

Summary (150)
Apicomplexan parasites rely on cyclic nucleotide-dependent kinases for host cell infection, yet the mechanisms that control their activation remain unknown. Here we show that an apically-localized guanylate cyclase (GC) controls microneme secretion and lytic growth in the model apicomplexan Toxoplasma gondii. Cell permeable cGMP reversed the block in microneme secretion seen in a knockdown of TgGC, linking its function to production of cGMP. TgGC possesses an N-terminal P-type-ATPase domain fused to a C-terminal heterodimeric guanylate cyclase domain, an architecture found only in Apicomplexa and related protists. Complementation with a panel of mutants revealed a critical requirement for the P-type ATPase domain for maximum GC function. We further demonstrate that knockdown of TgGC in vivo protects mice from lethal infection by blocking parasite expansion and dissemination. Collectively this work demonstrates that cGMP-mediated signaling in Toxoplasma relies on a multi-domain architecture, which may serve a conserved role in related parasites.
eTOC Blurb
Brown and Sibley demonstrate that essential cGMP signaling in Toxoplasma gondii is initiated by a conserved, apically-positioned P-type ATPase/guanylate cyclase enzyme (TgGC). Functional dissection of TgGC revealed a critical requirement for catalytically active enzymatic domains linked in the endogenous arrangement. A murine infection model revealed Toxoplasma virulence requires functional TgGC.
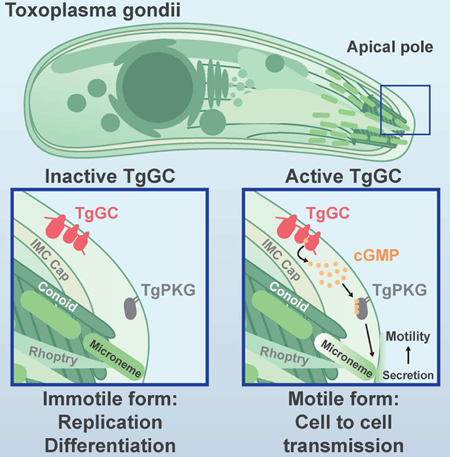
Introduction
Apicomplexan parasites are leading agents of infection in humans and animals worldwide. The diseases inflicted by these obligate-intracellular protozoans, including malaria and toxoplasmosis, are the result of tissue destruction and inflammation produced by lytic stages of parasite growth. The lytic lifecycle occurs in four general steps: host cell attachment, host cell invasion, parasite replication, and host cell egress. Other than replication, all steps in this lifecycle rely on timely secretion of proteins from apical organelles called micronemes (Carruthers and Tomley, 2008), presenting a functional bottleneck for parasite cell to cell transmission, yet it is unclear how microneme secretion is initiated.
The current understanding of how micronemes are secreted largely stems from investigating signaling pathways downstream of their origination. It is well-established that second messenger signaling drives microneme secretion, which include calcium (Ca2+) (Carruthers and Sibley, 1999), phosphatidic acid (PA) (Bullen and Soldati-Favre, 2016), and purine cyclic nucleotides (cGMP and cAMP) (Ono et al., 2008; Wiersma et al., 2004). Cytosolic Ca2+ is kept at low levels by organelle sequestration during immotile replicative stages but elevated during processes such as egress, migration, and invasion (Lourido and Moreno, 2015). It has been proposed that IP3 produced from phosphoinositide phospholipase C (PI-PLC) cleavage of PIP2 opens an IP3-sensitive Ca2+ channel (Lovett et al., 2002), though such channels have eluded identification in apicomplexans (Garcia et al., 2017). Once released, cytosolic Ca2+ activates apicomplexan calcium dependent protein kinases (CDPKs) (Billker et al., 2009; Lourido et al., 2010) and vesicle trafficking (Farrell et al., 2012) to promote microneme secretion. In addition to IP3/Ca2+, PI-PLC also produces DAG from PIP2 cleavage, which can be converted to PA by DAG kinase. PA is thought play an important role in microneme secretion by interacting with a PA receptor on the micronemes themselves called APH, priming micronemes for fusion with the plasma membrane (Bullen et al.,2016). PI-PLC function is regulated in part by inositol phosphate levels in Apicomplexa, which in turn are controlled by cGMP-dependent protein kinase (PKG) (Brochet et al., 2014). Importantly, PKG also controls a final step in microneme secretion as loss of PKG expression or activity cannot be overcome by systemically elevated Ca2+, PA, or cGMP (Brown et al., 2017; Brown et al., 2016; Bullen et al., 2016). For balance, cAMP- dependent protein kinase (PKA) has been proposed as a negative regulator of microneme secretion to prevent premature secretion and egress during parasite replication (Jia et al., 2017). Cyclic nucleotides are synthesized from nucleoside triphosphates by nucleotide cyclases. Genetic evidence exists for the importance of nucleotide cyclases in Plasmodium ookinetes and sporozoites (Hirai et al., 2006; Moon et al., 2009; Ono et al., 2008), yet it is unclear how these enzymes are regulated and whether they perform similar roles in other life stages or parasites.
Toxoplasma gondii is an exceptionally tractable apicomplexan parasite that expresses a limited yet representative subset of apicomplexan cyclase orthologs, making it an ideal model for investigating cyclic nucleotide signaling in Apicomplexa. Of the five putative nucleotide cyclases, only TgACα1 and TgACβ have been functionally characterized, both being classified as non-essential based on genetic disruption (Jia et al., 2017). The sole predicted guanylate cyclase (TgGC) awaits functional assignment but possesses an interesting domain structure that resembles a fusion of two seemingly unrelated eukaryotic genes: a P-type ATPase and a guanylate cyclase. P-type ATPases are a class of integral membrane proteins that utilize energy from ATP to transport ions, or flip lipids, across biological membranes (Palmgren and Nissen, 2011). The hybrid domain structure of TgGC is restricted to Apicomplexa and related protists but its functional significance has not been resolved in any organism (Gould and de Koning, 2011). Knowing which enzymes initiate microneme secretion for motility, and how they operate, is critical for understanding how these parasites sense and respond to their environment to transmit from one host cell to the next.
Here we performed a CRISPR knockout screen of all T. gondii cyclases, demonstrating that only TgGC was refractory to deletion. Conditional knockdown using an auxin-inducible degron (AID) system (Brown et al., 2017), revealed that TgGC is an initiator of motility in T. gondii by controlling microneme secretion. Transgenic complementation with mutant versions of TgGC also define an unexpected and important role for the P-type ATPase domain. Finally, we adapted the AID system for regulating parasite protein expression in mice, demonstrating a critical role for TgGC in vivo. Altogether, this work identifies a multi-domain GC that initiates essential biological adaptations in T. gondii, a role that may extend to other members of this deadly phylum of parasites.
Results
Reverse genetic screen for essential purine nucleotide cyclases in T. gondii
There are five nucleotide cyclases in the genome of T. gondii based on conserved cyclase homology domain (CHD; cd07302) sequences (Figure 1A). Four of the cyclases possess an adenylate cyclase (AC) signature (TgACα1, TgACα2, TgACα3, and TgACβ) whereas a guanylate cyclase (GC) signature is found in TgGC (Figure 1B). To identify essential cyclases, we used a CRISPR/Cas9 strategy to replace the coding region of each gene with a drug selection marker in a human isolate of T. gondii (Figure S1A). All four TgAC genes were amenable to individual deletion based on diagnostic PCR amplification of the modified loci (Figure S1B). The relative fitness of each knockout mutant was assessed by their ability to form plaques within host cell monolayers that are produced by successive rounds of lytic parasite growth. Parasites lacking TgACα3 had no apparent fitness defect, whereas loss of TgACα1, TgACα2, or TgACβ resulted in significantly fewer plaques than the parental line (Figure 1C). In contrast to the ease at which we knocked out the ACs, several attempts at deleting TgGC were unsuccessful, suggesting it was essential.
Figure 1-.
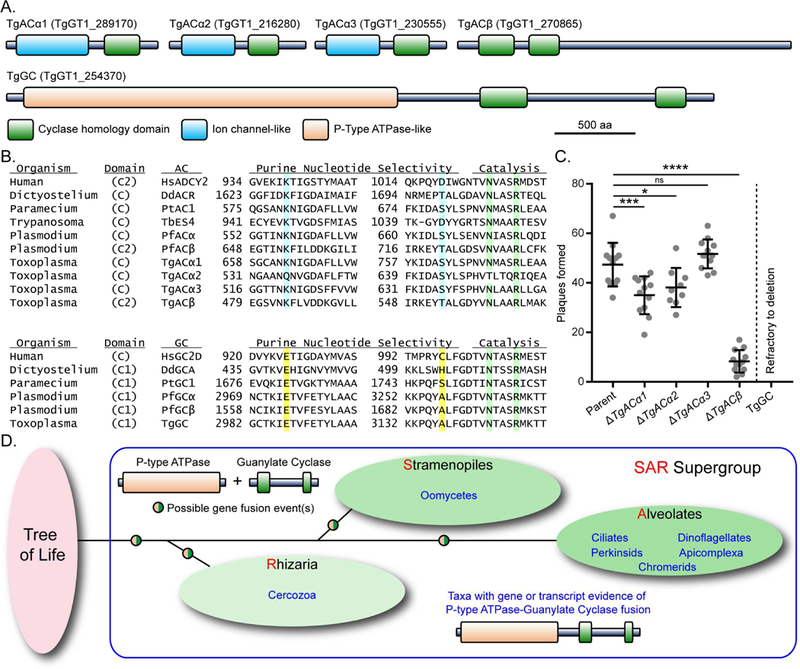
Reverse genetic screen for essential purine nucleotide cyclases in T. gondii (A) Predicted protein domain architectures of the five putative purine nucleotide cyclases in T. gondii based on NCBI CDD search.(B) Alignment of purine binding pockets of selected eukaryotic nucleotide cyclases. Residues important for purine selectivity (adenine = blue, guanine = yellow) and catalysis (green) are highlighted. The original alignment (Baker and Kelly, 2004) was modified here to include HuADCY2, PfGCα, and Toxoplasma cyclases. See Table S1 for protein accessions used in the alignment.(C) Plaques formed by wild type and knockout mutants on HFF monolayers at D8, 100 parasites / monolayer. Data represented as mean ± SD (n = 9-12 replicates combined from N = 3 or 4 trials). One-way ANOVA with Dunnett’s multiple comparisons test vs parent, Adjusted P values: * ≤ 0.05; *** ≤ 0.001; **** ≤ 0.0001. See also Figures S1A, S1B.(D) Schematic representation of taxa with gene or transcript evidence of a P-type ATPase/guanylate cyclase. See also Figure S1C and Table S1.
TgGC (4367 aa) is a 22 pass integral membrane protein featuring an N-terminal P- type ATPase domain with a C-terminal GC domain (catalytic heterodimer of CHDs C1 and C2) (Figure 1A). The C1 CHD contains the conserved glutamate residue (E2987) that dictates GTP selectivity of GCs (Figure 1B). Phylogenetic analysis of proteins with this particular GC architecture suggests that it evolved in a common ancestor of the Stramenopile, Alveolate, Rhizaria (SAR) clades of protists, although it is most commonly found in Alveolates (Figure 1D; Figure S1C). The conservation of this GC class is especially high among Apicomplexa, including the important human and animal pathogen genera Toxoplasma, Plasmodium, and Cryptosporidium.
TgGC is essential for the lytic lifecycle of T. gondii
To investigate TgGC function, we utilized the auxin-inducible degron (AID) system for conditional knockdown of proteins in T. gondii (Brown et al., 2017, 2018; Long et al.,2017). Starting with the auxin receptor (TIR1) expressing line RH TIR1-3FLAG, we used CRISPR/Cas9 genome editing to create epitope-tagged TgGC lines with 6Ty near the N-terminus and/or an AID-3HA (AID or mini-AID) at the C-terminus (Figure 2A, Figure S2A). TgGC protein was primarily found at the apical end of the parasite, as detected by immunofluorescence (IF) microscopy with antibodies to the N- and C-terminal epitope tags (Figure 2B). TgGC co-localized with the apical cap portion of the inner membrane complex as detected by co-staining with TgISP1 antisera in both intracellular and extracellular parasites (Figure S2B). Full length TgGC (477 kDa + tags) was detected along with smaller products by immunoblotting (Figure 2C, Figure S2C).
Figure 2-.
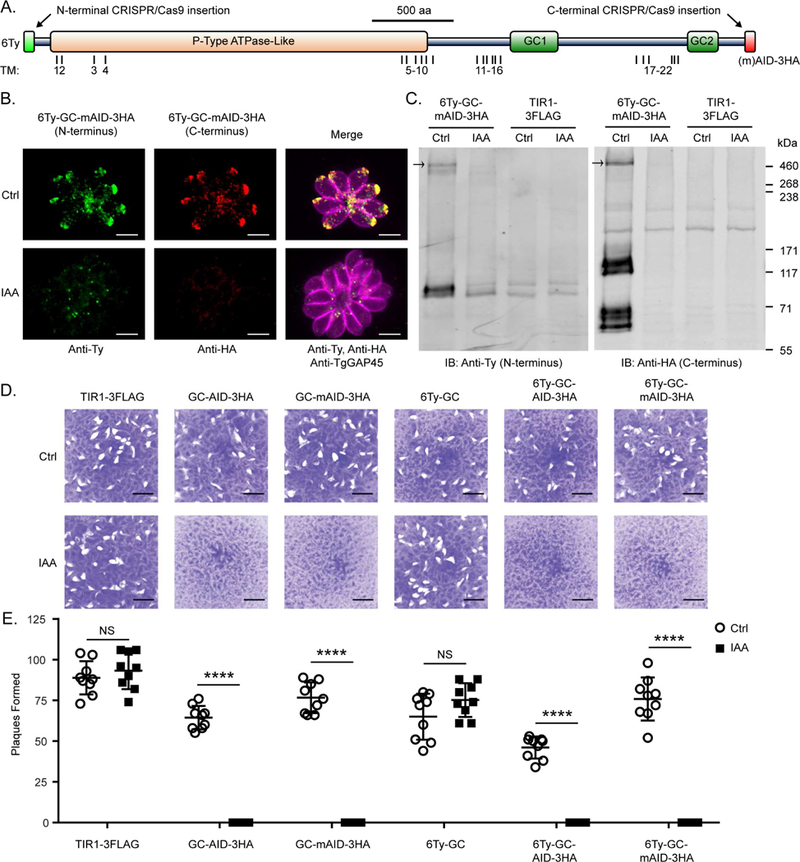
TgGC is essential for the lytic life cycle of T. gondii (A) Domain architecture of TgGC showing functional (NCBI CDD) and transmembrane domains (OCTOPUS) as well as insertion points for N- and C-terminal tags. See also Figure S2A. (B) IF microscopy of intracellular RH 6Ty-GC-mAID-3HA parasites treated with indole acetic acid (IAA) or vehicle for 14 h. Parasites were labeled with mouse anti-Ty and anti-mouse IgG Alexa Fluor 488 (green), rat anti-HA and anti-rat IgG Alexa Fluor 568 (red), and rabbit anti-TgGAP45 and anti-rabbit IgG Alexa Fluor 647 (magenta). Scale bar = 5 μm. See also Figure S2B. (C) Immunoblot of lysates from RH TIR1-3FLAG and derivative RH 6Ty-GC-mAID-3HA parasites treated with IAA or vehicle for 14 h. Blots were probed with rabbit anti-Ty and anti-rabbit IgG IRDye 680RD and mouse anti-HA and anti-mouse IgG IRDye 800CW. Separate channels of the same membrane scan are shown. See also Figure S2C. (D-E) Plaques formed by TIR1-3FLAG and derivative TgGC tagged lines on HFF monolayers with IAA or vehicle control (Ctrl) at D8 with 200 parasites/ monolayer. Scale bar = 5 mm. (E) Plaque data represented as mean ± SD (n = 9 replicates combined from N = 3 trials). Each parasite line was analyzed individually for statistical significance using an unpaired t test (IAA vs control), P values: **** ≤ 0.0001.
By fusing AID or mAID to TgGC, we were able to efficiently deplete TgGC by treating parasites with 3-indoleacetic acid (IAA or auxin) as detected by IF microscopy (Figure 2B) and immunoblotting (Figure 2C). Though the degrons were placed exclusively at the C-terminus of TgGC, depletion of TgGC was evident when probing for the protein from either terminus, providing further evidence that TgGC exists as a full- length protein in living parasites. Hence the breakdown products of TgGC seen by immunoblotting may reflect instability in parasite lysate. Importantly, conditional depletion of TgGC with either AID system completely blocked lytic parasite growth as measured by plaque formation on host cell monolayers (Figures 2D, E). Therefore TgGC is an apically-distributed cyclase that is essential for lytic parasite growth.
TgGC governs cell-to-cell transmission
To define the essential role TgGC provides for lytic parasite growth, we first examined parasite replication in host cells. Conditional depletion of TgGC did not prevent replication, as parasites achieved high numbers of parasite per vacuole (16-32) within 24 h, indicating that 4-5 rounds of cell division had occurred (Figure 3A, Figure S3A). Furthermore, there were no differences in the mean number of parasites per vacuole following loss of TgGC (Figure 3B), indicating it is not required for T. gondii replication.
Figure 3-.
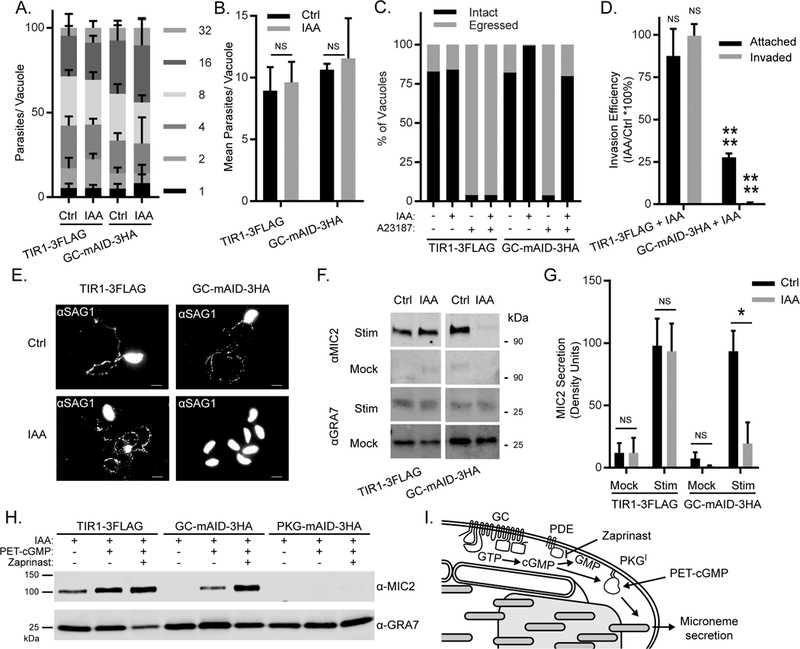
TgGC regulates PKG-dependent microneme secretion for motile processes (A-B) Replication of RH TIR1-3FLAG and derivative RH GC-mAID-3HA parasites grown in HFFs for 24 h in the presence of IAA or vehicle (added 1 h post-invasion). See also Figure S3A. (A) Mean percentages of parasite vacuoles containing indicated numbers of parasites ± SEM (N = 3). (B) Mean parasites per vacuole ± SEM (N = 3). Each parasite line was analyzed individually for statistical significance using an unpaired t test (IAA vs control). ns = not significant. (C) Egress assay of RH TIR1-3FLAG and RH GC-mAID-3HA parasites grown for 40 h in HFFs in the presence of IAA or vehicle (administered at 26 h post-infection). At 40 h post-infection, cultures were given a 5 min pulse with A23187 or vehicle. Shown are the percentages of intact and egressed vacuoles based on IF microscopy from a one of three trials (N = 3) with similar outcomes. See also Figure S3B. (D) Invasion of RH TIR1-3FLAG and RH GC-mAID-3HA parasites on HFF monolayers following 14 h treatment with IAA or vehicle. Data represented as mean ± SEM (N = 3). Each parasite line was analyzed individually for statistical significance using an unpaired t test (IAA vs control), P values: **** ≤ 0.0001. See also Figure (E) Motility of RH TIR1-3FLAG and RH GC-mAID-3HA parasites on glass coverslips following 14 h treatment with IAA or vehicle. Motility trails were detected by IF microscopy using rabbit anti-TgSAG1 and anti-rabbit IgG Alexa Fluor 488. Representative fields from one of three experiments with similar outcome are shown. (F-G) Microneme secretion of extracellular RH TIR1-3FLAG and RH GC-mAID-3HA parasites stimulated ± serum albumin/ethanol following 14 h treatment with IAA or vehicle. Stim = stimulated, mock = mock stimulation with vehicle. (F) Western blot of T. gondii excreted/secreted antigens probed for secreted micronemes (anti-MIC2) and constitutive dense granule secretion (anti-GRA7). Separate channels of each membrane scan (Stim, Mock) are shown. (G) Quantification of MIC2 secretion detected by Western blotting using densitometry. Data represented as mean ± SD (N = 3). Each parasite line and stimulation condition were analyzed individually for statistical significance using an unpaired t test (IAA vs control), P values: * < 0.01. (H) Microneme secretion of extracellular RH TIR1-3FLAG, RH GC-mAID-3HA, and RH PKG-mAID-3HA parasites stimulated ± PET-cGMP or PET-cGMP + Zaprinast following 14 h treatment with IAA. Western blot of T. gondii excreted/secreted antigens probed for secreted micronemes (anti-MIC2) and constitutive dense granule secretion (anti-GRA7). Separate channels of the same membrane scan are shown. Representative of four experiments with the same outcome. (I) Model of TgGC-dependent microneme secretion.
We next tested whether nascent parasites that form in the absence of TgGC could egress from their host cells and invade new ones. T. gondii egress occurs naturally over time but can also be triggered by flooding the parasite cytosol with Ca2+ using calcium ionophores (Endo et al., 1982). We examined both natural and Ca2+-stimulated parasite egress in host cell monolayers following treatment with vehicle or IAA to induce degradation of TgGC. Parasites depleted of TgGC by IAA treatment (14 h) showed severe defects in both natural egress (vehicle, 18% egress vs IAA, <1% egress) and A23187-stimulated egress (vehicle, 96% egress vs IAA, 20% egress) at 40 h post infection (Figure 3C, Figure S3B). To determine if parasites depleted of TgGC could invade host cells, we purified parasites that had been treated for 14 h with IAA or vehicle then added them to fresh host cell monolayers. Parasites depleted of TgGC displayed 80% fewer attached parasites and >99% fewer invaded parasites than vehicle control parasites (Figure 3D, Figure S3C). To test whether TgGC may control both invasion and egress by regulating parasite motility, we monitored the formation of parasite migration trails on serum-coated coverglass following TgGC depletion.
Migration trails were readily observable in the control parasites and the vehicle treatment group but were not observed in parasites depleted of TgGC (Figure 3E). Taken together, loss of TgGC paralyzes parasites, preventing them from transmitting from one cell to the next.
TgGC regulates PKG-dependent microneme secretion for motile processes
Since TgGC regulates parasite processes that depend on microneme secretion (i.e. motility, invasion, egress), we hypothesized that TgGC knockdown would also disrupt microneme secretion. Microneme secretion occurs basally at low levels but can be stimulated with bovine serum albumin (BSA) and ethanol, which are thought to elevate cGMP and Ca2+ respectively (Brown et al., 2016). To test whether TgGC participates in microneme secretion, we performed a microneme secretion assay using extracellular parasites that were pretreated with IAA or vehicle to regulate TgGC expression. By immunoblotting for secreted proteins in parasite-free supernatants, we found that loss of TgGC blocked both basal and BSA/ethanol-stimulated microneme secretion yet had no effect on dense granule protein secretion (Figures 3F, 3G).
The defining function of a guanylate cyclase is to produce cGMP from GTP. In T. gondii, cGMP is thought to function by activating TgPKGI, a cGMP-dependent kinase previously shown to be essential for microneme secretion (Brown et al., 2017). Measuring cGMP levels in T. gondii is complicated due to naturally low and/or transient levels cGMP that often fall below the limit of detection in standard cGMP ELISA assays using parasite lysates or in live parasites using genetically-encoded cGMP indicators (KMB, unpublished observations). Furthermore, T. gondii encodes an expanded repertoire of cyclic nucleotide phosphodiesterases (PDE)s (18 genes) that may degrade cGMP both in living parasites and following lysis, interfering with cGMP measurements. As an alternative approach, we reasoned that if the sole requirement of TgGC for microneme secretion is cGMP production, then a cell-permeable analog of cGMP should stimulate microneme secretion in the absence of TgGC. To test this hypothesis, we performed microneme secretion assays using extracellular parasites depleted of TgGC that were left unstimulated, or stimulated with cell-permeable cGMP ± a cGMP- PDE inhibitor called zaprinast. In parasites lacking TgGC, PET-cGMP ± zaprinast treatments rescued microneme secretion (Figure 3H). Importantly, PET-cGMP ± zaprinast had no stimulatory effect on microneme secretion in parasites lacking TgPKG (Figure 3H), confirming that cGMP operates downstream of TgGC but upstream of TgPKG. Taken together, these results point to a model of microneme secretion in which TgGC localizes to the apical pole where it is activated by extracellular signals (such as BSA) to produce cGMP for TgPKGI-dependent microneme secretion (Figure 3I).
Functional analysis of the multi-domain architecture of TgGC
TgGC initiates cGMP production and hence is necessary for microneme secretion (Figure 3). However, it is unclear how TgGC or other apicomplexan GCs operate as they possess a unique multi-domain architecture consisting of an N-terminal P-type ATPase domain fused to a heterodimeric guanylate cyclase domain (Figure 1D, Figure S1C). The N-terminal fusion may serve as a catalytically active regulatory domain or simply stabilize the GC domain and/or facilitate trafficking to the apical pole. To determine the functional significance of the unique domain structure of TgGC, we ectopically-expressed second copies of TgGC (wild-type or mutant versions fused to mCherry for detection) in the regulatable TgGC-mAID-3HA background. This functional analysis included 9 parasite lines (Figure S4A): 1. RH GC-mAID-3HA (parent, no complement), 2. mock complement (mCherry only), 3. wild-type complement (TgGC- mCherry). 4. P-type ATPase domain only (TgGC 1-2699-mCherry), 5. GC domain only (TgGC 2700-4367-mCherry), 6. P-type ATPase catalytic dead (TgGC D782A-mCherry), 7. GC catalytic dead (TgGC E2987A-mCherry), 8. Skip peptide (Tang et al., 2016) separating the P-type ATPase and GC domains (TgGC P2A-mCherry), 9. Non-skip peptide control (TgGC P2AGP/AA-mCherry). The second copies of TgGC were detected at similar rates based on RT-PCR (Figures S4B, S4C) and IF microscopy (Figures 4A, 4B), indicating that neither the domain architecture nor catalytic activities appear to influence the expression or stability of TgGC protein.
Figure 4-.
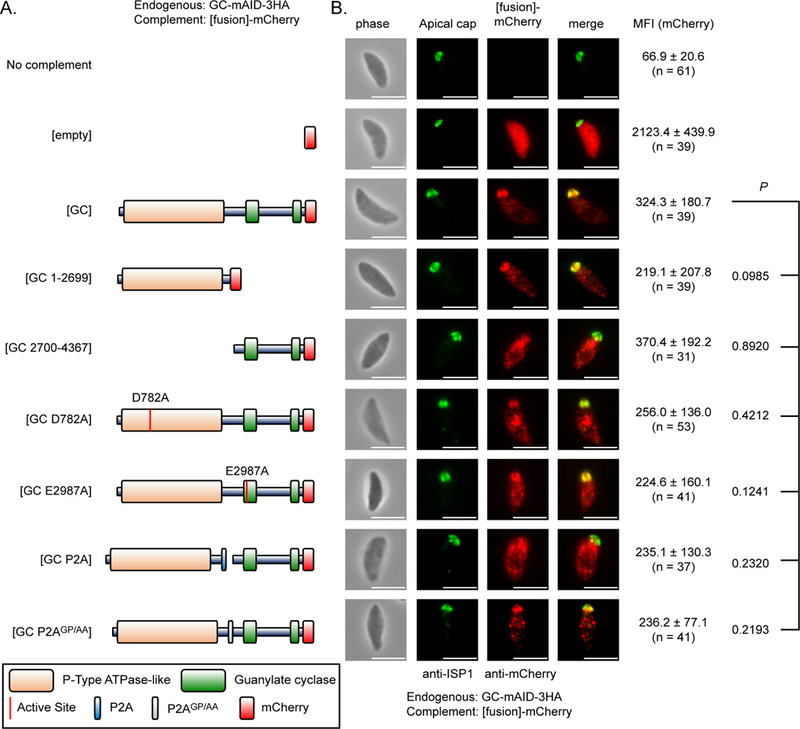
Expression and localization of mutant TgGC complementation constructs (A) Domain architectures of wild-type and mutant TgGC-mCherry fusion constructs. (B) IF microscopy of extracellular RH GC-mAID-3HA parasites stably expressing second copies of TgGC-mCherry fusions (depicted in Figure 4A, matched rows) probed for TgGC fusions (rat anti-mCherry and anti-rat IgG Alexa Fluor 568) and the apical cap (mouse anti-TgISP1 and anti-mouse IgG Alexa Fluor 488). Scale bar = 5 μm. Identical immunolabeling and imaging protocols were used for each line. The mean fluorescent intensities (MFI) of the raw mCherry signal data were averaged from at least 30 parasites (mean ± SD) for each line from 1 of 3 independent experiments with similar outcomes. Statistical differences in MFIs were assessed by one-way ANOVA with Dunnett’s multiple comparisons test (mutant vs wild-type control), adjusted P values are shown. See also Figures S4A-D.
Since the mutant forms of TgGC displayed equivalent expression, we next examined whether they also traffic and localize properly. TgGC-mCherry was largely distributed at the apical end of parasites near the apical cap marker TgISP1 (Figure 4B), with a portion showing an ER-like distribution, which is in agreement with endogenously tagged versions of TgGC (Figure S2B). The apical distribution was apparent in all mutants possessing the N-terminal P-type ATPase domain, including the P-type ATPase domain only construct (Figure 4B). However, the apical distribution was not apparent in the GC domain only construct (Figure 4B), suggesting that the P-type ATPase domain is important for subcellular targeting or trafficking of TgGC. We next examined the localization of the TgGC constructs where a P2A skip peptide or a nonskip p2AGP/AA peptide was inserted after D2699 between the two domains. Introduction of the P2A peptide separated the two domains, however they remain fused in the P2aGP/AA mutant, based on Western blotting for the full length ~500 kDa TgGC-mCherry product in (Figure S4D). Similar to the GC domain only construct, the P2A split C- terminal GC-mCherry fragment failed to localize to the apical end of the parasite (Figure 4B). Conversely, the mutant harboring the p2AGP/AA no skip peptide localized correctly to the apical pole (Figure 4B). Therefore, the N-terminal P-type ATPase is in part necessary for targeting TgGC to the apical plasma membrane where PKG-dependent microneme secretion occurs.
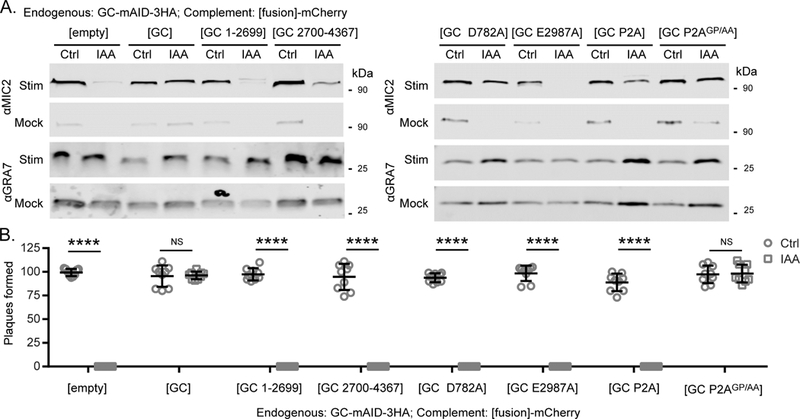
In the complementation lines, depletion of the endogenous copy of TgGC reveals the functional sufficiency of the second copy. Under basal and stimulated microneme secretion conditions, we found that the positive control (wild-type TgGC-mCherry) fully supported basal and stimulated secretion whereas the negative control (empty- mCherry) could not (Figure 5A). Next, we tested whether either domain TgGC could function independently to support microneme secretion. Similar to the negative control, the P-type ATPase domain only (TgGC 1-2699-mCherry) construct failed to support basal or stimulated microneme secretion whereas the GC domain only (TgGC 2700- 4367-mCherry) construct partially supported stimulated microneme secretion (Figure 5A).
Figure 5-.
Functional analysis of the multi-domain structure of TgGC (A) Microneme secretion of extracellular RH GC-mAID-3HA complement lines. Parasites were stimulated ± serum albumin/ethanol following 14 h treatment with IAA or vehicle. Western blot of T. gondii excreted/secreted antigens probed for secreted micronemes (anti-MIC2) and constitutive dense granule secretion (anti-GRA7). Stim = stimulated, mock = mock stimulation with vehicle. Representative blot shown (N = 2 similar experiments). (B) Plaques formed by RH GC-mAID-3HA complement lines on HFF monolayers with IAA or vehicle at D8, 200 parasites/ monolayer. Plaque data represented as mean ± SD (n = 9 replicates combined from N = 3 trials). Each parasite line was analyzed individually for statistical significance using an unpaired t test (IAA vs control), P values: **** ≤ 0.0001.
To determine whether TgGC requires active domains, we mutated conserved catalytic sites in each domain individually and tested their capacities to support microneme secretion. Mutating the invariant glutamate that governs GTP selectivity in CHD1 of the GC heterodimer (TgGC E2987A-mCherry) ablated the capacity of TgGC to support basal or stimulated microneme secretion (Figure 5A), consistent with cGMP synthesis being required. However, mutating the canonical aspartate that governs P- type ATPase activity (TgGC D782A-mCherry) prevented basal microneme secretion but only partially diminished stimulated microneme secretion (Figure 5B). These results indicate that TgGC requires catalytically active domains for maximum control of microneme secretion in T. gondii.
To investigate the importance of the P-type ATPase/GC polypeptide linkage, we inserted a P2A skip peptide between the two domains before position M2700 to coexpress them as separate proteins from the same mRNA transcript. Separating the two domains (TgGC P2A-mCherry) resulted in a microneme secretion defect that was similar to expression of the GC domain alone (Figure 5A). To ensure that the site chosen for P2A insertion was not critical for TgGC function, we also tested the non-skip P2A control mutant (TgGC P2AGP/AA-mCherry) which resulted in normal basal and stimulated microneme secretion (Figure 5A).
The ability of various TgGC functional mutants to partially support microneme secretion (Figure 5A) suggests that they may also be sufficient for lytic growth. To assess the overall contribution of different domains to parasite fitness, the complemented TgGC-mAID-3HA lines were subjected to a plaque assay in the presence or absence of IAA. In the absence of IAA allowing expression of wild-type TgGC-mAID-3HA, plaque formation was consistent in all lines. However, plaque formation in the absence of wild-type TgGC-mAID-3HA (plus IAA) required complementation with wild-type versions of TgGC (TgGC-mCherry or TgGC p2AGP/AA- mCherry) (Figure 5B). Surprisingly, none of the mutant forms of TgGC that could partially support microneme secretion were able to support lytic growth (Figure 5B). This phenotype is reminiscent of TgPKGII, a cytosolic PKG isoform that could partially support microneme secretion but ultimately failed to support lytic growth in T. gondii (Brown et al., 2017), emphasizing the importance of full cGMP signaling capacity and complete microneme secretion for parasite growth. Thus, TgGC requires linked, catalytically active domains to support efficient microneme secretion and lytic life cycle progression in vitro.
In vivo depletion of TgGC protects mice from lethal toxoplasmosis
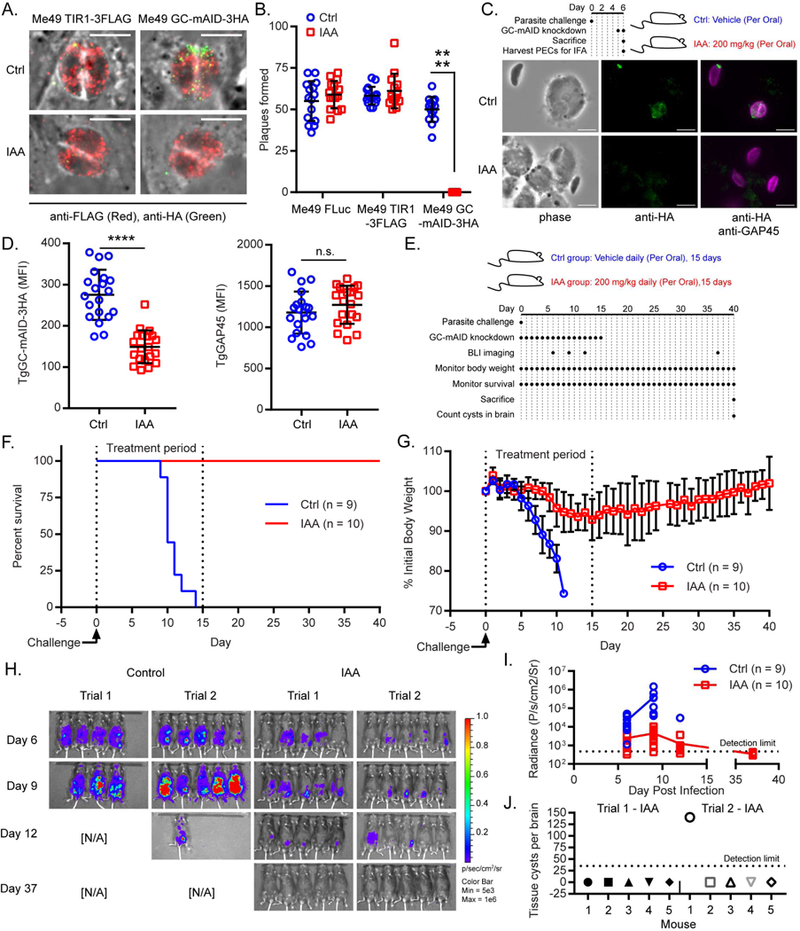
The requirements for parasite fitness in tissue culture are not likely to fully encapsulate the requirements for establishing an infection and virulence in the host. To study essential T. gondii proteins in vivo, we adapted the AID system for use in a luciferase- expressing type II line called Me49-FLuc (Tobin and Knoll, 2012), which has maintained both acute (tachyzoite) and chronic (bradyzoite) asexual life stages. In Me49-FLuc, we used CRISPR/Cas9 to replace TgKu80 with the auxin receptor TIR1-3FLAG (Figure S5A), as confirmed by diagnostic PCR of the modified locus (Figure S5B) and IF microscopy (Figure S5C). To determine whether auxin treatment affects the acute virulence of Me49 TIR1-3FLAG parasites, mice were challenged with Me49 TIR1- 3FLAG parasites the treated orally with 200 mg/kg/day IAA or vehicle alone. In both treatment groups, mice succumbed to the infection at equal rates (Figure S5D) indicating that IAA treatment does not promote susceptibility or resistance to Toxoplasma in mice. We then tagged TgGC with mAID-3HA in the Me49-Fluc TIR1- 3FLAG line and showed that it could be readily depleted with IAA (Figure 6A). Loss of TgGC prevented plaque formation, confirming its essentiality in the Me49 strain (Figure 6B). Next, we tested whether TgGC-mAID-3HA could be depleted in vivo. Mice were infected with Me49 GC-mAID-3HA parasites intraperitoneally, then treated orally with 200 mg/kg IAA (or vehicle control) at days 5 and 6 post-infection. At day 6 postinfection, mice were sacrificed and parasite-infected peritoneal exudate cells (PECs) were collected for IF microscopy (Figure 6C). Parasites from IAA treated mice displayed depleted TgGC-mAID-3HA levels but normal levels of a control protein TgGAP45 compared to parasites from the vehicle treated mice (Figures 6C, 6D).
Figure 6-.
Depletion of TgGC in vivo protects mice from lethal toxoplasmosis (A) IF microscopy of Me49 TIR1-3FLAG and Me49 GC-mAID-3HA treated with IAA or vehicle for 14 h. Parasites were labeled with rat anti-FLAG and anti-rat IgG Alexa Fluor 568 and mouse anti-HA and anti-mouse IgG Alexa Fluor 488. Scale bar = 5 μm. See also Figures S5A-D. (B) Plaques formed by Me49 FLuc, Me49 TIR1-3FLAG, and Me49 GC-mAID-3HA parasite lines on HFF monolayers with IAA or vehicle at D8, 200 parasites/ monolayer. Plaque data represented as mean ± SD (n = 15 replicates combined from N = 3 trials). Each parasite line was analyzed individually for statistical significance using an unpaired t test (IAA vs control), P values: **** ≤ 0.0001. (C) In vivo assessment of TgGC-mAID-3HA knockdown. C57Bl/6 mice were challenged with 1000 Me49 GC-mAID-3HA parasites intraperitoneally and treated with IAA or vehicle on days 5-6. On day 6, mice were sacrificed, and peritoneal exudate cells were collected for IF microscopy. Fixed cells were probed for parasites (rabbit anti-TgGAP45 and anti-rabbit IgG Alexa Fluor 647) and GC-mAID-3HA (mouse anti-HA and antimouse IgG Alexa Fluor 488). Scale bar = 5 μm. Identical immunolabeling and imaging protocols were used for each treatment group. (D) Quantification TgGC-mAID-3HA and TgGAP45 expression depicted in (C). The abundance of each protein (mean fluorescence intensity ± SD) was quantified and averaged from n = 20 parasites for each treatment group from one of two trials (N = 2). Statistical significance was assessed using an unpaired t test (IAA vs control), P values: **** ≤ 0.0001. n.s. = not significant. (E) Experimental design of in vivo test of TgGC essentiality using AID. Related to Figures 6F-J (n = 9 to 10 mice per group combined from the same N = 2 trials). (F) Survival curve of C57Bl/6 mice infected with 200 Me49 GC-mAID-3HA parasites intraperitoneally and treated with IAA or vehicle for 15 days. The Gehan-Breslow- Wilcoxon test was used to compare differences between the survival curves, P < 0.0001 (IAA vs Control). (G) Mean body weight ± SD of C57Bl/6 mice infected with 200 Me49 GC-mAID-3HA parasites intraperitoneally and treated with IAA or vehicle for 15 days. (H-I) Bioluminescence imaging of C57Bl/6 mice infected i.p. with 200 Me49 GC-mAID- 3HA parasites then treated with IAA or vehicle for 15 days. (H) Radiance overlay of luciferase-expressing Me49 GC-mAID-3HA in C57BI/6 mice at days 6, 9, 12, and 37 post-infection. (I) Quantification of whole body radiance shown in (H). Line indicates mean radiance. (J) Tissue cysts per brain from C57BI/6 mice infected with 200 Me49 GC-mAID-3HA parasites intraperitoneally and treated with IAA 15 days, then euthanized on day 40 post-infection for cyst determination in brain homogenates by microscopy. See also Figures S5E, S5F.
To determine the role of TgGC function in vivo, mice were challenged with Me49 GC-mAID-3HA parasites that express luciferase, then treated with IAA or vehicle orally for 15 days to deplete TgGC (Figure 6E). All mice receiving the vehicle control treatment succumbed to lethal toxoplasmosis by day 14 post-infection (Figure 6F). Conversely, IAA treatment (i.e. TgGC knockdown) rescued all animals from lethal toxoplasmosis, indicating that TgGC is necessary for acute virulence in T. gondii (Figure 6F). By day 5 post-infection, the control group began showing weight loss, a sign of illness, and continued to lose up to a quarter of their initial weights by the time of death (Figure 6G). The IAA treatment group showed both delayed and reduced weight loss that peaked at day 15 post infection (average loss = 7.1%) (Figure 6G). The severe morbidity and complete mortality seen in the control treatment group correlated with significantly higher parasite burdens compared to the IAA treatment group based on bioluminescence imaging for the luciferase-expressing parasites (Figures 6H, 6I). On days 6 and 9 post-infection, IAA treatment had reduced mean parasite burdens by 8.75- fold and 87.2-fold, respectively (Figure 6I). Suppressing TgGC expression during the acute phase of infection effectively blocked acute virulence of T. gondii since discontinuation of IAA treatment following day 15 did not result in morbidity (Figure 6G), mortality (Figure 6F), or parasite expansion (Figure 6H, 6I) as monitored for an additional 25 days.
To determine whether knockdown of TgGC during the acute infection prevented the establishment of a chronic infection, we examined tissue cyst formation in the brain. On day 37 post-infection, parasite burdens in the IAA treatment group were all below the limit of detection throughout the animals, as detected by bioluminescence (Figure 6H, 6I). Interestingly, 9 out of 10 mice that had received IAA treatment during the acute infection did not have a detectable number of tissue cysts (< 35 limit of detection) in their brains at day 40 post infection (Figure 6J). The one mouse that did contain quantifiable tissue cysts in its brain also had the highest parasite burden on day 12 (Trial 2, IAA mouse 1; Figure 6H), suggesting that TgGC knockdown was not as robust in this particular animal. To determine whether TgGC directly impacts tissue cyst formation, Me49 GC-mAID-3HA and Me49 TIR1-3FLAG parasites were grown in host cell monolayers in the presence of either IAA or vehicle, then stimulated to convert to bradyzoites tissue cysts using either pH 8.2/0.3 % CO2 (Bohne and Roos, 1997) or Compound 1, a trisubstituted pyrrole which is thought to stimulate bradyzoite development by modifying host cell transcription (Radke et al., 2006). Analogous to parasite replication (Figures 3A, 3B), loss of TgGC did not interfere with bradyzoite development under either condition tested based on a fluorescent labeling of tissue cyst walls (Figures S5E, S5F). Since loss of TgGC did not interfere with bradyzoite development in vitro, the failure of TgGC-depleted parasites to enact lethal acute infection or establish chronic brain infections in mice is most likely due to a defect in parasite dissemination as seen by bioluminescence imaging (Figure 6H).
Discussion
Apicomplexan parasites have adapted cyclic nucleotide signaling to precisely regulate the timing and amplitude of essential motile processes (Baker et al., 2017), yet it is unclear how these signals are actually initiated in these organisms. Here we performed a reverse genetic screen of enzymes predicted to synthesize cyclic nucleotides in the model apicomplexan T. gondii. From this screen, we found that a guanylate cyclase (TgGC) is essential for the lytic lifecycle of T. gondii by controlling motile processes including gliding motility, invasion, and egress. The defect in motility produced by loss of TgGC was explained by a profound defect in cGMP-dependent protein secretion that was reversed by supplementing cell-permeable cGMP. A systematic analysis of the multi-domain architecture of TgGC through genetic complementation with different domain and point mutants revealed a critical requirement for both P-type ATPase and guanylate cyclase activities. To determine if TgGC contributes to T. gondii infection of a mammalian host, we developed a system for investigating the function of essential T. gondii proteins in an animal model of toxoplasmosis. We found that auxin-induced depletion of TgGC in mice infected with T. gondii protected mice from lethal toxoplasmosis, indicating that TgGC is required for T. gondii fitness both in vitro and in vivo.
Conditional knockdown studies have demonstrated that T. gondii expresses two essential cyclic nucleotide-dependent kinases, TgPKAr/TgPKAc1 (Jia et al., 2017) and TgPKG (Brown et al., 2017), implying that both cAMP and cGMP are also essential in T. gondii. However, the enzymes responsible for producing cAMP and cGMP (i.e. cyclases) have yet to be identified and functionally described. Our ability to knock out each of the adenylate cyclases individually likely was the result of functional redundancy. Since loss of TgACα1, TgACα2, and TgACβ each had significant costs to parasite fitness, it is of interest to know whether loss of all three simultaneously would produce synthetic lethality. As our goal for this study was to identify essential cyclases, we focused our attention to the guanylate cyclase (TgGC) that was refractory to genetic deletion.
Since TgGC resisted several knockout attempts with CRISPR/Cas9, we utilized traditional epitope tagging and an auxin-inducible degron (AID) system (Brown et al., 2017, 2018; Long et al., 2017) to detect and regulate TgGC expression. TgGC localizes to the apical cap region of the plasma membrane in both intracellular and extracellular parasites. The apical end of apicomplexans is a defining feature of the phylum, demarcated by an apical cytoskeletal complex that focuses exocytosis of proteins from specialized organelles (e.g. micronemes and rhoptries) necessary for motile processes including extracellular migration, invasion, and egress (Carruthers and Tomley, 2008). Loss of TgGC effectively blocked motile processes in T. gondii, which was explained by a concomitant block in apical microneme secretion. We noted that loss of TgGC exactly phenocopies loss of TgPKG with regard to replication, microneme secretion, invasion, and egress (Brown et al., 2017), suggesting that they are functionally linked by cGMP signaling. In strong support of this possibility, cell-permeable cGMP could stimulate microneme secretion in parasites lacking TgGC but not in parasites lacking TgPKG. From this we conclude that the major role of TgGC is to produce cGMP for PKG- dependent microneme secretion, which cannot be compensated for by other cyclases or signaling pathways.
Perhaps the most prominent yet puzzling feature of TgGC is that it appears to be a fusion of two seemingly unrelated eukaryotic genes: a P-type ATPase and a guanylate cyclase. Once thought to have evolved in Alveolates (Biswas et al., 2009), our phylogenetic analysis suggests that hybrid P-type ATPase - guanylate cyclase genes may have evolved earlier in a common ancestor of the Stramenopile, Alveolate, Rhizaria (SAR) supergroup of protists or arisen separately within each superphyla.
Initial clues for the importance of this gene fusion have come from Plasmodium, which expresses two stage-specific guanylate cyclases (Carucci et al., 2000), where GCα is likely essential for asexual blood stages (Kenthirapalan et al., 2016) and GCβ regulates ookinete motility and invasion in the mosquito midgut (Hirai et al., 2006; Moon et al., 2009). An important distinction between Plasmodium GCα and GCβ lies in the P-type ATPase domain, which has degenerated in GCβ, suggesting that the two GCs may have modes of regulation and/or perform separate functions. Interestingly, purified recombinant guanylate cyclase domains of P. falciparum GCα and GCβ displayed disparate GC activities, where only the GC domain of PfGCβ was active (Carucci et al., 2000). Recombinant PfGCα could be intrinsically defective due to the heterologous expression system or require the P-type ATPase domain for GC activity. Also, the sufficiency of the GC domain of PfGCβ to produce cGMP does not exclude the possibility that the P-type ATPase domain acts as a regulatory module for GC activity. Curiously, it has yet to be demonstrated that any P-type ATPase/GC gene fusion can produce a full-length protein (Linder et al., 1999). A recent study examining TgGC expression by immunoblotting also failed to detect full length TgGC (Jia et al., 2017), which could represent an artifact of post-lysis proteolysis or imply that the P-type ATPase domain and GC domain are naturally processed to serve independent functions.
Here we presented five lines of evidence that TgGC functions as a full length, multidomain protein: 1) Full-length (477 kDa + tags) TgGC was detected by immunoblotting for epitope tags at either terminus, though breakdown products were also evident. 2) IF microscopy of 6Ty-GC-mAID-3HA protein in fixed parasites displayed overlapping staining patterns when probing for both termini, as expected for an intact protein. 3) When 6Ty-GC-mAID-3HA was depleted using the C-terminal degron, both N- and C- terminal staining was lost based on IF microscopy and immunoblotting which requires linked termini. 4) Ectopic expression of full-length TgGC cDNA, but not truncated or split forms, could rescue loss of endogenous TgGC. 5) Point mutations to conserved catalytic residues in either the P-type ATPase domain or the GC domain rendered TgGC non-functional based on genetic complementation. Taken together, these findings support a model where full-length TgGC requires dual catalytic activity with both domains linked in a single protein for proper function. We suspect the requirements for the co-expression of both domains in a single protein will also apply to orthologs among apicomplexans and their free-living relatives, but this point will require formal testing.
There are 5 subfamilies of P-type ATPases (I-V), which transport metal ions or phospholipids across membranes (Palmgren and Nissen, 2011). Although our studies indicate that the P-type ATPase domain is important for function, how it contributes to cGMP production is currently unknown. The P-type ATPase domain could feasibly regulate the GC domain activity by providing a metal cofactor such as Mg2+ or Mn2+ (Type II-like) or by promoting interaction of the two membrane-embedded CHDs by altering the surrounding lipid composition (Type IV-like). The P-type ATPase domain may also provide subcellular targeting information that positions TgGC near the site of PKG-dependent microneme exocytosis at the apical plasma membrane. The fusion of the two domains may also promote proper folding of TgGC needed for enzyme activity. The precise mechanism by which the P-type ATPase domain contributes to TgGC domain function warrants future investigation.
In summary, our findings, taken together with previous studies, define the mechanistic basis for the initiating step of cGMP signaling in T. gondii. Previous studies implicated TgGC in relaying extracellular signals (e.g. BSA), via the second messenger cGMP, to control essential parasite processes. Here we have uncovered TgGC as the upstream regulator of cGMP signaling and provided key functional analysis of the domain structure for this enzyme class, which revealed a critical requirement for both P- type ATPase and guanylate cyclase activities in controlling microneme secretion and parasite viability. The essential nature of TgGC was also extended in an animal model of infection, where loss of TgGC rendered T. gondii avirulent, reducing parasite expansion and dissemination, preventing lethal toxoplasmosis. Collectively these findings suggest that the P-type ATPase domain performs an essential function to activate or control the GC domain, thus generating cGMP to activate PKG. Given the conserved nature of PKG signaling in apicomplexans, the unique domain architecture of the GC is likely to control similar essential processes in related protists.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to the lead contact, David Sibley (sibley@wustl.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Parasite and host cell culture
T. gondii tachyzoites were maintained at 37°C in human fore skin fibroblast (HFF, infant male donor) monolayers cultured in D3 medium [Dulbecco’s modified Eagle’s medium (Invitrogen)] supplemented with 3% HyClone fetal bovine serum (GE Healthcare Life Sciences), 10 μg/ml gentamicin (Thermo Fisher Scientific), 10 mM glutamine (Thermo Fisher Scientific)]. All strains and host cell lines were determined to be mycoplasma negative with the e-Myco plus kit (Intron Biotechnology). All parasite lines used in this study are listed in Table S2. Parasite lines generated in other studies include RH TIR1- 3FLAG (Brown et al., 2017; Long et al., 2017), RH PKG-mAID-3HA (Brown et al., 2017), and Me49 FLuc (Tobin and Knoll, 2012).
Mice
Mice were purchased from Jackson Laboratories and maintained in an Association for Assessment and Accreditation of Laboratory Animal Care approved facility at the Washington University School of Medicine. All protocols were approved by the Institutional Animal Care and Use Committee at the Washington University School of Medicine. Female C57Bl/6 mice were aged 8-9 weeks at the start of the experiments and littermates were randomly assigned to experimental groups (n = 5 per group). Mice were challenged by intraperitoneal (i.p.) injection of 200 or 1000 T. gondii tachyzoites. Body weights and mortality were monitored daily for the duration of each experiment.
METHOD DETAILS
Phylogenetic analyses
Protein sequences were obtained from UniProt (www.uniprot.org) unless indicated otherwise and provided in Table S1. Clustal Omega was used for all protein alignments (https://www.ebi.ac.uk/Tools/msa/clustalo/). A bootstrap consensus tree of 51 P-type ATPase - GC protein sequences (Table S1) was generated with MEGA6 (http://www.megasoftware.net/) (Tamura et al., 2013) using Maximum Likelihood, tested by Bootstrap method with 1000 replications, JTT model for amino acid substitutions, with nearest-neighbor-interchange. The MEGA6 tree was re-drawn as a radial cladogram with Dendroscope (http://dendroscope.org/). Tree annotations and taxa illustrations were added in Adobe Illustrator (http://www.adobe.com).
Plasmids
All plasmids were generated by site-directed mutagenesis of existing plasmids or assembled from DNA fragments by the Gibson method. All plasmids used in this study are listed in Table S3. Plasmids generated in other studies include:
pSAG1:CAS9-GFP, U6:sgUPRT(Shen et al., 2014) pLoxP-DHFR-TS*-mCherry (Behnke et al., 2015) pEtHIS4:TgDHFR-TS*-YFP-P2A-RFP (Tang et al., 2016) p6Ty, DHFR-TS:HXGPRT(Brown et al., 2017) pTUB1:OsTIR1 −3FLAG, SAG1:CAT (Brown et al., 2017) pTUB1:OsTIR1 −3FLAG, SAG1:CAT (Brown et al., 2017) pTUB1:YFP-mAID-3HA, DHFR- TS: HXGPR T (Brown et al., 2017) pTUB1: YFP-AID-3HA, DHFR-TS:HXGPRT (Long et al., 2017)
Primers
All primers were synthesized by Integrated DNA Technologies and are listed in Table S4. Primers designed in other studies include: sgRNA_R (Shen et al., 2014)
pU6-sgRNA 2_F and pU6-sgRNA 2_R (Behnke et al., 2015)
Parasite transfection
Freshly harvested parasites were transfected as previously described (Soldati and Boothroyd, 1993). For each transfection, 10-20 million extracellular parasites in 200 μl cytomix buffer were mixed with up to 30 μl purified plasmid and/or amplicon DNA in a 4 mm gap BTX cuvette and electroporated with a BTX ECM 830 electroporator (Harvard Apparatus) using the following parameters: 1,700 V, 176 μs pulse length, 2 pulses, 100 msec interval between pulses. Transfected parasites were grown in HFF cultures for 24 h prior to drug selection for stable transformants.
CRISPR/Cas9 Gene Deletion
T. gondii strain RH was used for all knockout lines. See Figure S1A for knockout strategy. For each gene knockout attempt, a CRISPR/Cas9 plasmid with dual pU6- sgRNA cassettes was generated to cut the gene near the translation start and stop codons as described (Behnke et al., 2015). This plasmid was co-transfected with a gene-specific knockout plasmid that contained a floxed DHFR-mCherry selectable marker, flanked by two gene-specific 5’ and 3’ homology arms (1 kb each). Pyrimethamine (Sigma) was used at 3 μM for drug selection. Drug resistant isolates were examined for gene replacement (KO) by diagnostic PCR of the genomic locus (Figures S1A, S1B).
CRISPR/Cas9 Gene Tagging
T. gondii lines RH TIR1-3FLAG or Me49 TIR1-3FLAG were used for epitope tagging TgGC. For C-terminal tagging, a CRISPR/Cas9 + pU6-sgRNA plasmid was generated that creates a double stand nick in the 3’ UTR of TgGC near the translation stop codon. This plasmid was co-transfected with a plasmid containing (m)AID-3HA, floxed HXGPRT with 1 kb homology arms to TgGC (5’ arm = 1 kb upstream of translation stop codon, 3’ arm = 1 kb downstream of Cas9 break site). Transfected parasites were selected with mycophenolic acid (25 μg/ml) with xanthine (50 μg/ml). Tags were confirmed by PCR, IFA, and Western blotting for 3HA. For N-terminal tagging, a CRISPR/Cas9 + pU6-sgRNA plasmid was generated that creates a double strand nick in exon 2 of TgGC. This plasmid was co-transfected with a PCR amplicon of 6Ty containing 40 bp homology arms to TgGC (5’ arm = 40 bp upstream and including R14 of TgGC, 3’ arm = 40 bp downstream and including A15 of TgGC). Transfected parasites were FACS sorted with a FACSAria II (BD Biosciences) for Cas9-GFP expression and cloned by limiting dilution. Tags were confirmed by PCR, IFA, and Western blotting for 6Ty.
Conditional depletion of AID fusions in vitro
A stock of 500 mM IAA (Sigma) was prepared in 100% EtOH. For knockdown, IAA was used at 1:1,000 at a final concentration of 500 μM. Mock treatment consisted of an equivalent volume of 100% EtOH at a final concentration of 0.0789%, wt/vol.
Indirect immunofluorescence microscopy
For general IF microscopy of intracellular T. gondii, HFF monolayers grown on glass coverslips or 96-well glass-bottom plates (Greiner) were infected with T. gondii then fixed with 4% formaldehyde. For general extracellular IF microscopy, purified tachyzoites were fixed with 4% formaldehyde then spot-dried on to coverslips. For ex vivo IF microscopy, parasite-infected peritoneal exudate cells were allowed to settle on coverslips then fixed with 4% formaldehyde. The fixed samples were then permeabilized with 0.1% Triton X-100, blocked with 5% fetal bovine serum/ 5% normal goat serum, labeled with primary antibodies, and washed with phosphate-buffered saline (PBS). Antibody-labeled proteins were fluorescently labeled with highly cross adsorbed Alexa Fluor-conjugated secondary goat antibodies as indicated in the figure legends or associated method details. Nuclei were stained with Hoechst 33342 dye where indicated. Standard wide-field images were captured and analyzed with a 63× or 100× oil objective on an Axioskop 2 MOT Plus wide-field fluorescence microscope (Carl Zeiss, Inc.) running AxioVision LE64 software (Carl Zeiss, Inc.). High-content imaging and analysis were performed with a Cytation 3 (BioTek) multimode plate imager with a 20× objective running Gen5 software (BioTek). To maximize the clarity of qualitative image data, linear brightness and contrast adjustments were applied evenly across matching samples (variable(s) vs control) within each experiment. Fluorescent intensities were quantified using AxioVision LE64 or CellProfiler software using raw/unprocessed images.
Western blotting
Protein samples were prepared in Laemmli buffer containing 100 mM dithiothreitol, boiled for 5 min, separated on polyacrylamide gels by SDS-PAGE, and transferred to nitrocellulose membranes. The membranes were blocked with 5% (wt/vol) fat-free milk in PBS and then probed with primary antibodies diluted in blocking buffer containing 0.1% Tween 20. Membranes were washed with PBS + 0.1% Tween 20, then incubated with goat IR dye-conjugated secondary antibodies (LI-COR Biosciences) in blocking buffer as indicated in the figure legends or associated method details. Membranes were washed several times before scanning on a LiCor Odyssey imaging system (LI-COR Biosciences). Densitometry was performed using ImageJ (https://imagej.nih.gov/ij/).
Plaque assay
Freshly harvested parasites were counted, and 100 or 200 parasites, depending on the experiment, were added to six-well plates of confluent HFF monolayers in D3 medium. In some assays, cultures were treated with either vehicle (EtOH 1:1000) or 500 μM IAA to deplete (m)AID fusion proteins. The plaques were then given 8 days to develop. Plaque formation was assessed by counting the zones of clearance on EtOH-fixed, crystal violet-stained HFF monolayers.
Replication assay
Freshly harvested parasites were allowed to invade HFF monolayers grown on glass coverslips for 1 h, then washed to remove non-invaded parasites. Cultures were then treated with vehicle (EtOH 1:1,000) or 500 μM IAA to deplete mAID-3HA fusion proteins. At 24 h post-infection, infected monolayers were fixed and the number of parasites per vacuole was determined by IF microscopy using antibodies to HA and TgGAP45. In each experiment (N = 3), 10 image fields containing approximately 10 to 20 vacuoles were analyzed per treatment condition.
Egress assay
Freshly harvested parasites were allowed to invade HFF monolayers grown in 96 well glass bottom plates (2 × 104 parasites/well). At 26 h post-infection, cultures were treated with vehicle (EtOH 1:1000) or 500 μM IAA to deplete mAID-3HA fusion proteins. At 40 h post-infection, cultures were given a 5 min pulse with 2 μM A23187 or vehicle (DMSO 1:1000). The monolayers were then fixed and parasites were labeled with mouse anti- GRA5 (parasite vacuole) and anti-mouse IgG Alexa Fluor 488 and rabbit anti- TgAldolase (whole parasite marker) and anti-rabbit IgG Alexa Fluor 568. Images were captured and analyzed using a Cytation 3 running Gen5 software (BioTek Instruments). At least 193 vacuoles were counted per treatment condition per experiment (N = 3). Collapsed vacuoles surrounded by extracellular parasites were considered egressed. Vacuoles containing >2 parasites were considered intact. Vacuoles containing a single parasite were not counted to exclude the possibility of rapid reinvasion after egress.
Invasion assay
Parasites grown in HFF monolayers were treated with vehicle (EtOH 1:1,000) or 500 μM IAA for 14 h to deplete mAID-3HA fusion proteins. Parasites were then harvested, resuspended in D3, and added to HFF monolayers 96 well glass bottom plates (4 × 105 parasites/well) for 20 min at 37°C in the presence of the vehicle (EtOH 1:1,000) or 500 μM IAA. Invasion was stopped by formaldehyde fixation and invasion was analyzed by IF microscopy. Extracellular parasites were first labeled with mouse anti-TgSAG1 then washed with PBS. The monolayers were then permeabilized with 0.1% Triton X-100 followed by labeling all parasites with rabbit anti-TgSAG1. After washing with PBS, monolayers were labeled with goat Alexa Fluor-conjugated secondary antibodies (antimouse IgG Alexa Fluor 488 and anti-rabbit IgG Alexa Fluor 568). Images were captured and analyzed using a Cytation 3 running Gen5 software (BioTek Instruments). For each sample, thirty-six fields were analyzed per experiment. The relative efficiency of attachment and invasion of IAA-treated parasites was expressed as a mean percentage of the control treatment from N = 3 trials.
Motility assay
Parasites grown in HFF monolayers were treated with vehicle (EtOH 1:1,000) or 500 μM IAA for 14 h to deplete mAID-3HA fusion proteins. Parasites were then harvested, resuspended in EC buffer (5 mM KCl, 142 mM NaCI, 1 mM MgCl2, 1.8 mM CaCl2, 5.6 mM D-glucose, 25 mM HEPES, pH 7.4) containing vehicle (EtOH 1:1000) or 500 μM IAA and added to cell free coverslips (treated overnight with D3 medium) in 24 well plates. Parasites were incubated for 15 min at 37°C , then fixed with formaldehyde. Rabbit anti-TgSAG1 was used to label motility trails (Dobrowolski and Sibley, 1996) on the glass coverslips which were then detected with Alexa Fluor conjugated antibodies using IF microscopy. The presence or absence of motility trails was assessed for each sample in triplicate for each experiment (N = 2).
Microneme secretion assay
Parasites grown in HFF monolayers were treated with vehicle (EtOH 1:1,000) or 500 μM IAA for 14 h to deplete mAID-3HA fusion proteins. Parasites were then harvested, resuspended in EC buffer containing vehicle (EtOH 1:1,000) or 500 μM IAA. Parasites were then treated either with EC buffer alone or EC buffer containing secretagogue (indicated in Figure legends). Secretion was allowed to proceed for 10 min at 37°C. Following stimulation, parasites were chilled on ice and pelleted at 400g for 10 min. Excreted/secreted antigen (ESA) fractions were collected and centrifuged once more at 800g for 10 min. The ESA fractions were subjected to Western blotting to assess microneme secretion (mouse anti-TgMIC2 and anti-mouse IgG IRDye 800CW) and constitutive dense granule secretion (rabbit anti-TgGRA7 and anti-rabbit IgG IRDye 680RD). In each experiment, one replicate per treatment per strain was analyzed due to the limited number of lanes per gel. Where indicated, densitometry values were averaged from multiple experiments (N = 3) and assessed for statistical significance.
Generation of TgGC complemented lines
RH GC-mAID-3HA parasites were transfected with plasmids containing TgGC cDNA (full length or single domain mutants) fused to mCherry, driven by pGRA1. Stably transfected parasites were obtained by drug selection with 3 μM pyrimethamine using the DHFR-TS* drug selectable marker present on the complementation plasmids. To assess the relative expression of the TgGC-mCherry fusion transcripts, RNA from each parasite line was extracted from 5 × 107 tachyzoites with a RNeasy Kit (Qiagen) using the DNasel treatment step. One-hundred nanograms of RNA for each line was used as a template for One-Taq One-Step RT-PCR (New England Biolabs) reactions and according to manufacturer’s instructions. Amplicons from the TgGC-mCherry transcripts (as well as controls: TIR1-3FLAG and TgGC-mAID-3HA) were separated using agarose gel electrophoreses, imaged and quantified on a ChemiDoc MP (Bio-Rad) using Image Lab software (Bio-Rad).
Generation of the AID system in Me49 strain of T. gondii
In Me49 FLuc, CRISPR/Cas9 genome editing was used to replace TgKu80 with the auxin receptor TIR1-3FLAG based on CAT selection (see Figure S5A). This replacement allowed for both the homologous recombination needed for degron tagging as well as introducing the auxin receptor TIR1.
Conditional depletion of AID fusions in vivo
For survival studies, mice were challenged with 200 parasites intraperitoneally, then randomly assigned to one of two treatment groups in separate cages. Mice were administered either auxin (3-indoleacetic acid, IAA; Sigma) or vehicle orally in two forms. The IAA mouse group was supplied with unlimited 0.2 μm-filtered drinking water containing IAA (0.5 mg/ml), 3 mM NaOH, 5% sucrose (w/v), flavored with 2 mg/ml Cherry Kool-AID, pH 7.4 (adjusted with NaOH) whereas the control group received the same drinking water but lacking IAA. The drinking water was protected from light changed every 2-3 days. In addition, the IAA group received a daily 0.2 ml gavage of 0.2 μm-filtered water containing 12.5 mg/ml IAA, 3 mM NaOH, pH 7.4 (adjusted with HCl) whereas the control group received the same gavage but lacking IAA. The total combined daily dose of IAA for a 20 g mouse drinking 3 ml of IAA water (75 mg/kg) and receiving a 0.2 ml IAA gavage (125 mg/kg) was 200 mg/kg. This dose was selected since mice can tolerate up to 200 mg/kg/day IAA without obviously affecting health or body weight (Tupper et al., 2010). The number of mice per group (n) and experimental trials (N) was based on a historical precedent for establishing survival curves for virulent strains of T. gondii in mice (Su et al., 2002). A similar protocol was followed for the direct assessment of in vivo depletion of TgGC-mAID-3HA. In this case, n = 2 mice per group were treated orally with IAA (200 mg/kg/day as described above) or vehicle at days 5 and 6 post-infection. At day 6, mice were sacrificed by CO2 asphyxiation and peritoneal cells were examined by IF microscopy to measure the abundance of TgGC- mAID-3HA and the control protein TgGAP45 (N = 2).
Bioluminescence imaging
Parasite burdens in mice were determined based on firefly luciferase expression using a Xenogen IVIS200 machine, and analyzed with the Living Image software (Perkin- Elmer). Mice were anesthetized with 2% isoflurane and injected i.p. with D-luciferin (Biosynth AG) (150 mg/kg) prior to imaging.
Ex vivo bradyzoite tissue cyst counting
Mice were sacrificed at day 40 post-infection by CO2 asphyxiation for ex vivo analysis of chronic T. gondii infection. Whole brains were homogenized in PBS in a total volume of 1.4 ml, stained with FITC-conjugated Dolichos biflorus agglutinin (Vector laboratories), and DBA-positive bradyzoite tissue cysts were counted in duplicate 20 μl aliquots by fluorescence microscopy using a Zeiss Axioskop with a 10x objective.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data were collected and analyzed without blinding. Data was analyzed using Prism software (version 7.01; Graphpad). Parametric statistical tests were used when the data followed an approximately Gaussian distribution, non-parametric tests were used when populations were clearly not Gaussian. Comparisons were considered statistically significant when P values were less than 0.05. Experiment-specific statistical information is provided in the figure legends or associated method details including replicates (n), trials (N), standard error, and statistical test performed.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-Ty | In house hybridoma (Bastin et al., 1996) | mAB Clone BB2 |
| Rabbit anti-Ty | GenScript | Cat#A01004 |
| Mouse anti-HA.11 | BioLegend | Cat#901501 |
| Rabbit anti-HA | Thermo Fisher | Cat#71-5500 |
| Rat anti-HA | Roche, Millipore Sigma | Cat#11867423001 |
| Mouse anti-TgISP1 | Peter Bradley (Beck et al., 2010) | mAB Clone 7E8 |
| Rabbit anti-TgGAP45 | Dominique Soldati- Favre (Plattner et al., 2008) | Polyclonal |
| Mouse anti-TgGRA5 | In house hybridoma (Charif et al., 1990) | mAb Clone TG17-113 |
| Rabbit anti-TgAldolase | L. David Sibley (Starnes et al., 2006) | Polyclonal |
| Mouse anti-TgSAG1 | In house hybridoma (Burg et al., 1988) | mAb Clone DG52 |
| Rabbit anti-TgSAG1 | John Boothroyd | Polyclonal |
| Mouse anti-TgMIC2 | In house hybridoma (Carruthers et al., 2000) | mAb Clone 6D10 |
| Rabbit anti-TgGRA7 | L. David Sibley (Alaganan et al., 2013) | Polyclonal |
| Rabbit anti-TgACT1-GST | L. David Sibley (Dobrowolski et al., 1997) | Polyclonal |
| Rabbit anti-GFP | Thermo Fisher | Cat#A-6455 |
| Rat anti-mCherry | Thermo Fisher | Cat#M11217 |
| Rat anti-FLAG (DYKDDDDK Tag) | Biolegend | Cat#637301 |
| Alexa Fluor 488 Goat anti-mouse IgG (H+L) | Thermo Fisher | Cat#A-11029 |
| Alexa Fluor 488 Goat anti-rabbit IgG (H+L) | Thermo Fisher | Cat#A-11008 |
| Alexa Fluor 488 Goat anti-rat IgG (H+L) | Thermo Fisher | Cat#A-11006 |
| Alexa Fluor 568 Goat anti-mouse IgG (H+L) | Thermo Fisher | Cat#A-11031 |
| Alexa Fluor 568 Goat anti-rabbit IgG (H+L) | Thermo Fisher | Cat#A-11011 |
| Alexa Fluor 568 Goat anti-rat IgG (H+L) | Thermo Fisher | Cat#A-11077 |
| Alexa Fluor 647 Goat anti-rabbit IgG (H+L) | Thermo Fisher | Cat#A-11011 |
| IRDye 800CW Goat anti-mouse IgG (H+L) | LI-COR Biosciences |
Cat#925-32210 |
| IRDye 800CW Goat anti-rabbit IgG (H+L) | LI-COR Biosciences |
Cat#925-32211 |
| IRDye 800CW Goat anti-rat IgG (H+L) | LI-COR Biosciences |
Cat#925-32219 |
| IRDye 680RD Goat anti-mouse IgG (H+L) | LI-COR Biosciences |
Cat#925-68070 |
| IRDye 800CW Goat anti-rabbit IgG (H+L) | LI-COR Biosciences |
Cat#925-68071 |
| IRDye 800CW Goat anti-rat IgG (H+L) | LI-COR Biosciences |
Cat#925-68076 |
| Bacterial and Virus Strains | ||
| NEB5α | New England Biolabs | Cat#C2987I |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 3-indoleacetic acid (IAA/ auxin) | Millipore Sigma | Cat#I2SS6 |
| Bovine serum albumin | Millipore Sigma | Cat#B42S7 |
| Ethanol | Parmco-Aaper | Cat#11100020S |
| PET-cGMP | Axxora LLC | Cat#BLG-P001 −10 |
| Zaprinast | Milllipore Sigma | Cat#6S4500 |
| D-luciferin | Biosynth AG | Cat#L-S200 |
| Dolichos biflorus agglutinin - FITC conjugated | Vector Laboratories | Cat#FL-1031 |
| Critical Commercial Assays | ||
| Site-directed mutagenesis kit | New England Biolabs | Cat#E0554 |
| Gibson assembly cloning kit | New England Biolabs | Cat#E5510 |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| One-Taq One-Step RT-PCR kit | New England Biolabs | Cat#E5315 |
| Experimental Models: Cell Lines | ||
| Human Foreskin Fibroblasts | ATCC | Cat#CRL-1634 |
| Experimental Models: Organisms/Strains | ||
| Parasite lines in Table S2 | ||
| C57Bl/6 mice, female, aged 8-10 weeks | Jackson | Cat#000664 |
| Recombinant DNA | ||
| Plasmids listed in Table S3 | ||
| Oligonucleotides | ||
| Primers for PCR listed in Table S4 | ||
| Software and Algorithms | ||
| Clustal Omega | EMBL-EBI | www.ebi.ac.uk/Tools/msa/clustalo/ |
| MEGA6 | (Tamura et al., 2013) | www.Megasoftware.net/ |
| Dendroscope | Dendroscope | www.dendroscope.org/ |
| SnapGene | SnapGene | www.snapgene.com |
| OCTOPUS | Stockholm University |
http://octopus.cbr.su.se/index.php |
| AxioVision Se64 | Carl Zeiss Inc | www.zeiss.com |
| Gen5 | Biotek | www.biotek.com |
| Image Studio Lite | Li-Cor | www.licor.com |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Living Image | Perkin-Elmer | www.perkinelmer.com |
| Graphpad Prism | Graphpad Software | www.graphpad.com |
| CellProfiler | Broad Institute | http://cellprofiler.org/ |
| Image Lab | BIO-RAD | www.bio-rad.com |
Supplementary Material
Highlights.
TgGC is a conserved, apically-localized P-type ATPase / guanylate cyclase in Toxoplasma
TgGC regulates parasite motility by initiating cGMP-dependent microneme exocytosis
The ATPase and GC domains must be catalytically active and linked for TgGC function
TgGC is necessary for Toxoplasma fitness and virulence in vivo
Acknowledgments
Acknowledgements
We thank Qiuling Wang for technical assistance with mouse experiments and Jennifer Barks for technical assistance with tissue culture. Supported in part by grants from the National Institutes of Health to LDS (AI034036) and American Heart Association (15P0ST22130001) to KMB.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alaganan A, Fentress SJ, Tang K, Wang Q, and Sibley LD (2013). Toxoplasma GRA7 effector increases turnover of immunity-related GTPases and contributes to acute virulence in the mouse. Proc Natl Acad Sci (USA) 111, 1126–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DA, Drought LG, Flueck C, Nofal SD, Patel A, Penzo M, and Walker EM (2017). Cyclic nucleotide signalling in malaria parasites. Open Biol 7, 10.1098/rsob.170213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DA, and Kelly JM (2004). Structure, function and evolution of microbial adenylyl and guanylyl cyclases. Mol Microbiol 52, 1229–42. [DOI] [PubMed] [Google Scholar]
- Bastin P, Bagherzadeh Z, Matthews KR, and Gull K (1996). A novel epitope tag system to study protein targeting and organelle biogenesis in Trypanosoma brucei. Molec Biochem Parasitol 77, 235–239. [DOI] [PubMed] [Google Scholar]
- Beck JR, Rodriguez-Fernandez IA, de Leon JC, Huynh MH, Carruthers VB, Morrissette NS, and Bradley PJ (2010). A novel family of Toxoplasma IMC proteins displays a hierarchical organization and functions in coordinating parasite division. PLoS Pathog 6, e1001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnke MS, Khan A, Lauron EJ, Jimah JR, Wang Q, Tolia NH, and Sibley LD (2015). Rhoptry Proteins ROP5 and ROP18 Are Major Murine Virulence Factors in Genetically Divergent South American Strains of Toxoplasma gondii. PLoS genetics 11, e1005434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billker O, Lourido S, and Sibley LD (2009). Calcium-dependent signaling and kinases in apicomplexan parasites. Cell Host Microbe 5, 612–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas KH, Shenoy AR, Dutta A, and Visweswariah SS (2009). The evolution of guanylyl cyclases as multidomain proteins: conserved features of kinase-cyclase domain fusions. J Mol Evol 68, 587–602. [DOI] [PubMed] [Google Scholar]
- Bohne W, and Roos DS (1997). Stage-specific expression of a selectable marker in Toxoplasma gondii permits selective inhibition of either tachyzoites or bradyzoites. Mol Biochem Parasitol 88, 115–26. [DOI] [PubMed] [Google Scholar]
- Brochet M, Collins MO, Smith TK, Thompson E, Sebastian S, Volkmann K, Schwach F, Chappell L, Gomes AR, Berriman M, et al. (2014). Phosphoinositide metabolism links cGMP- dependent protein kinase G to essential Ca2+ signals at key decision points in the life cycle of malaria parasites. PLoS Biol 12, e1001806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KM, Long S, and Sibley LD (2017). Plasma Membrane Association by N-Acylation Governs PKG Function in Toxoplasma gondii. MBio 8, e00375–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KM, Long S, and Sibley LD (2018). Conditional Knockdown of Proteins Using Auxin-inducible Degron (AID) Fusions in Toxoplasma gondii. Bio Protoc 8, 10.21769/BioProtoc.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KM, Lourido S, and Sibley LD (2016). Serum Albumin Stimulates Protein Kinase G- dependent Microneme Secretion in Toxoplasma gondii. J Biol Chem 291, 9554–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullen HE, Jia Y, Yamaryo-Botte Y, Bisio H, Zhang O, Jemelin NK, Marq JB, Carruthers V, Botte CY, and Soldati-Favre D (2016). Phosphatidic Acid-Mediated Signaling Regulates Microneme Secretion in Toxoplasma. Cell Host Microbe 19, 349–60. [DOI] [PubMed] [Google Scholar]
- Bullen HE, and Soldati-Favre D (2016). A central role for phosphatidic acid as a lipid mediator of regulated exocytosis in apicomplexa. FEBS Lett 590, 2469–81. [DOI] [PubMed] [Google Scholar]
- Burg JL, Perelman D, Kasper LH, Ware PL, and Boothroyd JC (1988). Molecular analysis of the gene encoding the major surface antigen of Toxoplasma gondii. J Immunol 141, 3584–91. [PubMed] [Google Scholar]
- Carruthers VB, Sherman GD, and Sibley LD (2000). The Toxoplasma adhesive protein MIC2 is proteolytically processed at multiple sites by two parasite-derived proteases. J Biol Chem 275, 14346–53. [DOI] [PubMed] [Google Scholar]
- Carruthers VB, and Sibley LD (1999). Mobilization of intracellular calcium stimulates microneme discharge in Toxoplasma gondii. Mol Microbiol 31, 421–428. [DOI] [PubMed] [Google Scholar]
- Carruthers VB, and Tomley FM (2008). Microneme proteins in apicomplexans. Subcell Biochem 47, 33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carucci DJ, Witney AA, Muhia DK, Warhurst DC, Schaap P, Meima M, Li JL, Taylor MC, Kelly JM, and Baker DA (2000). Guanylyl cyclase activity associated with putative bifunctional integral membrane proteins in Plasmodium falciparum. J Biol Chem 275, 22147–56. [DOI] [PubMed] [Google Scholar]
- Charif H, Darcy F, Torpier G, Cesbron-Delauw MF, and Capron A (1990). Toxoplasma gondii: characterization and localization of antigens secreted from tachyzoites. Exp Parasitol 71, 114–24. [DOI] [PubMed] [Google Scholar]
- Dobrowolski JM, Niesman IR, and Sibley LD (1997). Actin in the parasite Toxoplasma gondii is encoded by a single copy gene, ACT1 and exists primarily in a globular form. Cell Motil Cytoskeleton 37, 253–62. [DOI] [PubMed] [Google Scholar]
- Dobrowolski JM, and Sibley LD (1996). Toxoplasma invasion of mammalian cells is powered by the actin cytoskeleton of the parasite. Cell 84, 933–939. [DOI] [PubMed] [Google Scholar]
- Endo T, Sethi KK, and Piekarski G (1982). Toxoplasma gondii: calcium ionophore A23187-mediated exit of trophozoites from infected murine macrophages. Exp Parasitol 53, 179–88. [DOI] [PubMed] [Google Scholar]
- Farrell A, Thirugnanam S, Lorestani A, Dvorin JD, Eidell KP, Ferguson DJ, Anderson-White BR, Duraisingh MT, Marth GT, and Gubbels MJ (2012). A DOC2 protein identified by mutational profiling is essential for apicomplexan parasite exocytosis. Science 335, 218–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia CR, Alves E, Pereira PH, Bartlett PJ, Thomas AP, Mikoshiba K, Plattner H, and Sibley LD (2017). InsP3 Signaling in Apicomplexan Parasites. Curr Top Med Chem, 2158–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould MK, and de Koning HP (2011). Cyclic-nucleotide signalling in protozoa. FEMS Microbiol Rev 35, 515–41. [DOI] [PubMed] [Google Scholar]
- Hirai M, Arai M, Kawai S, and Matsuoka H (2006). PbGCbeta is essential for Plasmodium ookinete motility to invade midgut cell and for successful completion of parasite life cycle in mosquitoes. J Biochem 140, 747–57. [DOI] [PubMed] [Google Scholar]
- Jia Y, Marq JB, Bisio H, Jacot D, Mueller C, Yu L, Choudhary J, Brochet M, and Soldati-Favre D (2017). Crosstalk between PKA and PKG controls pH-dependent host cell egress of Toxoplasma gondii. EMBO J 36, 3250–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenthirapalan S, Waters AP, Matuschewski K, and Kooij TW (2016). Functional profiles of orphan membrane transporters in the life cycle of the malaria parasite. Nat Commun 7, 10519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder JU, Engel P, Reimer A, Kruger T, Plattner H, Schultz A, and Schultz JE (1999). Guanylyl cyclases with the topology of mammalian adenylyl cyclases and an N-terminal P-type ATPase- like domain in Paramecium, Tetrahymena and Plasmodium. EMBO J 18, 4222–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long S, Brown KM, Drewry LL, Anthony B, Phan IQH, and Sibley LD (2017). Calmodulin-like proteins localized to the conoid regulate motility and cell invasion by Toxoplasma gondii. PLoS Pathog 13, e1006379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourido S, and Moreno SN (2015). The calcium signaling toolkit of the Apicomplexan parasites Toxoplasma gondii and Plasmodium spp. Cell Calcium 57, 186–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourido S, Shuman J, Zhang C, Shokat KM, Hui R, and Sibley LD (2010). Calcium-dependent protein kinase 1 is an essential regulator of exocytosis in Toxoplasma. Nature 465, 359–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett JL, Marchesini N, Moreno SN, and Sibley LD (2002). Toxoplasma gondii microneme secretion involves intracellular Ca2+ release from IP3 / ryanodine sensitive stores. J Biol Chem 277, 25870–25876. [DOI] [PubMed] [Google Scholar]
- Moon RW, Taylor CJ, Bex C, Schepers R, Goulding D, Janse CJ, Waters AP, Baker DA, and Billker O (2009). A cyclic GMP signalling module that regulates gliding motility in a malaria parasite. PLoS Pathog 5, e1000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono T, Cabrita-Santos L, Leitao R, Bettiol E, Purcell LA, Diaz-Pulido O, Andrews LB, Tadakuma T, Bhanot P, Mota MM, et al. (2008). Adenylyl cyclase alpha and cAMP signaling mediate Plasmodium sporozoite apical regulated exocytosis and hepatocyte infection. PLoS Pathog 4, e1000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmgren MG, and Nissen P (2011). P-type ATPases. Annu Rev Biophys 40, 243–66. [DOI] [PubMed] [Google Scholar]
- Plattner F, Yarovinsky F, Romero S, Didry D, Carlier MF, Sher A, and Soldati-Favre D (2008). Toxoplasma profilin is essential for host cell invasion and TLR11-dependent induction of an interleukin-12 response. Cell Host Microbe 3, 77–87. [DOI] [PubMed] [Google Scholar]
- Radke JR, Donald RG, Eibs A, Jerome ME, Behnke MS, Liberator P, and White MW (2006). Changes in the expression of human cell division autoantigen-1 influence Toxoplasma gondii growth and development. PLoS Pathog 2, e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Brown KM, Lee TD, and Sibley LD (2014). Efficient gene disruption in diverse strains of Toxoplasma gondii using CRISPR/CAS9. mBio 13;5(3):e01114–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldati D, and Boothroyd JC (1993). Transient transfection and expression in the obligate intracellular parasite Toxoplasma gondii. Science 260, 349–352. [DOI] [PubMed] [Google Scholar]
- Starnes GL, Jewett TJ, Carruthers VB, and Sibley LD (2006). Two separate, conserved acidic amino acid domains within the Toxoplasma gondii MIC2 cytoplasmic tail are required for parasite survival. J Biol Chem 281, 30745–54. [DOI] [PubMed] [Google Scholar]
- Su C, Howe DK, Dubey JP, Ajioka JW, and Sibley LD (2002). Identification of quantitative trait loci controlling acute virulence in Toxoplasma gondii. Proc Natl Acad Sci U S A 99, 10753–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, and Kumar S (2013). MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30, 2725–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Liu X, Tao G, Qin M, Yin G, Suo J, and Suo X (2016). “Self-cleaving” 2A peptide from porcine teschovirus-1 mediates cleavage of dual fluorescent proteins in transgenic Eimeria tenella. Vet Res 47, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin CM, and Knoll LJ (2012). A patatin-like protein protects Toxoplasma gondii from degradation in a nitric oxide-dependent manner. Infect Immun 80, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tupper J, Stratford MR, Hill S, Tozer GM, and Dachs GU (2010). In vivo characterization of horseradish peroxidase with indole-3-acetic acid and 5-bromoindole-3-acetic acid for gene therapy of cancer. Cancer Gene Ther 17, 420–8. [DOI] [PubMed] [Google Scholar]
- Wiersma HI, Galuska SE, Tomley FM, Sibley LD, Liberator PA, and Donald RGK (2004). A role for coccidian cGMP-dependent protein kinase in motility and invasion. Intl J Parasit 34, 369–380. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.