history of the disease is not clear, often due to heterogeneity. Clinical outcome measures for rare diseases have never or rarely been described or tested, and longitudinal follow‐up may be limited. Finally, the predictive biomarker measurements based on patient response, patient selection, or assessing activity may not be precise or reproducible. Hence, the need for developing a comprehensive natural history program involving the affected patients early in the developmental drug program is crucial. Meanwhile, precision medicine cancer trials such as “basket” trials with biomarker‐defined targets across histologic diagnoses should be considered (e.g., the trial used here). In exploring outcome measures when the treatment effect is unknown, it may be important to consider randomization in efficiently finding if there is a treatment effect. In this basket trial, the dose was reduced to 720 mg or 480 mg orally twice daily for all patients with ECD from the starting dose of 960 mg twice daily and one dose‐interruption due to an adverse reaction; however, the efficacy was maintained in these patients based on the ORR, and no differences in the minimum trough concentrations were observed regardless of disease type across clinical trials.
Given the rarity of ECD and the lack of available therapies, the FDA agreed to accept data from a small cohort of a basket trial for review. The rarity of this condition as well as the lack of an established standard therapy would have also prevented the conduct of a randomized clinical trial.
Conclusion
Prior to the approval of vemurafenib, there were no FDA approvals for the treatment of patients with ECD. When treated with single‐agent vemurafenib, patients with BRAFV600 mutation ECD experienced an ORR of 54.5% as assessed by the investigator. An exploratory evaluation of efficacy narratives identified supportive evidence that patients also reported to the treating clinician, improvements in disease‐related symptoms and physical function. The efficacy and safety results from trial MO28072 demonstrated an acceptable benefit‐risk (see Table 3) profile for vemurafenib for the treatment of adult patients with BRAFV600 mutation‐positive ECD. Evidence of a 54.5% ORR along with supportive evidence of clinician‐reported improvements in disease‐related symptoms and physical function permitted the FDA to grant regular approval for the proposed indication.
Table 3. U.S. Food and Drug Administration benefit‐risk assessment.

Abbreviations: ALT, alanine aminotransferase; CNS, central nervous system; DoR, duration of response; ECD, Erdheim‐Chester disease; ECG, electrocardiogram; ORR, overall response rate; QT, electrocardiogram QT corrected interval prolonged; sNDA, supplemental new drug application.
Author Contributions
Conception/design: Patricia A. Oneal, Virginia Kwitkowski
Collection and/or assembly of data: Patricia A. Oneal, Virginia Kwitkowski
Data analysis and interpretation: Patricia A. Oneal, Virginia Kwitkowski, Lola Luo, Yuan Li Shen, Sriram Subramaniam, Stacy Shord
Manuscript writing: Patricia A. Oneal, Virginia Kwitkowski
Final approval of manuscript: Patricia A. Oneal, Virginia Kwitkowski, Lola Luo, Yuan Li Shen, Sriram Subramaniam, Stacy Shord, Kirsten B. Goldberg, Amy E. McKee, Edvardas Kaminskas, Ann Farrell, Richard Pazdur
Disclosures
The authors indicated no financial relationships.
References
- 1.Haroche J, Charlotte F, Arnaud L et al. High prevalence of BRAF V600E mutations in Erdheim‐Chester disease but not in other non‐Langerhans cell histiocytoses. Blood 2012;120:2700–2703. [DOI] [PubMed] [Google Scholar]
- 2.Chester W. Uber Lipoidgranulomatose [in German]. Virchows Arch Pathol Anat Physiol Klin Med 1930:561–602. [Google Scholar]
- 3.Emile JF, Abla O, Fraitag S et al. Revised classification of histiocytoses and neoplasms of the macrophage‐dendritic cell lineages. Blood 2016;127:2672–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavalli G, Guglielmi B, Berti A et al. The multifaceted clinical presentations and manifestations of Erdheim‐Chester disease: Comprehensive review of the literature and of 10 new cases. Ann Rheum Dis 2013;72:1691–1695. [DOI] [PubMed] [Google Scholar]
- 5.Abdelfattah A, Arnaout K, Tabbara I. Erdheim‐Chester Disease: A comprehensive review. Anticancer Res 2014;34:3257–3262. [PubMed] [Google Scholar]
- 6.Campochiaro C, Tomelleri A, Cavalli G et al. Erdheim‐Chester disease. Eur J Int Med 2015;26:223–229. [DOI] [PubMed] [Google Scholar]
- 7.Haroche J, Cohen‐Aubart F, Emile JF et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim‐Chester disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood 2013;121:1495–1500. [DOI] [PubMed] [Google Scholar]
- 8.Hatzivassiliou G, Haling JR, Chen H et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS‐ versus BRAF‐driven cancers. Nature 2013;501:1–5. [DOI] [PubMed] [Google Scholar]
- 9.U.S. Food and Drug Administration . Zelboraf, Full Prescribing Information [PDF File]. Available at https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/202429s016lbl.pdf. Accessed March 2, 2018.