

Abstract
Key points
Ascent to high altitude imposes an acid‐base challenge in which renal compensation is integral for maintaining pH homeostasis, facilitating acclimatization and helping prevent mountain sicknesses.
The time‐course and extent of plasticity of this important renal response during incremental ascent to altitude is unclear.
We created a novel index that accurately quantifies renal acid‐base compensation, which may have laboratory, fieldwork and clinical applications.
Using this index, we found that renal compensation increased and plateaued after 5 days of incremental altitude exposure, suggesting plasticity in renal acid‐base compensation mechanisms.
The time‐course and extent of plasticity in renal responsiveness may predict severity of altitude illness or acclimatization at higher or more prolonged stays at altitude.
Abstract
Ascent to high altitude, and the associated hypoxic ventilatory response, imposes an acid‐base challenge, namely chronic hypocapnia and respiratory alkalosis. The kidneys impart a relative compensatory metabolic acidosis through the elimination of bicarbonate (HCO3 −) in urine. The time‐course and extent of plasticity of the renal response during incremental ascent is unclear. We developed an index of renal reactivity (RR), indexing the relative change in arterial bicarbonate concentration ([HCO3 −]a) (i.e. renal response) against the relative change in arterial pressure of CO2 () (i.e. renal stimulus) during incremental ascent to altitude (). We aimed to assess whether: (i) RR magnitude was inversely correlated with relative changes in arterial pH (ΔpHa) with ascent and (ii) RR increased over time and altitude exposure (i.e. plasticity). During ascent to 5160 m over 10 days in the Nepal Himalaya, arterial blood was drawn from the radial artery for measurement of blood gas/acid‐base variables in lowlanders at 1045/1400 m and after 1 night of sleep at 3440 m (day 3), 3820 m (day 5), 4240 m (day 7) and 5160 m (day 10) during ascent. At 3820 m and higher, RR significantly increased and plateaued compared to 3440 m (P < 0.04), suggesting plasticity in renal acid‐base compensations. At all altitudes, we observed a strong negative correlation (r ≤ −0.71; P < 0.001) between RR and ΔpHa from baseline. Renal compensation plateaued after 5 days of altitude exposure, despite subsequent exposure to higher altitudes. The time‐course, extent of plasticity and plateau in renal responsiveness may predict severity of altitude illness or acclimatization at higher or more prolonged stays at altitude.
Keywords: High Altitude, Acid‐Base Physiology, Renal Compensation, Respiratory Alkalosis, Metabolic Acidosis
Key points
Ascent to high altitude imposes an acid‐base challenge in which renal compensation is integral for maintaining pH homeostasis, facilitating acclimatization and helping prevent mountain sicknesses.
The time‐course and extent of plasticity of this important renal response during incremental ascent to altitude is unclear.
We created a novel index that accurately quantifies renal acid‐base compensation, which may have laboratory, fieldwork and clinical applications.
Using this index, we found that renal compensation increased and plateaued after 5 days of incremental altitude exposure, suggesting plasticity in renal acid‐base compensation mechanisms.
The time‐course and extent of plasticity in renal responsiveness may predict severity of altitude illness or acclimatization at higher or more prolonged stays at altitude.
Introduction
Exposure to high altitude (≥2500 m) is a potent physiological stressor on the human body, which elicits a series of integrated responses and adaptations. As a result of reductions in oxygen availability, there is an initial drop in arterial partial pressure of O2 () and arterial blood oxygen saturation (). Two of the most prominent responses and adaptations of the human body under conditions of steady‐state sustained hypoxia serve to elevate the and maintain normal blood pH. The key responses employed to maintain and acid‐base homeostasis in the context of chronic hypoxia are (i) the hypoxic ventilatory response (HVR) and (ii) renal excretion of bicarbonate (HCO3 −), respectively.
The hypoxic ventilatory response
Among the first responses elicited following exposure to acute hypoxic conditions is an increase in resting ventilation, known as the HVR (Dempsey & Forster, 1982; Teppema & Dahan, 2010). The HVR partially corrects levels for a given inspired ; however, a concomitant decrease in the partial pressure of arterial CO2 (; hypocapnia) results, which blunts both central and peripheral respiratory chemoreceptor activation and subsequent ventilatory drive. Thus, if the HVR is to be effective in increasing and in the context of high altitude, the imbalance associated with the blunting effects of hypocapnia must be countered, which involves increasing the HVR sensitivity through carotid body plasticity (i.e. ventilatory acclimatization) (Dempsey et al. 2014). Although this increase in the HVR improves oxygenation, further hypocapnia and acid‐base imbalance results. Because of the opposing effects ventilation has on (increases) and (decreases), and the concomitant respiratory alkalosis, a sole adjustment of the HVR would not be an adequate response mechanism in maintaining acid‐base homeostasis to chronic hypoxia; renal response mechanisms are also required.
Acid‐base responses
Sustained hypocapnia has two contradicting effects on acid‐base equilibrium (Krapf et al. 1991). Initially, a decrease in results in respiratory alkalosis, as described by the Henderson–Hasselbalch equation (Krapf et al. 1991):
| (1) |
where pKa = 6.1 at 37°C; [HCO3 −]a = arterial [HCO3 −]; s = 0.03 (mmol L–1) mmHg–1.
With the persistence of hypocapnia, a compensatory mechanism through acid retention and increased HCO3 − excretion in the urine is elicited (Krapf et al. 1991). This compensatory process results in a decrease in arterial blood pH (pHa) towards normal levels (pHa ∼7.4), known as a relative compensatory metabolic acidosis (Swenson, 2016). An inadequate renal response to respiratory alkalosis may result in a sustained increase in blood pH, blunted HVR, decreased oxygen saturation and decreased cerebral blood flow, and all of these factors have been shown to affect acclimatization, as well as facilitate the onset and severity of acute mountain sickness symptoms (Forester et al. 1975; Krapf et al. 1991; Cumbo et al. 2002, 2006; Gilmartin et al. 2006; Fan et al. 2010; Richalet et al. 2012; Swenson, 2016).
Index of renal reactivity (RR)
Although the importance of the renal responsiveness through acid‐base modifications is well‐defined, the time course of the response during incremental ascent and extent of plasticity in this response is unclear. In addition, although there are clear definitions of the HVR, there is a lack of a simple and accurate index to quantify the renal response during exposure to chronic hypobaric hypoxia and persistent hypocapnia experienced during ascent to high altitude. The Henderson–Hasselbalch equation (Eqn (1)) can be used to calculate blood pH at a given instant (i.e. a fixed equilibrium). However, the Henderson–Hasselbalch equation cannot be used to identify the gain of this response or for predictive purposes when assessing changes over time. In clinical settings, standard tests exist to assess renal function such as a calculation of renal clearance (Traynor et al. 2006). However, these tests require anaerobic arterial blood and urine samples, as well as measurements of urine flow, and are thus are difficult to employ in the context of high altitude. In accordance with general measurements of physiological reflexes, the HVR can be quantified by indexing the change in response (i.e. Δ ventilation) against a change in stimulus (e.g. Δ) (Teppema & Dahan, 2010; Pfoh et al. 2016). Given that a change in in sustained hypoxia is the renal stimulus (which probably impacts intracellular [H+] in renal tubule luminal cells) and a responding compensation occurs via a reduction in [HCO3 −]a, we propose to characterize an index of RR as .
Aims and hypotheses
Although acid‐base regulation at high altitude has been previously investigated, the majority of the available data consist of lumbar cerebrospinal fluid values (Severinghaus et al. 1963; Forster et al. 1975), venous values (Cumbo et al. 2006) and/or rely on simulated altitudes in the laboratory (Ge et al. 2006) that do not take into account the realistic conditions encountered upon exposure to high altitude. Thus, the present study aimed to assess renal acid‐base responses during an incremental ascent to high altitude in the Everest region in the Nepal Himalaya using arterial blood values. Based on the well‐described relationships between and [HCO3 −]a, we developed a simple index of RR, indexing the change in [HCO3 −]a against the relative change in during incremental ascent to altitude () and applied it to a model of incremental ascent to 5160 m of altitude exposure over 10 days. We aimed to determine whether RR increases with incremental ascent over time, suggesting renal plasticity, and whether RR magnitude was correlated with relative changes in arterial pH (ΔpHa) during ascent.
Previous reports using simulated altitude in humans showed that the full renal compensatory response is slow to elicit (Ge et al. 2006) and studies in animal models show evidence of molecular plasticity in response to experimental acid‐base disturbances (Nee et al. 1982; Wagner et al. 2002; de Seigneux et al. 2007). Thus, we hypothesized that, during incremental ascent to high altitude, RR would increase with incremental ascent. In addition, we hypothesized that individuals with larger RR would exhibit a smaller deviation from their baseline pHa throughout ascent.
Methods
Participant recruitment and ethics
Participants were recruited on a voluntary basis to undergo serial arterial blood gas measurements during a research expedition to Everest base camp (5300 m) in the Nepal Himalaya. Inclusion criteria included adult participants over 18 years of age who planned to trek the entire journey and those willing to provide free, verbal and written informed and ongoing consent. We pre‐screened and recruited 20 healthy participants for inclusion (10 females) with no known cardiovascular, respiratory, renal or metabolic disorders. One male participant was taking anti‐hypertensive medication. This study abided by the Canadian Government Tri‐Council policy on research ethics with human participants (TCPS2) and conformed with the standards set by the latest revision of the Declaration of Helsinki, except for registration in a database. Ethical approval was received in advance through the Mount Royal University Human Research Ethics Board (100012 and 101012), University of Alberta Health Research Ethics Board Biomedical Panel (00064195) and was harmonized with the Nepal Health Research Council (109‐2017). Although this study was taking place in the context of a large research expedition to high altitude, the specific study design, research question and data collection were planned a priori.
Study protocol and ascent profile
Participants had baseline measurements performed at a low altitude of 1045 m (Calgary) or 1400 m (Kathmandu, Nepal). They were then flown to 2800 m (Lukla) before beginning the trek (Fig. 1). During the trek, all measurements were made on rest days (i.e. no altitude gain) following one night at each altitude, which occurred on rest days between 09.00 h and 17.00 h at altitudes of 3440 m (Namche), 3820 m (Debuche), 4240 m (Pheriche) and 5160 m (Gorak Shep). Prior to measurements at 5160 m, participants trekked to ∼5300 m (Everest base camp) and back (∼5 h of walking), after which measurements were taken at rest between in the afternoon in a lodge.
Figure 1. Ascent profile of expedition in the Everest region in Nepal.
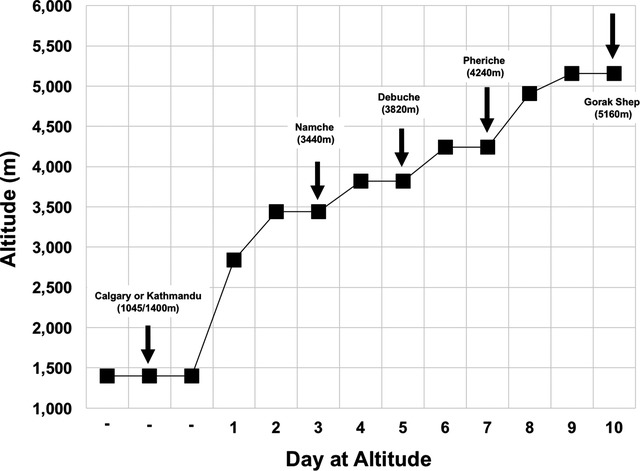
Arrows indicate data collection points and locations. *Baseline data were either collected at 1400 m (as shown) or ∼1 week earlier, prior to departure, at 1045 m.
Measurements
Arterial blood samples were obtained from the radial artery by a trained, registered and experienced respiratory therapist (HEN) when the participant was at rest, in supine position. Samples were analysed for measurement of (mmHg), (%), (mmHg), [HCO3 −]a (mmol L–1), arterial [creatinine] (μmol L–1), haematocrit (%), pHa, strong ion difference (SID; mmol L–1), base excess (mmol L–1) and osmolality (mosmol kg–1) using a portable blood gas/electrolyte analyser (iSTAT, CG4+ and CHEM 8+ cartridges; Abbott, Mississauga, Ontario, Canada; all samples were subject to thermal correction and pressure calibration). Blood was drawn when the participant was at rest, in supine position in a heated lodge. Ancillary measurements included portable pulse oximetry (; %) and capnography [pressure of end‐tidal ; mmHg; adjusted for atmospheric pressure].
Analysis
SID was calculated in accordance with the approach of Stewart (1983). Serum osmolality was calculated in accordance with the recommended formula reported in Martín‐Calderón et al. (2015).
Relative delta values were calculated with respect to baseline (1045/1400 m) values using:
| (2) |
where x = any variable
RR was calculated for each altitude using:
| (3) |
where [HCO3 −]a is in mmol L–1 and is in mmHg.
Individual arterial blood variables and RR values were plotted using Excel, version 14.7.3 (Microsoft Corp., Redmond, WA, USA) with mean values specified. For each of the altitudes, calculated RR was correlated with relative ΔpHa.
To further analyse and visualize the acid‐base disturbances, Davenport diagrams were created for each altitude by plotting points that incorporate the arterial variables: (mmHg), [HCO3 −]a and pHa. Davenport diagrams were also generated using Excel.
Statistical analysis
A Sharpiro–Wilk test was used to confirm normal distribution of variables. The Brown–Forsythe test was used as an equal variance test, where necessary. One‐way repeated‐measures ANOVA tests were performed for arterial blood gas and ancillary data analysed at different altitudes. The Student–Newman–Keuls post hoc test was used for multiple comparisons between the various altitudes. A Pearson product moment correlation test was used to assess relationships between RR vs. ΔpHa. Values are reported as the mean ± SD. P < 0.05 was considered statistically significant using SigmaPlot, version 14 (Systat, San Jose, CA, USA).
Results
Participants
Twenty participants (27.5 ± 9.5 years; range 19–49 years; 26.1 ± 4.4 kg m–2) were recruited with baseline measures at either 1045 or 1400 m, and all 20 were included in analysis. Both baseline locations are considered low altitude and therefore the data were pooled. All participants were native lowlanders who were not exposed to high altitude for at least 1 year prior to the study. No participants included in the study took acetazolamide at any time for the duration of the study. Sample size decreases with ascent were a result of time constraints, illness, device error (n = 1) and/or voluntary withdrawal.
Ancillary and arterial blood variables
Table 1 reports instantaneous ancillary and arterial blood values at each altitude. Figure 2 graphically demonstrates changes in arterial blood variables with ascent. Altitude gain was associated with significant decreases in , , and compared to baseline measures (P < 0.001) (Fig. 2 and Table 1), as expected. Haematocrit showed a statistically significant increase compared to baseline values (1045/1400 m) at 5160 m (day 10; P < 0.001) (Table 1). We found no significant differences in serum [creatinine] with ascent (Table 1) and these values were within normal ranges, suggesting that kidney function was not impaired during ascent.
Table 1.
Mean changes in arterial blood and ancillary variables with ascent
| Variable (mean ± SD) | 0 m (sea level) | 1045/1400 m (n = 20) | 3440 m (n = 18) | 3820 m (n = 17) | 4240 m (n = 15) | 5160 m (n = 14) |
|---|---|---|---|---|---|---|
| Atm pressure (mmHg) | 760 | 675/648 | 509 | 486 | 454 | 411 |
| P iO 2 (mmHg) | 160 | 141/136 | 107 | 102 | 95 | 86 |
| (mmHg) | 75–1001 | 80.2 ± 8.3 | 48.9 ± 7.1* | 52.4 ± 5.7* | 47.9 ± 5.0* | 35.9 ± 4.8*† |
| (%) | 96–1001 | 96.0 ± 1.5 | 85.4 ± 6.4* | 87.6 ± 3.4* | 85.2 ± 3.9* | 72.9 ± 7.2*† |
| (%) | 94–1001 | 97.5 ± 1.2a | 91.9 ± 4.6* | 92.5 ± 3.0* | 88.1 ± 5.0*† | 79.4 ± 4.2*† |
| (mmHg) | 30–432 | 32.9 ± 4.9 | 27.4 ± 2.8* | 25.6 ± 2.5*† | 21.6 ± 2.6*† | 18.9 ± 2.4*† |
| Creatinine (μmol L–1) | 44–1051 | 73.4 ± 21.4 | 72.4 ± 13.3 | 69.9 ± 11.2 | 71.9 ± 13.6 | 75.1 ± 18.6 |
| Haematocrit (%) | 35–521 | 42.6 ± 3.6 | 42.9 ± 3.4b | 42.9 ± 3.2 | 43.5 ± 3.2 | 45.1 ± 3.5*† |
| SID (mmol L–1) | 403 | 39.8 ± 2.0a | 38.3 ± 2.7*b | 38.1 ± 2.4* | 38.2 ± 2.3* | 36.0 ± 2.1*† |
| Base excess (mmol L–1) | (−2)–(+2)4 | −0.8 ± 2.2 | −2.6 ± 2.9* | −4.1 ± 2.0*† | −4.4 ± 2.4* | −6.3 ± 3.3*† |
| Osmolality (mosmol kg–1) | 285–2905 | 294.6 ± 3.8 | 290.6 ± 4.3* b | 292.7 ± 3.3* | 291.5 ± 3.4* | 292.9 ± 2.1 |
| (mmHg) | 35–451 | 35.7 ± 3.8 | 30.7 ± 3.8* | 30.3 ± 3.3* | 29.0 ± 3.2*† | 25.8 ± 2.4*†c |
| [HCO3 −]a (mmol L–1) | 22–261 | 23.7 ± 2.1 | 21.4 ± 2.7* | 20.2 ± 2.1*† | 19.7 ± 2.2* | 18.4 ± 1.8*†c |
| pHa | 7.35–7.451 | 7.430 ± 0.020 | 7.451 ± 0.025* | 7.432 ± 0.022† | 7.441 ± 0.022 | 7.459 ± 0.018* |
1Chernecky & Berger (2004). 2Respironics (2004). 3Chawla & Drummond (2008). 4Berg & Meyer (2008). 5Gennari (1984).
P iO 2, partial pressure of inspired oxygen; , partial pressure of arterial oxygen; , arterial oxygen saturation; , peripheral oxygen saturation; , partial pressure of end tidal carbon dioxide; , partial pressure of arterial carbon dioxide; [HCO3 −]a, concentration of arterial bicarbonate; pHa, arterial pH; SID, strong ion difference. Values at 0 m (sea level) are normative values. *Difference in mean from baseline (1045 /1400 m), P < 0.05. †Difference in mean from prior altitude, P < 0.05. a n = 19. b n = 17. c n = 13.
Figure 2. Changes in arterial blood variables with ascent to high altitude.
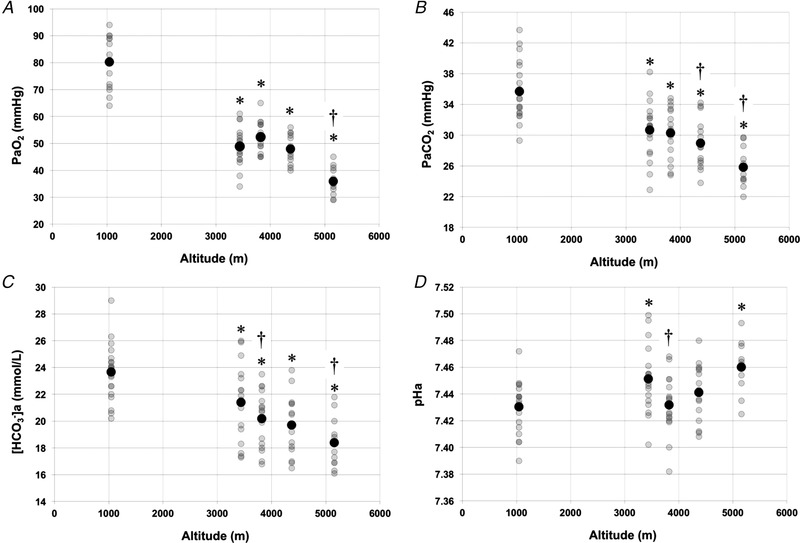
A, partial pressure of arterial O2 (; mmHg). B, partial pressure of arterial CO2 (; mmHg). C, concentration of arterial bicarbonate ([HCO3 −]a; mmol L–1). D, arterial pH (pHa). Note that baseline data (1045 and 1400 m) were pooled. A black circle indicates mean values. *Statistically significant difference from baseline (1045 /1400 m) (P < 0.05). †Statistically significant difference from prior altitude (P < 0.05).
SID decreased significantly at all altitudes compared to baseline (P < 0.05) (Table 1) with a further decrease observed at 5160 m compared to the previous altitude (4240 m; P = 0.009). Base excess was significantly decreased at all altitudes compared to baseline (P < 0.01) (Table 1) with additional significant decreases compared to the previous altitude observed at 3820 m (P = 0.022) and 5160 m (P = 0.027). Osmolality was significantly decreased at 3440, 3820 and 4240 m compared to baseline (P < 0.05) (Table 1). At 5160 m, osmolality approached baseline values (P = 0.09). These differences were within 5 mosmol kg–1 of baseline values, highlighting the normal hydration status of the participants with ascent.
Acid‐base variables
Mean was significantly decreased (P < 0.001) (Fig. 2 and Table 1) at all altitudes compared to baseline values. Values decreased from a baseline mean of 35.7 ± 3.8 mmHg (mean±SD) to 25.8 ± 2.4 mmHg at 5160 m, demonstrating incremental respiratory alkalosis.
There were statistically significant decreases in mean [HCO3 −]a at all altitudes compared to baseline values (P < 0.001) (Fig. 2 and Table 1). Mean [HCO3 −]a values decreased from a baseline of 23.7 ± 2.1 mmol L–1 to 18.4 ± 1.8 mmol L–1 at 5160 m, demonstrating the development of a relative compensatory metabolic acidosis.
There were statistically significant differences in mean pHa compared to baseline at 3440 m (day 3; P < 0.05) and 5160 m (day 10; P < 0.001). At 3820 m (day 5) and 4240 m (day 7), no significant difference were observed between the corresponding pHa values and baseline pHa, indicating that compensatory measures were successful in maintaining pHa at baseline levels at these altitudes (Fig. 2 and Table 1).
RR and related variables
Figure 3 and Table 2 report RR and delta variables at each altitude. Delta and [HCO3 −]a values became more negative with ascent (Fig. 3 and Table 2). Delta pHa values showed variability with ascent, although the changes were mirrored by changes in RR (Fig. 3). RR mean values were 0.39 ± 0.37, 0.61 ± 0.24, 0.57 ± 0.22 and 0.54 ± 0.13 at 3440, 3820, 4240 and 5160 m, respectively. Therefore, RR increased from 3440 m (day 3) to 3820 m (day 5) (P < 0.001) and, above 3820 m (and following 5 days of altitude exposure), RR plateaued compared to the first measurement at 3440 m (i.e. no further statistical changes were observed) (Fig. 3).
Figure 3. The relationship between ΔpHa and RR with ascent to high altitude.
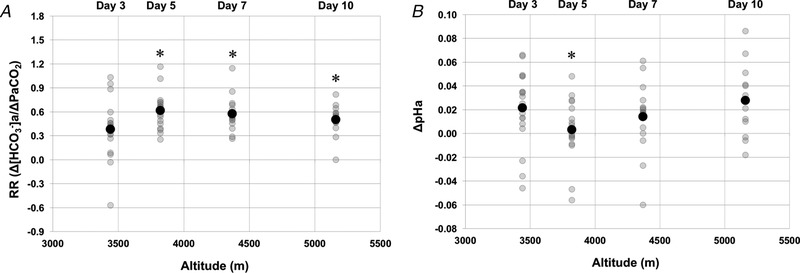
A, RR (Δ[HCO3 −]a)/Δ). B, ΔpHa. A black circle indicates mean values. Delta values at each altitude are compared to baseline values (see Eqn (2)). *Statistically significant difference from 3440 m (P < 0.05). Days reported represent days of altitude exposure.
Table 2.
Mean changes in RR and corresponding delta variables
| Variable (mean ± SD) | 3440 m (n = 18) | 3820 m (n = 17) | 4240 m (n = 15) | 5160 m (n = 13) |
|---|---|---|---|---|
| Δ (mmHg) | −5.1 ± 2.8 | −5.6 ± 2.0 | −7.4 ± 2.2*† | −10.0 ± 2.5*† |
| Δ[HCO3 −]a (mmol L–1) | −2.3 ± 2.2 | −3.5 ± 2.0* | −4.2 ± 1.8* | −5.3 ± 1.8*† |
| ΔpHa | 0.021 ± 0.032 | 0.003 ± 0.026* | 0.014 ± 0.030 | 0.028 ± 0.0291 |
| RR () | 0.385 ± 0.373 | 0.614 ± 0.235* | 0.574 ± 0.221* | 0.539 ± 0.133* |
change in partial pressure of arterial carbon dioxide; Δ[HCO3 −]a, change in concentration of arterial bicarbonate; ΔpHa, change in arterial pH. Note: all changes are with respect to baseline values (see Eqn (2)). *Difference in mean from 3440 m (P < 0.05). †Difference in mean from prior altitude (P < 0.05).
Correlation between RR and ΔpHa
Figure 4 demonstrates correlations between RR and ΔpHa. Strong negative correlations were found between these two variables at all altitudes. Correlation coefficients (r) were −0.71, −0.98, −0.96 and −0.97 at 3440, 3820, 4240 and 5160 m, respectively (all P < 0.001). These results indicate a strong negative relationship between RR and ΔpHa. Interestingly, the slope of this linear relationship increased with incremental ascent. Specifically, the slopes were −0.06, −0.12, −0.13 and −0.20 at 3440, 3820, 4370 and 5160 m, respectively.
Figure 4. Correlations between ΔpHa and RR (Δ[HCO3 −]a)/) with ascent to high altitude.
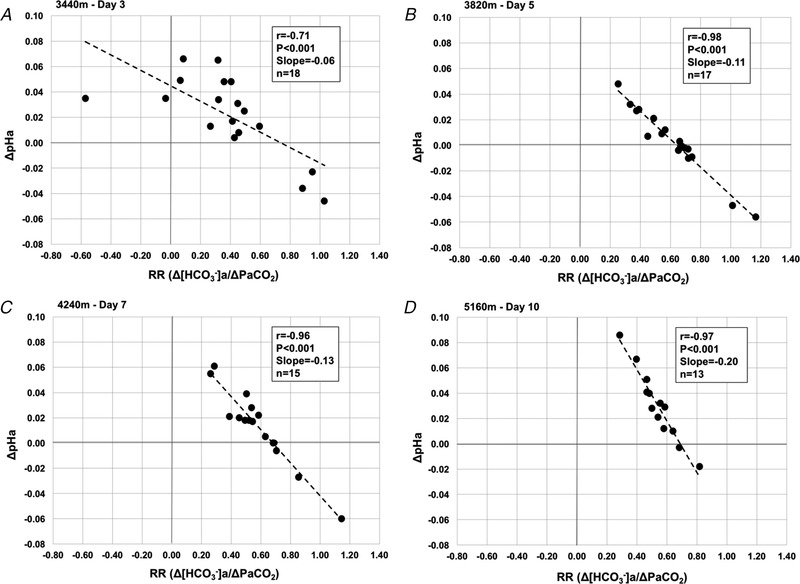
A, 3440 m, day 3. B, 3820 m, day 5. C, 4240 m, day 7. D, 5160 m, day 10. Correlation coefficients (r), P values, slope of the linear response and n are reported in each case. Delta values at each altitude are compared to baseline values (see Eqn (2)). Days reported represent days of altitude exposure.
Davenport diagrams
Figure 5 consists of Davenport diagrams for each altitude with individual points for each participant plotted. Figure 5B presents baseline values at 1045/1400 m. At 3440 m (Fig. 5 C), an initial rightward and downward shift from baseline provides evidence of the development of respiratory alkalosis. At 3820 m (Fig. 5 D), a leftward and downward shift is observed, demonstrating almost complete metabolic compensation. At 4240 m (Fig. 5 E) and 5160 m (Fig. 5 F), further rightward and downward shifts are observed, indicating subsequent progression toward respiratory alkalosis, and only partial compensation.
Figure 5. Davenport acid‐base diagrams during incremental ascent to high altitude.
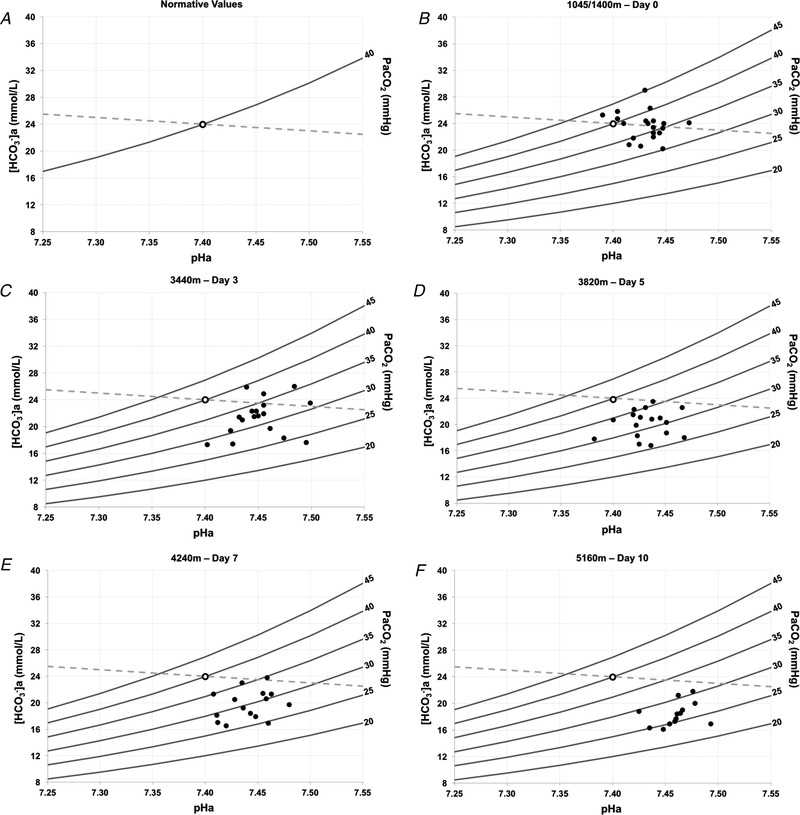
Demonstration of the Henderson‐Hasselbalch relationship depicting acid‐base disturbances and their corresponding compensations, including arterial blood pHa (x‐axis), [HCO3 − a] (y‐axis), isopleths and the [non‐HCO3 − buffer] slope. Solid grey lines represent partial pressure of isopleths. The dashed grey line represents the standard non‐HCO3 − buffer slope. A, template Davenport diagram illustrating the CO2 isopleths and the position of the reference baseline value, where arterial HCO3 − is 24 mmol L–1 (y‐axis), arterial pH (pHa) is 7.4 (x‐axis) and partial pressure of arterial is 40 mmHg. B, 1045/1400 m low altitude baseline for comparison (n = 20). C, 3440 m on day 3 of ascent (n = 18). D, 3820 m on day 5 of ascent (n = 17). E, 4240 m on day 7 of ascent (n = 15). F, 5160 m on day 10 of ascent (n = 13). Days reported represent days of altitude exposure.
Discussion
The present study aimed to assess acid‐base responses during an incremental ascent to high altitude in the Everest region in Nepal. Specifically, we aimed to assess the time course and plasticity of these responses through development and testing of a novel index of ‘renal reactivity’ with ascent. The principal findings include: (i) RR increased over 5 days of ascent, suggesting plasticity in renal acid‐base compensations; (ii) RR plateaued after 5 days, despite further increases in altitude and concomitant hypocapnia; and (iii) the derived RR index was strongly correlated to pHa compensation.
Evidence of hypoxia and hypocapnia and corresponding effects
As expected, we observed the development of hypoxia and hypocapnia with ascent, as indicated by arterial blood and ancillary values. The effects of hypocapnia can be assessed by assessing the carbonic acid/bicarbonate buffer system reaction:
| (4) |
Relative hypocapnia causes a leftward shift in the above reaction in the blood, resulting in decreased H+ production and an increase in blood pH (i.e. respiratory alkalosis). Compensatory metabolic acidosis develops more slowly to bring blood pH toward normal values via renal excretion of HCO3 − in urine. In the present study, the development of a relative compensatory metabolic acidosis was confirmed via the observed decrease in [HCO3 −]a, base excess and SID. The latter takes into consideration electrolyte and acid‐base status in a comprehensive model (Stewart, 1983).
RR
The characteristic development of a primary respiratory alkalosis and relative compensatory metabolic acidosis in response to hypoxia and hypocapnia has been well‐documented (Forster et al. 1975; Gledhill et al. 1975; Krapf et al. 1991; Singh et al. 2003; Cumbo et al. 2006; Ge et al. 2006). However, questions about the time course and extent of plasticity in these acid‐base disturbances and their respective renal response mechanisms remain. Gledhill et al. (1975) previously reported the onset of renal responses through evidence of decreased [HCO3 −]a and increased HCO3 − excretion within 2 h of exposure to a simulated altitude of 3100 m, with pHa compensation incomplete after 26 h. More recently, Ge et al. (2006) provided evidence of the onset of renal compensation within 6 h of exposure to simulated altitudes and demonstrated pHa compensation completion within 24 h at low to moderate altitudes, whereas there was incomplete compensation at higher altitudes (2445 and 2800 m). Generally, at higher altitudes, renal compensation for respiratory alkalosis has been reported to take several days or longer. This was demonstrated by Forster et al. (1975), where evidence of elevated arterial and cerebral spinal fluid pH persisted at 5 and 10 days following exposure to 4300 m. Taken together, these findings suggest that renal acid‐base compensation is more rapid and complete at lower altitudes compared to higher altitudes; however, many of these studies involved altitude simulation and/or evaluation of responses at a single altitude. These questions are further complicated when a realistic altitude exposure scenario is encountered, such as the incremental ascent profile described in our study.
When assessing renal responses using the derived RR index () during incremental ascent to high altitude, we observed an increase in RR over the initial 5 days of altitude exposure up to 3820 m, which suggests plasticity in renal acid‐base compensations. Following the fifth day of altitude exposure (>3820 m), RR plateaued despite further increases in altitude and more pronounced hypocapnia. This was in contrast to our initial hypothesis that RR would continue to increase with ascent. The RR plateau was not associated with significant pHa increases (i.e. additional alkalosis) until day 10 at 5160 m.
Another principle finding was that the RR was strongly correlated to ∆pHa compensation at all altitudes during incremental ascent. Our hypothesis was confirmed by the observation that individuals with larger RR exhibited less relative deviation from their baseline (i.e., low altitude) pHa. At 3440 m (day 3), we observed the lowest correlation between ΔpHa and RR (r = −0.71), whereas all other altitude measurements exhibited correlations of r ≥ −0.96. The weaker correlation at 3440 m probably suggests greater inter‐individual variability in renal response onset and/or magnitude during the initial 3 days of altitude exposure compared to the rest of the ascent. These linear relationships also increased in slope, suggesting (i) that this relationship becomes more pronounced with the elimination of bicarbonate buffer in the blood and (ii) the acid‐base consequences of an attenuated renal reactivity are more profound.
Davenport diagrams
The Davenport diagram (Fig. 5) is a graphical demonstration of the Henderson–Hasselbalch relationship that depicts acid‐base disturbances and their corresponding compensations, which include pHa, , [HCO3 −]a and the [non‐HCO3 − buffer] slope (Davenport, 1974). Davenport diagrams are often used as a teaching tool because they provide a comprehensive model that depicts the steady‐state relationship between various arterial blood variables and their corresponding influence in acid‐base regulation in real time. In Fig. 5, the observed shifts from baseline provide evidence of the respiratory alkalosis and compensatory metabolic acidosis elicited during incremental ascent to high altitude. Furthermore, the location of the points allows for differentiation between partial and complete compensation when referenced to baseline values. Thus, our Davenport diagram representation under hypobaric hypoxic conditions clarifies our further understanding of acid‐base disturbances and their renal compensatory responses during ascent, including the extent of variability between participants. To our knowledge, this is the first demonstration of acid‐base deviations and their associated compensations via Davenport diagrams at various altitudes during incremental ascent to high altitude.
Possible mechanisms underlying changes in renal reactivity
Physiological mechanisms
Although the potential mechanism for the RR plateau cannot be explained from our results, some possibilities can be explored from the published literature. First, altitude exposure is associated with initial hypovolaemia and the hypoxic diuretic response (Goldfarb‐Rumyantzev & Alper, 2013). Additionally, the increase in HCO3 − excretion that characterizes the compensatory metabolic acidosis can contribute to further volume depletion (Cumbo et al. 2002). Volume depletion has been associated with slower alkalosis correction by the kidneys as a result of a reluctance to excrete HCO3 − and accompanying cations to promote fluid retention (West et al. 2012). Furthermore, volume reduction stimulates aldosterone and anti‐diuretic hormone release, which could result in a further reduction of sodium and HCO3 − excretion (Bartsch et al. 1991; Khan et al. 1996). These possibilities, which would all lead to HCO3 − retention for a given decrease, are characteristic of a decreased RR. In the present study, a decrease in osmolality with ascent was observed, suggesting that participants did not experience dehydration at 3440, 3820 and 4240 m. However, osmolality began to increase again at 5160 m; thus, hydration status may have contributed to the observed RR plateau at this higher altitude.
Second, another possibility for the RR plateau could be related to a reduction of effective renal plasma flow. Singh et al. (2003) reported a significant reduction in effective renal plasma flow on day 10 of altitude exposure at 3500 m, with further decreases evident on day 60 and at 5800 m on day 90. Increased haematocrit and blood viscosity, which was only evident at day 10, were associated with the reduction in effective renal plasma flow (Singh et al. 2003). In the present study, we also observed significant increases in haematocrit only after longer altitude exposure when at a higher altitude (day 10 at 5160 m) and therefore our participants could have experienced similar effects of decreased effective renal plasma flow. A decrease in effective renal plasma flow over time could result in less HCO3 − being filtered and excreted in the urine, thus decreasing RR.
Finally, ventilatory acclimatization may contribute to the platuae observed in RR. Forester et al. (1975) proposed that the persistence of alkalosis may be the result of a gradual increase in the sensitivity of peripheral respiratory chemoreceptors during acclimatization. Ge et al. (2006) further speculated that chemosensitivity may change when decreases into the steep portion of the haemoglobin dissociation curve because that is where O2 content is very sensitive to small changes in ventilation and . Thus, increases in chemoreceptor gain may also contribute to RR changes because of its association with respiratory alkalosis at higher altitudes.
Molecular mechanisms
Molecular modifications and the time course of such modifications may have contributed to the observed increase in RR (i.e. plasticity), as well as the observed RR plateau. Although the present study cannot provide direct evidence of the underlying molecular mechanisms, we provide a number of speculations consistent with laboratory studies. The majority of renal HCO3 − reabsorption occurs in the proximal tubule, predominantly by proton secretion at the apical membrane via the Na+‐H+ exchanger (NHE3) (Bartsch et al. 1991). The compensatory increase in HCO3 − excretion in response to respiratory alkalosis has been partially attributed to a decrease in hydrogen ion secretion (Gledhill et al. 1975; Ge et al. 2006; Hamm et al. 2015). The NHE3 found in proximal tubule cells is particularly responsive to acute pH changes, where proton availability allosterically modulates the exchangers resulting in activity changes (Nee et al. 1982; Hamm et al. 2015). Thus, during the development of compensatory metabolic acidosis, associated pH changes may have influenced the activity and/or abundance of such transporters resulting in alterations of HCO3 − excretion. Changes in the levels and activity of the intracellular catalyst carbonic anhydrase (CAII) may also influence HCO3 − excretion, possibly contributing to the observed RR changes.
The remainder of HCO3 − reabsorption occurs in the collecting duct, chiefly by intercalated cells (ICs). Specifically, type A ICs are responsible for H+ secretion and HCO3 − reabsorption, whereas type B ICs have the ability to secrete HCO3 − (Hamm et al. 2015). Within the collecting duct, laboratory studies have demonstrated remodelling and alteration of the number of type A and/or B ICs in response to experimental acid‐base disturbances (Bagnis et al. 2001; Schwartz et al. 2002; Wagner et al. 2002; Al‐Awqati, 2003; Welsh‐Bacic et al. 2011). Additionally, changes in the abundance of transporters found in these cells has been demonstrated in response to experimental acid‐base disturbances (Schwartz et al. 2002; Wagner et al. 2002; de Seigneux et al. 2007; Welsh‐Bacic et al. 2011). Thus, the plasticity demonstrated by these cells may also be relevant in the acid‐base disturbances experienced at high altitude and may contribute to the observed RR increase and subsequent plateau in the present study.
Methodological considerations
Two different altitudes and dates were used for baseline measurements, which was necessary given our recruitment of participants as a part of a larger research expedition. Although this may represent a limitation, we consider that any differences in baseline data are negligible as a result of the low altitude and very small difference between the PIO2 (141 mmHg at 1045 m; 136 mmHg at 1400 m).
There was also a sample size decrease with ascent from n = 20 at baseline to n = 13 at 5160 m. Because of the logistical difficulties of data collection during high altitude field studies, we were satisfied to have obtained any number of arterial blood draws above 5000 m, and very few studies have done so. Regardless, the existence of statistical differences in (stimulus) and HCO3 − (response) at 5160 m lead us to assume that these data are reliable despite the decrease in sample size.
Another consideration is the additional altitude gain on day 10 to ∼5300 m and back prior to data collection at 5160 m. The altitude gain was small and the difference in PIO2 negligible. However, the ∼5 h of walking may have been associated with changes in ventilation and hydration status. This may have contributed to the acid‐base responses observed at 5160 m, such as the sustained respiratory alkalosis. However, measurements were taken following completion of the activity later in the day with the participants having eaten and being hydrated, as well as completely at rest in a supine position. Additionally, numerous studies have demonstrated persistent alkalosis at higher altitudes and over time; thus, these caveats associated with our highest altitude probably play a minimal role.
Potential significance, applications and future directions
The Sagarmatha National Park, where the Everest Base Camp trek is located, is visited by ∼45,000 trekkers per year (Sagarmatha National Park Office, 2017). With the increasing popularity of high altitude trekking and climbing, it is necessary to continue to gain further understanding regarding the integrated responses to incremental hypoxia in healthy lowlanders. Our incremental ascent model represents a ‘real world’ ascent profile that is applicable to large numbers of trekkers, many of whom experience acute mountain sickness, pulmonary oedema and cerebral oedema. Acid‐base regulation has been demonstrated to be important in maintaining health and avoiding maladaptive responses that are associated with the development of mountain sicknesses (Forester et al. 1975; Krapf et al. 1991; Cumbo et al. 2002, 2006; Gilmartin et al. 2006; Fan et al. 2010; Richalet et al. 2012; Swenson, 2016). Thus, a simple measure of renal reactivity during acid‐base perturbations could be useful to integrative physiologists in laboratory or fieldwork contexts. Our novel index may also have utility in assessing renal acid‐base compensations in clinical context where respiratory acidosis (e.g. chronic obstructive pulmonary disease) (Bruno & Valenti, 2012) or alkalosis (e.g. congestive heart failure) (Milionis et al. 2002) is present.
Future research directions could include testing the RR index and associated correlations at higher altitudes and/or exploring RR changes over time at a single altitude. Additionally, correlations between the RR index and other variables involved in the high altitude adaptation and acclimatization (e.g. cerebral blood flow, central sleep apnoea) could be investigated. The integrative assessment and management of acid‐base homeostasis is of significant importance during chronic hypoxic and/or hypo‐ and hypercapnic blood gas challenges, whether from high altitude exposure or clinical disorders.
Conclusions
We developed and characterized an index of renal reactivity during incremental ascent to high altitude. The principle findings of the present study were (i) RR increased over 5 days of ascent, suggesting plasticity in renal acid‐base compensations; (ii) RR plateaued after 5 days, despite further increases in altitude and more profound hypocapnia; and (iii) the derived RR index was strongly and inversely correlated to relative pHa compensation. We consider that the RR index accurately quantified acid‐base responsiveness and renal compensatory mechanisms during incremental exposure to high altitude hypobaric hypoxia and resulting hypocapnia up to an altitude of 5160 m. This novel RR index may be useful to integrative physiologists in the laboratory or field and may have important clinical utility with respect to tracking changes in acid‐base status over time.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
SMZ, HCL, KDO, TDB, CDS and TAD were responsible for intellectual contributions. MTS was responsible for the Nepalese collaboration. CDS was responsible for assistance with ethics. MTS was responsible for assistance with ethics in Nepal. TAD was responsible for ethical clearance. TAD was responsible for organizing the expedition. TAD was responsible for the study design. SMZ was responsible for first draft of manuscript. TDB and HEN were responsible for data collection. CEN was responsible for data organization. SMZ and TAD were responsible for data analysis. SMZ, KDO, TDB, HEN, CEN, CDS, MTS and TAD were responsible for manuscript editing. TAD was responsible for funding. All listed co‐authors approved the final version of the manuscript submitted for publication, agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved, all persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
Funding for this study was provided by (i) Alberta Government Student Temporary Employment Program (SMZ); (ii) Alberta Innovates Health Solutions Summer Studentship (CEN); (iii) Natural Sciences and Engineering Research Council of Canada (NSERC) Undergraduate Research Student Assistantship (SMZ; HCL); and (iv) NSERC Discovery grant (TAD; RGPIN‐2016‐04915).
Acknowledgements
We are grateful to our research participants for their time and effort, and also for the efforts of our Sherpa guide team. We wish to thank (alphabetical by last name) Garrick Chan, Jason Chan, Alexandra Chiew, Tegen Jones, Andrea Linares, Carli Mann, Joel Peltonen, Alexander Rimke, Rupinder Sandhu and Gary Saran for helping to obtain ancillary measures at each altitude.
Biography
Shaelynn Zouboules is a recent graduate from Mount Royal University in Calgary, Alberta, Canada, where she obtained a Bachelor of Science degree. Her research interests include acid‐base physiology, cerebrovascular physiology and high‐altitude physiology. She was recently a recipient of the Barbara A. Horwitz and John M. Horowitz Outstanding Undergraduate Abstract Award and the Barbara A. Horwitz and John M. Horowitz Excellence in Undergraduate Research Award from the American Physiological Society, presented at the Experimental Biology meeting in 2018.

Edited by: Harold Schultz & Scott Powers
This is an Editor's Choice article from the 15 December 2018 issue.
References
- Al‐Awqati Q (2003). Terminal differentiation of intercalated cells: the role of hensin. Annu Rev Physiol 65, 567–583. [DOI] [PubMed] [Google Scholar]
- Bagnis C, Marshansky V, Breton S & Brown D (2001). Remodeling the cellular profile of collecting ducts by chronic carbonic anhydrase inhibition. Am J Physiol Renal Physiol 280, F437–F448. [DOI] [PubMed] [Google Scholar]
- Bartsch P, Maggiorini M, Schobersberger W, Shaw S, Rascher W, Girard J, Weidmann P & Oelz O (1991). Enhanced exercise‐induced rise of aldosterone and vasopressin preceding mountain sickness. J Appl Physiol 71, 136–143. [DOI] [PubMed] [Google Scholar]
- Berg MD & Meyer RJ (2008). Gas exchange and acid‐base physiology In Pediatric Respiratory Medicine, 2nd edn, ed. Taussig LM. & Landau LI, pp. 179–200. Mosby, Philadelphia, PA. [Google Scholar]
- Bruno CM & Valenti M (2012). Acid‐base disorders in patients with chronic obstructive pulmonary disease: a pathophysiological review. J Biomed Biotechnol 2012, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla G & Drummond G (2008). Water, strong ions, and weak ions. BJA Educ 8, 108–112. [Google Scholar]
- Chernecky CC & Berger BJ (2004). Laboratory Tests & Diagnostic Procedures. Elsevier Inc, Philadelphia, PA. [Google Scholar]
- Cumbo TA, Basnyat B, Graham J, Lescano AG & Gambert S (2002). Acute mountain sickness, dehydration, and bicarbonate clearance: preliminary field data from the Nepal Himalaya. Aviat Space Environ Med 73, 898. [PubMed] [Google Scholar]
- Cumbo TA, Braude D, Basnyat B, Rabinowitz L, Lescano AG, Shah MB, Radder DJ, Bashyal G & Gambert SR (2006). Higher venous bicarbonate concentration associated with hypoxemia, not acute mountain sickness, after ascent to moderate altitude. J Travel Med 12, 184–189. [DOI] [PubMed] [Google Scholar]
- Davenport HW (1974). The ABC of Acid‐Base Chemistry: The Elements of Physiological Blood‐Gas Chemistry for Medical Students and Physicians. University of Chicago Press, Chicago, IL. [Google Scholar]
- de Seigneux S, Malte H, Dimke H, Frøkiaer J, Nielsen S & Frische S (2007). Renal compensation to chronic hypoxic hypercapnia: downregulation of pendrin and adaptation of the proximal tubule. Am J Physiol Renal Physiol 292, F1256–F1266. [DOI] [PubMed] [Google Scholar]
- Dempsey JA & Forster HV (1982). Mediation of ventilatory adaptations. Physiol Rev 62, 262–346. [DOI] [PubMed] [Google Scholar]
- Dempsey JA, Powell FL, Bisgard GE, Blain GM, Poulin MJ & Smith CA (2014). Role of chemoreception in cardiorespiratory acclimatization to, and deacclimatization from, hypoxia. J Appl Physiol. 116, 858–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan JL, Burgess KR, Basnyat R, Thomas KN, Peebles KC, Lucas SJE, Lucas RAI, Donnelly J, Cotter JD & Ainslie PN (2010). Influence of high altitude on cerebrovascular and ventilatory responsiveness to CO2 . J Physiol 588, 539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster HV, Dempsey JA & Chosy LW (1975). Incomplete compensation of CSF [H+] in man during acclimatization to high altitude (4300 M). J Appl Physiol 38, 1067. [DOI] [PubMed] [Google Scholar]
- Ge R, Babb TG, Sivieri M, Resaland GK, Karlsen T, Stray‐Gundersen J & Levine BD (2006). Urine acid–base compensation at simulated moderate altitude. High Alt Med Biol 7, 64–71. [DOI] [PubMed] [Google Scholar]
- Gennari F (1984). Current concepts: serum osmolality: uses and limitations. N Engl J Med 310, 102. [DOI] [PubMed] [Google Scholar]
- Gilmartin G, Tamisier R, Anand A, Cunnington D & Weiss JW (2006). Evidence of impaired hypoxic vasodilation after intermediate‐duration hypoxic exposure in humans. Am J Physiol Heart Circ Physiol 291, H2173–H2180. [DOI] [PubMed] [Google Scholar]
- Gledhill N, Beirne GJ & Dempsey JA (1975). Renal response to short‐term hypocapnia in man. Kidney Int 8, 376–384. [DOI] [PubMed] [Google Scholar]
- Goldfarb‐Rumyantzev AS & Alper SL (2013). Short‐term responses of the kidney to high altitude in mountain climbers. Nephro Dial Transplant 29, 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamm LL, Nakhoul N & Hering‐Smith KS (2015). Acid‐base homeostasis. Clin J Am Soc Nephrol 10, 2232–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan DA, Aslam M & Khan ZU (1996). Changes in plasma electrolytes during acclimatization at high altitude. J Pak Med Assoc 46, 128. [PubMed] [Google Scholar]
- Krapf R, Beeler I, Hertner D & Hulter HN (1991). Chronic respiratory alkalosis. The effect of sustained hyperventilation on renal regulation of acid‐base equilibrium. N Engl J Med 324, 1394–1401. [DOI] [PubMed] [Google Scholar]
- Martín‐Calderón JL, Bustos F, Tuesta‐Reina LR, Varona JM, Caballero L & Solano F (2015). Choice of the best equation for plasma osmolality calculation: comparison of fourteen formulae. Clin Biochem 48, 529–533. [DOI] [PubMed] [Google Scholar]
- Milionis HJ, Alexandrides GE, Liberopoulos EN, Bairaktari ET, Goudevenos J & Elisaf MS (2002). Hypomagnesemia and concurrent acid‐base and electrolyte abnormalities in patients with congestive heart failure. Eur J Heart Fail 4, 167–173. [DOI] [PubMed] [Google Scholar]
- Nee J, Suhm MA & Aronson PS (1982). Modifier role of internal H+ in activating the Na+‐H+ exchanger in renal microvillus membrane vesicles. Nature 299, 161–163. [DOI] [PubMed] [Google Scholar]
- Pfoh JR, Tymko MM, Abrosimova M, Boulet LM, Foster GE, Bain AR, Ainslie PN, Steinback CD, Bruce CD & Day TA (2016). Comparing and characterizing transient and steady‐state tests of the peripheral chemoreflex in humans. Exp Physiol 101, 432–447. [DOI] [PubMed] [Google Scholar]
- Richalet JP, Larmignat P, Poitrine E, Letournel M & Canouï‐Poitrine F (2012). Physiological risk factors for severe high‐altitude illness: a prospective cohort study. Am J Respir Crit Care Med 185, 192–198. [DOI] [PubMed] [Google Scholar]
- Respironics (2004). Capnography Reference Handbook. Respironics Inc., Wallingford. [Google Scholar]
- Sagarmatha National Park Office (2017). Sagarmatha National Park Fact Sheet. Sagarmatha National Park Office, Namche Bazaar. [Google Scholar]
- Schwartz G, Tsuruoka S, Vijayakumar S, Petrovic S, Mian A & Al‐Awqati Q (2002). Acid incubation reverses the polarity of intercalated cell transporters, an effect mediated by hensin. J Clin Invest 109, 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severinghaus JW, Mitchell RA, Richardson BW & Singer MM (1963). Respiratory suggesting control at high altitude active transport regulation of CSF pH. J Appl Physiol 18, 1155–1166 [DOI] [PubMed] [Google Scholar]
- Singh MV, Salhan AK, Rawal SB, Tyagi AK, Kumar N, Verma SS & Selvamurthy W (2003). Blood gases, hematology, and renal blood flow during prolonged mountain sojourns at 3500 and 5800 m. Aviat Space Environ Med 74, 533–536. [PubMed] [Google Scholar]
- Stewart PA (1983). Modern quantitative acid–base chemistry. Can J Physiol Pharmacol 61, 1444–1461. [DOI] [PubMed] [Google Scholar]
- Swenson ER (2016). Hypoxia and its acid–base consequences: from mountains to malignancy. Adv Exp Med Biol 903, 301–323. [DOI] [PubMed] [Google Scholar]
- Teppema L & Dahan A (2010). The ventilatory response to hypoxia in mammals: mechanisms, measurement, and analysis. Physiol Rev 90, 675–754. [DOI] [PubMed] [Google Scholar]
- Traynor J, Mactier R, Geddes CC & Fox JG (2006). How to measure renal function in clinical practice. BMJ 333, 733–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner CA, Finberg KE, Stehberger PA, Lifton RP, Giebisch GH, Aronson PS, Geibel JP (2002). Regulation of the expression of the Cl–/anion exchanger pendrin in mouse kidney by acid‐base status. Kidney Int 62, 2109–2117. [DOI] [PubMed] [Google Scholar]
- Welsh‐Bacic D, Nowik M, Kaissling B & Wagner CA (2011). Proliferation of acid‐secretory cells in the kidney during adaptive remodelling of the collecting duct. PLoS ONE 6, e25240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JB, Schoene RB, Luks A & Milledge JS (2012). High Altitude Medicine and Physiology. CRC Press, Boca Raton, FL. [Google Scholar]