

Abstract
Key points
Recurrent periods of over‐excitation in the paraventricular nucleus (PVN) of the hypothalamus could contribute to chronic over‐activation of this nucleus and thus enhanced sympathetic drive.
Stimulation of the PVN glutamatergic population utilizing channelrhodopsin‐2 leads to an immediate frequency‐dependent increase in baseline blood pressure.
Partial lesions of glutamatergic neurons of the PVN (39.3%) result in an attenuated rise in blood pressure following Deoxycorticosterone acetate (DOCA)‐salt treatment and reduced index of sympathetic activity.
These data suggest that stimulation of PVN glutamatergic neurons is sufficient to cause autonomic dysfunction and drive the increase in blood pressure during hypertension.
Abstract
Neuro‐cardiovascular dysregulation leads to increased sympathetic activity and neurogenic hypertension. The paraventricular nucleus (PVN) of the hypothalamus is a key hub for blood pressure (BP) control, producing or relaying the increased sympathetic tone in hypertension. We hypothesize that increased central sympathetic drive is caused by chronic over‐excitation of glutamatergic PVN neurons. We tested how stimulation or lesioning of excitatory PVN neurons in conscious mice affects BP, baroreflex and sympathetic activity. Glutamatergic PVN neurons were unilaterally transduced with channelrhodopsin‐2 using an adeno‐associated virus (CamKII‐ChR2‐eYFP‐AAV2) in wildtype mice (n = 7) to assess the impact of acute stimulation of excitatory PVN neurons selectively on resting BP in conscious mice. Stimulation of the PVN glutamatergic population resulted in an immediate frequency‐dependent (2, 10 and 20 Hz) increase in BP from baseline by ∼9 mmHg at 20 Hz stimulation (P < 0.001). Additionally, in vGlut2‐cre mice glutamatergic neurons of the PVN were bilaterally lesioned utilizing a cre‐dependent caspase (AAV2‐flex‐taCASP3‐TEVp). Resting BP and urinary noradrenaline (norepinephrine) levels were then recorded in conscious mice before and after DOCA‐salt hypertension. Partial lesions of glutamatergic neurons of the PVN (39.3%, P < 0.05) resulted in an attenuated rise in BP following DOCA‐salt treatment (P < 0.05 at 7 day time point, n = 8). Noradrenaline levels as an index of sympathetic activity between the lesion and wildtype groups showed a significant reduction after DOCA‐salt treatment in the lesioned animals (P < 0.05). These experiments suggest that stimulation of PVN glutamatergic neurons is sufficient to cause autonomic dysfunction and drive the increase in BP.
Keywords: Hypertension, Hypothalamic neurone, Glutamate, Optogenetics, Sympathetic nervous system, Autonomic Nervous system, blood pressure
Key points
Recurrent periods of over‐excitation in the paraventricular nucleus (PVN) of the hypothalamus could contribute to chronic over‐activation of this nucleus and thus enhanced sympathetic drive.
Stimulation of the PVN glutamatergic population utilizing channelrhodopsin‐2 leads to an immediate frequency‐dependent increase in baseline blood pressure.
Partial lesions of glutamatergic neurons of the PVN (39.3%) result in an attenuated rise in blood pressure following Deoxycorticosterone acetate (DOCA)‐salt treatment and reduced index of sympathetic activity.
These data suggest that stimulation of PVN glutamatergic neurons is sufficient to cause autonomic dysfunction and drive the increase in blood pressure during hypertension.
Introduction
Hypertension is one of the most prevalent conditions observed in health care, with 46% of the US adult population affected by the disease, showing complex aetiology including an overactive sympathetic tone (Whelton et al. 2018). Blood pressure (BP) and cardiac output are continuously adjusted by the central nervous system (CNS) to match the fluctuating metabolic requirements of behaviour. These rapid adjustments occur almost entirely via changes in sympathetic and cardio‐vagal efferent activity (Guyenet, 2006). Mean (24 h average) BP, however, is maintained within relatively narrow limits by the kidneys via blood volume control and by the properties of blood vessels. Mean BP is also influenced by the sympathetic nervous system and many forms of hypertension are currently presumed to be partly caused by excessive sympathetic tone (Esler et al. 2001). Sympathetic drive is determined by a vast network of neurons that converge on a limited number of pre‐sympathetic neurons including those in the hypothalamic paraventricular nucleus (PVN), brainstem and spinal cord that directly innervate sympathetic preganglionic neurons (Swanson, 1977; Samson et al. 2007; Marina et al. 2011; Guyenet et al. 2013). The hypothalamus modulates a myriad of body functions, including cardiovascular control, metabolic regulation, body temperature, and sexual and feeding behaviours (Saper & Lowell, 2014). The role of the PVN in the central regulation of BP and autonomic function is well established (Zhang et al. 2002; Shi et al. 2010; Sriramula et al. 2011). The PVN assimilates inputs from circumventricular nuclei that contain osmo‐sensitive neurons (e.g. subfornical organ) and the nucleus of the solitary tract (NTS) relaying them down to multiple areas including the rostral ventrolateral medulla (RVLM) in the brainstem (Geerling et al. 2010; Stocker et al. 2013). The RVLM, an integral part of sympathetic output relay, is thought to be the main recipient of these PVN outputs (Esler et al. 2001).
The degree to which the PVN contributes to BP both at baseline and in hypertensive conditions in conscious mammals still requires investigation. The vast majority of studies up to this point focus on the hypothalamic‐hormonal contribution from neurons in this part of the brain (Geraldes et al. 2014; Vivas et al. 2014; Brown, 2016; Lozic et al. 2018). However, there is a growing body of evidence suggesting a role for the PVN in more immediate sympathetic nervous system regulation (Guyenet, 2006). It has been shown that in anaesthetized and reduced preparations, i.e. experiments that isolate one part of the system/network being studied to decrease the variables involved, the PVN plays an important role in BP regulation (Kc & Dick, 2010; Gomes da Silva et al. 2012). Pharmacological interventions activating PVN neurons or inhibiting GABAergic neurons of the PVN cause an increase in BP and an observed increase in sympathetic nerve activity (Li et al. 2001; Gabor & Leenen, 2012; Gomes da Silva et al. 2012). This activation is presumably functioning through the glutamatergic neuronal population in the PVN projecting to the RVLM but this remains to be validated in conscious animals (Geerling et al. 2010). Glutamate, a well‐known excitatory neurotransmitter in the CNS, has been studied and found to be functionally relevant in the PVN (Miyawaki et al. 1996; Butcher & Cechetto, 1998). More specifically, in pharmacological studies where NMDA receptors of the PVN are activated unambiguously, sympathetic nerve activity increases, thus affecting the BP (Li et al. 2001). Accordingly, we hypothesize that increased central sympathetic drive is caused by chronic over‐excitation of glutamatergic PVN neurons during the development of neurogenic hypertension. This was examined in two ways using conscious mice, through selective activation of this neuronal population with channelrhodopsin‐2 (ChR2) and secondly, through selective destruction of the same population. We find that unilateral activation of the glutamate population of PVN neurons is sufficient to drive an increase in BP similar to that seen during hypertension. Moreover, loss‐of‐function experiments demonstrate impaired baroreflex function, attenuated BP rise and decreased sympathetic outflow during DOCA‐salt hypertension.
Methods
Animals
Experiments were performed on male and female C57Bl6/J mice (n = 35; 20–35 g) or vGlut2‐Cre mice (B6J.129S6(FVB)‐Slc17a6tm2(cre)Lowl/MwarJ that were mated with CAG‐tdTomato reporter mice (B6.Cg‐Gt(ROSA)26Sortm14(CAG‐tdTomato)Hze/J (Jackson Laboratories, Bar Harbor, ME, USA). All mice were on a C57Bl6/J background. All procedures conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Louisiana State University Health Sciences Center Animal Care and Use Committee. Animals were housed under a standard 12 h light/dark cycle with ad libitum access to food and water.
Viral constructs and virus preparation
For experiments on conscious mice, two different adeno‐associated viral vectors (AAVs) were used. The first expresses the photoactivatable cation channel channelrhodopsin‐2 (ChR2) under the calcium/calmodulin‐dependent protein kinase type II α (CaMKIIα) promoter (AAV‐CamKIIα‐ChR2‐eYFP serotype 2) (courtesy of Dr Karl Deisseroth, Stanford Univ., CA, USA). The CamKIIα promoter has been shown to be selective for glutamate neurons in the thalamic neuronal populations (Liu & Jones, 1996; Wang et al. 2013; Watakabe et al. 2015). The AAV‐flex‐taCasp3‐TEVp (courtesy of Dr N. Shah, Univ. of Arkansas, AR, USA) was used for lesion experiments, performed on vesicular glutamate2 cre‐recombinase (vGlut2‐Cre) mice. Viral vectors were obtained from the University of North Carolina virus core. The ChR2 and Caspase AAV2s were used at concentrations of 5.2 × 1012 and 1.8 × 1012 viral particles/ml, respectively.
Injections of vectors and mouse instrumentation
Mice were anaesthetized with 1.5–2% isoflurane in an oxygen flow (1 l/min). Depth of anaesthesia was assessed by absence of corneal and hindpaw withdrawal reflexes. Body temperature was kept near 37°C with a heating pad (WPI Inc. Sarasota, FL, USA) and blanket.
The AAVs were microinjected into the PVN using a stereotaxic approach. The mouse was placed prone on a Stoelting stereotaxic frame making sure the skull lay flat. A 1.5 mm‐diameter hole was drilled into the parietal plate on the left or both sides caudal to bregma. Unilateral microinjections were performed on mice receiving the AAV‐CamKIIα‐ChR2‐eYFP and bilaterally on those receiving the AAV‐flex‐taCasp3‐TEVp. Viral vectors were loaded into a 1.2 mm internal diameter glass pipette broken to 25 μm tip (external diameter). Each AAV was injected into three rostro‐caudally aligned sites separated by 200 μm resulting in a total volume of 400–600 nl/side (∼200 nl/injection site). Stereotaxic coordinates of PVN microinjections were 0.3 mm lateral, 0.6 mm caudal and 4.5 mm ventral. In animals that received the unilateral microinjections of AAV‐CamKIIα‐ChR2‐eYFP, an optical fibre was then implanted for light stimulation. Optical fibres (125 μm; Thorlabs, Newton, NJ, USA) were fitted with ferrules, as previously described (Sparta et al. 2011) and implanted into the PVN with the tip 0.5 mm dorsal to the vector injection site during the same surgery period. The optic‐ferrule assembly was secured to the skull using a two‐part epoxy adhesive (Loctite). Incisions were then closed with absorbable sutures and Vetbond adhesive. Mice received postoperative Buprenorphine SR (0.5 mg/kg subcutaneously) and penicillin (600–1000 UI/10 g i.m.) and were monitored daily. No sham operations were performed for this study. Previous work shows injections of AAV alone do not affect BP (Basting et al. 2015). Mice were given gel food, allowed to recover for a minimum of 5 weeks for the ChR2 and 2 weeks for the caspase and then implanted with a radio‐telemetry probe (PA‐C10, Data Sciences International (DSI), St Paul, MN, USA) to record BP via the left carotid artery. Spontaneous baroreceptor reflex sensitivity (SBRS), reflecting the baroreflex control of heart rate, was calculated using the sequence method as described previously (Stauss et al. 2006). These mice were allowed to recover for an additional week before physiological experiments began.
Optogenetic experiments in conscious mice
On the day of the experiment, mice were lightly anaesthetized with isoflurane (2% in an oxygen flow (1 l/min) for <1 min) to allow cleaning of hardware and connection of the ferrule to the laser. A 125 μm‐thick multimode optical fibre, terminated with a ferrule, was mated to the implanted ferrule with a zirconium sleeve. Optical matching gel (Fibre Instrument Sales, Oriskany, NY, USA) was applied at the junction of the ferrules to reduce light loss. A minimum of 45 min was allowed for recovery from anaesthesia.
Photoexcitation of ChR2‐expressing PVN neurons was achieved with a blue laser (473 nm, Shanghai Laser and Optics Century, Shanghai, China). The Laser was controlled using an A300 model stimulator (WPI Inc., Sarasota, FL, USA). Blue light was pulsed for 10 s at 2, 10 or 20 Hz, in a randomized fashion, with a pulse‐width of 20 ms at ∼9 mW of light output at the tip. The transmission efficiency of each implanted optical fibre was tested before implantation with a light power meter (Thorlabs, Newton, NJ, USA). A minimum of five photoactivation trials were conducted in each mouse, at each stimulation frequency level. Average BP values were extracted from all trials not influenced by body movements, as indicated by BP recording and observation. Cardiovascular parameters (BP, heart rate) were averaged during the 10 s photostimulus. Baseline values were measured during the 20 s preceding light delivery.
DOCA‐salt protocol
For each surgery, the mouse was anaesthetized with isoflurane, placed on a heating pad to maintain body temperature and received postoperative care as described above.
Baseline BP was recorded, using DSI Ponemah software and radio telemetry probes (PA‐C10), in uni‐nephrectomized mice after a week of recovery (Xia et al. 2013). Mice undergoing the DOCA‐salt treatment were implanted subcutaneously with a DOCA‐silicone pellet (DOCA‐silicone = 1:3; DOCA: 1 mg/g body weight) a week after being uni‐nephrectomized. Drinking water was switched to 1% NaCl solution. BP was recorded for three additional weeks. All recordings were done for a continuous hour‐long period between 09.00 and 11.00 h depending on the day.
Western blot
After the animal was killed by ketamine/xylazine overdose, the brain was rapidly extracted and frozen on dry ice. The RVLM was identified (−7.0 mm to −6.0 mm relative to bregma) and dissected grossly (Franklin & Paxinos, 1997). Tissues were suspended in 200 μl lysis buffer per sample (Cell Lysis Buffer 1, cat. no. 890713; R&D Systems, Minneapolis, MN, USA) containing a protease inhibitor cocktail (cat. no. 4693159001; Sigma, St Louis, MO, USA). Tissue was sonicated and centrifuged at 4°C and whole‐protein supernatant was collected. Protein concentration was measured via bicinchoninic acid (BCA) assay (cat. no. 23225; Pierce, Rockford, IL, USA). Lysate samples with equal amounts of protein were prepared via dilution in lysis buffer. Lysate samples were mixed with SDS–PAGE sample buffer and boiled at 95°C for 5 min. Samples were loaded on a 4–15% SDS–polyacrylamide gel for electrophoresis at 80 V for 40 min, followed by 120 V for 30 min (cat. no. 456‐1086; Bio‐Rad, Hercules, CA, USA). Proteins were transferred to a nitrocellulose membrane on ice at 100 V for 1 h (cat. no. 162‐0115; Bio‐Rad). The membrane was washed once for 15 min and 3 times for 5 min each in Tris buffered saline with Tween 20 (TBST) prior to 1 h incubation in a 5% non‐fat milk and 0.01% w/v thimerosal in TBST (cat. no. AAJ77500K8; Fisher Scientific, Houston, TX, USA) prior to overnight incubation with anti‐tumor necrosis factor receptor 1 (anti‐TNF‐R1) (1:1000, cat. no. sc‐8436; Santa Cruz Biotechnology, Santa Cruz, CA, USA). Membranes were washed once for 15 min and 3 times for 5 min in TBST, then incubated with goat anti‐mouse immunoglobulin G (IgG)–horseradish peroxidase (HRP) in blocking buffer (1:10,000, cat. no. NEF822001EA; Perkin Elmer, Waltham, MA, USA) for 1 h at room temperature. Bands were developed via chemiluminescence after HRP activation with an ECL reagent (cat. no. RPN2232; GE Healthcare UK, Little Chalfont, UK). Bands were detected and densitized using the commercial Amersham Imager 600 hardware and software (cat. no. 29083461; GE Healthcare Bio‐Sciences AB, Uppsala, Sweden).
For the loading control, the membrane was washed 1 × 15 min and 3 × 5 min in TBST before re‐probing with a mixed primary/secondary/blocking buffer solution (2 h incubation with 1:1000 rabbit‐anti‐GAPDH, cat. no. 14C10; Cell Signaling, Danvers, MA, USA and 1:5000 HRP‐conjugated goat‐anti‐rabbit, cat. no. NA934vs; GE Healthcare UK, Little Chalfont, UK). Loading control bands were imaged on the same Amersham imaging system listed above.
Urine noradrenaline, gene expression and serum copeptin
Urine was collected from both controls and mice with bilateral PVN lesions before and after the DOCA‐salt protocol. Noradrenaline (norepinephrine; NE) levels were measured as an index of sympathetic activity, using a noradrenaline enzyme‐linked immunosorbent assay (ELISA) kit (IB89552; Immuno‐Biological Laboratories, Minneapolis, MN, USA) according to the manufacturer's instructions. The optical density was measured with a photometer at 405 nm (reference wavelength: 620–650 nm). Each 10 μl sample was measured in duplicate.
Total RNA was isolated from the RVLM using RNeasy Plus Micro Kit (Qiagen, Waltham, MA, USA). Real time PCR amplification reactions were performed with SYBR Green Master mix (Roche, Mannheim, Germany) using a Real time PCR machine (Roche LightCycler 480). Data were normalized to β‐actin expression by the comparative method.
Blood serum copeptin levels were measured in controls and mice with bilateral PVN lesions (Cloud‐Clone Corp., Katy, TX, USA, ELISA kit: CEA365Mu). Each sample was collected by sub‐mandibular puncture of the facial vein.
Histology
Animals were deeply anaesthetized with a ketamine (100 mg/kg)/xylazine (10 mg/kg) mix and transcardially perfused with 4% paraformaldehyde in PBS. The brain was removed and sectioned at 20 μm‐thick slices of the PVN, collected in a cryostat and mounted for further analysis. The PVN was defined according to Paxinos and Franklin's Mouse Brain Atlas from anterior to posterior relative to bregma (+0.5 to −1.22 mm) (Franklin & Paxinos, 1997). Cell mapping, counting and photography were done using the Leica LASX software with a Leica DMi8 microscope. A gold/yellow filter was used (Fig. 4) to aid in cell counting, emphasizing red cell bodies in tdTomato‐positive neurons.
Figure 4. Virally mediated lesions of glutamate neurons in the PVN.
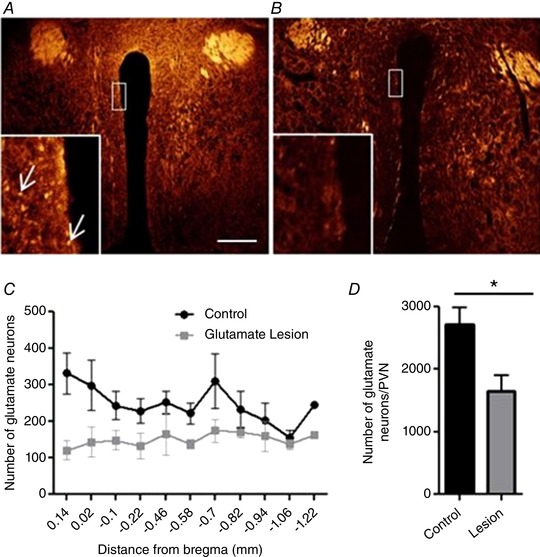
A, example of a vGlut2‐cre tdTomato control coronal cross‐section of the PVN (pseudo colour). Red/yellow neurons (indicated by arrows) are glutamate‐ and td‐Tomato‐positive with filter adjustment. B, example of the PVN following bilateral injections of AAV‐flex‐taCasp3‐TEVp, partially lesioning the glutamatergic population of neurons. C, rostro‐caudal quantification of distribution of glutamate neurons in the PVN. D, total number of glutamate neurons counted in control vs. lesioned animals throughout the PVN. * P < 0.05, n = 3–4/group (Student's t test), scale bar 200 μm. [Color figure can be viewed at wileyonlinelibrary.com]
Statistics
Data are expressed as means ± SEM. Data were analysed by Student's t test, Kruskal–Wallis test and Dunn's multiple comparison test, one‐way ANOVA (Tukey's post hoc tests to compare replicate means) or two‐way ANOVA (with Bonferroni's post hoc test) when appropriate. Statistical comparisons were performed using Prism5 (GraphPad Software, San Diego, CA, USA).
Results
ChR2 stimulation of the PVN in conscious mice
To determine unequivocally whether activation of PVN neurons is sufficient to drive a change of BP in conscious, freely moving mice, this region was transfected with an AAV expressing ChR2. Five to 6 weeks after injection, the PVN neurons were activated in conscious freely moving mice. Stimulations were performed in the home cage when the mice were calm and quietly awake. The stimulation did not have an observable effect on behaviour. Figure 1 shows that ChR2 stimulation with blue light (473 nm) at 2 Hz (20 ms pulses) had no effect on BP (P > 0.05). However, when the frequency of stimulation increased, the BP rose significantly at both 10 Hz, with systolic BP rise equal to 4.7 ± 1.9 mmHg (P < 0.01), and 20 Hz, with systolic BP rise equal to 8.5 ± 2.2 mmHg (P < 0.001). Quantitative results recorded from seven mice undergoing PVN neuron stimulation showed a frequency‐dependent effect (Fig. 1) with BP rising as frequency of stimulation increased.
Figure 1. Increased blood pressure elicited by PVN excitation in a frequency‐dependent manner in conscious mice.
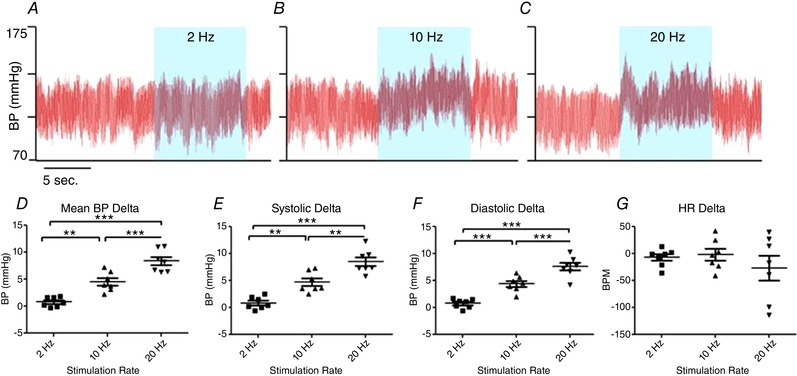
A–C, representative BP recordings from a mouse previously injected with AAV‐CamKIIα‐ChR2‐eYFP. Unilateral photo‐excitation of PVN neurons (blue areas) had no effect on BP at 2 Hz but produced robust BP elevations at 10 and 20 Hz stimulations. D, quantified data, from 7 mice, confirm increased mean, systolic and diastolic BP at 2, 10 and 20 Hz. There was no statistically significant bradycardia observed at any frequency of PVN stimulation (BPM, beats per minute). ** P < 0.01, *** P < 0.001 (one‐way repeated measures ANOVA). [Color figure can be viewed at wileyonlinelibrary.com]
Resting heart rate did not change statistically (2, 10 and 20 Hz, P > 0.05) although two out of the seven mice exhibited a dramatic bradycardia following PVN activation at the highest frequency of light stimulation.
Immunohistochemistry
In order to confirm the location of the ChR2‐transfected CamKIIα neurons to the PVN, immunohistochemistry was performed, taking advantage of the AAV2‐CamKIIα‐ChR2 fused with enhanced yellow fluorescent protein (eYFP) (Fig. 2). The data shown in Fig. 2 are from a mouse in the same group of animals used in the optogenetic stimulation experiments. Figure 2 A and B shows transfection to the PVN while surrounding nuclei, such as the subfornical organ (SFO) (Fig. 2 C) and arcuate nucleus (Fig. 2 D), were not transfected with the viral construct, providing evidence of successful stereotaxic injections specifically to the PVN.
Figure 2. Location of CamKIIα‐transduced neurons.
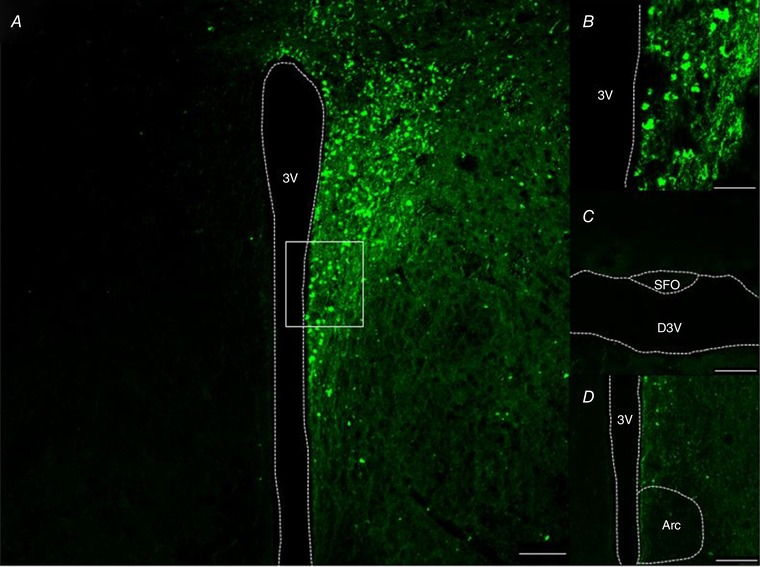
A, example of CamKIIα‐eYFP‐expressing PVN neurons in a coronal cross‐section through the hypothalamus of a mouse injected unilaterally with AAV‐CamKIIα‐ChR2‐eYFP (0.6 mm caudal to bregma). B, high magnification (63×) of PVN neurons from the white box highlighted in A. C and D, no transduced neurons in subfornical organ (SFO) or arcuate nucleus. 3V, 3rd ventricle; D3V, dorsal 3rd ventricle; Arc, arcuate nucleus. A, C and D, scale bar = 100 μm; B, scale bar = 25 μm. [Color figure can be viewed at wileyonlinelibrary.com]
To assess the specificity of the CamKIIα promoter targeting a majority of glutamate neurons, injections of the AAV2‐CamKIIα‐ChR2‐eYFP were performed in vGlut2‐Cre mice bred with tdTomato reporter animals. Wenker et al. (2017) quantified and characterized an AAV2 transfection with the CamKIIα promoter in the C1 population of the RVLM and found it to be 60% glutamate neurons, 40% GABA and 1% glutamate/GABA coexpressing neurons (Wenker et al. 2017). As shown in Fig. 3, the large majority of CamKIIα‐ChR2‐eYFP (green) colocalized with the glutamate population (red) of PVN neurons. These data suggest that most, if not all, PVN glutamatergic neurons were transfected by AAV2‐CamKIIα‐ChR2‐eYFP.
Figure 3. CamKIIα‐transduced neurons colocalize with glutamate neurons.
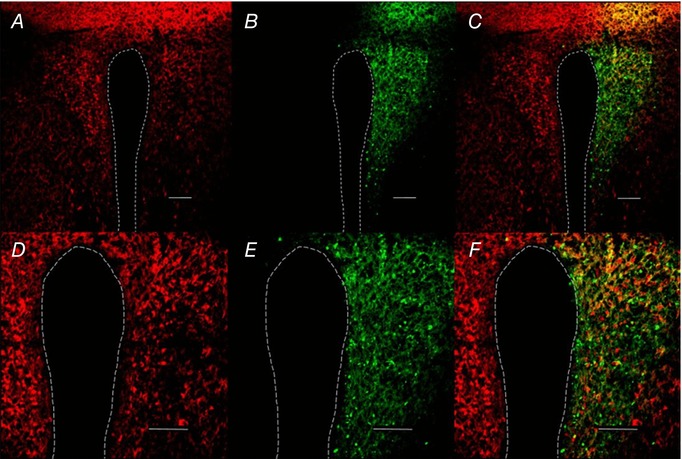
A, example of vGlut2‐Cre‐tdTomato‐positive neurons in a coronal cross‐section through the hypothalamus of a mouse (0.6 mm caudal to bregma). B, AAV2‐CamKIIα‐ChR2‐eYFP‐expressing PVN neurons. C, merge. A–C, scale bar = 100 μm. D–F, higher magnification (20×) of PVN CamKII‐transduced neurons; scale bar = 75 μm.
Lesions of PVN glutamatergic neurons
To assess the contribution of the glutamatergic population of PVN neurons to the regulation of BP at baseline and during DOCA‐salt hypertension, vGlut2‐Cre mice were bred with a tdTomato reporter mouse (Vong et al. 2011). vGlut2‐cre+/−/tdTomatofl/fl mice received bilateral Cre‐dependent caspase (AAV‐flex‐taCasp3‐TEVp) injections to destroy the glutamate population of neurons within the PVN. Expression of the tdTomato reporter (i.e. red fluorescence) on glutamate‐positive neurons allowed quantification of lesioned neurons in experimental vs. control animals (Fig. 4). There was a 39.3% decrease in glutamatergic neurons of the PVN relative to controls (Fig. 4 A and B; P < 0.05), indicating a partial but significant lesion of the glutamatergic neuronal population (Fig. 4). Figure 3 C shows the distribution of glutamatergic neurons at baseline and post‐lesion with the result demonstrating a greater number of glutamate neurons in the rostral portion of the nucleus than the caudal and a steady deletion throughout the PVN.
Acknowledging the fact that lesioning the glutamate population of the PVN may impact the neuro‐hormonal axis and glutamatergic PVN projections (primarily to the RVLM), we performed the following experiments. First, we tested circulating copeptin levels in control and lesioned groups of mice. Copeptin is the COOH‐terminal fragment of the full arginine vasopressin (AVP) pro‐protein. Due to its longer half‐life, copeptin is more reliable as a measure of chronic AVP release (Szinnai et al. 2007). A disruption in copeptin levels has been used as an indicator of endocrine imbalance. However, Fig. 5 A shows that copeptin levels remain unchanged between lesioned and control groups, suggesting that the lesion did not alter hormonal secretion from the surrounding neurons. Additionally, Fig. 5 B shows mRNA levels for genes associated with glutamatergic and GABAergic signalling. There was a small but significant rise in GluR1 mRNA expression in the RVLM (P = 0.017) after lesioning of the PVN, a gene involved in AMPA receptor expression. This increase in mRNA could suggest an up‐regulation of glutamate AMPA receptors within the RVLM to offset the loss of glutamate activity in the PVN. Gene expression for Grin1, a gene involved in NMDA receptor expression, GAD67, a gene expressed in GABA neurons and the gene involved in GABAa receptor expression, remained unchanged in the RVLM between control and lesioned animals.
Figure 5. Copeptin and gene expression.
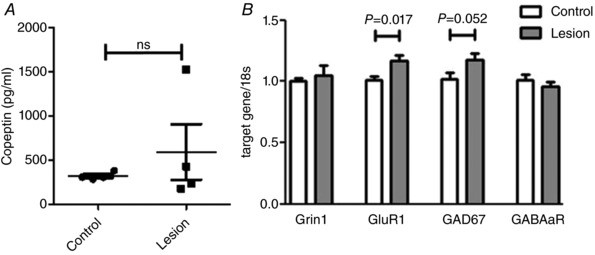
A, copeptin expression in lesioned mice at baseline. Mice with reduced number of PVN glutamate neurons show no difference in serum copeptin levels. B, gene expression in the RVLM of control and lesioned animals. Grin1, gene involved in NMDA receptor expression; GluR1, gene involved in AMPA receptor expression; GAD67, Glutamic Acid Decarboxylase gene expressed in GABA neurons; GABAaR, gene involved in GABAa receptor expression. n = 4/group run in duplicate (Student's t test).
NE release from peripheral nerves into the circulation which is then excreted through urine can be measured as an index of sympathetic tone. To further clarify the involvement of the glutamatergic PVN neurons in the regulation of sympathetic activity at baseline and during DOCA‐salt hypertension, urinary NE levels were assessed (Fig. 6 A). Lesioning of glutamatergic neurons in the PVN was associated with an unexpected rise in NE levels, suggesting that in baseline conditions, some of these PVN neurons might be involved in the inhibition of NE release. Alternatively, this increase in NE in the lesioned group at baseline could be due to compensation by other parts of the network (e.g. RVLM). As we reported previously, NE levels rose in control mice following DOCA‐salt treatment (P < 0.01) supporting an increase in sympathetic activity associated with hypertension (control = 152 ± 13 ng/ml vs. DOCA = 265 ± 21 ng/ml). Interestingly, in mice with reduced numbers of glutamatergic neurons in the PVN, there was a decrease in NE with DOCA‐salt treatment (115 ± 17 ng/ml, P < 0.05) compared to hypertensive controls.
Figure 6. Urine noradrenaline (NE) levels.
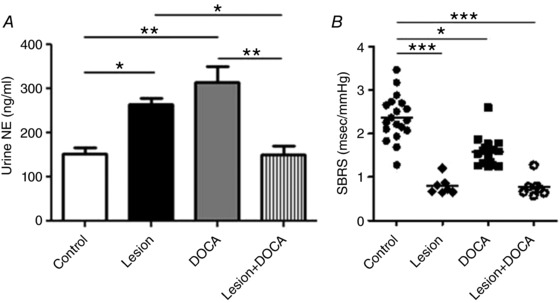
A, urine was collected from vGlut2‐Cre mice that had previously received bilateral AAV‐flex‐taCasp3‐TEVp to the PVN (lesioned mice) and wildtype controls before and 21 days after DOCA‐salt treatment. DOCA treatment significantly increased NE levels in control mice. Lesioned mice had higher NE at baseline and lower values after DOCA treatment. There was no difference between control mice at baseline and Lesion + DOCA mice. B, spontaneous baroreceptor reflex sensitivity (SBRS) was measured in control or lesioned animals at baseline and after DOCA‐salt treatment. A, n = 16 control, n = 8 lesion; B, n = 19 control, n = 7 lesion; * P < 0.05, ** P < 0.01, *** P < 0.001 (Kruskal–Wallis test and Dunn's multiple comparison test).
The lesioned mice also showed a dramatic reduction of baroreflex sensitivity at baseline, beyond the reduction induced by DOCA‐salt (P < 0.001; Fig. 6 B) supporting a role for PVN glutamatergic neurons in baroreflex modulation (Xu et al. 2015; Holbein et al. 2018). As a possible cause for this large baroreflex sensitivity change, we assessed whether the glutamatergic PVN lesion led to inflammation in the RVLM. We chose the RVLM because, in addition to being a large target of PVN projections, it is a critical region involved in the baroreceptor reflex (Wu et al. 2012). However, although changes in TNFα signalling are commonly associated with inflammation in the CNS, our western blot analysis showed that the inflammatory marker TNFα‐R1 did not change in the RVLM between control and lesion cohorts of mice (P = 0.29), suggesting that inflammation might not be responsible for the altered baroreflex sensitivity (Fig. 7).
Figure 7. TNF‐R1 is not induced in the RVLM by AAV‐flex‐taCasp3‐TEVp injections in the PVN.
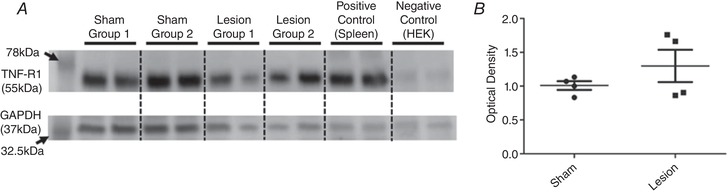
A, raw protein levels determined by western blot targeting TNF‐R1 (top) and GAPDH (bottom). B, quantification of optical density in A. Band density was not significantly different based on lesion vs. sham (n = 4/group run in duplicate, Student's t test, P = 0.29). HEK: human embryonic kidney cells.
In control mice, DOCA‐salt treatment induced a robust hypertension with a BP that remained elevated over 2 weeks (MAP increase = 36.3 ± 1.9 mmHg, P < 0.001). In mice with lesioned glutamatergic neurons in the PVN, baseline BP showed a trend to rise but this failed to reach significance. Moreover, DOCA‐salt treatment only produced an attenuated pressor response (Fig. 8 A) in these mice suggesting that PVN glutamatergic neurons are necessary for the development of DOCA‐salt hypertension.
Figure 8. DOCA‐salt hypertension in mice with lesioned glutamatergic neurons in the PVN.
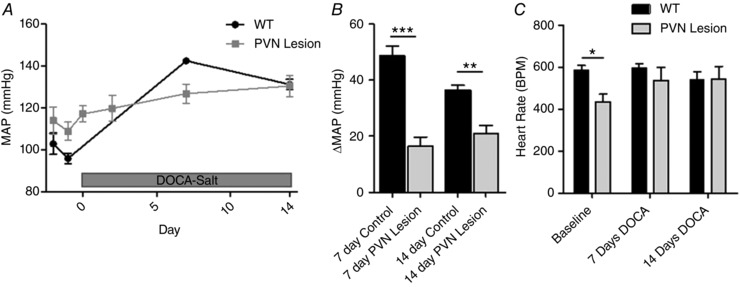
A, BP in vGlut2‐Cre mice that had previously received bilateral AAV‐flex‐taCasp3‐TEVp in the PVN. B, change in BP from baseline BP to day 7 and day 14 of DOCA‐salt treatment. C, heart rate in beats per minute (BPM) at days −1, 7 and 14. n = 9 control, n = 8 lesion; * P < 0.05, ** P < 0.01, *** P < 0.001 (two‐way ANOVA).
Discussion
The current study provides new evidence for the requirement of PVN glutamatergic neurons in the development of neurogenic hypertension in conscious mice. When these PVN neurons are directly photo‐activated in conscious mice there is a frequency‐dependent increase in BP. Additionally, in a separate experiment, mice with glutamate neurons lesioned from the PVN show an attenuated hypertension and no increase in urinary noradrenaline levels under DOCA‐salt treatment.
The stimulation of PVN neurons at baseline causing an immediate increase in BP indicates this neuronal population is sufficient to promote an elevation of BP (Fig. 1). This rise in BP is likely caused by an increase in sympathetic drive. ChR2 stimulation causes a rapid BP increase (<0.5 s) indicating a CNS‐mediated activation. The speed of BP increase rules out a hormonal (i.e. vasopressin) source of BP rise, which would act in the order of minutes (Yamaguchi, 2015). Secondly, partial lesioning of the glutamate population within the PVN resulted in a less dramatic rise in BP and prevented the increase of NE levels during hypertension, indicating that this excitatory neuronal population may be overactive during neurogenic hypertension.
PVN and its role in BP regulation
It has long been hypothesized that the convergence of inputs from multiple brain regions involved in BP regulation, ranging from the forebrain to the hindbrain, onto the PVN and its projections to the RVLM and intermediolateral (IML) column makes this nucleus an ideal hub for sympathetic nervous system integration and regulation. Whether PVN neurons contribute to BP regulation at baseline and to what extent in conscious animals is still debated (Callahan et al. 1992; Ramchandra et al. 2013; Steckelings et al. 2017). However, it is generally accepted that the pre‐sympathetic glutamate population of PVN neurons is tonically inhibited by upstream GABAergic inputs from other regions (Cullinan et al. 2008). Martins‐Pinge et al. (2012) showed in conscious rats that had received microinjections of kynurenic acid a marginal BP decrease and a significantly increased BP during GABA blockade (Martins‐Pinge et al. 2012). Moreover, Park et al. (2007) showed through patch‐clamp experiments that there is the ‘presence of a persistent GABA(A)‐mediated inhibitory modality in pre‐sympathetic PVN neurons, which plays a major role in modulating their excitability and firing activity’(Park et al. 2007).
The role of the glutamatergic neuronal population in the PVN during the development and maintenance of hypertension has become increasingly evident in recent studies. Li & Pan (2017) recently reviewed the increased activation of glutamate receptors in the PVN both pre‐ and postsynaptically in multiple hypertension models. These authors emphasized the importance of plasticity of the glutamatergic population, specifically the NMDA receptors, within the PVN, during the development of hypertension. These changes result in an increased sympathetic drive that potentiates neurogenic hypertension. Our findings, examining the role of the glutamatergic population of the PVN before and during neurogenic hypertension, support this theory. Here we show for the first time that in conscious mice the glutamatergic population of neurons of the PVN is sufficient to drive BP when stimulated and it may be through this chronic over‐activation that neurogenic hypertension is produced and maintained.
Unlike other approaches, ChR2 measures the instantaneous contribution of the transfected neuronal population with little to no homeostatic compensation. ChR2 allowed us to entrain this population of neurons with stimulations ranging from 2–20 Hz, obtaining the full range of physiological levels of firing for these neurons (Chen & Toney, 2010; Abbott et al. 2014). Our results show that unilateral stimulation of the CamKIIα‐expressing neuronal population of the PVN, which we highlighted as being mostly glutamatergic (Fig. 3), is enough to cause a significant rise in BP in a frequency‐dependent manner. Wang et al. (2013) showed in recent studies, and we confirm here with current histological analysis, that glutamatergic neurons of the PVN can be selectively transfected (Wang et al. 2013; Watakabe et al. 2015). Through stereotaxic injection and histological analysis, we confirm that the PVN alone is transfected with the AAV2‐CamKII‐ChR2‐eYFP and not surrounding nuclei (e.g. SFO and arcuate nuclei). We speculate that this transfected population of neurons has the potential to cause an even greater increase in BP if the entire bilateral glutamatergic population were to be chronically over‐excited, as is hypothesized in neurogenic hypertension.
Disease state‐dependent control of BP by the PVN
Increased BP can be elicited by stimulation of the PVN or its downstream targets, such as the RVLM (Abbott et al. 2009; Martins‐Pinge et al. 2012; Burke et al. 2014; Li & Pan, 2017). When sympathetic outflow from the RVLM is inhibited through optogenetics, BP decreases (Basting et al. 2015). The present study adds three novel insights into this sympatho‐excitatory pathway. First, stimulation of a subset of the excitatory pre‐sympathetic neurons of the PVN in conscious mice causes an immediate rise in BP (Liu & Jones, 1996; Watakabe et al. 2015). Second, partial lesioning of the glutamatergic population of the PVN does not cause a significant change of baseline BP though NE levels do rise. However, throughout the development of hypertension these changes are reversed. Within the first week, mice on the DOCA‐salt protocol, that had received bilateral lesions of the glutamatergic neurons of the PVN, exhibited a blunted rise in BP compared to control animals. Additionally, urinary NE levels in lesioned mice dropped down to levels equivalent to control animals at baseline. Third, through vGlut2‐tdTomato‐positive neuronal cell counts, we documented that there is an uneven distribution of vGlut2‐positive neurons throughout the PVN, with the rostral PVN containing more vGlut2‐positive neurons than that of the caudal portion (Fig. 4). Further investigation is needed to identify the functional connectome of these cells.
The PVN sends reciprocal connections to the RVLM in addition to innervating the IML (Chen & Toney, 2010). This connection has been shown to play multiple roles, one of which is maintaining a healthy baroreflex (Holbein et al. 2018). After lesioning this population of neurons, we observed a severe impairment of this reflex (Fig. 6 B) that has been shown to exacerbate the pathogenesis of neurogenic hypertension (Ketch et al. 2002). Also, lesioning the glutamatergic population of the PVN resulted in an increased mRNA expression of GluR1, a gene involved in AMPA receptor regulation in the RVLM suggesting a possible compensatory action by the brainstem. The increased activity of the PVN observed in both ChR2 experiments and throughout the DOCA‐salt protocol is reasonably causing these populations of neurons to become more excitable themselves. These connections contribute to the pronounced increase of sympathetic nerve activity in hypertension (Guyenet, 2006; Ye et al. 2013; Basting & Lazartigues, 2017).
Experimental limitations
AAV2‐CamKIIα‐ChR2 transduces a mixed population of neurons though preferentially targets glutamatergic neurons, as described previously (Liu & Jones, 1996; Watakabe et al. 2015) and confirmed here. However, in the ChR2 experiments, selective stimulation of the transduced cell population resulted in an immediate rise in BP, returned to pre‐stimulated values within 30 s of the laser being turned off, and did not cause any observable behavioural changes, indicating that the stimulatory effects were most plausibly neurogenic, as opposed to hormonal.
For both the stimulation and the lesion experiments utilizing the CamKIIα‐ChR‐eYFP and the flex‐taCasp3‐TEVp, respectively, our aim was to target the PVN specifically. There is a chance that the viruses could have been taken up by nerve terminals and been expressed in afferent connections. After histology and quantification of the lesions it is clear that specific transfection to the PVN was achieved through the combination of precise stereotaxic injections and optrode placement but did not transduce every neuron of interest. It is highly probable that if 100% of glutamatergic neurons were transfected bilaterally the effects seen would increase significantly.
Novelty and significance
To measure the contribution of excitatory neurons of the PVN to BP in conscious mice, we used two different approaches. First, gain‐of‐function optogenetics experiments utilizing ChR2 were performed. PVN neurons were transduced unilaterally to express the photoactivatable cation channels allowing us to observe the instantaneous contribution of the transfected cell population in conscious animals. Secondly, we used a Cre‐dependent caspase that was virally delivered to selectively lesion the glutamatergic neuronal population of the PVN. These animals then underwent a DOCA‐salt protocol that is a well‐established model of neurogenic hypertension (Basting & Lazartigues, 2017). Studies shown here allowed us to investigate the roles of similar sets of neurons in the PVN under different experimental conditions through monitoring BP, heart rate, baroreflex, copeptin and NE levels.
Perspectives
The present study could help explain why many forms of hypertension are exacerbated by high‐salt diets, dehydration and chronic stress. During periods of prolonged exposure to high‐salt diets, dehydration and stressors, animals and humans show an increase in BP (Vivas et al. 2014; Holbein & Toney, 2015). Salt‐sensing and thirst are both mediated either directly or indirectly by the PVN or areas that are relaying that information to the PVN (e.g. SFO and median preoptic nucleus) (Speth et al. 2014; Vivas et al. 2014). Stress has been shown to increase components of the renin–angiotensin system, such as angiotensin II and decrease beneficial components, such as angiotensin‐converting enzyme 2 (ACE2) in the hypothalamus (Sriramula et al. 2011; Martins Lima et al. 2013; Xia et al. 2015). Recurrent periods of over‐excitation in the PVN could contribute to chronic over‐activation of this nucleus and thus enhanced sympathetic drive.
Additional information
Competing interests
None.
Author contributions
T.B., J.X., S.M., J.E., R.F. and E.L. contributed to the conception and design of the study, acquisition, analysis and interpretation of the data, and drafting the study. S.S. contributed to conception and design of the study, interpretation of the data, and revising the draft critically for important intellectual content. The study was conducted in the laboratory at Louisiana State University. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the study. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This study was supported by research grants from the National Institutes of Health (HL140865 to T.B., HL093178 to E.L. and COBRE P30GM106392) and from the American Heart Association (15SDG25720021 to S.S.).
Acknowledgements
The authors would like to thank Ms Kathryn Earnest and Ms Charlotte Pearson for excellent technical support and Dr Luis Marrero, Director of the Morphology and Imaging Core.
Biography
Tyler Basting’s overall interests centre on studying the role of the CNS in autonomic function in the healthy and diseased nervous system. He firmly believes that groundbreaking discoveries emerge directly from development of new methods and approaches. To this end he has continued his graduate career in the Guyenet Lab at the University of Virginia, and postdoctoral training in the Lazartigues Lab at the Louisiana State University Health Sciences Center. Developing comprehensive novel approaches for CNS optogenetic excitation and inhibition in anaesthetized and conscious animals has been instrumental in his findings thus far. Building an unprecedented set of tools has now placed him in a unique position to be able to tackle questions never before accessible in the intact live brain. He envisions using these tools as a solid foundation to study multiple aspects of nervous system biology in the immediate future and beyond.

Edited by: Harold Schultz & Julie Chan
Linked articles This article is highlighted in a Perspectives article by Becker. To read this article, visit https://doi.org/10.1113/JP277043.
References
- Abbott SB, Holloway BB, Viar KE & Guyenet PG (2014). Vesicular glutamate transporter 2 is required for the respiratory and parasympathetic activation produced by optogenetic stimulation of catecholaminergic neurons in the rostral ventrolateral medulla of mice in vivo. Eur J Neurosci 39, 98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott SB, Stornetta RL, Socolovsky CS, West GH & Guyenet PG (2009). Photostimulation of channelrhodopsin‐2 expressing ventrolateral medullary neurons increases sympathetic nerve activity and blood pressure in rats. J Physiol 587, 5613–5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basting T & Lazartigues E (2017). DOCA‐salt hypertension: an update. Curr Hypertens Rep 19, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basting TM, Burke PG, Kanbar R, Viar KE, Stornetta DS, Stornetta RL & Guyenet PG (2015). Hypoxia silences retrotrapezoid nucleus respiratory chemoreceptors via alkalosis. J Neurosci 35, 527–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CH (2016). Magnocellular neurons and posterior pituitary function. Compr Physiol 6, 1701–1741. [DOI] [PubMed] [Google Scholar]
- Burke PG, Abbott SB, Coates MB, Viar KE, Stornetta RL & Guyenet PG (2014). Optogenetic stimulation of adrenergic C1 neurons causes sleep state‐dependent cardiorespiratory stimulation and arousal with sighs in rats. Am J Respir Crit Care Med 190, 1301–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher KS & Cechetto DF (1998). Receptors in lateral hypothalamic area involved in insular cortex sympathetic responses. Am J Physiol 275, H689–H696. [DOI] [PubMed] [Google Scholar]
- Callahan MF, Thore CR, Sundberg DK, Gruber KA, O'Steen K & Morris M (1992). Excitotoxin paraventricular nucleus lesions: stress and endocrine reactivity and oxytocin mRNA levels. Brain Res 597, 8–15. [DOI] [PubMed] [Google Scholar]
- Chen QH & Toney GM (2010). In vivo discharge properties of hypothalamic paraventricular nucleus neurons with axonal projections to the rostral ventrolateral medulla. J Neurophysiol 103, 4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullinan WE, Ziegler DR & Herman JP (2008). Functional role of local GABAergic influences on the HPA axis. Brain Struct Funct 213, 63–72. [DOI] [PubMed] [Google Scholar]
- Esler M, Rumantir M, Kaye D & Lambert G (2001). The sympathetic neurobiology of essential hypertension: disparate influences of obesity, stress, and noradrenaline transporter dysfunction? Am J Hypertens 14, 139S–146S. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ & Paxinos G (1997). The Mouse Brain in Stereotaxic Coordinates. Academic Press, Inc., San Diego. [Google Scholar]
- Gabor A & Leenen FH (2012). Cardiovascular effects of angiotensin II and glutamate in the PVN of Dahl salt‐sensitive rats. Brain Res 1447, 28–37. [DOI] [PubMed] [Google Scholar]
- Geerling JC, Shin JW, Chimenti PC & Loewy AD (2010). Paraventricular hypothalamic nucleus: axonal projections to the brainstem. J Comp Neurol 518, 1460–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraldes V, Goncalves‐Rosa N, Liu B, Paton JF & Rocha I (2014). Chronic depression of hypothalamic paraventricular neuronal activity produces sustained hypotension in hypertensive rats. Exp Physiol 99, 89–100. [DOI] [PubMed] [Google Scholar]
- Gomes da Silva AQ, Xavier CH, Campagnole‐Santos MJ, Caligiorne SM, Baltatu OC, Bader M, Santos RA & Fontes MA (2012). Cardiovascular responses evoked by activation or blockade of GABAA receptors in the hypothalamic PVN are attenuated in transgenic rats with low brain angiotensinogen. Brain Res 1448, 101–110. [DOI] [PubMed] [Google Scholar]
- Guyenet PG (2006). The sympathetic control of blood pressure. Nat Rev Neurosci 7, 335–346. [DOI] [PubMed] [Google Scholar]
- Guyenet PG, Stornetta RL, Bochorishvili G, Depuy SD, Burke PG & Abbott SB (2013). C1 neurons: the body's EMTs. Am J Physiol Regul Integr Comp Physiol 305, R187–R204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbein WW, Blackburn MB, Andrade MA & Toney GM (2018). Burst patterning of hypothalamic paraventricular nucleus‐driven sympathetic nerve activity in ANG II‐salt hypertension. Am J Physiol Heart Circ Physiol 314, H530–H541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbein WW & Toney GM (2015). Activation of the hypothalamic paraventricular nucleus by forebrain hypertonicity selectively increases tonic vasomotor sympathetic nerve activity. Am J Physiol Regul Integr Comp Physiol 308, R351–R359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kc P & Dick TE (2010). Modulation of cardiorespiratory function mediated by the paraventricular nucleus. Respir Physiol Neurobiol 174, 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketch T, Biaggioni I, Robertson R & Robertson D (2002). Four faces of baroreflex failure: hypertensive crisis, volatile hypertension, orthostatic tachycardia, and malignant vagotonia. Circulation 105, 2518–2523. [DOI] [PubMed] [Google Scholar]
- Li DP & Pan HL (2017). Glutamatergic regulation of hypothalamic presympathetic neurons in hypertension. Curr Hypertens Rep 19, 78. [DOI] [PubMed] [Google Scholar]
- Li YF, Mayhan WG & Patel KP (2001). NMDA‐mediated increase in renal sympathetic nerve discharge within the PVN: role of nitric oxide. Am J Physiol Heart Circ Physiol 281, H2328–H2336. [DOI] [PubMed] [Google Scholar]
- Liu XB & Jones EG (1996). Localization of alpha type II calcium calmodulin‐dependent protein kinase at glutamatergic but not gamma‐aminobutyric acid (GABAergic) synapses in thalamus and cerebral cortex. Proc Natl Acad Sci U S A 93, 7332–7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozic M, Sarenac O, Murphy D & Japundzic‐Zigon N (2018). Vasopressin, central autonomic control and blood pressure regulation. Curr Hypertens Rep 20, 11. [DOI] [PubMed] [Google Scholar]
- Marina N, Abdala AP, Korsak A, Simms AE, Allen AM, Paton JF & Gourine AV (2011). Control of sympathetic vasomotor tone by catecholaminergic C1 neurones of the rostral ventrolateral medulla oblongata. Cardiovasc Res 91, 703–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins Lima A, Xavier CH, Ferreira AJ, Raizada MK, Wallukat G, Velloso EP, dos Santos RA & Fontes MA (2013). Activation of angiotensin‐converting enzyme 2/angiotensin‐(1–7)/Mas axis attenuates the cardiac reactivity to acute emotional stress. Am J Physiol Heart Circ Physiol 305, H1057–H1067. [DOI] [PubMed] [Google Scholar]
- Martins‐Pinge MC, Mueller PJ, Foley CM, Heesch CM & Hasser EM (2012). Regulation of arterial pressure by the paraventricular nucleus in conscious rats: interactions among glutamate, GABA, and nitric oxide. Front Physiol 3, 490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki T, Minson J, Arnolda L, Chalmers J, Llewellyn‐Smith I & Pilowsky P (1996). Role of excitatory amino acid receptors in cardiorespiratory coupling in ventrolateral medulla. Am J Physiol 271, R1221–R1230. [DOI] [PubMed] [Google Scholar]
- Park JB, Skalska S, Son S & Stern JE (2007). Dual GABAA receptor‐mediated inhibition in rat presympathetic paraventricular nucleus neurons. J Physiol 582, 539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramchandra R, Hood SG, Frithiof R, McKinley MJ & May CN (2013). The role of the paraventricular nucleus of the hypothalamus in the regulation of cardiac and renal sympathetic nerve activity in conscious normal and heart failure sheep. J Physiol 591, 93–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson WK, Bagley SL, Ferguson AV & White MM (2007). Hypocretin/orexin type 1 receptor in brain: role in cardiovascular control and the neuroendocrine response to immobilization stress. Am J Physiol Regul Integr Comp Physiol 292, R382–R387. [DOI] [PubMed] [Google Scholar]
- Saper CB & Lowell BB (2014). The hypothalamus. Curr Biol 24, R1111–R1116. [DOI] [PubMed] [Google Scholar]
- Shi P, Diez‐Freire C, Jun JY, Qi Y, Katovich MJ, Li Q, Sriramula S, Francis J, Sumners C & Raizada MK (2010). Brain microglial cytokines in neurogenic hypertension. Hypertension 56, 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparta DR, Stamatakis AM, Phillips JL, Hovelso N, van Zessen R & Stuber GD (2011). Construction of implantable optical fibers for long‐term optogenetic manipulation of neural circuits. Nat Protoc 7, 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speth RC, Vento PJ, Carrera EJ, Gonzalez‐Reily L, Linares A, Santos K, Swindle JD & Daniels D (2014). Acute repeated intracerebroventricular injections of angiotensin II reduce agonist and antagonist radioligand binding in the paraventricular nucleus of the hypothalamus and median preoptic nucleus in the rat brain. Brain Res 1583, 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriramula S, Cardinale JP, Lazartigues E & Francis J (2011). ACE2 overexpression in the paraventricular nucleus attenuates angiotensin II‐induced hypertension. Cardiovasc Res 92, 401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauss HM, Moffitt JA, Chapleau MW, Abboud FM & Johnson AK (2006). Baroreceptor reflex sensitivity estimated by the sequence technique is reliable in rats. Am J Physiol Heart Circ Physiol 291, H482–H483. [DOI] [PubMed] [Google Scholar]
- Steckelings UM, Kloet A & Sumners C (2017). Centrally mediated cardiovascular actions of the angiotensin II type 2 receptor. Trends Endocrinol Metab 28, 684–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker SD, Monahan KD & Browning KN (2013). Neurogenic and sympathoexcitatory actions of NaCl in hypertension. Curr Hypertens Rep 15, 538–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson LW (1977). Immunohistochemical evidence for a neurophysin‐containing autonomic pathway arising in the paraventricular nucleus of the hypothalamus. Brain Res 128, 346–353. [DOI] [PubMed] [Google Scholar]
- Szinnai G, Morgenthaler NG, Berneis K, Struck J, Muller B, Keller U & Christ‐Crain M (2007). Changes in plasma copeptin, the c‐terminal portion of arginine vasopressin during water deprivation and excess in healthy subjects. J Clin Endocrinol Metab 92, 3973–3978. [DOI] [PubMed] [Google Scholar]
- Vivas L, Godino A, Dalmasso C, Caeiro XE, Macchione AF & Cambiasso MJ (2014). Neurochemical circuits subserving fluid balance and baroreflex: a role for serotonin, oxytocin, and gonadal steroids In Neurobiology of Body Fluid Homeostasis: Transduction and Integration, ed. De Luca LA, Jr, Menani JV. & Johnson AK. CRC Press /Taylor and Francis, Boca Raton, FL, USA. [PubMed] [Google Scholar]
- Vong L, Ye C, Yang Z, Choi B, Chua S Jr & Lowell BB (2011). Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 71, 142–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhang C, Szabo G & Sun QQ (2013). Distribution of CaMKIIα expression in the brain in vivo, studied by CaMKIIα‐GFP mice. Brain Res 1518, 9–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watakabe A, Ohtsuka M, Kinoshita M, Takaji M, Isa K, Mizukami H, Ozawa K, Isa T & Yamamori T (2015). Comparative analyses of adeno‐associated viral vector serotypes 1, 2, 5, 8 and 9 in marmoset, mouse and macaque cerebral cortex. Neurosci Res 93, 144–157. [DOI] [PubMed] [Google Scholar]
- Wenker IC, Abe C, Viar KE, Stornetta DS, Stornetta RL & Guyenet PG (2017). Blood pressure regulation by the rostral ventrolateral medulla in conscious rats: effects of hypoxia, hypercapnia, baroreceptor denervation, and anesthesia. J Neurosci 37, 4565–4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ, Dennison Himmelfarb C, DePalma SM, Gidding S, Jamerson KA, Jones DW, MacLaughlin EJ, Muntner P, Ovbiagele B, Smith SC Jr, Spencer CC, Stafford RS, Taler SJ, Thomas RJ, Williams KA Sr, Williamson JD & Wright JT Jr (2018). 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension 71, e13–e115. [DOI] [PubMed] [Google Scholar]
- Wu KL, Chan SH & Chan JY (2012). Neuroinflammation and oxidative stress in rostral ventrolateral medulla contribute to neurogenic hypertension induced by systemic inflammation. J Neuroinflammation 9, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H, Queiroz TM, Sriramula S, Feng Y, Johnson T, Mungrue IN & Lazartigues E (2015). Brain ACE2 overexpression reduces DOCA‐salt hypertension independently of endoplasmic reticulum stress. Am J Physiol Regul Integr Comp Physiol 308, R370–R378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H, Sriramula S, Chhabra K & Lazartigues E (2013). Brain ACE2 shedding contributes to the development of neurogenic hypertension. Circ Res 113, 1087–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Zheng H, Liu X & Patel KP (2015). Activation of afferent renal nerves modulates RVLM‐projecting PVN neurons. Am J Physiol Heart Circ Physiol 308, H1103–H1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi K (2015). Evaluation for roles of neurosteroids in modulating forebrain mechanisms controlling vasopressin secretion and related phenomena in conscious rats. Neurosci Res 95, 38–50. [DOI] [PubMed] [Google Scholar]
- Ye ZY, Li DP & Pan HL (2013). Regulation of hypothalamic presympathetic neurons and sympathetic outflow by group II metabotropic glutamate receptors in spontaneously hypertensive rats. Hypertension 62, 255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZH, Francis J, Weiss RM & Felder RB (2002). The renin‐angiotensin‐aldosterone system excites hypothalamic paraventricular nucleus neurons in heart failure. Am J Physiol Heart Circ Physiol 283, H423–H433. [DOI] [PubMed] [Google Scholar]