

Profiling olfactory neuronal cells from many individuals reveals variations in epigenetic signatures.
Abstract
As part of PsychENCODE, we developed a three-dimensional (3D) epigenomic map of primary cultured neuronal cells derived from olfactory neuroepithelium (CNON). We mapped topologically associating domains and high-resolution chromatin interactions using Hi-C and identified regulatory elements using chromatin immunoprecipitation and nucleosome positioning assays. Using epigenomic datasets from biopsies of 63 living individuals, we found that epigenetic marks at distal regulatory elements are more variable than marks at proximal regulatory elements. By integrating genotype and metadata, we identified enhancers that have different levels corresponding to differences in genetic variation, gender, smoking, and schizophrenia. Motif searches revealed that many CNON enhancers are bound by neuronal-related transcription factors. Last, we combined 3D epigenomic maps and gene expression profiles to predict enhancer-target gene interactions on a genome-wide scale. This study not only provides a framework for understanding individual epigenetic variation using a primary cell model system but also contributes valuable data resources for epigenomic studies of neuronal epithelium.
INTRODUCTION
Many epigenetic studies of neuronal cells use postmortem brain samples. However, the limited numbers of samples due to the requirement for postmortem tissue collection, the intermixed cell types in the tissues (such as postmitotic neurons and glial cells), the variable quality of the samples, and the relatively small numbers of cells per sample (which limits biochemical analyses) point to a need for neuronal cell culture systems, especially systems related to neurodevelopment. The olfactory neuroepithelium is one of the few regions in the human adult nervous system that displays continuous regeneration of nerve cells, and a relatively noninvasive nasal biopsy can be used to collect these cells. Although there are several types of cells in a nasal biopsy, pure populations of primary, nontransformed, proliferating neuronal progenitors can be obtained through specific cell culture conditions (1). These cultured neuronal cells derived from olfactory neuroepithelium (CNON) have transcriptomes similar to those of fetal brain tissues (2). Because CNON can be subjected to a large number of passages, they are ideal for genomic or epigenomic assays that require large cell numbers. In addition, as these cells are neural progenitors (1), they may serve as a good model for epigenetic studies of the neurodevelopmental component of certain brain disorders.
Cellular phenotype is determined by the collective contributions of all genes expressed in a given cell type. The level at which any individual gene is transcribed is determined by the combination of proximal and distal regulatory elements that increase or decrease gene expression. Recent studies from the ENCODE Consortium have identified more than 1.31 million regulatory elements with a small percentage of these elements active in any given cell type (www.encodeproject.org). This cell-type specificity in the active state of regulatory elements points to an absolute requirement that regulatory elements be mapped in each cell model system. Two types of DNA elements involved in gene activation include promoters and enhancers. Promoters are defined as genomic regions near transcription start sites (TSSs), and active promoters display a region of open chromatin spanning the TSS that is flanked on either side by a nucleosome containing histone H3 trimethylated on lysine 4 (H3K4me3). Enhancers have a smaller region of open chromatin flanked on either side by one or more nucleosomes containing histone H3 acetylated on lysine 27 (H3K27ac) and/or histone H3 monomethylated on lysine 4 (H3K4me1). In contrast, repressor elements are marked by histone H3 trimethylated on lysine 27 or lysine 9 (H3K27me3 or H3K9me3). All of these “regulatory signposts” can be identified using the technique of chromatin immunoprecipitation followed by sequencing (ChIP-seq). Further refinement of regulatory regions identified by ChIP-seq can be accomplished using nucleosome occupancy and methylome sequencing (NOMe-seq), a method that identifies nucleosome-depleted regions (NDRs) within regulatory elements and provides information concerning positions of the flanking nucleosomes at single-molecule resolution (3–5).
Regulatory elements can be located at quite far distances from target genes, but the current model in the field is that most regulatory interactions are contained within the same chromatin domain, known as a topologically associating domain (TAD), as the gene itself. Therefore, to develop a comprehensive regulatory map of a cell type, one can identify activation and repression elements genome wide and then assign each element to a specific TAD. TADs, which are ~500 kb to 1 Mb in size, are thought to be created by long-range chromatin loops mediated by the insulator protein CTCF, with one CTCF binding to each anchor and then interacting to form a loop. CTCF is also thought to influence enhancer-mediated gene regulation, functioning in both positive and negative ways. For example, CTCF may help to bring an enhancer closer [in three-dimensional (3D) space] to a target promoter via its ability to form loops with other CTCF sites. In contrast, binding of CTCF at a site between an enhancer and promoter can, in some cases, block long-range regulation (6).
To develop a comprehensive 3D epigenomic regulatory map of CNON, we began with nasal biopsies from 63 individuals. Neuroepithelial cells from the biopsied tissues were enriched using specific culture conditions (see the Supplementary Materials), and the resultant CNON were grown on plastic dishes coated with Matrigel for all assays. Using these primary CNON, we identified chromatin TADs and interaction loops, profiled regulatory elements genome wide, and identified NDRs of open chromatin. Furthermore, we identified individual variations (associated with genotypic or phenotypic classifications) in epigenetic signatures at regulatory elements, and using 3D chromatin maps, we predicted enhancer-target gene interactions (Fig. 1).
Fig. 1. Project overview.
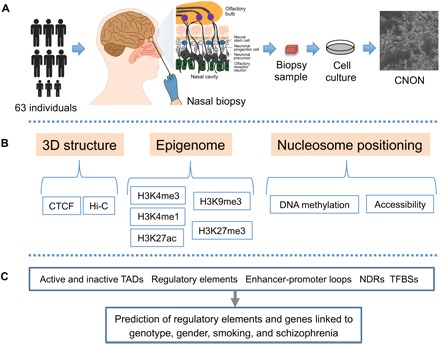
(A) To develop a comprehensive regulatory map of CNON, we began with nasal biopsies from 63 individuals. Neuroepithelial cells from the biopsied tissues were enriched using specific culture conditions, and the resultant CNON were grown on plastic dishes coated with Matrigel for all assays. (B) Using primary CNON, we performed in situ Hi-C (n = 2) and CTCF ChIP-seq (n = 33) to develop a 3D chromatin map, epigenomic profiling using ChIP-seq for modified histones (n = 56, 47, 4, 22, and 11 for H3K4me3, H3K27ac, H3K4me1, H3K27me3, and H3K9me3, respectively), and NOMe-seq (n = 5) to provide information concerning nucleosome positioning. (C) Using comprehensive epigenomic and chromatin maps, we identified TADs, chromatin interaction loops, and regulatory elements and defined NDRs of open chromatin [including transcription factor (TF) binding sites (TFBSs)]. Last, we identified regulatory elements having variation linked to genotype, gender, smoking, and schizophrenia (SCZ), as well as observed and predicted enhancer-target gene interactions for these olfactory neuroepithelial cells.
RESULTS
3D epigenomic maps of CNON that identify active and heterochromatic TADs
To develop a genome-wide 3D map of the chromatin structure of CNON, we performed in situ Hi-C using primary CNON from two different individuals (see table S1 for information concerning all genomic and epigenomic datasets). We first analyzed the SEP044 and SEP045 Hi-C datasets individually and identified TADs (fig. S1). Consistent with previous studies of other cell types (7–9), we identified 6800 TADs in CNON, having an average size of 405 kb (table S2). The boundaries of several TADs and a chromatin interaction map for a 700-kb region of chromosome 19 are shown in Fig. 2A.
Fig. 2. Creation of a 3D epigenomic map of active and heterochromatic TADs in CNON.
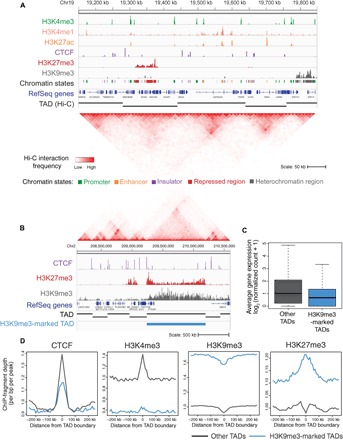
(A) A browser snapshot of the H3K4me3, H3K4me1, H3K27ac, CTCF, H3K27me3, and H3K9me3 patterns for a 700-kb region of chromosome (Chr) 19p13.11. All datasets shown are from CNON taken from an individual designated SEP044. A track indicating chromatin state, the RefSeq gene track, a track indicating the identified TADs, and a Hi-C chromatin interaction browser snapshot for the region are also shown. (B) A browser snapshot of the CTCF, H3K27me3, and H3K9me3 patterns for a 2.5-Mb region of chromosome 2 that harbors H3K9me3-marked TAD. The Hi-C interaction data, the RefSeq gene track, a track indicating all TADs within the region, and a track indicating an H3K9me3-marked TAD are shown. (C) The average expression level of genes in H3K9me3-marked TADs (blue) and all other TADs (dark gray). (D) The ChIP fragment depth for CTCF, H3K4me3, H3K9me3, and H3K27me3 datasets, centered on TAD boundaries. Profiles of H3K9me3-marked TADs are shown in blue, and profiles of the other TADs are shown in dark gray.
Although the boundaries between TADs are often conserved across cell types from different origins (8), the epigenetic chromatin states of TADs can differ between cell types (10), with dynamic expression levels of genes in the same TAD showing similar changes during cell differentiation (11, 12). These previous findings highlight the importance of characterizing the epigenomic profiles of CNON TADs. To develop epigenomic maps in CNON, we performed ChIP-seq for histone modifications associated with promoters and enhancers (H3K4me3, H3K27ac, and H3K4me1), for repressed and heterochromatic regions (H3K27me3 and H3K9me3), and for a DNA binding protein associated with establishment of chromatin structural domains (CTCF). To ensure that the ChIP-seq datasets were of high quality, we performed experiments using CNON from SEP044 and SEP045, and reproducibility was confirmed using the ENCODE3 ChIP-seq pipeline (https://github.com/ENCODE-DCC/chip-seq-pipeline). After identifying regions enriched for each of the histone modifications and/or CTCF in SEP044 and SEP045, a sequential classification scheme (see the Supplementary Materials) was used to sort the elements into five different chromatin states (table S3). An example signal track for each type of ChIP-seq dataset is shown in Fig. 2A; details concerning the ChIP-seq datasets used in this study can be found in table S1.
Although most TADs were associated with active chromatin, we identified 830 TADs that have more than 50% of their genomic region covered by the heterochromatic mark H3K9me3 (H3K9me3-marked TADs; Fig. 2B and table S2); these TADs have a median of five genes per TAD (as compared to a median of seven genes per TAD in the other 5970 TADs). On average, genes within H3K9me3-marked TADs are expressed at considerably lower levels than are genes in other TADs (Fig. 2C). Plotting the ChIP-seq signals at the boundaries of the H3K9me3-marked TADs (Fig. 2D), we found that, as with most TADs, the boundaries are flanked by CTCF. As expected, we found that H3K4me3 signals are lower and H3K9me3 signals are higher in the H3K9me3-marked TADs. There is a dip of H3K9me3 and a peak of H3K27me3 at the boundaries of the H3K9me3-marked TADs (Fig. 2D). Peaks of H3K27me3 were not found at the boundaries of other TADs (see Fig. 2B for an example of an H3K9me3-marked TAD that is covered by H3K9me3 with H3K27me3 signals at the borders). Although different protein complexes, such as G9a/GLP and polycomb repressive complex 2 (PRC2), create H3K9me3 and H3K27me3, respectively, it has been suggested that a cross-talk between H3K9 and H3K27 methylation can cooperate to maintain silencing (13). Thus, PRC2 may assist in creating or stabilizing the loops (14, 15) in the inactive H3K9me3-marked CNON TADs.
Predicting target genes of CNON enhancers using 3D epigenomic maps
After identifying promoters, enhancers, insulators, and repressed and heterochromatic regions in CNON (Fig. 3A and table S3), our next goal was to associate target genes with these regulatory elements. It is easy to assign a target gene to regulatory elements near a TSS. However, many H3K27ac marks and CTCF sites are far from a TSS and are thought to interact with target promoters via long-range loops. Therefore, we used the Hi-C chromatin interaction data to assist in linking distal CNON regulatory elements to target genes. First, we called intrachromosomal chromatin interactions from each of the Hi-C datasets at 10-kb resolution, after adjusting the local background using the Fit-Hi-C program (q < 1 × 10–12; table S4) (16). Next, to gain insight into the function of the identified chromatin interactions, we classified the chromatin interactions according to the type of regulatory elements at one or both ends (Fig. 3B and table S5). For these analyses, we used a merged Hi-C dataset to capture as many as possible promoter-enhancer chromatin interactions at high resolution (5-kb resolution). We found that the most frequent chromatin interactions have active regulatory elements such as CTCF at one or both ends (Fig. 3B) and that chromatin interactions that have repressed or heterochromatic anchors are less frequent than ones that have active regions at anchors (fig. S2). We identified 115,039 interactions (5-kb resolution) that have at least one end mapping to an active promoter (q < 0.05). These interactions encompassed 79% of promoters marked by H3K4me3 in CNON cells (13,738 of 17,427 active promoters), with a median number of four chromatin interactions per promoter. However, among the promoter-anchored interactions, only a small number of interactions (12,970 of 115,039) were between a promoter and an enhancer, with a median number of two interactions for 9276 different promoters.
Fig. 3. Identification of target genes of enhancers active in CNON cells.
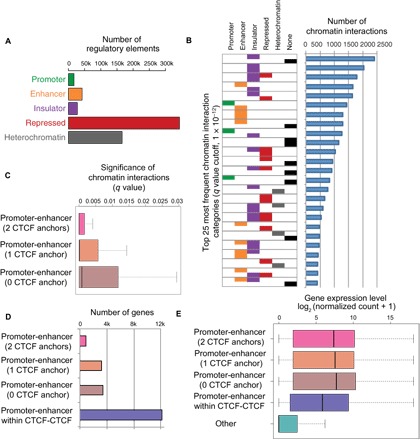
(A) The number of regulatory elements identified. (B) The 25 most frequent chromatin interaction categories. Left: For each category, each of the two ends of the interactions is identified by one or more of the five types of regulatory elements described in (A) (none indicates that one or both ends of the interaction did not correspond to a regulatory region). Right: The number of interactions in each category, using a q value cutoff of 1 × 10−12 (see also fig. S2). (C) The chromatin interaction frequencies of promoter-enhancer interactions that have 2, 1, or 0 CTCF anchors. (D) The number of genes in each category of promoter-enhancer interactions and the number of promoter-enhancer predicted pairs (not identified by direct chromatin interaction data) within CTCF-CTCF loops. (E) The expression level of genes in each category from (D). The expression level of all other genes in CNON is shown.
To further study the association of chromatin interactions and gene activation, we selected the subset of interactions identified in the Hi-C datasets that occur between promoters and enhancers. We next determined whether the presence of a CTCF peak at one or both anchors strongly influenced the promoter-enhancer interaction strength. We found that among the 12,970 promoter-enhancer interactions, 1063 had CTCF at both anchors, 5731 had CTCF at one anchor, and 6176 did not have CTCF at either anchor. The q values of each of these subgroups are shown in Fig. 3C. Similar to studies of other cell types (17, 18), the promoter-enhancer interactions identified by the CNON Hi-C datasets that have a CTCF peak at both anchors have the lowest q values, suggesting that these are the strongest type of promoter-enhancer interactions. However, the median q value for all of the promoter-enhancer loop categories is quite low, providing strong support for this set of experimentally observed interactions.
The studies described above provide direct chromatin interaction evidence that allows determination of target genes of a set of CNON enhancers. However, an additional set of target genes of enhancers can also be predicted using chromatin interaction data. For example, it is thought that most enhancers do not cross loop boundaries but rather regulate promoters that are within the same chromatin loop (19). Specifically, previous studies (20, 21) have identified promoter-enhancer regulatory pairs that occur within strong CTCF-CTCF interaction loops but which were not identified by direct observation of chromatin loops between the promoter and enhancer. Therefore, we further analyzed the CTCF-CTCF interaction loops that do not have promoters or enhancers at their anchors. We identified all active promoters (as determined by the presence of H3K4me3 at the TSS) and all active enhancers (as determined by the presence of H3K27ac at a nonpromoter site) within CTCF-CTCF interaction loops. These analyses identified ~17,000 active genes that reside within a CTCF-CTCF interaction loop that also contains at least one active enhancer. Some of these genes had already been identified in the set of experimentally observed promoter-enhancer pairs described above; however, the intraloop analysis identified an additional 12,120 unique genes that could be putatively paired with a distal enhancer. Thus, in total, we identified 5623 genes involved in direct promoter-enhancer chromatin interactions and predicted 12,120 additional enhancer-regulated genes within CTCF-CTCF interaction loops (Fig. 3D and table S6). Notably, all categories of genes involved in direct promoter-enhancer interactions had higher expression levels than did the genes in the set of predicted promoter-enhancer pairs (for which no direct interactions were observed in the Hi-C data; adjusted P < 3.1 × 10−7; Fig. 3E).
Assessing individual variation in epigenomic profiles of active regulatory elements
In the analyses above, we used ChIP-seq peaks from CNON obtained from two individuals. Although this approach provides a high-confidence peak set, many regulatory elements could be missed because of individual-to-individual variation and uncontrollable technical issues in ChIP assays. Therefore, to obtain a comprehensive view of the CNON epigenome, we performed H3K4me3, H3K27ac, and CTCF ChIP-seq using CNON derived from 63 individuals in an attempt to identify all possible promoters, enhancers, and insulators active in this cell type. The set of ChIP-seq peaks for each mark was subjected to a cluster analysis to ensure that outlier datasets were not included in our analyses (fig. S3); the final datasets include 56 H3K4me3, 47 H3K27ac, and 33 CTCF ChIP-seq samples (tables S1 and S7).
To determine how many additional peaks can be identified using a larger set of individuals, we plotted the number of newly found peaks in each subsequently analyzed dataset, with promoter (defined as ±2 kb from a TSS) and nonpromoter peaks in each dataset being separately analyzed. We identified ~13,000 to 18,000 sites for each mark at promoter regions for a single individual; in addition, we identified ~52,000 to 69,000 nonpromoter sites for each mark for a single individual (table S7). H3K4me3 is normally associated with promoter regions. The nonpromoter H3K4me3 signals may be due to the presence of previously uncharacterized (alternative) promoters, to H3K4me3 spreading over very broad domains (22), to RNA polymerase II initiating transcription at an enhancer, or to capturing the H3K4me3 mark at a looped enhancer region (23, 24). Many other studies have shown that both proximal and distal CTCF peaks can be robust and involved in chromatin interactions (17, 25); therefore, it was expected to identify promoter and nonpromoter CTCF peaks. In addition, of course, H3K27ac can be found at both promoters and distal enhancers. We note that the vast majority of the peaks in the individual sets of H3K4me3, CTCF, or H3K27ac peaks located in promoter regions are common across samples. However, nonpromoter peaks for each mark were more variable (Fig. 4A); we found that only 1613 nonpromoter H3K4me3 peaks are common to all 56 individuals, 16,295 nonpromoter CTCF peaks are common to all 33 individuals, and 10,962 nonpromoter H3K27ac peaks are common to all 47 individuals. When we analyzed only those sites that are found in at least 20% of the individuals (Fig. 4B), we identified 16,711 H3K4me3, 12,871 CTCF, and 16,100 H3K27ac promoter peaks and 40,125 H3K4me3, 50,314 CTCF, and 61,419 H3K27ac nonpromoter peaks (table S7). We note that, for each mark, the set of nonpromoter peaks that were in less than 20% of the individuals was, on average, considerably smaller than the other sets of peaks (Fig. 4C). In addition, when we selected peaks that are found in only one individual, we found that the signals of those peaks were very low. Therefore, it is possible that some of the elements found in only one or two samples could be false positives.
Fig. 4. Assessing individual variation in epigenomic profiles of active regulatory elements.
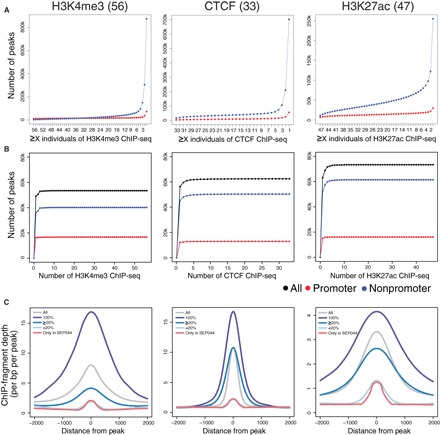
ChIP-seq was performed from 63 individuals; after quality assessment (see fig. S3), peak sets were retained for 56 H3K4me3 datasets, 33 CTCF datasets, and 47 H3K27ac datasets. (A) The number of peaks shared in the different sets of H3K4me3, CTCF, and H3K27ac ChIP-seq samples. (B) The cumulative number of unique peaks in the merged datasets with increasing numbers of different ChIP-seq samples. Only peaks found in at least 20% of individuals are plotted. (C) A comparison of the size of the SEP044 peaks in the set of H3K4me3, CTCF, and H3K27ac peaks present in every individual ChIP-seq peak sets (100%), in at least 20% of all samples (≥20%), in less than 20% of all samples (<20%), and only in the SEP044 sample. The tag density plot is centered on the middle of the peak (x axis), and the read depth is indicated by y axis.
Enrichment of neuronal-related transcription factor motifs in CNON regulatory regions
To gain insight into the TFs that establish the transcriptome of a given cell type, one can perform motif analysis of the regulatory regions. Because enhancers are thought to be responsible for cell identity (26, 27), we have focused on CNON enhancers for motif studies. H3K27ac ChIP-seq peaks are quite broad, at minimum spanning two nucleosomes. Therefore, motif analysis using the entire genomic span of the H3K27ac peaks could identify TF motifs that are at the edges of a ChIP signal; these would likely be false positives (not actually occupied by a TF). Therefore, we have used NOMe-seq to identify NDRs within the large H3K27ac-marked regions. NOMe-seq is a genome-wide method that uses endogenous CpG DNA methylation levels along with accessibility of GpC dinucleotides to exogenous methylation to precisely identify nucleosome-free regions at single-molecule resolution (3–5). Regions of at least 140 bp that have high levels of GpCm and low levels of CmpG are defined as NDRs (see the Supplementary Materials). By intersecting the NOMe-seq data with the larger H3K27ac ChIP-seq peaks, we can precisely define the TF binding platforms in active enhancer regions (Fig. 5A).
Fig. 5. Motifs enriched in CNON regulatory regions identify neuronal-related TFs.
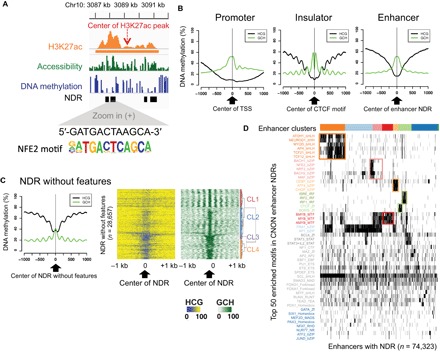
(A) An example of a single H3K27ac ChIP-seq peak and the position of the NDRs within the peak. One NDR identifies an NFE2 motif. (B) The average endogenous DNA methylation level [HCG (where H = A, C, or T): black] and the accessibility [GCH (where H = A, C, or T): green] of NDRs at promoters (16,711 H3K4me3 peaks identified in ≥20% of all samples) centered on the TSS, insulators (62,433 CTCF peaks identified in ≥20% of all samples) centered on the CTCF motif, and enhancers (61,419 H3K27ac peaks identified in ≥20% of all samples) centered to the middle of the NDR. (C) Left: The average endogenous DNA methylation level (HCG: black) and the accessibility (GCH: green) for the NDRs without features. Right: A cluster analysis of the NDRs without features based on levels of accessibility. Endogenous DNA methylation for the same NDRs is shown in the middle. (D) A cluster analysis of the top 50 TF binding motifs identified using the 74,323 NDR regions contained within distal CNON H3K27ac peaks.
Because the number of NDRs identified is highly correlated with sequencing depth (fig. S4), we generated NOMe-seq datasets from CNON from five different individuals (table S1), sequencing between 700 million and 1.1 billion read pairs for each library. To select robust NDRs, we first applied stringent cutoffs (P < 10−15) for called NDRs from each NOMe-seq dataset. We next took the union of the NDRs from the most sequenced library (from SEP030; sequenced at 1.1 billion read pairs) and the NDRs found in at least two of the other libraries, identifying 166,240 NDRs (table S8). We then classified the NDRs as overlapping a promoter, an active enhancer, or an insulator, using the ChIP-seq peaks that were found in more than 20% of the samples in each set of regulatory elements (table S7). We note that 82% of the NDRs overlap with these three classes of regulatory elements; 45,793 NDRs overlap promoter regions, 58,059 NDRs overlap insulator regions, and 74,323 NDRs overlap enhancer regions (table S8). As expected, the opposite patterns of CpG and GpC methylation can be seen at promoters, insulators, and enhancers (Fig. 5B). The promoter NDRs have a large region of open chromatin, as defined by a broad region that has low levels of endogenous CpG methylation, a peak of nucleosome-free chromatin surrounding the TSS, and phased nucleosomes downstream of the TSS. The insulator NDRs have a narrow region of open chromatin with low levels of endogenous CpG methylation and highly phased nucleosomes on either side of the bound CTCF (the position of the tightly bound CTCF can be visualized by the dip in open chromatin centered at the CTCF motif). The enhancer NDRs also have a narrow region of open chromatin with low levels of endogenous CpG methylation; the phasing of nucleosome in the enhancer NDRs is not as prominent as the phasing of nucleosomes surrounding CTCF sites.
We identified 28,657 NDRs that do not overlap with the three classes of active regulatory elements (designated as “NDRs without features”). These NDRs have two notable characteristics (Fig. 5C). First, they have slightly higher levels of endogenous DNA methylation than active regions and smaller peaks of open chromatin. Second, they have very robust nucleosome phasing on either side of the NDR, suggesting that these NDRs are not false positives but instead are highly structured regulatory regions. Plotting the endogenous CpG methylation and the exogenous GpC methylation reveals four clusters of these NDRs. The tightly phased nucleosomes suggest that the NDRs in cluster 1 may be binding sites for CTCF or pioneer TFs that can access nucleosomes (28). The methylation patterns of the NDRs in cluster 2 suggest that they may be poised enhancers or distal repressor elements (which are not marked by H3K27ac). The NDRs in clusters 3 and 4 have similar nucleosome phasing patterns as promoters, but the region of low DNA methylation is narrower than at active promoters.
We next performed motif analysis using the 74,323 NDRs contained within the distal H3K27ac peaks found in more than 20% of individuals (Fig. 5D). We found that the enhancer NDRs are enriched for several different clusters of motifs. For example, the orange cluster is enriched for motifs for basic helix-loop-helix (bHLH) TFs, such as ATOH1, NEUROD1, and MYOD1; motifs for bHLH TFs have been previously identified in brain-specific enhancers (29). Another group of enhancer NDRs (the pink cluster) is enriched for motifs for basic leucine zipper domain TFs, such as BACH1, BACH2, NFE2, and MAF; BACH2, NFE2, and MAF are involved in neuronal processes (30, 31). The group of enhancers identified by the red cluster is enriched for motifs for the MYB family; MYB family members are involved in regulating stem cell populations (32, 33). The green cluster includes motifs for CTCF family members, as well as interferon regulatory factor and signal transducers and activators of transcription family members, which are known to cooperate in transcriptional regulation (34, 35).
Characterization of factors that affect individual variation in enhancers
To further investigate variability in distal regulatory elements, we correlated changes in the strength of the H3K27ac peaks in individual CNON samples with variables such as genotype, gender, smoking, and disease. We first examined genetic variation in the set of H3K27ac sites using data from genotyping arrays (table S1) (2). We found that of the 254,705 nonpromoter H3K27ac peaks that are found at least once among the 47 samples, 165,482 H3K27ac peaks had at least one single-nucleotide polymorphism (SNP) underneath the peak. Of these 165,482 SNP-containing peaks, 4167 showed a statistically significant correlation between genotype and peak strength (adjusted P < 0.05; table S9A); the 4167 unique H3K27ac peaks showed a correlation of peak strength with 8847 unique SNPs, giving 8847 peak SNP pairs. Furthermore, as a SNP may affect the binding of TFs that recruit histone acetyltransferases, which could affect H3K27ac peak signals, we determined whether any of the genotype variation that shows a correlation with H3K27ac peak size alters a TF motif that lies within an NDR. An example of an H3K27ac site that is located in a gene desert region of chromosome 10 and that shows a correlation of peak strength with the genotype of rs1757069 is shown in Fig. 6A (top); the H3K27ac peak shows a correlation of signal strength that varies according to the sequence of the SOX3 motif at the two alleles at rs1757069 (Fig. 6A, bottom).
Fig. 6. Characterization of factors that affect individual variation in enhancers.
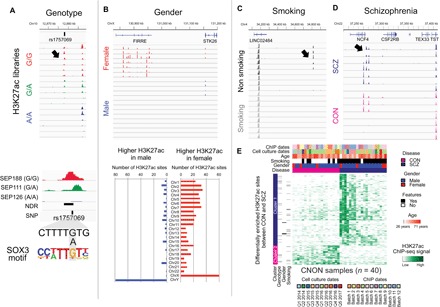
(A) Top: A browser snapshot of an H3K27ac peak that shows peak strength variation as a correlation with genotype of rs1757069. Bottom: A correlation between the peak height and the making or breaking of a SOX3 motif by rs1757069. (B) Top: A browser snapshot of a cluster of H3K27ac peaks that show peak strength variation as a correlation with gender. Bottom: The number of H3K27ac peaks on each chromosome that are differentially enriched in male or female CNON samples. (C and D) A browser snapshot of an H3K27ac peak that shows peak strength variation as a correlation with smoking (C) or SCZ (D). (E) A heatmap representing unsupervised clustering of differentially enriched H3K27ac sites between SCZ and CON. Cluster 1 is higher in SCZ, and cluster 2 is higher in CON. Individual sample metadata and ChIP-seq data information are on the top of the heatmap, whereas the H3K27ac sites linked to genotype, gender, and smoking that are among the H3K27ac differentially associated with disease state are indicated on the left.
We next determined whether we could identify factors other than genotype that correlate with H3K27ac ChIP-seq signal strength. We found 619 H3K27ac peaks that showed a signal difference between male (n = 33) and female (n = 7) samples [false discovery rate (FDR) < 0.05, empirical P < 0.05; table S9B]. Among these 619 sites, 150 sites had a stronger signal in males than in females (with 79 of these 150 sites being located on ChrY), and 469 sites had a stronger signal in females than in males (with 60 sites of these 469 sites being located on ChrX). As an example, female-linked H3K27ac sites include ones in the long noncoding RNA FIRRE locus (Fig. 6B), which helps maintain an important epigenetic feature of the inactive X chromosome in female (36). We also identified 114 H3K27ac peaks that showed different signal strengths between smoking and nonsmoking individuals (FDR < 0.05, empirical P < 0.05; Fig. 6C and table S9C). We attempted to identify H3K27ac peaks that correlated with age but could not do so due to the fact that most of the individuals that provided the biopsies were of similar ages. SCZ is a heritable psychiatric disorder often associated with defects in olfactory perception; because CNON cells are derived from the olfactory neuroepithelium, it has been suggested that CNON may be a good source for studying SCZ (1). When the CNON biopsies were taken, the subjects were classified as having a diagnosis of SCZ or as nonschizophrenic controls (CON). To investigate whether any of the variable H3K27ac peaks were associated with a diagnosis of SCZ, we selected the H3K27ac datasets from 20 SCZ and 20 CON biopsies and confirmed that the samples were correctly classified by genotyping the ChIP-seq datasets. Then, we compared the H3K27ac signals between CON and SCZ samples. We identified 607 H3K27ac sites that are differentially enriched (FDR < 0.05, empirical P < 0.05; table S9D), with 479 sites having a higher signal in SCZ than in CON samples and 128 sites having a higher signal in CON than in SCZ samples. For example, an H3K27ac site that is located near the NCF4 gene shows a higher signal in a subset of SCZ samples than of CON samples (Fig. 6D). NCF4 has previously been suggested to be associated with SCZ and bipolar disorder (37, 38). We note that most of the disease-associated sites we identified were not identified in the correlations analyzing genotype, gender, or smoking status (Fig. 6E).
Identification of SCZ risk-associated regulatory elements that are active in CNON
SCZ is highly heritable, but few genes have been linked to the disease. To gain insight into the genetic mechanisms of SCZ, several groups have used genome-wide association studies (GWAS) to identify more than 100 genomic loci significantly associated with increased risk for the disease (39, 40). However, GWAS is performed using arrays, which only allow identification of an index variant for each of the identified loci. Functional characterization of the risk-associated loci requires analysis not only of the index variants but also of all variants inherited with the index variants. We, and others, have taken the approach of focusing on variants that fall within regulatory elements (7, 41–45), under the assumption that these variants are more likely to be the causal variants. Several previous studies have characterized SCZ-associated risk loci using epigenomic profiles (46, 47). However, these studies used primary tissues; thus, further follow-up experiments were not practical. In contrast, CNON represent neuronal tissues that can be expanded in culture for functional studies. Therefore, as a complementary study to other post-GWAS studies performed in different cell types and tissues, we identified SCZ risk-associated variants located in regulatory regions of CNON (Fig. 7). We began with 224 SCZ-related GWAS index variants, located at 132 different genomic loci (39, 40), and then selected index and high LD (linkage disequilibrium) variants (R2 > 0.5) that fell within CNON regulatory elements (H3K4me3, H3K27ac, and CTCF ChIP-seq peaks and NDRs) using the FunciSNP tool (48). Then, we further refined the set of regulatory variants to only include those variants that are both in high LD with the index variant and located within the same TAD or CTCF-CTCF interaction loop as the index variant, under the assumption that variants, even if in high LD with an index variant, would not influence expression of the same genes as the index variants unless they were in the same TAD or CTCF-CTCF interaction loop. Using this classification scheme, we identified 2891 variants from 118 SCZ risk-associated loci located in regulatory elements active in CNON (table S10).
Fig. 7. A workflow for identifying SCZ risk-related variants using CNON epigenomic and Hi-C data.
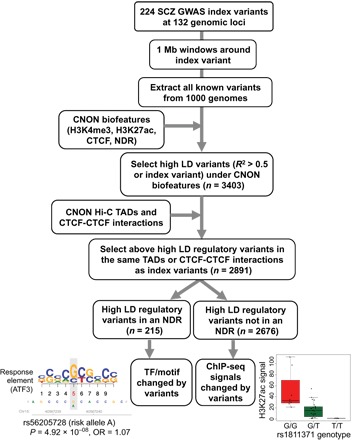
Upon overlapping CNON active regulatory elements found from 63 individuals with SCZ GWAS risk-associated high LD variants, 3403 high LD variants were identified. Among those, 2891 high LD variants are located either within the same TAD or CTCF-CTCF interaction loop as the index variant; these were used for further analysis. The high LD regulatory variants located in an NDR (n = 215) were used to identify TF motifs changed by the variants, whereas the other 2676 high LD regulatory variants were examined for correlation with ChIP-seq signal strength; examples of altered motifs and peak strength between risk and nonrisk alleles are shown. OR, odds ratio.
One explanation for how a SNP located outside of a coding region can influence disease risk is that it may alter TF binding potential in a regulatory element. As noted above, the TF binding platform of a regulatory element identified by ChIP-seq can be further refined by intersection with the set of NDRs. Of the set of 2891 identified SCZ risk-associated variants, 215 fell within an NDR, with most of these variants being located in promoters and enhancers. Further analysis identified a subset of these variants that alter the motif of a known TF or TF binding, as determined using ENCODE TF ChIP-seq data (table S10). We also found a subset of SCZ risk-associated variants that are highly correlated with ChIP-seq signals (table S10).
Functional significance of individual variation in distal regulatory elements
We have identified individual variation in H3K27ac peak strength that can be correlated with genotype, sex, smoking status, disease diagnosis, and SCZ-risk associated SNPs. It is possible that the variation in the H3K27ac peak strength may, in some cases, result in differences in expression levels of target genes in different individuals. To identify genes that may be regulated by these variable H3K27ac sites, we selected (i) the set of target genes determined by the direct promoter-enhancer chromatin interaction data, (ii) the set of target genes predicted using the CTCF-CTCF looping data, and (iii) the set of target genes predicted using TAD information (Fig. 8A and table S6). When we correlated expression levels of putative target genes with enhancer signals, we identified 12,620 promoter-enhancer pairs for 5115 genes that had a statistically significant correlation between the strength of the H3K27ac signal and the expression level of the predicted target gene (adjusted P < 0.05; table S11). As an example, expression of the CASK gene is positively correlated with the strength of the H3K27ac signal in ChIP-seq datasets taken from many different individuals; the correlated enhancer is located 170 kb from the promoter of the CASK gene (Fig. 8A). Gene ontology analyses identified genes involved in hormone receptor binding in the potential target genes of enhancers linked to gender, genes involved in flavonoid and drug metabolic processes as potential target genes of enhancers linked to smoking, and genes involved in regulation of neurodevelopmental processes in the set of SCZ-linked enhancers (Fig. 8B).
Fig. 8. Finding target genes of enhancers using 3D epigenomic datasets from many individuals.
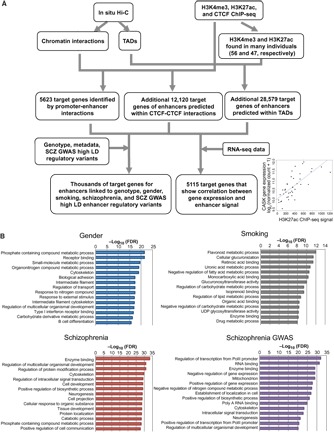
(A) Schematic overview of the process used to identify target genes of CNON enhancers. In situ Hi-C datasets from two individuals (SEP044 and SEP045) were used to reveal chromatin interactions and TADs. Using ChIP-seq peaks from the same individuals, 5623 target genes were identified by direct promoter-enhancer chromatin interactions, and an additional 12,120 target genes of enhancers were predicted. In addition, using H3K4me3 and H3K27ac ChIP-seq data found from many individuals, additional enhancer target genes were predicted on the basis of colocalization within the same TADs. Last, the subsets of target genes of enhancers linked to gender, smoking, and SCZ, as well as 5115 target genes that show a correlation between gene expression and H3K27ac peak strength, was identified. (B) Gene ontology analyses for the target genes of enhancers linked to gender or smoking, differentially present in SCZ samples, or identified by SCZ GWAS variants. UDP, uridine diphosphate.
DISCUSSION
We have performed epigenomic profiling of CNON using biopsies from 63 individuals, identifying hundreds of thousands of regulatory elements and NDRs. Using Hi-C data, we identified TADs and regulatory elements involved in chromatin interactions, predicting a large set of enhancer-target gene interactions for CNON. By integrating genotype and metadata, we identified enhancers having epigenetic patterns linked to genetic variation, gender, smoking, and SCZ. Motif searches within NDRs revealed that many CNON enhancers are bound by neuronal-related TFs and that target genes of enhancers linked to SCZ are related to development and neurogenesis. These findings suggest that CNON may be a useful model system for understanding neurodevelopment. In summary, we provide comprehensive epigenomic and 3D chromatin maps of an understudied primary neuroepithelial cell model.
A current model in the field is that long-distance gene regulation is achieved by the interaction of distal enhancers with the promoter regions of their target genes. To study promoter-enhancer chromatin interactions, we developed a high-resolution 3D epigenomic map of CNON by combining in situ Hi-C chromatin interaction data with epigenomic ChIP-seq data (using ChIP-seq data from the same individuals as used for Hi-C), identifying ~13,000 chromatin interactions that directly link an active enhancer to an active promoter; this represents ~53% of promoters with H3K4me3 mark and ~30% of distal H3K27ac sites in CNON cells. These results suggest that only a subset of promoters and enhancers that are active in CNON engage in chromatin interactions that are detectable by Hi-C. It is possible that some promoter-enhancer interactions are too weak or transient to detect using Hi-C data. Therefore, we also identified pairs of active promoters and enhancers located within CTCF-CTCF interaction loops. The target genes directly linked to active enhancers via chromatin interaction data are expressed higher than other genes in CNON cells.
Although it is recognized that there may be individual differences in active enhancers in a given cell type, few studies have examined genome-wide enhancer profiles using primary biopsy samples from many individuals. Because CNON are obtained from a relatively noninvasive procedure (and can be grown to large cell numbers), we were able to compare genome-wide enhancer profiles from 47 people. Although only ~11,000 active enhancers were common to all 47 individuals, we identified a union set of 110,000 active enhancers that were detected in two or more individuals. By integrating metadata of the samples, we identified distinct sets of enhancers linked to genetic variation, gender, and smoking.
We have also taken advantage of our comprehensive epigenomic datasets to study SCZ, a neurodevelopmental psychiatric disorder. We identified 607 enhancers differentially enriched between SCZ and CON groups, which clustered into different subgroups of individuals with SCZ; further studies with increased sample sizes may reveal unique characteristics of these different subgroups of patients with SCZ. Moreover, using GWAS SNPs linked to SCZ, we also identified 2891 SCZ risk-associated variants in promoters, enhancers, or insulators that are active in CNON. We note that other studies (46), as well as colleagues in the PsychENCODE Consortium (49, 50), have used different neuronal cell types to classify SCZ GWAS SNPs. However, because enhancers are very cell type specific (even when comparing different cell types from primary brain tissues), each additional neuronal cell type studied adds new information regarding the genetic heritability of SCZ. Last, by integrating ChIP-seq, NOMe-seq, Hi-C, RNA sequencing (RNA-seq), and genotype data, we predicted target genes of enhancers enriched in SCZ ChIP-seq datasets and of enhancers harboring SCZ risk-associated SNPs. The identification of neurogenesis as an enriched category in both sets of target genes is consistent with the classification of SCZ as a developmental disease.
In conclusion, we provide a comprehensive genomic and 3D epigenomic profile of CNON, which are a renewable source of primary human neuroepithelial cells that can be grown to large cell numbers. We have capitalized on these unique characteristics of CNON to study individual variation of regulatory elements. The 3D epigenomic map of cultured neuronal cells derived from olfactory epithelium biopsies from many living individuals, generated in this study, will serve as a great resource for future studies that investigate regulatory elements in primary neuroepithelial cells.
MATERIALS AND METHODS
CNON were derived from nasal biopsy tissue samples obtained from 63 individuals. The study was approved by the University of Southern California Institutional Review Board, and informed consent was obtained from all individuals. Most patients and CON subjects were recruited from participants of the National Institutes of Health–funded GPC (Genomic Psychiatry Cohort) study (1R01MH085548), and some patients were recruited through Los Angeles County/University of Southern California outpatient psychiatric clinics.
In situ Hi-C experiments were performed following the original protocol by Rao et al. (17) with minor modifications, such as the use of a four-cutter restriction enzyme (MboI) instead of a six-cutter restriction enzyme (7). Hi-C datasets were processed using the HiC-Pro (51), TADs were identified using the TopDom program (9), and intrachromosomal loops were selected using Fit-Hi-C (16). ChIP-seq data were processed using the ENCODE3 ChIP-seq pipeline (www.encodeproject.org/chip-seq/). All sequencing data were mapped to hg19 (see table S1). Using the DiffBind R package (52), ChIP-seq signals were normalized and compared across samples. HOMER (http://homer.ucsd.edu/homer/) was used to perform motif analyses and generate plots. NOMe-seq data were aligned to a bisulfite-converted genome using BSMAP (53) and processed to call NDRs as previously described (3). CNON RNA-seq and genotype data were processed as previously described (2). The FunciSNP R package (48) was used to find high LD regulatory variants active in CNON. The motifbreakR R package (54) was used to search for TF motifs overlapping the SCZ-related variants, and ENCODE TF ChIP-seq data were used to investigate TF binding. Detailed description of methods can be found in the Supplementary Materials.
Supplementary Material
Acknowledgments
Data were generated as part of the PsychENCODE Consortium, supported by U01MH103392, U01MH103365, U01MH103346, U01MH103340, U01MH103339, R21MH109956, R21MH105881, R21MH105853, R21MH103877, R21MH102791, R01MH111721, R01MH110928, R01MH110927, R01MH110926, R01MH110921, R01MH110920, R01MH110905, R01MH109715, R01MH109677, R01MH105898, R01MH105898, R01MH094714, and P50MH106934 awarded to S. Akbarian (Icahn School of Medicine at Mount Sinai), G. Crawford (Duke University), S. Dracheva (Icahn School of Medicine at Mount Sinai), P. Farnham (University of Southern California), M. Gerstein (Yale University), D. Geschwind (University of California, Los Angeles), F. Goes (Johns Hopkins University), T. M. Hyde (Lieber Institute for Brain Development), A. Jaffe (Lieber Institute for Brain Development), J. A. Knowles (University of Southern California), C. Liu (SUNY Upstate Medical University), D. Pinto (Icahn School of Medicine at Mount Sinai), P. Roussos (Icahn School of Medicine at Mount Sinai), S. Sanders (University of California, San Francisco), N. Sestan (Yale University), P. Sklar (Icahn School of Medicine at Mount Sinai), M. State (University of California, San Francisco), P. Sullivan (University of North Carolina), F. Vaccarino (Yale University), D. Weinberger (Lieber Institute for Brain Development), S. Weissman (Yale University), K. White (University of Chicago), J. Willsey (University of California, San Francisco), and P. Zandi (Johns Hopkins University). We thank the ENCODE Project Consortium and Roadmap Epigenomics Mapping Consortium for data access, the USC Center for High-Performance Computing (https://hpcc.usc.edu/), the UCLA Technology Center for Genomics and Bioinformatics, and Huy Dinh for advice on bioinformatics analysis. Funding: This work was supported by the following grants from the NIH: NIH U01MH103346A, NIH R01CA136924, NIH K01CA229995, and NIH R01MH086874. Author contributions: S.K.R.: all ChIP-seq, NOMe-seq, Hi-C, RNA-seq, genotype datasets, GWAS analyses, and writing manuscript; S.S.: production of ChIP-seq and NOMe-seq datasets; H.W.: production of ChIP-seq datasets; C.A.: genotyping of genomic datasets and processing of RNA-seq datasets; F.D.L.: production of Hi-C datasets; A.C.: sequencing genomic datasets; V.N.S.: cell culture; Y.G.: functional assay guidance; B.P.B.: development of the NOMe-seq pipeline; O.V.E.: sequencing guidance; J.A.K.: overall project guidance; and P.J.F.: overall project guidance and writing manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data were produced as part of PsychENCODE Consortium, and data used in this study can be accessed at psychencode.org. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/12/eaav8550/DC1
Supplementary Materials and Methods
Fig. S1. CNON Hi-C datasets.
Fig. S2. Chromatin interactions with different q value cutoffs.
Fig. S3. H3K27ac ChIP-seq clustering results.
Fig. S4. NOMe-seq depth and number of NDRs.
Table S1. Information about the CNON ChIP-seq, NOMe-seq, and Hi-C datasets.
Table S2. CNON TADs identified by Hi-C.
Table S3. CNON ChIP-seq peaks classified as promoters, enhancers, insulators, repressed, or heterochromatin regions.
Table S4. CNON chromatin interactions identified by Hi-C.
Table S5. Epigenomic classification of CNON Hi-C chromatin interactions.
Table S6. Predicted target genes of enhancers using CNON Hi-C.
Table S7. Coordinates of promoter H3K4me3, non-promoter H3K27ac, and CTCF peaks found in many individuals of CNON datasets.
Table S8. Identification and classification of NDRs identified by CNON NOMe-seq.
Table S9. Enhancers linked to genotype, gender, smoking status, and schizophrenia diagnosis.
Table S10. Classification and characterization of SCZ risk-associated variants.
Table S11. Predicted target genes of enhancers linked to individual variations.
REFERENCES AND NOTES
- 1.Evgrafov O. V., Wrobel B. B., Kang X., Simpson G., Malaspina D., Knowles J. A., Olfactory neuroepithelium-derived neural progenitor cells as a model system for investigating the molecular mechanisms of neuropsychiatric disorders. Psychiatr. Genet. 21, 217–228 (2011). [DOI] [PubMed] [Google Scholar]
- 2.O. V. Evgrafov, C. Armoskus, B. B. Wrobel, V. N. Spitsyna, T. Souaiaia, J. S. Herstein, C. P. Walker, J. D. Nguyen, A. Camarena, J. R. Weitz, J. M. H. Kim, E. L. Duarte, K. Wang, G. M. Simpson, J. L. Sobell, H. Medeiros, M. T. Pato, C. N. Pato, J. A. Knowles, Gene expression in patient-derived neural progenitors provide insights into neurodevelopmental aspects of schizophrenia. bioRxiv 209197 [Preprint]. 26 October 2017. 10.1101/209197. [DOI]
- 3.Rhie S. K., Schreiner S., Farnham P. J., Defining regulatory elements in the human genome using nucleosome occupancy and methylome sequencing (NOMe-Seq). Methods Mol. Biol. 1766, 209–229 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelly T. K., Liu Y., Lay F. D., Liang G., Berman B. P., Jones P. A., Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 22, 2497–2506 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rhie S. K., Yao L., Luo Z., Witt H., Schreiner S., Guo Y., Perez A. A., Farnham P. J., ZFX acts as a transcriptional activator in multiple types of human tumors by binding downstream of transcription start sites at the majority of CpG island promoters. Genome Res. 28, 310–320 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farrell C. M., West A. G., Felsenfeld G., Conserved CTCF insulator elements flank the mouse and human β-globin loci. Mol. Cell. Biol. 22, 3820–3831 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luo Z., Rhie S. K., Lay F. D., Farnham P. J., A prostate cancer risk element functions as a repressive loop that regulates HOXA13. Cell Rep. 21, 1411–1417 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dixon J. R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J. S., Ren B., Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shin H., Shi Y., Dai C., Tjong H., Gong K., Alber F., Zhou X. J., TopDom: An efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res. 44, e70 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixon J. R., Jung I., Selvaraj S., Shen Y., Antosiewicz-Bourget J. E., Lee A. Y., Ye Z., Kim A., Rajagopal N., Xie W., Diao Y., Liang J., Zhao H., Lobanenkov V. V., Ecker J. R., Thomson J. A., Ren B., Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nora E. P., Lajoie B. R., Schulz E. G., Giorgetti L., Okamoto I., Servant N., Piolot T., van Berkum N. L., Meisig J., Sedat J., Gribnau J., Barillot E., Blüthgen N., Dekker J., Heard E., Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhan Y., Mariani L., Barozzi I., Schulz E. G., Blüthgen N., Stadler M., Tiana G., Giorgetti L., Reciprocal insulation analysis of Hi-C data shows that TADs represent a functionally but not structurally privileged scale in the hierarchical folding of chromosomes. Genome Res. 27, 479–490 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mozzetta C., Pontis J., Fritsch L., Robin P., Portoso M., Proux C., Margueron R., Ait-Si-Ali S., The histone H3 lysine 9 methyltransferases G9a and GLP regulate polycomb repressive complex 2-mediated gene silencing. Mol. Cell 53, 277–289 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Cavalli G., Misteli T., Functional implications of genome topology. Nat. Struct. Mol. Biol. 20, 290–299 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Le Dily F., Beato M., TADs as modular and dynamic units for gene regulation by hormones. FEBS Lett. 589, 2885–2892 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Ay F., Bailey T. L., Noble W. S., Statistical confidence estimation for Hi-C data reveals regulatory chromatin contacts. Genome Res. 24, 999–1011 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rao S. S. P., Huntley M. H., Durand N. C., Stamenova E. K., Bochkov I. D., Robinson J. T., Sanborn A., Machol I., Omer A. D., Lander E. S., Aiden E. L., A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ren G., Jin W., Cui K., Rodrigez J., Hu G., Zhang Z., Larson D. R., Zhao K., CTCF-mediated enhancer-promoter interaction is a critical regulator of cell-to-cell variation of gene expression. Mol. Cell 67, 1049–1058.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hnisz D., Day D. S., Young R. A., Insulated neighborhoods: Structural and functional units of mammalian gene control. Cell 167, 1188–1200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ji X., Dadon D. B., Powell B. E., Fan Z. P., Borges-Rivera D., Shachar S., Weintraub A. S., Hnisz D., Pegoraro G., Lee T. I., Misteli T., Jaenisch R., Young R. A., 3D chromosome regulatory landscape of human pluripotent cells. Cell Stem Cell 18, 262–275 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hnisz D., Weintraub A. S., Day D. S., Valton A.-L., Bak R. O., Li C. H., Goldmann J., Lajoie B. R., Fan Z. P., Sigova A. A., Reddy J., Borges-Rivera D., Lee T. I., Jaenisch R., Porteus M. H., Dekker J., Young R. A., Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 351, 1454–1458 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benayoun B. A., Pollina E. A., Uçar D., Mahmoudi S., Karra K., Wong E. D., Devarajan K., Daugherty A. C., Kundaje A. B., Mancini E., Hitz B. C., Gupta R., Rando T. A., Baker J. C., Snyder M. P., Cherry J. M., Brunet A., H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell 158, 673–688 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pekowska A., Benoukraf T., Zacarias-Cabeza J., Belhocine M., Koch F., Holota H., Imbert J., Andrau J.-C., Ferrier P., Spicuglia S., H3K4 tri-methylation provides an epigenetic signature of active enhancers. EMBO J. 30, 4198–4210 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russ B. E., Olshansky M., Li J., Nguyen M. L. T., Gearing L. J., Nguyen T. H. O., Olson M. R., Mc Quilton Hayley A., Nüssing S., Khoury G., Purcell D. F. J., Hertzog P. J., Rao S., Turner S. J., Regulation of H3K4me3 at transcriptional enhancers characterizes acquisition of virus-specific CD8+ T Cell-lineage-specific function. Cell Rep. 21, 3624–3636 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Kim T. H., Abdullaev Z. K., Smith A. D., Ching K. A., Loukinov D. I., Green R. D., Zhang M. Q., Lobanenkov V. V., Ren B., Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell 128, 1231–1245 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rhie S. K., Hazelett D. J., Coetzee S. G., Yan C., Noushmehr H., Coetzee G. A., Nucleosome positioning and histone modifications define relationships between regulatory elements and nearby gene expression in breast epithelial cells. BMC Genomics 15, 331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rhie S. K., Guo Y., Tak Y. G., Yao L., Shen H., Coetzee G. A., Laird P. W., Farnham P. J., Identification of activated enhancers and linked transcription factors in breast, prostate, and kidney tumors by tracing enhancer networks using epigenetic traits. Epigenetics Chromatin 9, 50 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zaret K. S., Carroll J. S., Pioneer transcription factors: Establishing competence for gene expression. Genes Dev. 25, 2227–2241 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roadmap Epigenomics Consortium; Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M. J., Amin V., Whitaker J. W., Schultz M. D., Ward L. D., Sarkar A., Quon G., Sandstrom R. S., Eaton M. L., Wu Y.-C., Pfenning A. R., Wang X., Claussnitzer M., Liu Y., Coarfa C., Harris R. A., Shoresh N., Epstein C. B., Gjoneska E., Leung D., Xie W., Hawkins R. D., Lister R., Hong C., Gascard P., Mungall A. J., Moore R., Chuah E., Tam A., Canfield T. K., Hansen R. S., Kaul R., Sabo P. J., Bansal M. S., Carles A., Dixon J. R., Farh K.-H., Feizi S., Karlic R., Kim A.-R., Kulkarni A., Li D., Lowdon R., Elliott GiNell, Mercer T. R., Neph S. J., Onuchic V., Polak P., Rajagopal N., Ray P., Sallari R. C., Siebenthall K. T., Sinnott-Armstrong N. A., Stevens M., Thurman R. E., Wu J., Zhang B., Zhou X., Beaudet A. E., Boyer L. A., De Jager P. L., Farnham P. J., Fisher S. J., Haussler D., Jones S. J. M., Li W., Marra M. A., McManus M. T., Sunyaev S., Thomson J. A., Tlsty T. D., Tsai L.-H., Wang W., Waterland R. A., Zhang M. Q., Chadwick L. H., Bernstein B. E., Costello J. F., Ecker J. R., Hirst M., Meissner A., Milosavljevic A., Ren B., Stamatoyannopoulos J. A., Wang T., Kellis M., Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oyake T., Itoh K., Motohashi H., Hayashi N., Hoshino H., Nishizawa M., Yamamoto M., Igarashi K., Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol. Cell. Biol. 16, 6083–6095 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kannan M. B., Solovieva V., Blank V., The small MAF transcription factors MAFF, MAFG and MAFK: Current knowledge and perspectives. Biochim. Biophys. Acta 1823, 1841–1846 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Baker S. J., Ma’ayan A., Lieu Y. K., John P., Reddy M. V. R., Chen E. Y., Duan Q., Snoeck H.-W., Reddy E. P., B-myb is an essential regulator of hematopoietic stem cell and myeloid progenitor cell development. Proc. Natl. Acad. Sci. U.S.A. 111, 3122–3127 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheasley D., Pereira L., Lightowler S., Vincan E., Malaterre J., Ramsay R. G., Myb controls intestinal stem cell genes and self-renewal. Stem Cells 29, 2042–2050 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Lehtonen A., Matikainen S., Julkunen I., Interferons up-regulate STAT1, STAT2, and IRF family transcription factor gene expression in human peripheral blood mononuclear cells and macrophages. J. Immunol. 159, 794–803 (1997). [PubMed] [Google Scholar]
- 35.Kumatori A., Yang D., Suzuki S., Nakamura M., Cooperation of STAT-1 and IRF-1 in interferon-γ-induced transcription of the gp91phox gene. J. Biol. Chem. 277, 9103–9111 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Yang F., Deng X., Ma W., Berletch J. B., Rabaia N., Wei G., Moore J. M., Filippova G. N., Xu J., Liu Y., Noble W. S., Shendure J., Disteche C. M., The lncRNA Firre anchors the inactive X chromosome to the nucleolus by binding CTCF and maintains H3K27me3 methylation. Genome Biol. 16, 52 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walton E., Hass J., Liu J., Roffman Joshua L., Bernardoni F., Roessner V., Kirsch M., Schackert G., Calhoun V., Ehrlich Stefan, Correspondence of DNA methylation between blood and brain tissue and its application to schizophrenia research. Schizophr. Bull. 42, 406–414 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Glatt S. J., Chandler S. D., Bousman C. A., Chana G., Lucero G. R., Tatro E., May T., Lohr J. B., Kremen W. S., Everall I. P., Tsuang M. T., Alternatively spliced genes as biomarkers for schizophrenia, bipolar disorder and psychosis: A blood-based spliceome-profiling exploratory study. Curr. Pharmacogenomics Pers. Med. 7, 164–188 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schizophrenia Working Group of the Psychiatric Genomics Consortium , Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Z., Chen J., Yu H., He L., Xu Y., Zhang D., Yi Q., Li C., Li X., Shen J., Song Z., Ji W., Wang M., Zhou J., Chen B., Liu Y., Wang J., Wang P., Yang P., Wang Q., Feng G., Liu B., Sun W., Li B., He G., Li W., Wan C., Xu Q., Li W., Wen Z., Liu K., Huang F., Ji J., Ripke S., Yue W., Sullivan P. F., O’Donovan M. C., Shi Y., Genome-wide association analysis identifies 30 new susceptibility loci for schizophrenia. Nat. Genet. 49, 1576–1583 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Tak Y. G., Farnham P. J., Making sense of GWAS: Using epigenomics and genome engineering to understand the functional relevance of SNPs in non-coding regions of the human genome. Epigenetics Chromatin 8, 57 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yao L., Tak Y. G., Berman B. P., Farnham P. J., Functional annotation of colon cancer risk SNPs. Nat. Commun. 5, 5114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rhie S. K., Coetzee S. G., Noushmehr H., Yan C., Kim J. M., Haiman C. A., Coetzee G. A., Comprehensive functional annotation of seventy-one breast cancer risk loci. PLOS ONE 8, e63925 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hazelett D. J., Rhie S. K., Gaddis M., Yan C., Lakeland D. L., Coetzee S. G.; Ellipse/GAME-ON Consortium; Practical consoritum, Henderson B. E., Noushmehr H., Cozen W., Kote-Jarai Z., Eeles R. A., Easton D. F., Haiman C. A., Lu W., Farnham P. J., Coetzee G. A., Comprehensive functional annotation of 77 prostate cancer risk loci. PLOS Genet. 10, e1004102 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feng Y., Rhie S. K., Huo D., Ruiz-Narvaez E. A., Haddad S. A., Ambrosone C. B., John E. M., Bernstein L., Zheng W., Hu J. J., Ziegler R. G., Nyante S., Bandera E. V., Ingles S. A., Press M. F., Deming S. L., Rodriguez-Gil J. L., Zheng Y., Yao S., Han Y. J., Ogundiran T. O., Rebbeck T. R., Adebamowo C., Ojengbede O., Falusi A. G., Hennis A., Nemesure B., Ambs S., Blot W., Cai Q., Signorello L., Nathanson K. L., Lunetta K. L., Sucheston-Campbell L. E., Bensen J. T., Chanock S. J., Marchand L. L., Olshan A. F., Kolonel L. N., Conti D. V., Coetzee G. A., Stram D. O., Olopade O. I., Palmer J. R., Haiman C. A., Characterizing genetic susceptibility to breast cancer in women of African ancestry. Cancer Epidemiol. Biomarkers Prev. 26, 1016–1026 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Won H., de la Torre-Ubieta L., Stein J. L., Parikshak N. N., Huang J., Opland C. K., Gandal M. J., Sutton G. J., Hormozdiari F., Lu D., Lee C., Eskin E., Voineagu I., Ernst J., Geschwind D. H., Chromosome conformation elucidates regulatory relationships in developing human brain. Nature 538, 523–527 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tansey K. E., Hill M. J., Enrichment of schizophrenia heritability in both neuronal and glia cell regulatory elements. Transl. Psychiatry 8, 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coetzee S. G., Rhie S. K., Berman B. P., Coetzee G. A., Noushmehr H., FunciSNP: An R/bioconductor tool integrating functional non-coding data sets with genetic association studies to identify candidate regulatory SNPs. Nucleic Acids Res. 40, e139 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Giusti-Rodriguez P., Lu L., Yang Y., Crowley C. A., Liu X., Bryois J., Juric I., Martin J. S., Allred S. C., Ancalade N., Crowley J. J., Guintivano J., Jansen P. R., Jurjus G. J., Mahajan G., Rajkowska G., Overholser J. C., Pochareddy S., Santpere G., Savage J. E., Shin Y., Pardiñas A. F., O’Donovan M. C., Owen M. J., Posthuma D. Sestan N., Stockmeier C. A., Walters J. T. R., Crawford G. E., Li Y., Jin F., Hu M., Li Y., Sullivan P. F., Schizophrenia and a high-resolution map of the three-dimensional chromatin interactome of adult and fetal cortex. bioRxiv, 10.1101/406330 (2018). [DOI] [Google Scholar]
- 50.Rajarajan P., Borrman T., Liao W., Schrode N., Flaherty E., Casiño C., Powell S., Yashaswini C., LaMarca E. A., Kassim B., Javidfar B., Espeso-Gil S., Li A., Won H., Geschwind D. H., Ho S.-M., MacDonald M., Hoffman G. E., Roussos P., Zhang B., Hahn C.-G., Weng Z., Brennand K. J., Akbarian S., Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science 362, eaat4311 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Servant N., Varoquaux N., Lajoie B. R., Viara E., Chen C. J., Vert J. P., Heard E., Dekker J., Barillot E., HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ross-Innes C. S., Stark R., Teschendorff A. E., Holmes K. A., Ali H. R., Dunning M. J., Brown G. D., Gojis O., Ellis I. O., Green A. R., Ali S., Chin S. F., Palmieri C., Caldas C., Carroll J. S., Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xi Y., Li W., BSMAP: Whole genome bisulfite sequence MAPping program. BMC Bioinf. 10, 232 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Coetzee S. G., Coetzee G. A., Hazelett D. J., motifbreakR: An R/bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics 31, 3847–3849 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wrobel B. B., Mazza J. M., Evgrafov O. V., Knowles J. A., Assessing the efficacy of endoscopic office olfactory biopsy sites to produce neural progenitor cell cultures for the study of neuropsychiatric disorders. Int. Forum Allergy Rhinol. 3, 133–138 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.A. Simon, FastQC: A quality control tool for high throughput sequence data (2010); www.bioinformatics.babraham.ac.uk/projects/fastqc.
- 58.Wingett S., Ewels P., Furlan-Magaril M., Nagano T., Schoenfelder S., Fraser P., Andrews S., HiCUP: Pipeline for mapping and processing Hi-C data. F1000Res 4, 1310 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., Glass C. K., Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Servant N., Lajoie B. R., Nora E. P., Giorgetti L., Chen C. J., Heard E., Dekker J., Barillot E., HiTC: Exploration of high-throughput ‘C’ experiments. Bioinformatics 28, 2843–2844 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu Y., Siegmund K. D., Laird P. W., Berman B. P., Bis-SNP: Combined DNA methylation and SNP calling for bisulfite-seq data. Genome Biol. 13, R61 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lay F. D., Liu Y., Kelly T. K., Witt H., Farnham P. J., Jones P. A., Berman B. P., The role of DNA methylation in directing the functional organization of the cancer epigenome. Genome Res. 25, 467–477 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thorvaldsdóttir H., Robinson J. T., Mesirov J. P., Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kheradpour P., Kellis M., Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res. 42, 2976–2987 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang J., Zhuang J., Iyer S., Lin X. Y., Greven M. C., Kim B. H., Moore J., Pierce B. G., Dong X., Virgil D., Birney E., Hung J. H., Weng Z., Factorbook.org: A Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res. 41, D171–D176 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kulakovskiy I. V., Medvedeva Y. A., Schaefer U., Kasianov A. S., Vorontsov I. E., Bajic V. B., Makeev V. J., HOCOMOCO: A comprehensive collection of human transcription factor binding sites models. Nucleic Acids Res. 41, D195–D202 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.ENCODE Project Consortium , An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/12/eaav8550/DC1
Supplementary Materials and Methods
Fig. S1. CNON Hi-C datasets.
Fig. S2. Chromatin interactions with different q value cutoffs.
Fig. S3. H3K27ac ChIP-seq clustering results.
Fig. S4. NOMe-seq depth and number of NDRs.
Table S1. Information about the CNON ChIP-seq, NOMe-seq, and Hi-C datasets.
Table S2. CNON TADs identified by Hi-C.
Table S3. CNON ChIP-seq peaks classified as promoters, enhancers, insulators, repressed, or heterochromatin regions.
Table S4. CNON chromatin interactions identified by Hi-C.
Table S5. Epigenomic classification of CNON Hi-C chromatin interactions.
Table S6. Predicted target genes of enhancers using CNON Hi-C.
Table S7. Coordinates of promoter H3K4me3, non-promoter H3K27ac, and CTCF peaks found in many individuals of CNON datasets.
Table S8. Identification and classification of NDRs identified by CNON NOMe-seq.
Table S9. Enhancers linked to genotype, gender, smoking status, and schizophrenia diagnosis.
Table S10. Classification and characterization of SCZ risk-associated variants.
Table S11. Predicted target genes of enhancers linked to individual variations.